

Abstract
CRISPR-based genome editing technologies are poised to enable countless new therapies to prevent, treat or cure diseases with a genetic basis. However, the safe and effective delivery of genome editing enzymes represents a substantial challenge that must be tackled to enable the next generation of genetic therapies. In this perspective we summarize recent progress in developing enzymatic tools to combat genetic disease and examine current efforts to deliver these enzymes to the cells in need of correction. Viral vectors already in use for traditional gene therapy are being applied to enable in vivo CRISPR-based therapeutics, as are emerging technologies such as nanoparticle-based delivery of CRISPR components and direct delivery of pre-assembled RNA-protein complexes. Success in these areas will allow CRISPR-based genome editing therapeutics to reach their full potential.
Introduction
A major barrier to treatment or prevention of many diseases is the delivery of an effective medicine to a target. Barriers to delivery of medicines exist at the level of the organism, organ, cell and organelle. More than 100 years of modern medical research has given us vaccines, antibiotics, anti-cancer drugs and many other lifesaving preventive, therapeutic or curative medicines based on biologics or small molecule pharmaceuticals. Despite this we continue to battle both old and new diseases that require new forms of medicine.
With the advent of robust genome engineering technologies we are rapidly entering a new era of biologics in which genome therapies and new types of cellular therapies that rely on genome engineering will reshape our ability to prevent, treat or cure disease. The discovery and application of CRISPR systems as programmable gene editors have revolutionized our ability to use gene editing in biomedical research. Many excellent articles have reviewed recent progress in the field of therapeutic genome editing1–7 and so here we will focus on the delivery of current gene editing platforms in vivo as genome therapeutics. This perspectives piece is motivated by the fact that in many diseases the successful application of genome therapies to treat human diseases will be limited by effective in vivo delivery platforms. In this perspective, we will discuss the current state of the art and next generation options for in vivo delivery of genome therapies. We will focus on physiologic barriers to gene editor delivery and then highlight viral, nanoparticle and engineered protein delivery options for genome therapeutics (Figure 1).
Figure 1.
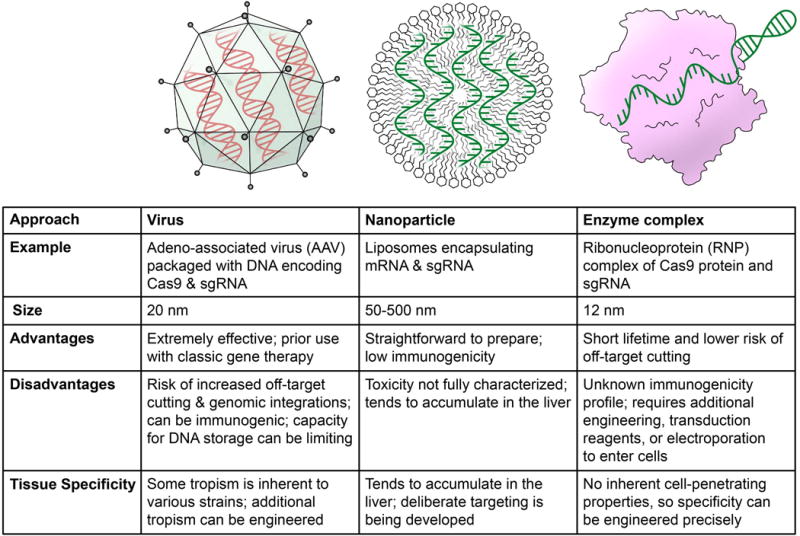
Three strategies for in vivo genome-editing enzyme delivery and their key properties.
The challenges to implementing any genome therapeutic as a medicine in humans are perhaps most similar to those faced for pioneering gene therapy efforts. Gene therapy platforms generally seek to deliver a transgene to restore the function of cells that are defective due to a genetic mutation. Although this idea is conceptually quite simple, in practice implementing gene therapy in humans is enormously challenging. In gene therapy a common mantra is that the three most important challenges for gene therapy are delivery, delivery and delivery. A second and perhaps less glib challenge is for gene therapy to be a safe form of medicine in humans. Initial gene therapy trials demonstrated that delivery methods such as early lentiviral platforms can induce a risk of cancer due to integration near proto-oncogenes which is an unacceptable safety risk in many diseases8. The acceptable risks of a particular therapy are related to the severity of the condition being treated, and genome editing will only reach its full therapeutic potential if it is safe enough to warrant treatment of non-life-threatening diseases.
Genome therapeutics correct, prevent or alleviate disease by targeting inherited or spontaneous pathogenic alleles or mutations in the human genome. In contrast, gene therapies correct diseases through delivery of an artificial transgene. Initially, genome therapeutics were thought to face three main challenges which were gene editing activity, gene editing specificity and gene editor delivery. However, due to the incredibly rapid progress of CRISPR gene editing platforms it is increasingly apparent that the major challenge for genome therapeutics is delivery. It is however important to note that the first genome therapy trials in humans are being carried out using a Zinc Finger Nuclease platform9. These first trials will provide a roadmap for how regulatory agencies will measure efficacy and safety for genome therapeutics in humans. Gene therapy and genome therapeutics largely face the same delivery challenges, which have been approached using similar means but with important distinctions. For example, gene therapies are delivered as DNA or RNA while CRISPR genome therapeutics can be delivered as DNA, RNA or ribonucleoprotein complexes (RNP). Most gene therapies are designed to permanently change a disease state but some that utilize RNA delivery platforms must be continually applied. In contrast by definition a gene editor applied as a DNA, RNA or RNP genome therapeutic will induce a permanent change. In the simplest case, genome therapeutics are designed to disrupt dominant negative or gain-of-function pathogenic mutations. To correct a loss-of-function mutation a genome therapeutic must consist of both a gene editor system and a homology repair donor. Homology repair donors can either be single or double stranded DNA. To create a plan for how any genome therapeutic should be applied in humans we must consider more generally the principles of pharmacology for biologic therapeutics.
The pharmacology of genomic therapeutics
Pharmacology is the study of drug action. More specifically pharmacology is frequently described as the science of drug absorption, distribution, metabolism and excretion. These topics seek to describe drug activity on the physiology of different organs and cell types within the body. Historically, pharmacology has focused on small molecule drugs, although many of the principles of pharmacology apply to biologics and more specifically to genomic therapies. Small molecule drugs are the most widely used type of medicine in part because it is relatively simple to design a small molecule drug that is permeable across cell membranes and orally bioavailable. Many widely used small molecule drugs broadly follow classic rules which maximize favorable small molecule pharmacology in humans. Most medicinal chemistry campaigns identify a lead small molecule compound and then hundreds of derivative small molecules are created to search for an optimal molecule with both high affinity for a target and favorable pharmacology. In contrast, biopharmaceuticals are not orally bioavailable and often are highly engineered to maximize their pharmacological properties. Biologics such as recombinant proteins or antibodies engage their target to therapeutic effect at the cell surface and thus delivery considerations are limited to issues such as stability in the body and tissue distribution. In contrast, genome therapeutics must be delivered to the target cell, enter the cell and deliver a gene editing reagent to the nucleus. Traditional biological therapeutics that must enter a cell have had limited success, as the intracellular delivery of most macromolecular drugs is generally too inefficient to durably inhibit the target protein or RNA. In contrast, genomic therapeutics enact a permanent change to the target sequence of DNA and as such are likely to have more success even with low delivery efficiency. However, the viability of this concept in a therapeutic context remains to be determined for most diseases.
The delivery of both small molecule and biologic therapeutics is not equal across the body. Some organs such as the liver are highly accessible to both small molecules and biologics. The liver is the main organ responsible for detoxifying small molecules or toxins from the blood through the action of cytochrome C P450 enzymes. The liver also is responsible for regulation of cholesterol through the release and absorption of chylomicrons which have similar properties to biologics such as nanoparticles and some viruses. Due to these properties, in practice most small molecules and many biologics accumulate at high concentrations in the liver where they are absorbed and either cleared or have some activity. This property has been exploited for gene therapy and dozens of clinical trials are currently in progress for gene therapy in the liver10. However, for many diseases the liver’s ability to act as a sponge for macromolecules and viruses represents a major barrier to effective delivery of therapeutics to other organs.
Tissues such as skeletal muscle and the brain have other unique physiology that must be specifically considered in the context of genome therapeutics, both when considering means of delivery and required payload. Mature muscle fibers form from fusion of immature myoblasts and as such are polyploid. In diseases that arise from muscle dysfunction, this likely means that editing of causative disease genes is only required in some fraction of nuclei within a muscle fiber in order to regenerate muscle function. Indeed, it has been estimated for Duchenne’s muscular dystrophy that restoration of the causative DMD gene to as little as 4% of wild type levels or function will rescue many of the worst clinical consequences of the disease11. Therefore, this feature of muscle physiology may in some cases simplify application of genome therapeutics for treatment of disease of muscle that currently lack therapeutic alternatives. For most diseases experimental models will be required to determine the level of therapeutic genome therapy necessary for clinical benefit.
Perhaps the most difficult organ for delivery is the brain. The brain is naturally protected from small molecules, biologic and cellular therapeutics by the blood brain barrier. The blood brain barrier is the interface of endothelial and peri-endothelial cells between the peripheral circulatory system and brain tissue. Some progress has been made on predicting or designing small molecules that penetrate the blood brain barrier but many drugs fail to accumulate at high levels in the brain12,13. Of course, this barrier function is useful to protect the brain against pathogens and overactive immune cells, but for brain diseases such as glioblastoma this feature of the brain is one factor that has limited our ability to treat this type of cancer. Exciting progress has recently been made through sophisticated protein engineering on protein biologics that can bypass through the blood brain barrier14. Such examples should serve to inform genome therapeutic efforts. Other organs such as the eye or testis also have unique physical barriers that will need to be considered in the context of genome therapeutics. The eye is additionally an immune privileged organ, so there is more leeway to treat patients with genome therapeutic platforms such as viruses for which pre-existing immunity exists in the human population.
Preclinical modeling of genome therapeutics
Genome therapeutics are a complicated form of medicine resulting in a permanent change to the genome and as such will require the highest level of confidence for nominating gene targets as well as rigorous preclinical evidence. Preclinical validation will be required for both gene editor reagents and for gene editor delivery mechanisms. Preclinical validation for genome therapeutics may be in part informed by current experience with the zinc finger nuclease platform of gene editors but also by biologics such as humanized antibodies, as these therapies require additional layers of preclinical testing. Genome therapy using CRISPR technology has been modeled in animals for diseases of the liver15,16, muscle11,17,18, hematopoietic system19, eye20, brain21 and as a cellular therapeutic in embryos22, patient derived stem cells23, patient derived organoids24 and T cell immunotherapy25, and cardiovascular disease26,27.
For some diseases we have robust animal models of human biology. For example, we can construct humanized mice in which the immune system of the mouse is replaced with a human immune system28. This enables us to test genome therapeutics and delivery mechanisms against human diseases such as sickle cell anemia or severe combined immunodeficiency disorder. Recent changes to regulatory policy in some countries have made possible the construction of additional human disease models for diseases such as diabetes through the use of new interspecies chimeras29. However, despite the ability to model gene editing in preclinical models we lack models which enable us to test delivery principles in humans. Most preclinical validation in academic science is conducted in rodent models with a small amount of validation in larger mammals such as pigs, dogs and monkeys but in any model there will be substantial differences between even the best animal models and humans that will impact the delivery of genome therapeutics and thereby the activity and specificity of genome therapeutics30–33. In summary, modeling the implementation of genome therapeutics in human cell culture models is relatively straightforward even in advanced models such as induced pluripotent stem cells or organoids, while modeling the delivery and activity of a human genome therapeutic in a preclinical model of human biology remains extremely challenging.
Delivery of human genome therapeutics
Three main platforms currently exist for in vivo delivery of genome therapeutics. Recombinant viral delivery platforms are the most widely used delivery platform and have a long history in the context of gene therapy trials. Nanoparticle delivery is a newer and highly attractive synthetic delivery option but to date a lack of efficacy combined with high cost have limited adoption of nanoparticles as a delivery platform in human clinical trials. Engineered protein and RNA delivery platforms are also a promising new option for delivery of genome therapies.
Recombinant viral delivery platforms for in vivo genome therapy
Viruses have evolved to infect human cells and propagate across cells, tissues and individuals in a highly effective manner. Beginning in the 1990’s researchers realized that the properties of a viral infection might be harnessed as a delivery platform for gene therapy. It was hypothesized that a virus could be recombinantly engineered as a safe programmable delivery platform capable of homing to a target tissue, infecting target cells and efficiently delivering a payload. Several types of viruses have been modified and deployed as gene delivery platforms including adenovirus, adeno-associated virus, herpes simplex virus, lentivirus, retroviruses, and vaccinia virus. Engineering viral vectors is practically challenging but substantial progress has been made and to date more than 2300 clinical trials using recombinant viruses have demonstrated that for select diseases recombinant viral platforms are an effective means of delivery34–37. Lentiviral and the adeno-associated viral vectors are two recombinant viral platforms currently in wide use for gene delivery. Both viruses are useful for genome therapy because they are relatively safe, easy to work with and have low immunogenicity unlike some other viral platforms such as adenovirus.
The lentivirus most commonly used for clinical purposes is a modified form of the human immunodeficiency virus (HIV) that can infect both dividing and quiescent cells. Lentivirus is relatively easy to work with and as such it is straightforward and inexpensive to scale up production to generate the high titers required for clinical medicine. Modified HIV is widely used for this purpose, as a decade of research demonstrated that removal of HIV genes and modification of viral genome elements render HIV a robust and relatively safe viral gene delivery platform. HIV is a retrovirus with an RNA genome. Upon infection the viral RNA genome is reverse transcribed to DNA and then randomly integrated into the host genome. Following integration, the genes encoded in a lentiviral genome are stably and permanently expressed in the infected cell from exogenous or engineered promoters encoded in the modified viral genome. Thus, there is still some risk associated with this approach because the lentiviral DNA genome is semi-randomly integrated into the host genome and at some frequency this integration can activate aberrant expression of a proximal proto-oncogene resulting in cellular transformation.
Lentiviral systems package two copies of an RNA genome that is ~10 kilobases, enabling one to use this platform to co-deliver most commonly used CRISPR/Cas proteins such as Streptococcus pyogenes Cas9 and an sgRNA expression cassette with a single viral infection event. An additional advantage to lentiviral systems is their low intrinsic immunogenicity, although the adaptive immune response to any genome therapy will also depend on which genes are being delivered by the lentivirus. To date more than 300 gene therapy trials using lentiviral vectors have been carried out resulting in a large amount of clinical expertise for the design, production and safety concerns of a lentiviral medicine.
Adeno-associated virus (AAV) is a small ssDNA virus that is non-pathogenic and weakly immunogenic. The virus is thought to generally persist in infected cells in an extrachromosomal state although it can be integrated. If viral integration occurs there is preferentially integration at the human AAVS1 locus. Because AAV either does not integrate or integrates preferentially at a neutral site in the human genome AAV genome therapy platforms are thought to have a major safety advantage over lentiviral genome therapy platforms. It has been demonstrated that because the AAV genome is ssDNA one can use the AAV genome as a homology repair donor for knock-in mutations38. This ability to encode both CRISPR-Cas9 and a homology donor in a single system is a key feature of AAV for many applications. Another major advantage to AAV over lentivirus is that multiple serotypes of AAV exist with different intrinsic or engineered tropism for various tissues such as lung epithelial cells, skeletal muscle cell, neurons or heart, which will significantly help with tissue-specific delivery. New engineered AAV systems are an extremely promising platform for encoding tissue specificity for genome therapies39–42.
One major disadvantage to AAV is that this virus has a small genome and AAV delivery platform genomes must be 4.8 kilobases or smaller. Unfortunately, commonly used CRISPR/Cas gene editors such as the S. pyogenes Cas9 gene are 4.1kb; as such one must encode Cas9 and an sgRNA on separate AAV vectors and rely on viral co-infection for genome therapy, which can greatly reduce therapeutic editing potential. This incompatibility between AAV systems and early CRISPR/Cas9 systems has spurred the search for and use of small CRISPR/Cas9 effector proteins enabling one to encode both a CRISPR/Cas9 protein and an sgRNA in one AAV genome therapy vector. Currently, most advanced system that uses AAV to deliver a genome therapeutic for in vivo genome editing is the Staphylococcus aureus Cas9 system43. The S. aureus Cas9 gene is 3159 nucleotides enabling construction of an AAV encoding both the S. aureus Cas9 and an sgRNA. New even smaller CRISPR proteins such as CasX, which is 2940 nucleotides, may further facilitate use of AAV as a genome therapy platform44. Another study partitioned the two halves of the large S. pyogenes Cas9 gene into parallel AAV particles, effectively sidestepping the size limit45.
More than 110 gene therapy trials have been performed using AAV which should translate into rapid adoption of this platform for genome therapy trials. Currently patients must be pre-screened for immunity to AAV serotypes to increase delivery and safety in a clinical trial. Additionally, in most applications patients generate immunity after a single round of AAV therapy which presents a problem for some applications. This may not be such a concern for genome therapies that require only one period of activity to induce durable or permanent effects. It is also possible that engineered artificial AAV serotypes can be created to avoid natural immunity and enable repeat application of an AAV medicine.
Engineering nanoparticles for in vivo genome therapy
In theory, we should be able to engineer an artificial strategy to deliver any protein or nucleic acid to any cell in the human body. One route towards this goal is to use engineered small particles with tunable size, surface properties and cargo content that are non-immunogenic and highly efficient at delivering a cargo into cells in vivo. Such a delivery platform broadly has been described as a nanoparticle or protocell. Many different types of nanoparticles have been created and these platforms are excellently reviewed so we will focus on application of nanoparticles as delivery mechanisms for genome therapy. Genome therapies have benefited from more than a decade of intense research in this field enabling the rapid creation and preclinical application of the first nanoparticle CRISPR genome therapeutics46,47. These studies demonstrated nanoparticle-mediated gene disruption with efficiency up to 30% in tissue culture and 13% in vivo.
In preclinical models, a nanoparticle delivery platform has been used to deliver CRISPR genome therapeutics to the liver15. In principle, all tissues in the body are accessible for nanoparticle delivery but in practice it can be difficult to deliver high levels of nanoparticle encapsulated cargo to many tissues and most nanoparticle delivery platforms directed to other tissues also co-target the liver. Nanoparticle systems can deliver Cas9 in vivo as an mRNA or possibly as a ribonucleoprotein while the sgRNA has either been delivered as a synthetic RNA co-packaged together with Cas9 or encoded on an AAV vector delivered in trans. The first descriptions of nanoparticle delivery of CRISPR/Cas9 for genome therapy utilized a platform previously optimized for either RNA interference or mRNA delivery and so it may be possible to use custom nanoparticle delivery platforms to deliver RNPs in vivo or to co-deliver a DNA donor together with mRNA, sgRNA or RNPs. In preclinical data nanoparticle delivery platforms have enabled gene editing in the liver of mice without obvious or substantial liver toxicity suggesting this may be a relatively safe delivery platform but additional data will be needed in larger mammals. There are relatively few published papers reporting the use of nanoparticles for CRISPR mediated genome therapy but given that pioneering companies have publicly disclosed interest in such platforms we expect that this field will be explored as a delivery option for human genome therapy in the coming years. Despite the enormous promise of engineered nanoparticles expectations should be tempered by more than two decades of nanoparticle research48–50.
Engineered protein and RNA delivery platforms for in vivo genome therapy
In principle, a reductionist and elegant approach for in vivo application of genome therapeutics could be an engineered CRISPR/Cas9 RNP or RNA system capable of directly delivering gene editing activity to any cell in the body without need for a viral or nanoparticle delivery platform. The allure of such a straightforward approach has been reviewed for RNA based gene therapies but the emergence of CRISPR genome therapeutics has expanded interest in this type of medicine51.
Two main challenges exist to apply RNP or RNA CRISPR genome therapeutics to a disease. First, one must stabilize the delivered reagent to achieve a sufficient in vivo dosage to result in cytosolic delivery to target cells. One route to circumventing this challenge is to apply CRISPR genome therapies locally rather than systemically52,53. Another way to reduce the required dose is by targeting delivery to a specific cell, tissue, or organ using an attached targeting agent such as a ligand, antibody, or aptamer. Such an approach may provide an advantage over delivery using AAV or LNP, as those platforms have some inherent cellular or physiological tropism but it is inherent to each platform and thus less versatile. The second challenge is to deliver the editing reagent across the cell membrane and ultimately into the nucleus. This is a major hurdle in macromolecular delivery, estimated to block 99.99% of cargo in the case of siRNAs54. Cas9 protein or RNPs can be engineered to increase translocation across the cell membrane by modification of the electrostatic charge of the protein55. CRISPR/Cas9 RNP components can also be co-delivered with small peptides that increase translocation across the membrane56. These studies have shown editing of ~15% in tissue culture, while in vivo editing was observed but not quantified. As with nanoparticles, with additional engineering an RNP-based delivery platform may be amenable to co-delivery of DNA donor for homologous recombination.
One promising strategy combined the benefits of RNP use with those of nanoparticle delivery, complexing an intact Cas9 RNP with cationic lipids that promoted cell penetration57. This approach produces a short-lived complex, which is expected to reduce the number of deleterious, spurious “off-target” edits58. This RNP-based platform cut genomic DNA with 40% editing efficiency in tissue culture and 20% efficiency in vivo. Although these results are somewhat encouraging, the cationic lipid delivery platforms such as those used in this study are known to be toxic, limiting their therapeutic potential59. Another recent study has reported the use of gold nanoparticles to co-localize Cas9 RNP, donor DNA and endosome-disrupting peptides to perform genetic correction in DMD model mice60. This strategy produced 3% correction of the disease-causing mutation and improved muscle function in the treated mice, though the approach may require improvement since delivery was limited to the local area of injection.
The presence of S. pyogenes Cas9 protein in the serum may pose an immunogenic challenge, since most humans have been exposed to the bacterium. If that is the case, identifying epitopes of S. pyogenes Cas9 might serve to create a stealthy RNP that isn’t recognized by the human immune system. The prospect of direct delivery of genome editing enzymes holds incredible promise but further efforts are needed to establish our ability to safely and effectively deliver such reagents in pre-clinical models and in humans.
Ex vivo delivery and autologous transplantation
Ex vivo editing refers to genetic modification of a patient’s cells outside their body, and corrected cells can subsequently be returned to the patient to complete a process known as autologous transplantation. This process avoids some of the technical barriers associated with in vivo delivery of genome editing enzymes and thus there will likely be an initial enrichment of ex vivo clinical trials as CRISPR-based therapies are being developed. For ex vivo therapy, CRISPR-Cas9 can be delivered to the patient’s cultured cells in the form of an RNP via electroporation, which has been demonstrated for human hematopoietic stem/progenitor cells19 and T cells61,62 in experiments that demonstrate the foundation for sickle-cell anemia therapy and anti-cancer immunotherapies. Ex vivo delivery via virus has been demonstrated in cultured mouse muscle stem cells63, and nanoparticle-based delivery would also be anticipated to work for some cell types. Although these avenues are extremely promising, new technologies for ex vivo delivery would be beneficial since electroporation can be harsh, impacting cell viability.
Conclusion
Current genomic therapy trials herald a new era in medicine. In the short term, cellular medicines that can be easily modified ex vivo by CRISPR genome therapeutics and then transplanted back into the body will robustly and rapidly enter the clinic. Examples of such modified cellular therapeutics include hematopoietic stem cell therapies, immune cell therapies and regenerative medicines derived from induced pluripotent stem cells. Genome therapeutics applied systemically or locally to intact tissues will inevitably produce enormous clinical benefit but delivery of such genome therapeutics is challenging. It is possible that none of the three delivery platforms described above can surmount the many challenges to in vivo genome editing and some more elaborate form of protein engineering will be needed to create a delivery platform capable of efficiently and specifically delivering a gene editing reagent to any desired tissue in the body64. One can imagine construction of an engineered artificial virus that would have all of the desirable properties of a virus without limits to the delivery cargo or safety concerns. Our ability to construct or manipulate complex biological systems is rapidly advancing for biological discovery and bioengineering but application to medicine has lagged behind. With heightened interest garnered by the potential of CRISPR-based genome therapeutics, this may not be true for much longer.
Acknowledgments
LAG is supported by NIH/NCI Pathway to Independence Award K99/R00 CA204602.
References
- 1.Yi L, Li J. CRISPR-Cas9 therapeutics in cancer: promising strategies and present challenges. Bioch Biophys Acta. 2016;1866:197–207. doi: 10.1016/j.bbcan.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Li X, Wu R, Ventura A. The present and future of genome editing in cancer research. Hum Genet. 2016;135:1083–92. doi: 10.1007/s00439-016-1713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell. 2017;8:634–643. doi: 10.1007/s13238-017-0410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ely A, Moyo B, Arbuthnot P. Progress With Developing Use of Gene Editing To Cure Chronic Infection With Hepatitis B Virus. Mol Ther. 2016;24:671–677. doi: 10.1038/mt.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deleidi M, Yu C. Genome editing in pluripotent stem cells: research and therapeutic applications. Biochem Biophys Res Commun. 2016;473:665–74. doi: 10.1016/j.bbrc.2016.02.113. [DOI] [PubMed] [Google Scholar]
- 6.Pankowicz FP, Jarrett KE, Lagor WR, Bissig K. CRISPR/Cas9: at the cutting edge of hepatology. Gut. 2017;66:1329–1340. doi: 10.1136/gutjnl-2016-313565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strong A, Musunuru K. Genome editing in cardiovascular diseases. Nat Rev Cardiol. 2017;14:11–20. doi: 10.1038/nrcardio.2016.139. [DOI] [PubMed] [Google Scholar]
- 8.Schlimgen R, Howard J, Wooley D, Thompson M, Baden LR, Yang OO, Christiani DC, Mostoslavsky G, Diamond DV, Duane EG, Byers K, Winters T, Gelfand JA, Fujimoto G, Hudson TW, Vyas JM. Risks Associated With Lentiviral Vector Exposures and Prevention Strategies. J Occup Environ Med. 2016;58:1159–1166. doi: 10.1097/JOM.0000000000000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haworth KG, Peterson CW, Kiem H. CCR5-edited gene therapies for HIV cure: Closing the door to viral entry. Cytotherapy. 2017 doi: 10.1016/j.jcyt.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Kattenhorn LM, Tipper CH, Stoica L, Geraghty DS, Wright TL, Clark KR, Wadsworth SC. Adeno-Associated Virus Gene Therapy for Liver Disease. Hum Gene Ther. 2016;27:947–961. doi: 10.1089/hum.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long C, Amoasii L, Mireault A, McAnally J, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton J, Bassel-Duby R, Olson E. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi CR, Labhasetwar V, Ghorpade A. Destination Brain: the Past, Present, and Future of Therapeutic Gene Delivery. J Neuroimmune Pharmacol. 2017;12:51–83. doi: 10.1007/s11481-016-9724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walters B, Azam A, Gillon C, Josselyn S, Zovkic I. Advanced In vivo Use of CRISPR/Cas9 and Anti-sense DNA Inhibition for Gene Manipulation in the Brain. Front Genet. 2016;6 doi: 10.3389/fgene.2015.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discovery. 2016;15:275–292. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 15.Yin H, Song C, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, Gupta A, Bolukbasi MF, Walsh S, Bogorad RL, Gao G, Weng Z, Dong Y, Koteliansky V, Wolfe SA, Langer R, Xue W, Anderson DG. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, Yu H, Xu C, Morizono H, Musunuru K, Batshaw ML, Wilson JM. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–348. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong Le, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeWitt M, Magis W, Bray N, Wang T, Berman J, Urbinati F, Heo S, Mitros T, Muñoz D, Boffelli D, Kohn D, Walters M, Carroll D, Martin D, Corn J. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruan G, Barry E, Yu D, Lukason M, Cheng SH, Scaria A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, Sun X, Qin Z, Jin P, Li S, Li X. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest. 2017;127:2719–2724. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H, Marti-Gutierrez N, Park S, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Dyken C, Li Y, Kang E, Park A, Kim D, Kim S, Gong J, Gu Y, Xu X, Battaglia D, Krieg S, Lee D, Wu D, Wolf D, Heitner S, Belmonte J, Amato P, Kim J, Kaul S, Mitalipov S. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- 23.Dever D, Bak R, Reinisch A, Camarena J, Washington G, Nicolas C, Pavel-Dinu M, Saxena N, Wilkens A, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg K, Porteus M. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 25.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chadwick AC, Wang X, Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler, Thromb, Vasc Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Q, Strong A, Patel KM, Ng S, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walsh NC, Kenney LL, Jangalwe S, Aryee K, Greiner DL, Brehm MA, Shultz LD. Humanized Mouse Models of Clinical Disease. Annu Rev Pathol. 2017;12:187–215. doi: 10.1146/annurev-pathol-052016-100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Platero-Luengo A, Sakurai M, Sugawara A, Gil MA, Yamauchi T, Suzuki K, Bogliotti YS, Cuello C, Valencia MM, Okumura D, Luo J, Vilariño M, Parrilla I, Soto DA, Martinez CA, Hishida T, Sánchez-Bautista S, Martinez-Martinez ML, Wang H, Nohalez A, Aizawa E, Martinez-Redondo P, Ocampo A, Reddy P, Roca J, Maga EA, Esteban CR, Berggren WT, Delicado EN, Lajara J, Guillen I, Guillen P, Campistol JM, Martinez EA, Ross PJ, Belmonte JC. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell. 2017;168:473–486e15. doi: 10.1016/j.cell.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandamme TF. Use of rodents as models of human diseases. J Pharm BioAllied Sci. 2014;6:2–9. doi: 10.4103/0975-7406.124301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- 32.Sánchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387–395. doi: 10.1038/nrc3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol (Amsterdam, Neth) 2014;87:162–171. doi: 10.1016/j.bcp.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Bender E. Gene therapy: Industrial strength. Nature. 2016;537:S57–59. doi: 10.1038/537S57a. [DOI] [PubMed] [Google Scholar]
- 35.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keeler AM, ElMallah MK, Flotte TR. Gene Therapy 2017: Progress and Future Directions. Clin Transl Sci. 2017;10:242–248. doi: 10.1111/cts.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 – an update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 38.Gaj T, Staahl BT, Rodrigues GM, Limsirichai P, Ekman FK, Doudna JA, Schaffer DV. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017;45:e98. doi: 10.1093/nar/gkx154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 40.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 42.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ran FA, Cong Le, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC, Doudna JA, Banfield JF. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–241. doi: 10.1038/nature21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chew WL, Tabebordbar M, Cheng JK, Mali P, Wu EY, Ng AH, Zhu K, Wagers AJ, Church GM. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mout R, Ray M, Tonga GY, Lee Y, Tay T, Sasaki K, Rotello VM. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS nano. 2017;11:2452–2458. doi: 10.1021/acsnano.6b07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang C, Mei M, Bin Li, Zhu X, Zu W, Tian Y, Wang Q, Guo Y, Dong Y, Tan X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017;27:440–443. doi: 10.1038/cr.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park K. Facing the truth about nanotechnology in drug delivery. ACS nano. 2013;7:7442–7447. doi: 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venditto VJ, Szoka FC., Jr Cancer nanomedicines: so many papers and so few drugs! Adv Drug Delivery Rev. 2013;65:80–88. doi: 10.1016/j.addr.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics–developing a new class of drugs. Nature reviews Drug discovery. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 52.Kim K, Park SW, Kim JH, Lee SH, Kim D, Koo T, Kim K, Kim JH, Kim J. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Research. 2017;27:419–426. doi: 10.1101/gr.219089.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu W, Lu Z, Li F, Wang W, Qian N, Duan J, Zhang Y, Wang F, Chen T. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:1660–1665. doi: 10.1073/pnas.1614775114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–229. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 55.Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, Urnes C, Munares GA, Ghosh A, Doudna JA. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramakrishna S, Dad AK, Beloor J, Gopalappa R, Lee S, Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zuris J, Thompson D, Shu Y, Guilinger J, Bessen J, Hu J, Maeder M, Joung J, Chen Z, Liu D. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Kim D, Cho SW, Kim J, Kim J. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. The J Clin Invest. 2007;117:3623–3632. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang W, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017 doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, Marson A. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu P, Wu F, Mosenson J, Zhang H, He T, Wu W. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol Ther–Nucleic Acids. 2017;7:31–41. doi: 10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bale JB, Gonen S, Liu Y, Sheffler W, Ellis D, Thomas C, Cascio D, Yeates TO, Gonen T, King NP, Baker D. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science. 2016;353:389–394. doi: 10.1126/science.aaf8818. [DOI] [PMC free article] [PubMed] [Google Scholar]