

We report a new nanoparticle system for chemoimmunotherapy that elicits robust antitumor immunity against established tumors.
Abstract
Although immune checkpoint blockade has shown initial success for various cancers, only a small subset of patients benefits from this therapy. Some chemotherapeutic drugs have been reported to induce antitumor T cell responses, prompting a number of clinical trials on combination chemoimmunotherapy. However, how to achieve potent immune activation with traditional chemotherapeutics in a manner that is safe, effective, and compatible with immunotherapy remains unclear. We show that high-density lipoprotein–mimicking nanodiscs loaded with doxorubicin (DOX), a widely used chemotherapeutic agent, can potentiate immune checkpoint blockade in murine tumor models. Delivery of DOX via nanodiscs triggered immunogenic cell death of cancer cells and exerted antitumor efficacy without any overt off-target side effects. “Priming” tumors with DOX-carrying nanodiscs elicited robust antitumor CD8+ T cell responses while broadening their epitope recognition to tumor-associated antigens, neoantigens, and intact whole tumor cells. Combination chemoimmunotherapy with nanodiscs plus anti–programmed death 1 therapy induced complete regression of established CT26 and MC38 colon carcinoma tumors in 80 to 88% of animals and protected survivors against tumor recurrence. Our work provides a new, generalizable framework for using nanoparticle-based chemotherapy to initiate antitumor immunity and sensitize tumors to immune checkpoint blockade.
INTRODUCTION
Cancer immunotherapy aims to harness the host’s own immune system to fight cancer, and immune checkpoint blockers (ICBs) have shown marked initial success in the past few years, as exemplified by the clinical success of anti–cytotoxic T lymphocyte-associated antigen 4 (αCTLA-4), anti–programmed death 1 (αPD-1), and recently U.S. Food and Drug Administration–approved anti–PD-L1 (programmed death ligand 1) antibodies (1–4). However, despite their potential, ICBs currently benefit only a subset of patients, generally with 10 to 40% response rates reported in the clinic (2, 5). Because their therapeutic efficacy depends largely on licensing pre-existing antitumor T cells to kill their target tumor cells, the majority of patients bearing “cold” tumors with a low number of tumor antigen–specific T cells respond poorly to ICBs (6, 7). Hence, there has been considerable interest in developing complementary approaches, including therapeutic vaccines (8–10), radiation therapy (11–13), and chemotherapy (14–16), which could increase the repertoire and abundance of antitumor T cells so that combination immunotherapy with ICBs might exert strong antitumor immunity against cancer cells.
Notably, recent studies have shown that certain chemotherapeutic drugs, such as doxorubicin (DOX), may contribute to antitumor T cell responses by inducing a special form of tumor-cell killing, known as immunogenic cell death (ICD) (17–22). Tumor cells undergoing ICD up-regulate “eat me” and “danger” signals. The eat me signals, such as calreticulin (CRT) exposed on the surfaces of immunogenically dying tumor cells, enable dendritic cells (DCs) to phagocytose those tumor cells and present tumor antigen epitopes in the context of major histocompatibility complex (MHC) class I or II (18, 22). In turn, the danger signals, such as high-mobility group box 1 (HMGB1) released by immunogenically dying tumor cells, promote activation of DCs and trigger antigen-specific T cell responses (17, 21, 23). Thus, the use of ICD-inducing chemotherapeutic agents may offer a convenient and universal strategy for killing cancer cells while simultaneously eliciting broad antitumor T cell responses.
Recent studies have shown promising preclinical results reporting antitumor immune responses induced by free DOX treatment either as a monotherapy or combined with immunotherapy (14, 24–26). These findings have led to multiple ongoing clinical trials in phases 1, 2, and 3 that aim to investigate DOX therapy combined with ICBs (27–30). While results from these clinical trials are yet to be reported, how to achieve an optimal therapeutic outcome with chemoimmunotherapy remains to be seen, especially because there are concerns of inadequate circulation half-life and limited intratumoral accumulation of DOX as well as its off-target toxicities, including its widely documented cardiotoxicity (31), which may exacerbate toxicities of ICBs (32).
To address these challenges, here we have sought to develop a general strategy to improve the delivery of chemotherapeutics in a way that is safe, effective, and compatible with immune activation for combination immunotherapy. We have chosen to work with DOX because it is a widely used anticancer therapeutic agent and has ICD-inducing properties (18, 21, 22, 24). Delivery of DOX via nanosystems has been investigated intensively with a wide range of biomaterials, including liposomes, synthetic polymers, micelles, and inorganic nanostructures, in various stages of development (33–37). However, it is not yet clear how to apply these drug delivery systems to achieve immune activation in a manner compatible with cancer immunotherapy, while, at the same time, addressing the long-standing issues of industrial scale-up and clinical safety associated with various classes of nanomedicine.
Here, we have developed synthetic high-density lipoprotein (sHDL)–like nanodiscs, composed of an apolipoprotein A1 (ApoA1) mimetic peptide and phospholipids, for stimuli-responsive delivery of chemotherapy and demonstrated their potency for combination chemoimmunotherapy in vivo (Fig. 1). In particular, in recent clinical trials for cardiovascular applications, the sHDL platform has been successfully scaled up and demonstrated to be safe (38–40), thus prompting us to evaluate sHDL as the delivery platform for chemoimmunotherapy. In that effort, we have achieved efficient loading of DOX in sHDL and its stimuli-dependent release in the acidic pH of endosomes/lysosomes of tumor cells. Treatment with sHDL covalently attached with DOX (sHDL-DOX) induced ICD of tumor cells, improved pharmacokinetic profiles and tumor targeting of DOX, and exhibited significant antitumor efficacy without causing any overt off-target side effects. Tumor-bearing mice treated with sHDL-DOX elicited robust T cell responses directed against live tumor cells, tumor-associated antigens, and neoantigens, which are a class of patient-specific mutant epitopes encoded by somatic mutations in cancerous cells (41) and shown to dictate patient responses to immune checkpoint blockade (42–44). We report that sHDL-DOX markedly potentiated antitumor T cell responses and therapeutic efficacy of αPD-1 immunotherapy, leading to elimination of established CT26 and MC38 colon carcinoma in 80 to 88% of mice, inhibition of CT26 liver metastasis, and induction of long-term immunity against tumor cell re-challenge. Overall, these results demonstrated a generalizable strategy to induce robust antitumor immunity with nanoparticle-based chemotherapy that can sensitize tumors to immune checkpoint blockade.
Fig. 1. Schematic of doxorubicin-loaded sHDL (sHDL-DOX) for chemo-immunotherapy.
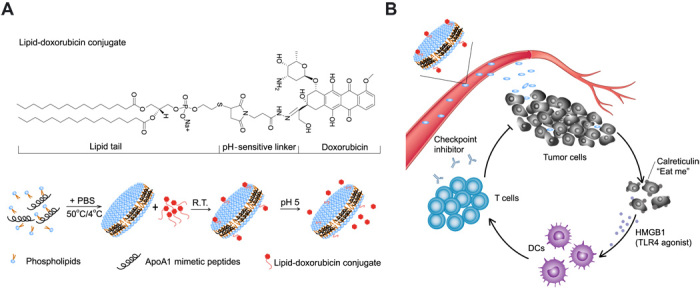
(A) sHDL-DOX is formulated by incubation of lipid-DOX with preformed sHDL. (B) The ultrasmall size and prolonged circulation of sHDL enable intratumoral delivery of DOX, followed by internalization by tumor cells and pH-responsive release of DOX in the endosomes/lysosomes. Released DOX kills tumor cells and triggers ICD, promoting up-regulation of CRT (the “eat me” signal) and release of danger signals such as HMGB1. DCs recruited to the immunogenically dying tumor cells phagocytose them, process tumor antigens, and cross-prime tumor antigen–specific T cells. Antitumor immunity primed with sHDL-DOX synergizes with immune checkpoint blockade, leading to efficient elimination of established tumors and prevention of tumor relapse. R.T., room temperature.
RESULTS
Intracellular delivery of DOX with HDL-mimicking nanodiscs
We prepared sHDL nanodiscs composed of an ApoA1 mimetic 37-mer peptide and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) using a thermal-cycling method, as we have reported previously (40, 45, 46). To promote loading and pH-triggered release of DOX from sHDL, we tethered DOX to a hydrophobic anchor with a hydrazone linker (33), which allowed for stable drug incorporation into sHDL at pH 7.4 but rapid drug release at pH 5. We conjugated DOX to 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (PTD) with an N-β-maleimidopropionic acid hydrazide (BMPH) cross-linker (Fig. 1A and fig. S1) and confirmed the formation of a lipid-DOX conjugate by mass spectrometry (fig. S2). To load lipid-DOX into the lipid layers of sHDL, preformed sHDL was simply admixed with lipid-DOX and incubated for 5 min at 37°C, resulting in efficient incorporation of lipid-DOX into sHDL (80 ± 2% encapsulation efficiency and 2.0 ± 0.2% w/w loading), as demonstrated by co-elution of sHDL and DOX (maximum absorbance at 220 and 485 nm, respectively) in gel permeation chromatography (GPC) (Fig. 2A). In contrast, when free DOX without the lipid tail was incubated with preformed sHDL, <1% of DOX was incorporated into sHDL (Fig. 2A), suggesting that the intermolecular interaction between the hydrophobic anchor of lipid-DOX and sHDL lipid layers is the major factor that drives drug loading. Transmission electron microscopy (TEM) and dynamic laser scattering (DLS) showed a homogeneous hydrodynamic size of ~10 nm for both “blank” sHDL and sHDL-DOX, indicating the minimal impact of drug loading on the formation and homogeneity of sHDL-DOX (Fig. 2, B and C). Notably, sHDL-DOX stored in a lyophilized powder form for at least 2 months was readily reconstituted with water to form homogeneous sHDL-DOX with a hydrodynamic size, polydispersity index, and GPC chromatogram similar to those of freshly prepared sHDL-DOX (Fig. 2C and fig. S3). We then examined pH-sensitive release of DOX from sHDL-DOX. Whereas sHDL-DOX incubated at pH 7.4 in phosphate-buffered saline (PBS) released less than 5% of DOX over 24 hours, sHDL-DOX incubated at pH 5 rapidly released ~60% of DOX within 24 hours (Fig. 2D), indicating pH-responsive drug release from sHDL in acidic pH of endosomes/lysosomes encountered upon internalization into tumor cells.
Fig. 2. Preparation and characterization of sHDL-DOX.
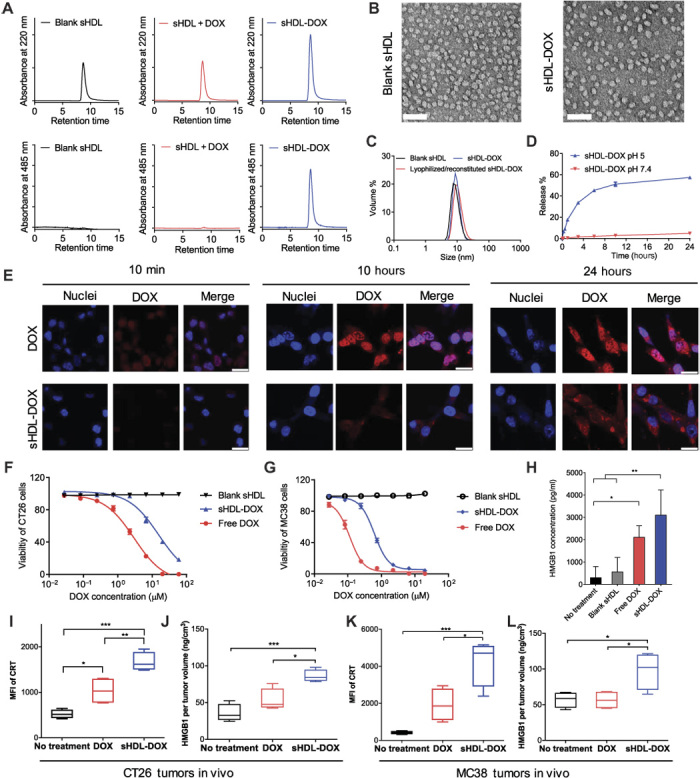
(A) GPC of blank sHDL, the physical mixture of sHDL + DOX, and sHDL covalently attached with DOX (sHDL-DOX) at 220 and 485 nm. (B) TEM of blank sHDL and sHDL-DOX. Scale bars, 50 nm. (C) Sizes of sHDL-DOX before and after lyophilization/reconstitution measured by DLS. (D) Release of DOX from sHDL at pH 5 and pH 7.5. Data represent mean ± SD (n = 3). (E) CT26 cells were incubated with 40 μM free DOX or sHDL-DOX for indicated lengths of time, and the intracellular distribution of DOX was imaged by confocal microscopy. Scale bars, 20 μm. (F to H) CT26 tumor cells (F) or MC38 tumor cells (G) were incubated with serial dilutions of free DOX or sHDL-DOX for 72 hours, and cellular viability was measured by the cell counting kit. (H) Release of HMGB1 was quantified by enzyme-linked immunosorbent assay (ELISA) after CT26 tumor cells were treated with indicated formulations (equivalent to 50 μM DOX). (I and J) BALB/c mice or (K and L) C57BL/6 mice were subcutaneously inoculated with 2 × 105 CT26 (I and J) or 2 × 105 MC38 cells (K and L) on day 0 and treated with DOX (4 mg/kg) in the indicated formulations on days 8 and 11. On day 15, the animals were euthanized and tumor tissues were harvested for analyses of ICD markers. Shown are (I and K) the levels of CRT on tumor cells (DAPI−CD45−) and (J and L) the amount of released HMGB1 per tumor volume. *P < 0.05, **P < 0.01, and ***P < 0.001 analyzed by one-way analysis of variance (ANOVA) (H to L) with Tukey’s multiple comparisons post test. Data in (D) and (F) to (H) represent mean ± SD (n = 3), and data in (I) to (L) are represented as box plots (whiskers, 5th to 95th percentile; n = 4) from a representative experiment from two to three independent experiments. MFI, mean fluorescence intensity.
We next investigated the intracellular delivery of DOX and sHDL-DOX and examined their impact on danger signals (for example, HMGB1 and CRT) implicated in ICD (20, 21). We treated CT26 colon cancer cells, a widely used murine model of colon adenocarcinoma, with DOX formulations and visualized DOX fluorescence with confocal microscopy. Within 10 min of treatment, CT26 cancer cells internalized free DOX as shown by dim, diffuse DOX fluorescence signals detected throughout the cells, and uptake of free DOX was further increased until 10 hours after incubation (Fig. 2E and fig. S4). In contrast, sHDL-DOX treatment slowed down DOX uptake with the nanodiscs first internalized into endolysosomes and the intracellular DOX signal steadily increasing over 24 hours (Fig. 2E and figs. S4 and S5A). The IC50 (half maximal inhibitory concentration) value of free DOX was slightly lower than that of sHDL-DOX in vitro in CT26 cells (3 and 15 μM, respectively; Fig. 2F) as well as in another murine colon carcinoma cell line, MC38 cells (0.11 and 0.62 μM, respectively; Fig. 2G). The reduction in cytotoxicity of sHDL-DOX may be attributed to the delayed cellular uptake and drug release from sHDL-DOX in vitro. Despite delayed sHDL-DOX internalization, sHDL-DOX treatment mediated up-regulation of CRT (fig. S5B) and triggered robust release of HMGB1 from CT26 cells (P < 0.01, compared to the no-treatment control; Fig. 2H) to a similar degree as free DOX treatment. Notably, sHDL-DOX treatment also strongly induced markers associated with ICD in vivo. Specifically, we inoculated 2 × 105 CT26 cells or MC38 colon carcinoma cells subcutaneously in the flank of syngeneic BALB/c or C57BL/6 mice, respectively, and on days 8 and 11, mice were administered intravenously with DOX (4 mg/kg) in the free soluble or sHDL form. Analyses of tumors on day 15 indicated that compared with free soluble DOX, sHDL-DOX treatment significantly increased the expression levels of CRT on the surfaces of CT26 cells (P < 0.01; Fig. 2I) and MC38 cells (P < 0.05; Fig. 2K) while enhancing intratumoral release of HMGB1 (P < 0.05; Fig. 2, J and L).
Together, we have successfully synthesized an sHDL-DOX nanoformulation with the attractive features of efficient drug loading, homogeneity, long-term stability, and stimuli-responsive drug release tailored to the endolysosomal condition (Fig. 2, A to D). Furthermore, we have demonstrated that sHDL-DOX killed cancer cells while effectively triggering ICD-associated danger signals in tumors (Fig. 2, E to L).
In vivo chemotherapy with sHDL-DOX
We next examined the in vivo distribution and efficacy of the sHDL formulations in tumor-bearing mice. We inoculated BALB/c mice subcutaneously in the flank with CT26 colon carcinoma cells on day 0 and performed intravenous administration on day 11 with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR), a model fluorescent tracer with a hydrophobic anchor as in the lipid-DOX conjugate, formulated in either sHDL, liposomes, or PEGylated liposomes. Noninvasive whole-animal imaging over time revealed that mice administered with sHDL had the intratumoral fluorescence signal peaking at 24 hours and lasting up to 72 hours (Fig. 3A), with markedly enhanced radiant efficiency (defined as fluorescence intensity/area/time), compared with liposomes (3.6-fold increase, P < 0.0001) and PEGylated liposomes (2.0-fold increase, P < 0.0001) (fig. S6). After sHDL-DiR administration, we also detected at least sevenfold greater DiR radiant efficiency in tumor tissues, compared with those in spleens, lungs, or kidneys (Fig. 3, B and C). As expected, the sHDL-DiR signal was strong in the liver, which is the major site for elimination of HDL (47). Given these results, we focused on sHDL-DOX to understand how sHDL affects the pharmacokinetics of DOX. We quantified the serum concentrations of DOX after intravenous administration and fitted the results into a two-compartment model. The area under the curve (AUC) for sHDL-DOX was 27-fold greater than that of free DOX (217.5 ± 15.2 μg/ml·hour for sHDL-DOX and 7.9 ± 0.1 μg/ml·hour for DOX; Fig. 3D). Consistent with the improved AUC, sHDL-DOX treatment resulted in a 2.8-fold increase in the cellular uptake of DOX within tumors, compared with free DOX treatment (P < 0.01; fig. S7).
Fig. 3. Antitumor efficacy and T cell immunity exerted by sHDL-DOX monotherapy.

(A) CT26 tumor–bearing mice were intravenously (iv) injected with sHDL-DiR, and the biodistribution of sHDL-DiR at different time points was imaged by the IVIS optical imaging system. (B) At 72 hours after injection, major organs were harvested and imaged ex vivo, and (C) fluorescence signal was quantified. (D) BALB/c mice were intravenously injected with free DOX or sHDL-DOX at DOX (4 mg/kg). Shown are the serum concentrations of DOX fitted to the two-compartment model. Data represent mean ± SD (n = 3) from a representative experiment from two to three independent experiments. (E) BALB/c mice were subcutaneously inoculated with 2 × 105 CT26 cells on day 0. On days 8, 11, and 14, tumor-bearing mice were treated with indicated formulations at DOX (4 mg/kg). (F and G) The average and individual CT26 tumor growth curves for mice treated with indicated formulations. CR, complete tumor regression. (H) Body weights of CT26 tumor–bearing mice treated with indicated formulations. (I) Hematoxylin and eosin (H&E) staining of the hearts and livers harvested on day 20 from tumor-bearing mice treated with indicated formulations. (J and K) The percentage of tumor cell-reactive T cells (IFN-γ+CD8+) among PBMCs on day 20 was measured by ICS. Shown are (J) the percentage of IFN-γ+CD8+ among PBMCs on day 20, and (K) the representative scatterplots. (L) The percentage of CT26 tumor antigen peptide AH1-specific CD8+ T cells among PBMC on day 20, and (M) the representative scatterplots. Data in (J) and (L) are represented as box plots (whiskers, 5th to 95th percentile; n = 5) from a representative experiment from two independent experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001 analyzed by one-way ANOVA (J and L) or two-way ANOVA (G and H) with Tukey’s multiple comparisons post test.
Having shown increased accumulation of sHDL in tumors as well as improved pharmacokinetics of sHDL-DOX, we next examined the therapeutic potential of sHDL-DOX and its effect on antitumor immune responses in vivo. BALB/c mice were inoculated subcutaneously with CT26 cells, and when the tumor size reached ~80 mm3 on day 8, the animals were treated three times with DOX (4 mg/kg) in either the free soluble or sHDL form (Fig. 3E). At this limited dose, free DOX treatment had no discernable impact on the overall tumor growth, compared with the no-treatment control group (Fig. 3, F and G). In contrast, sHDL-DOX treatment significantly slowed tumor growth, compared with free DOX or no-treatment groups (P < 0.0001; Fig. 3, F and G). Notably, as widely reported in the literature (31), free DOX treatment triggered adverse side effects, including body weight decrease and vacuolization of cardiomyocytes (Fig. 3, H and I). However, mice treated up to three times with the equivalent amount of DOX (4 mg/kg) in sHDL-DOX exhibited no overt signs of toxicity, weight loss, or cardiac or liver tissue damage (Fig. 3, H and I).
Robust antitumor T cell responses induced by sHDL-DOX therapy
We next examined the impact of sHDL-mediated delivery of DOX on antitumor immune responses. We first aimed to evaluate broad antitumor cellular immune responses induced by sHDL-DOX versus free DOX treatment. Briefly, we treated CT26 tumor–bearing BALB/c mice with DOX formulations as indicated above and examined induction of functional CD8α+ T cells against whole CT26 tumor cells by co-culturing peripheral blood mononuclear cells (PBMCs) with live CT26 tumor cells and performing intracellular cytokine staining (ICS) for interferon-γ (IFN-γ). Mice that received free DOX treatment failed to expand any CT26-specific IFN-γ+CD8α+ T cells beyond the basal level, whereas sHDL-DOX treatment generated a sevenfold higher frequency of IFN-γ+CD8α+ T cells that recognized intact CT26 tumor cells (P < 0.01; Fig. 3, J and K).
To gain insight into antigen specificity of cellular immune responses, we used the MHC-I minimal epitope of CT26 gp70 (AH1) (H-2Ld–restricted SPSYVYHQF) as the surrogate marker of tumor-specific antigen and quantitated the frequency of AH1-specific CD8α+ T cells among PBMCs. Whereas CT26 tumor–bearing mice that received free DOX treatment had the basal frequency of AH1-specific CD8α+ T cells among PBMCs, sHDL-DOX treatment induced 3.9-fold and 3.1-fold higher AH1-specific CD8α+ T cell responses, relative to the free DOX and no-treatment groups, respectively (P < 0.001 and P < 0.01, respectively; Fig. 3, L and M). Notably, compared with mice treated with free DOX, sHDL-DOX–treated animals had a higher frequency of CD11c+CD11b+Ly6c+ DCs within the tumor-draining lymph nodes (TDLNs) (P < 0.05; fig. S8A). These CD11c+CD11b+Ly6c+ DCs, which are a subset of antigen-presenting cells (APCs) that play crucial roles in the presentation of tumor antigens (48), also exhibited increased expression of a co-stimulatory marker CD86 within TDLNs (fig. S8B).
Potent antitumor efficacy of sHDL-DOX + αPD-1
Having confirmed immune responses triggered by sHDL-DOX monotherapy, we asked whether we could further amplify antitumor immunity and improve the therapeutic efficacy of sHDL-DOX therapy by combining this treatment with immune checkpoint blockade. Specifically, we chose to inhibit the immunosuppressive PD-1/PD-L1 pathway (5, 49) with αPD-1 immunoglobulin G (IgG) therapy to reflect ongoing combination immunotherapy clinical trials. BALB/c mice were inoculated subcutaneously with 2 × 105 CT26 tumor cells, and when the tumor size reached ~80 mm3 on day 8, the animals were treated intravenously on days 8, 11, and 14 with DOX (4 mg/kg) in either soluble or sHDL formulation, each supplemented with intraperitoneal administrations of αPD-1 (100 μg per dose) (Fig. 4A). Compared with the αPD-1 monotherapy, the combination of free DOX and αPD-1 therapy did not lead to significant expansion of AH1-specific CD8α+ T cells (Fig. 4, B and C). In stark contrast, the combination therapy of sHDL-DOX + αPD-1 led to remarkable expansion of AH1-specific CD8α+ T cells, reaching the peak frequency of 5 to 18% AH1-specific CD8α+ T cells among PBMCs on day 20 (eightfold greater than the αPD-1 monotherapy on average, P < 0.01, and fourfold greater than the dual free DOX + αPD-1 therapy, P < 0.05; Fig. 4, B and C).
Fig. 4. Potentiation of αPD-1 immunotherapy with sHDL-DOX for treatment of CT26 tumors.
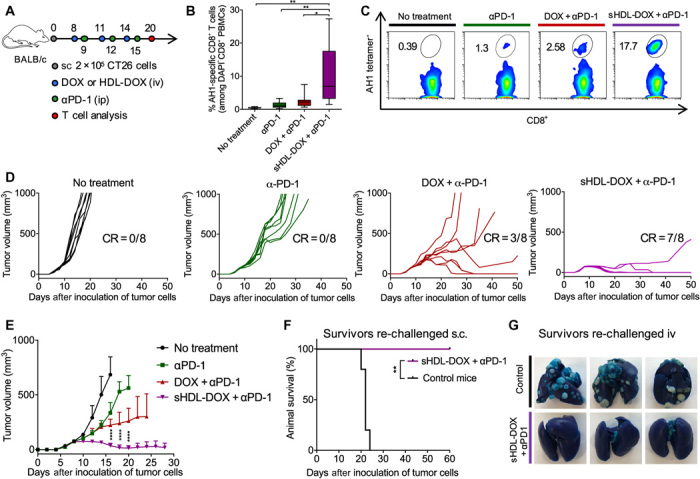
(A) BALB/c mice were subcutaneously (sc) inoculated with 2 × 105 CT26 cells on day 0. On days 8, 11, and 14, tumor-bearing mice were treated with indicated formulations at DOX (4 mg/kg). αPD-1 was injected intraperitoneally (ip) at 100 μg per dose on days 9, 12, and 15. (B) The percentage of CT26 tumor antigen AH1-specific CD8+ T cells among PBMCs on day 20, and (C) the representative scatterplots. Data are represented as box plots (whiskers, 5th to 95th percentile). n = 5 from a representative experiment from two independent experiments. (D) Individual growth curves for mice treated with indicated formulations. (E) The average tumor growth curves for mice treated with indicated formulations. Data represent mean ± SD (n = 8) from a representative experiment from two independent experiments. (F and G) On day 60, sHDL-DOX + αPD-1–treated animals in (E) were re-challenged by subcutaneous or intravenous injection of 2 × 105 CT26 cells. For the control groups, naïve BALB/c mice were re-challenged with the same number of CT26 cells. Shown are the animal survival (F) and lung metastasis (G) of CT26 cells on day 22 after re-challenge. Naïve mice were used as control and inoculated with the same number of tumor cells. *P < 0.05, **P < 0.01, and ****P < 0.0001 analyzed by one-way ANOVA (B) or two-way ANOVA (E) with Tukey’s multiple comparisons post test or log rank (Mantel-Cox) test (F).
Consistent with the enhanced antitumor immune responses, the combination chemoimmunotherapy with sHDL-DOX and αPD-1 exerted dramatic antitumor efficacy, eliminating established tumors (~80 mm3 at the initiation of treatment on day 8) in 88% of animals after three cycles of dual sHDL-DOX + αPD-1 therapy (P < 0.0001; Fig. 4, D and E). This is in stark contrast to the αPD-1 monotherapy or free DOX + αPD-1 dual therapy that failed to inhibit the average tumor growth at this low-dose/low-frequency regimen (P < 0.0001; Fig. 4, D and E). One hundred percent of the surviving animals from the sHDL-DOX + αPD-1 treatment group were protected against the subsequent re-challenge performed on day 60, as demonstrated by complete tumor-free survival for another 60 days after subcutaneous administration of 2 × 105 CT26 cells (Fig. 4F) or by the absence of lung tumor nodules 22 days after intravenous administration of 2 × 105 CT26 cells (Fig. 4G). These results suggest the establishment of durable immunity against tumor relapse. Moreover, throughout our studies, we did not observe any signs of weight loss, toxicity, or autoimmunity in animals treated up to three times with sHDL-DOX + αPD-1 dual therapy. Collectively, these results demonstrated that the sHDL-DOX therapy combined with αPD-1 therapy elicited potent antitumor CD8α+ T cell responses in vivo, thereby exerting robust antitumor efficacy against established tumors and tumor relapse.
T cell responses in the tumor microenvironment
Because tumor-infiltrating lymphocytes are one of the key determinants for the outcome of immunotherapy, we sought to profile antitumor T lymphocytes in the tumor microenvironment of animals undergoing chemoimmunotherapy. BALB/c mice were inoculated with 2 × 105 CT26 tumor cells and treated with various formulations, as stated above. Notably, among various formulations tested, the dual sHDL-DOX + αPD-1 therapy recruited the highest frequency of CD8α+ T cells into the tumor microenvironment (threefold greater than the free DOX treatment, P < 0.05; fig. S9A). The sHDL-DOX + αPD-1 therapy also promoted the highest frequency and the highest absolute number of tumor-infiltrating CD8α+ T cells recognizing the CT26 AH1 antigen (fivefold greater than the nontreated control group, P < 0.05; fig. S9, B and C). In general, these results reflected the patterns of systemic antigen-specific CD8α+ T cell responses induced after the combination sHDL-DOX + αPD-1 therapy (Fig. 4, B and C).
Neoantigen-specific CD8+ T cell responses induced by chemoimmunotherapy
Recent studies have shown that the antitumor efficacy of immune checkpoint blockade is strongly correlated with T cell responses against neoantigens, which are antigens encoded by somatic gene mutations only found in cancerous cells (41, 43, 50). Here, we studied the impact of sHDL-mediated DOX delivery on the generation of neoantigen-specific T cell responses and also sought to validate our results using another murine colon carcinoma model of MC38 tumor cells syngeneic to C57BL/6 mice. Animals were inoculated with 2 × 105 MC38 tumor cells via subcutaneous administration, and when the average tumor size reached ~60 mm3 on day 8, we performed intravenous administration of free DOX or sHDL-DOX with the equivalent amount of DOX (4 mg/kg), supplemented with αPD-1 therapy or PBS (Fig. 5A). To monitor neoantigen-specific T cell responses, we used a recently reported mutated neo-epitope within the Adpgk protein (ASMTNRELM → ASMTNMELM mutation), which is presented on MC38 tumor cells in the context of H-2Db molecules (51). The dual sHDL-DOX + αPD-1 chemoimmunotherapy generated a 2.4-fold greater expansion of neoantigen-specific CD8α+ T cells among PBMCs, compared with free DOX or sHDL-DOX treatment (P < 0.05; Fig. 5, B and C). sHDL-DOX + αPD-1 dual therapy exerted potent antitumor efficacy, leading to complete regression of established tumors (~60 mm3 at the initiation of therapy on day 8) in 80% of animals (P < 0.0001; Fig. 5, D and E). This is in contrast to all other treatment groups that exhibited increasing average tumor sizes over time. Overall, two cycles of sHDL-DOX + αPD-1 chemoimmunotherapy led to complete tumor response in 80% of animals (Fig. 5E). On the other hand, αPD-1 monotherapy and free DOX + αPD-1 dual therapy mediated tumor regression in ~40% of animals. None of the animals treated with DOX or sHDL-DOX chemotherapy had tumor regression. Mice cured of the primary MC38 tumors with sHDL-DOX + αPD-1 dual therapy were also completely protected against the re-challenge performed on day 60 with 2 × 105 MC38 tumor cells inoculated by either subcutaneous or intravenous routes (P < 0.01; Fig. 5, F and G), thus indicating long-term protection against tumor relapse.
Fig. 5. Chemo-immunotherapy for induction of neoantigen-specific T cell responses and elimination of MC38 tumors.
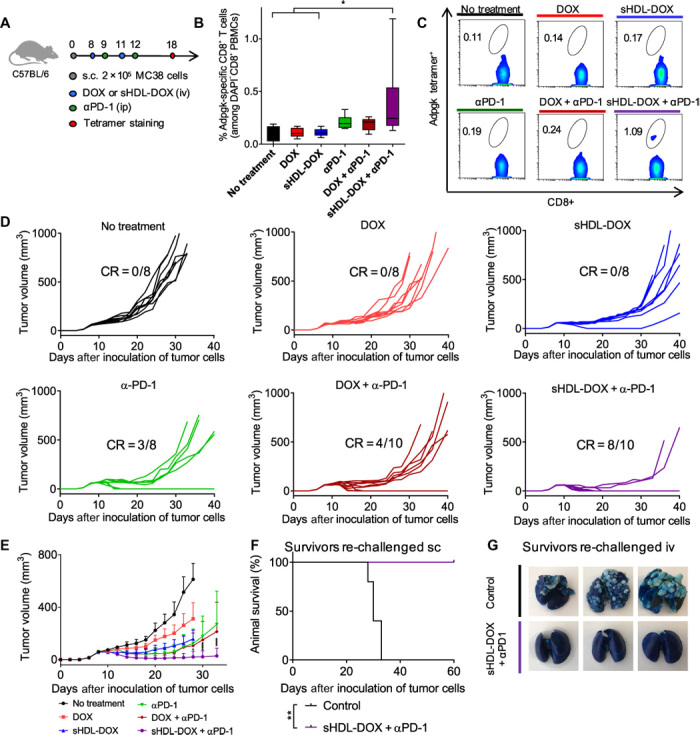
(A) C57BL/6 mice were inoculated subcutaneously with 2 × 105 MC38 cells on day 0. On days 8 and 11, tumor-bearing mice were treated with indicated DOX-containing formulations at DOX (4 mg/kg). For the combination immunotherapy, αPD-1 was injected intraperitoneally at 100 μg per dose on days 9 and 12. On day 18, the percentage of Adpgk-specific CD8+ T cells among PBMCs was measured. Data are represented as box plots (whiskers, 5th to 95th percentile). n = 5 for no treatment and n = 8 for other groups, from a representative experiment from two independent experiments. (B and C) The percentage of Adpgk-specific CD8+ T cells among PBMCs (B) and the representative scatterplots (C). (D) Individual tumor growth curves of mice treated with indicated formulations. (E) The average tumor growth curves of mice treated with indicated formulations. Data represent mean ± SD. n = 8 to 10, from a representative experiment from two independent experiments. (F and G) On day 60, sHDL-DOX + αPD-1–treated animals in (E) were re-challenged by subcutaneous or intravenous injection of 2 × 105 MC38 cells. For the control groups, naïve C57BL/6 mice were re-challenged with the same number of MC38 cells. Shown are the survival (F) and lung metastasis of MC38 cells (G) on day 26 after re-challenge. Naïve mice were used as control and inoculated with the same number of tumor cells. *P < 0.05, **P < 0.01, and ****P < 0.0001 analyzed by one-way ANOVA (B) or two-way ANOVA (D) with Tukey’s multiple comparisons post test or log rank (Mantel-Cox) test (F).
Therapeutic efficacy in advanced tumor models
We have also evaluated our strategy in advanced murine tumor models that respond poorly to immune checkpoint blockade. First, we have used an orthotopic colon carcinoma model, where 2 × 106 CT26-FL3-Luc cells were injected into the cecum wall, establishing highly aggressive tumors that formed liver metastases within several weeks (52, 53). After confirming establishment of tumors on day 8 with an in vivo imaging system (IVIS) (Fig. 6A), three cycles of dual sHDL-DOX + αPD-1 therapy were administered as described above. By day 20, sHDL-DOX + αPD-1 treatment markedly reduced the bioluminescence signal from whole animals (P < 0.0001; Fig. 6, A and B), compared with the αPD-1 group or the free DOX + αPD-1 group that exhibited a similar bioluminescence signal to the nontreated animals. Ex vivo imaging confirmed the drastic decrease of the CT26-FL3-Luc bioluminescence signal from colon after sHDL-DOX + αPD-1 therapy (1740-fold decrease from the αPD-1 group, P < 0.0001, and 1160-fold decrease from the DOX + αPD-1 group, P < 0.05), with nondetectable liver metastasis (Fig. 6, C and D). Overall, the sHDL-DOX + αPD-1 dual therapy produced a robust response rate of 88% (Fig. 6E), which is in stark contrast to the poor response rates (<13%) observed after αPD-1 monotherapy (P < 0.001) or free DOX + αPD-1 therapy (P < 0.01). Last, we have also examined the antitumor efficacy of chemoimmunotherapy in the MCA205 fibrosarcoma model that responds poorly to αPD-1 therapy. In C57BL/6 mice bearing MCA205 tumors, three cycles of sHDL-DOX + αPD-1 therapy significantly inhibited the overall tumor growth (P < 0.0001) and extended animal survival (P < 0.01), whereas αPD-1 monotherapy or free DOX + αPD-1 dual therapy yielded no survival benefit (fig. S10). Together, these studies have demonstrated the potency and wide applicability of sHDL-DOX–based chemoimmunotherapy in multiple murine tumor models.
Fig. 6. Chemo-immunotherapy for orthotopic colon cancer.

(A to E) BABL/c mice were inoculated with 2 × 106 CT26-FL3-Luc cells in the cecum wall on day 0. On days 8, 11, and 14, mice were injected intravenously with DOX (4 mg/kg) in the indicated formulations. On days 9, 12, and 15, mice were injected intraperitoneally with 100 μg per dose of αPD-1. Shown are (A) the whole-animal in vivo imaging over time and (B) quantification of the bioluminescence signal. (C and D) On day 22, major organs were harvested and imaged ex vivo. Shown are (C) representative bioluminescence images and (D) quantification of the signal in each organ. Data in (D) are represented as box plots (whiskers, 5th to 95th percentile; n = 4) from a representative experiment from two independent experiments. (E) Shown are the animal survival curves with n = 8 combined from two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 analyzed by two-way ANOVA (B and D) with Tukey’s multiple comparisons post test or log rank (Mantel-Cox) test (E). avg, average.
DISCUSSION
Here, we have used sHDL nanodiscs to deliver an ICD inducer, DOX, and achieved potent antitumor efficacy in combination with ICBs. Specifically, we have demonstrated that sHDL nanodiscs, composed of the 37–amino acid ApoA1 mimetic peptide and phospholipids, serve as a promising platform for chemoimmunotherapy. Compared with free DOX therapy, sHDL-DOX showed a 27-fold increase in pharmacokinetic profiles in animals and increased tumor accumulation without any targeting moiety (Fig. 3, A to D). Monotherapy with sHDL-DOX significantly delayed tumor growth without any overt off-target side effects (Fig. 3, F to I). sHDL-DOX treatment triggered robust expression of danger signals associated with ICD within tumors (Fig. 2, I to L), generating potent antitumor T cell responses and broadening their epitope recognition to tumor-associated antigens, neoantigens, and intact whole tumor cells (Fig. 3, J to M). Elicitation of neoantigen-specific T cell responses is quite notable because this class of tumor antigens is under intense investigation for personalized cancer vaccines, as we and others have reported (10, 51, 54). Furthermore, the therapeutic efficacy of ICBs was recently shown to be directly correlated with neoantigen-specific T cell responses (42–44), thus raising the prospect that “priming” tumors with sHDL-DOX therapy may potentiate ICBs—even without a priori knowledge of tumor antigens. The combination of sHDL-DOX and ICB therapy elicited strong antitumor immune responses (Figs. 4, B and C, and 5, B and C, and fig. S9) and markedly augmented their therapeutic efficacy; co-treatment with sHDL-DOX + αPD-1 IgG antibody induced complete regression of established colon carcinoma in 80 to 88% of animals (CT26 and MC38 tumors in BALB/c and C57BL/6 mice, respectively) while protecting all survivors against tumor cell re-challenge (Figs. 4, D to G, and 5, D to G). We have also demonstrated the strong therapeutic benefits of sHDL-DOX + αPD-1 chemoimmunotherapy in advanced colon carcinoma and fibrosarcoma models (Fig. 6 and fig. S10). This is in stark contrast to the αPD-1 monotherapy or free DOX + αPD-1 dual therapy that yielded poor response rates in these tumor models.
An extensive list of nanoparticle systems, such as liposomes, synthetic polymers, micelles, and inorganic nanostructures, has been examined for delivery of DOX with varying levels of success (33–37), but their impact on antitumor immunity and, hence, their potential as a platform for chemoimmunotherapy remain to be explored. The work presented here is, to the best of our knowledge, the first report of chemo-nanotechnology designed to trigger ICD of tumor cells and elicit T cell immunity against a broad range of tumor antigens, including neoantigens, thereby potentiating immune checkpoint blockade. Although other conventional nanoformulations may also be applicable to this approach, we believe that the sHDL system is particularly attractive for translation because of the ease of synthesis, established large-scale manufacturing, proven human safety, and nonimmunogenicity of the blank sHDL, as demonstrated in a number of clinical trials (38–40). In addition, the cardioprotective effect of HDL (55–57) may further alleviate the cardiotoxicity associated with sHDL-DOX treatment. Moreover, conventional nanoparticles typically require PEGylation for sufficient circulation half-life and drug accumulation in tumors; however, repeated administrations of PEGylated materials can cause chronic hand-foot syndrome (58) and anti-PEG (polyethylene glycol) antibody responses (59), thus potentially complicating their application in immunotherapy. In contrast, sHDL nanodiscs mimicking endogenous HDL do not require PEGylation for efficient DOX delivery. Our current study and a previous study (46) have indicated more efficient intratumoral accumulation of sHDL, compared with liposomes or PEGylated liposomes. Although the precise mechanisms are under investigation, we speculate that sHDL-mediated intratumoral delivery is facilitated in part by their ultrasmall particle size (~10 nm), extended pharmacokinetics, and extensive uptake by metabolically highly active cancer cells that require a large amount of lipids and cholesterol for proliferation (40, 46, 60).
In conclusion, we have produced a new, generalizable framework for chemoimmunotherapy. By delivering chemotherapeutic agents via nanocarriers in a manner that sensitizes tumor cells to immune activation and subsequent immune checkpoint blockade, we have achieved potent antitumor efficacy, leading to elimination of established tumors in 80 to 88% of animals. Our approach may be readily applied to other chemotherapeutic agents known to induce ICD of tumor cells (17, 18, 21). Because there is intense interest in improving the patient response rate and therapeutic efficacy of immune checkpoint blockade, our strategy presented here may have a wide-ranging impact in the field of drug delivery, nanotechnology, and cancer immunotherapy.
MATERIALS AND METHODS
Preparation and characterization of sHDL-DOX
sHDL was prepared by the lyophilization method that we have previously developed (40, 61, 62). Briefly, DPPC and an ApoA1 mimetic peptide, DWLKAFYDKVAEKLKEAFPDWAKAAYDKAAEKAKEAA (37A), were mixed at a 1.5:1 weight ratio in acetic acid, followed by lyophilization. The lyophilized powder was hydrated in PBS (pH 7.4) and cycled between 55°C and room temperature to obtain sHDL. We synthesized a pH-sensitive lipid-DOX conjugate for efficient loading and pH-triggered release of DOX from sHDL. Briefly, DOX was activated with BMPH (Thermo Fisher Scientific) in anhydrous methanol containing trifluoroacetic acid (33). The mixture was allowed to react for 24 hours at room temperature, followed by rotary evaporation. Activated DOX was then reacted with PTD in chloroform containing 10% triethylamine for 24 hours in the dark. After rotary evaporation, the resulting lipid-DOX conjugate was kept at −20°C until further use. The molecular weight of the conjugate was confirmed by electrospray ionization mass spectrometry. To load DOX in sHDL, the lipid-DOX conjugate was dissolved in dimethyl sulfoxide and then incubated with preformed sHDL suspension in PBS (pH 7.4) for 5 min at 37°C on an orbital shaker. The resulting sHDL-DOX was passed through a desalting column (Pierce) to remove any unincorporated DOX.
The concentration of DOX loaded in sHDL-DOX was measured by a fluorescence-based method. sHDL-DOX (10 μl) diluted in water was incubated with 240 μl of 1% Triton X-100 solution for 30 min at room temperature in the dark, and the fluorescence signal from DOX was detected using a microplate reader with Ex = 470 nm and Em = 590 nm. Homogeneity of sHDL-DOX was analyzed by GPC using a Shimadzu HPLC system equipped with a TSKgel G2000SWxl column [7.8 mm (internal diameter) × 30 cm, Tosoh Bioscience LLC], and the detection wavelengths were set at 220 and 485 nm for the quantification of ApoA1 mimetic peptide 37A and DOX, respectively. The particle size of sHDL-DOX was measured by DLS on a Malvern Zetasizer. The sHDL morphology was assessed by TEM after proper dilution of the original samples. Then, 3 μl of the diluted sample solution was deposited on a carbon film–coated 400-mesh copper grid (Electron Microscopy Sciences) and dried for 1 min. Samples were then negatively stained with 1% (w/v) uranyl formate, and the grid was dried before TEM observation. All specimens were imaged on a 100-kV Morgagni TEM equipped with a Gatan Orius CCD.
Measurements of intracellular delivery, cytotoxicity, and ICD in tumor cells treated with sHDL-DOX
To examine sHDL-DOX for its pattern of intracellular delivery, 100,000 CT26 tumor cells were seeded in 35-mm petri dishes (MatTek Corp.) and cultured overnight. Cells were incubated with 40 μM sHDL-DOX or free DOX for predetermined durations (10 min, 10 hours, and 24 hours). After incubation, cells were washed with PBS, fixed with 4% paraformaldehyde, and stained with 4′,6-diamidino-2-phenylindole (DAPI) before imaging with a confocal microscope (Nikon A1). Cytotoxicity of sHDL-DOX was measured using Cell Counting Kit-8 (Dojindo Molecular Technologies) following the manufacturer’s instructions. Markers of ICD, such as CRT and HMGB1, were analyzed following published reports (17, 18). Briefly, 100,000 CT26 cells were seeded on 35-mm petri dishes (MatTek Corp.) precoated with polylysine. After overnight incubation, cells were treated with 50 μM DOX or sHDL-DOX for 24 hours, washed twice with fluorescence-activated cell sorting (FACS) buffer [1% bovine serum albumin (BSA) in PBS] followed by incubation with CD16/32 for 10 min and rabbit anti-mouse CRT antibody (1:100 dilution) for 30 min. Cells were washed and then incubated with Hoechst 33342 and anti-rabbit antibody labeled with allophycocyanin (APC) for 20 min and then observed under a confocal microscope. To measure the release of HMGB1 from dying tumor cells, 50,000 CT26 cells seeded in 96-well plates were incubated with 50 μM DOX or sHDL-DOX for 72 hours. After incubation, each supernatant was collected and centrifuged at 1000g for 20 min before HMGB1 measurement using a mouse HMGB1 ELISA kit (LifeSpan BioSciences Inc.).
Biodistribution and pharmacokinetic studies in vivo
sHDL was loaded with a near-infrared fluorescent dye, DiR, for the biodistribution study (46). Briefly, DiR [0.1 mole percent (mol %)] was mixed with DPPC and 37A in acetic acid, followed by lyophilization and hydration in PBS to form sHDL-DiR, as described above. For the preparation of DiR-loaded liposomes (Lipo-DiR) or DiR-loaded PEGylated liposomes (PEG-Lipo-DiR), DiR (0.1 mol %) was mixed with DPPC or DPPC with 5 mol % DSPE-PEG2000, followed by hydration in PBS and extrusion through 100-nm polycarbonate membranes. BALB/c mice inoculated with 200,000 CT26 tumor cells on the right flank were injected intravenously with sHDL-DiR, Lipo-DiR, or PEG-Lipo-DiR [DiR (20 μg/ml)] on day 10. At predetermined time points after injection, whole-body imaging was performed using the IVIS optical imaging system. At the 72-hour time point, the tumor-bearing mice were euthanized, and major organs (brain, heart, liver, spleen, lung, kidney, and tumor) were harvested for ex vivo imaging. For the pharmacokinetic analysis, mice were administered with DOX (4 mg/kg) or sHDL-DOX (4 mg/kg) intravenously. Following drug treatment, at each time point (15 min, 1 hour, 3 hours, 7 hours, and 24 hours), 50 μl of blood was collected in Microvette 500 Z-gel tubes by submandibular bleeding and kept on ice. The samples were centrifuged at 10,000g for 5 min at room temperature, and 10 μl of the serum was added with 10 μl of PBS and incubated with 480 μl of acidified isopropanol (75 mM HCl, 10% water, 90% isopropanol) overnight at 4°C in the dark to extract DOX. The isopropanol extract was centrifuged at 14,000 rpm for 10 min, and 125 μl of the supernatant was used for fluorescence detection of DOX on a microplate reader with Ex = 485 and Em = 590 nm. The standard curve was generated with DOX spiked in normal serum and measured following the same protocol. DOX serum concentration–time curves were fitted with a two-compartment model by GraphPad Prism 6 to determine the AUC values (60).
Study of therapeutics in tumor-bearing animals
Mice were cared for following federal, state, and local guidelines. All work performed on animals was in accordance with and approved by the University Committee on Use and Care of Animals at the University of Michigan, Ann Arbor. For treatment studies involving animals injected with CT26 cells, BALB/c mice were inoculated with 2 × 105 CT26 cells per mouse on the right flank by subcutaneous injection on day 0 and intravenously injected with DOX (4 mg/kg) in sHDL or free soluble form on days 8, 11, and 14. For the combinatorial chemoimmunotherapy, anti-mouse PD-1 (100 μg per mouse) was administered intraperitoneally on days 9, 12, and 15 in addition to the intravenous injection of DOX-containing formulations. For the orthotopic colon carcinoma studies, we followed a previously described procedure (52, 53). Briefly, 2 × 106 CT26-FL3-Luc cells were injected into the cecum subserosa after exteriorizing the cecum in anesthetized BALB/c mice. A similar treatment regimen was given as above. We visualized the bioluminescence signal from CT26-FL3-Luc tumors in either whole animals or harvested organs using an IVIS imaging system.
For studies with MC38 colon carcinoma or MCA205 fibrosarcoma cells, C57BL/6 mice were inoculated subcutaneously with 2 × 105 MC38 cells or MCA205 cells on day 0 and intravenously injected with DOX (4 mg/kg) in sHDL or free soluble form on days 8 and 11 with or without intraperitoneal administration of anti-mouse PD-1 (100 μg per mouse) on days 9 and 12. Tumor growth was monitored every other day, and the tumor volume was calculated by the following equation: tumor volume = length × width2 × 0.52. When individual tumor masses reached 15 mm in diameter in any dimension or when animals became moribund with severe weight loss or active ulceration, animals were euthanized. On day 20, some CT26 tumor–bearing mice were euthanized and the hearts and livers were collected and fixed in 10% Buffered Formalin (Fisher Diagnostics). A series of 5-μm sections were stained with H&E. Stained slides were then observed by microscopy (PerkinElmer Mantra). On day 60, mice cured of primary CT26 or MC38 tumors were re-challenged by subcutaneous injection of 2 × 105 of the same tumor cells, and subsequent tumor growth was monitored as described above. Alternatively, some mice, cured of primary CT26 or MC38 tumors, were re-challenged by intravenous injection of the same 2 × 105 tumor cells, and the lung metastasis of CT26 or MC38 tumor cells was visualized by injecting India ink (1:10 dilution in PBS) into the lungs via the trachea and fixing the lungs in Fekete’s solution (54). Naïve mice were used as controls and re-challenged in the same way.
For a subset of studies, tumor tissues and TDLNs harvested on indicated time points were cut into small pieces of 2 to 4 mm, and cells were dissociated in digestion buffer [collagenase type IV (1 mg/ml) and deoxyribonuclease I (100 U/ml) in serum-free RPMI] for 20 to 30 min at 37°C with gentle shaking (10). This cell suspension was passed through a 70-μm nylon strainer and washed with FACS buffer. Cells were then incubated with CD16/32 for 10 min and then stained with antibodies against CD8α (53-6.7), CD11c (HL3), CD11b (M1/70), Ly6c (AL-21), CD86 (GL1), CD45.2 (104), and CRT (EPR3924) on ice before flow cytometry (Cyan 5, Beckman Coulter). In some experiments, cells were incubated with AH1 peptide-MHC tetramer (H-2Ld–restricted SPSYVYHQF) or Adpgk peptide-MHC tetramer (H-2Db–restricted ASMTNMELM) to label the antigen-specific T cells at room temperature for 30 min before incubation with the above antibodies. In some experiments, after dissociating the tumor tissue into single-cell suspension, the concentration of HMGB1 in digestion buffer was measured using a mouse HMGB1 ELISA kit (LifeSpan BioSciences Inc.).
Tetramer staining and ICS
The percentages of tumor antigen–specific CD8α+ T cells among PBMCs were analyzed using the tetramer staining assay, as described previously (10). Briefly, 100 μl of blood was collected from each mouse on indicated days by submandibular bleeding, and red blood cells were lysed using Ammonium-Chloride-Potassium (ACK) lysis buffer. PBMCs were then washed with FACS buffer and blocked by anti-CD16/32 antibody and incubated with peptide-MHC tetramer (for example, H-2Ld–restricted SPSYVYHQF or H-2Db–restricted ASMTNMELM) for 30 min at room temperature. Samples were then incubated with anti-mouse CD8α-APC for 20 min on ice. Cells were washed twice with FACS buffer and resuspended in DAPI solution (2 μg/ml) for analysis by flow cytometry.
For ICS assay (10), 100 to 150 μl of peripheral blood collected from mice was lysed with ACK lysis buffer, washed with PBS, and plated at ~10 million cells/ml in 50 μl of T cell media [RPMI 1640 supplemented with 10% FBS, 2 mM l-glutamine, 55 μM β-mercaptoethanol, 1 mM pyruvate, penicillin (100 U/ml), streptomycin (100 μg/ml), 10 mM Hepes, and nonessential amino acids] in 96-well U-bottom plates. These PBMCs were incubated with 0.1 million CT26 cells per well for 16 hours in the presence of the protein transport inhibitor, brefeldin A (BD Biosciences). Cells were then washed twice with ice-cold FACS buffer (1% BSA in PBS), followed by incubation with anti-CD16/32 for at least 10 min and anti-CD8α for 20 min on ice. Cells were then fixed/permeabilized for 20 min on ice and then stained with anti–IFN-γ–phycoerythrin (PE) for 30 min on ice. After extensive washing, cells were analyzed by flow cytometry.
Statistical analysis
Sample sizes were chosen based on preliminary data from pilot experiments and previously published results in the literature. All animal studies were performed after randomization. Data were analyzed by one-way or two-way ANOVA, followed by Tukey’s multiple comparisons post test or log rank (Mantel-Cox) test with Prism 6.0 (GraphPad Software). Data were normally distributed and variance between groups was similar. P values less than 0.05 were considered statistically significant. All values are reported as means ± SD with the indicated sample size. No samples were excluded from analysis.
Acknowledgments
We acknowledge the University of Michigan Consulting for Statistics, Computing, and Analytics Research for help with statistical analyses, the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of MHC-I tetramers, D. Patra and G. Skiniotis for help with TEM imaging, W. Zou (University of Michigan, Ann Arbor) for MC38 cells, L. Huang (University of North Carolina, Chapel Hill) for CT26-FL3-Luc cells, A. D. Weinberg (Portland Providence Medical Center) for MCA205 cells, and K. Moynihan and D. J. Irvine (Massachusetts Institute of Technology) for technical help with ICS with whole tumor cells.
Funding: This work was supported in part by NIH (R01EB022563, R01AI127070, and R01CA210273 to J.J.M. and R21NS091555 and R01HL134569 to A.S.), Michigan Translational Research and Commercialization for Life Sciences Hub, Emerald Foundation, and University of Michigan Forbes Institute for Cancer Discovery Pilot Grant. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), U.S. Department of Defense (DOD)/Congressionally Directed Medical Research Programs Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), and NSF CAREER Award (1553831). R.K. was supported by the Broomfield International Student Fellowship and the American Heart Association (AHA) Predoctoral Fellowship (15PRE25090050). W.Y. was supported by the AHA Postdoctoral Fellowship (16POST27760002). Y.F. was supported by the Rackham Predoctoral Fellowship. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the DOD.
Author contributions: R.K., W.Y., A.S., and J.J.M. designed the experiments. R.K. and W.Y. performed the experiments. J.N. and S.S. contributed to the establishment of the orthotopic colon carcinoma model. Y.X. contributed to the IVIS imaging and flow cytometry. Y.F. contributed to the measurement of ICD markers. R.K., W.Y., A.S., and J.J.M. analyzed the data. R.K. and J.J.M. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data used to obtain the conclusions in this paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaao1736/DC1
fig. S1. Schematic for the synthesis of the lipid-DOX conjugate.
fig. S2. Mass spectroscopy confirmed the conjugation of DOX to PTD.
fig. S3. sHDL-DOX was lyophilized and stored for 2 months before reconstitution by adding water to the lyophilized powder.
fig. S4. Cellular uptake of DOX or sHDL-DOX by CT26 cells in vitro.
fig. S5. Confocal images of CT26 cells and their CRT staining after sHDL-DOX treatment.
fig. S6. Biodistribution of Lipo-DiR, PEG-Lipo-DiR, and sHDL-DiR in CT26 tumors in vivo.
fig. S7. Intratumoral uptake of DOX or sHDL-DOX in CT26 tumors in vivo.
fig. S8. Analysis of DCs in TDLNs after treatment of CT26 tumor-bearing mice with DOX or sHDL-DOX.
fig. S9. Antitumor immune responses in the tumor microenvironment.
fig. S10. Efficacy of sHDL-DOX + αPD-1 therapy against MCA205 fibrosarcoma.
REFERENCES AND NOTES
- 1.Hodi F. S., O’Day S. J., McDermott D. F., Weber R. W., Sosman J. A., Haanen J. B., Gonzalez R., Robert C., Schadendorf D., Hassel J. C., Akerley W., van den Eertwegh A. J., Lutzky J., Lorigan P., Vaubel J. M., Linette G. P., Hogg D., Ottensmeier C. H., Lebbé C., Peschel C., Quirt I., Clark J. I., Wolchok J. D., Weber J. S., Tian J., Yellin M. J., Nichol G. M., Hoos A., Urba W. J., Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C., Schachter J., Long G. V., Arance A., Grob J. J., Mortier L., Daud A., Carlino M. S., McNeil C., Lotem M., Larkin J., Lorigan P., Neyns B., Blank C. U., Hamid O., Mateus C., Shapira-Frommer R., Kosh M., Zhou H., Ibrahim N., Ebbinghaus S., Ribas A.; KEYNOTE-006 investigators , Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Garon E. B., Rizvi N. A., Hui R., Leighl N., Balmanoukian A. S., Eder J. P., Patnaik A., Aggarwal C., Gubens M., Horn L., Carcereny E., Ahn M. J., Felip E., Lee J. S., Hellmann M. D., Hamid O., Goldman J. W., Soria J. C., Dolled-Filhart M., Rutledge R. Z., Zhang J., Lunceford J. K., Rangwala R., Lubiniecki G. M., Roach C., Emancipator K., Gandhi L., Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg J. E., Hoffman-Censits J., Powles T., van der Heijden M. S., Balar A. V., Necchi A., Dawson N., O’Donnell P. H., Balmanoukian A., Loriot Y., Srinivas S., Retz M. M., Grivas P., Joseph R. W., Galsky M. D., Fleming M. T., Petrylak D. P., Perez-Gracia J. L., Burris H. A., Castellano D., Canil C., Bellmunt J., Bajorin D., Nickles D., Bourgon R., Frampton G. M., Cui N., Mariathasan S., Abidoye O., Fine G. D., Dreicer R., Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian S. L., Hodi F. S., Brahmer J. R., Gettinger S. N., Smith D. C., McDermott D. F., Powderly J. D., Carvajal R. D., Sosman J. A., Atkins M. B., Leming P. D., Spigel D. R., Antonia S. J., Horn L., Drake C. G., Pardoll D. M., Chen L., Sharfman W. H., Anders R. A., Taube J. M., McMiller T. L., Xu H., Korman A. J., Jure-Kunkel M., Agrawal S., McDonald D., Kollia G. D., Gupta A., Wigginton J. M., Sznol M., Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellman I., Coukos G., Dranoff G., Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardoll D. M., The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho H.-I., Barrios K., Lee Y.-R., Linowski A. K., Celis E., BiVax: A peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol. Immunother. 62, 787–799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duraiswamy J., Kaluza K. M., Freeman G. J., Coukos G., Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demaria S., Kawashima N., Yang A. M., Devitt M. L., Babb J. S., Allison J. P., Formenti S. C., Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 11, 728–734 (2005). [PubMed] [Google Scholar]
- 12.Belcaid Z., Phallen J. A., Zeng J., See A. P., Mathios D., Gottschalk C., Nicholas S., Kellett M., Ruzevick J., Jackson C., Albesiano E., Durham N. M., Ye X., Tran P. T., Tyler B., Wong J. W., Brem H., Pardoll D. M., Drake C. G., Lim M., Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLOS ONE 9, e101764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twyman-Saint Victor C., Rech A. J., Maity A., Rengan R., Pauken K. E., Stelekati E., Benci J. L., Xu B., Dada H., Odorizzi P. M., Herati R. S., Mansfield K. D., Patsch D., Amaravadi R. K., Schuchter L. M., Ishwaran H., Mick R., Pryma D. A., Xu X., Feldman M. D., Gangadhar T. C., Hahn S. M., Wherry E. J., Vonderheide R. H., Minn A. J., Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang T. H., Mao C.-P., Lee S. Y., Chen A., Lee J.-H., Kim T. W., Alvarez R. D., Roden R. B. S., Pardoll D., Hung C.-F., Wu T.-C., Chemotherapy acts as an adjuvant to convert the tumor microenvironment into a highly permissive state for vaccination-induced antitumor immunity. Cancer Res. 73, 2493–2504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfirschke C., Engblom C., Rickelt S., Cortez-Retamozo V., Garris C., Pucci F., Yamazaki T., Poirier-Colame V., Newton A., Redouane Y., Lin Y.-J., Wojtkiewicz G., Iwamoto Y., Mino-Kenudson M., Huynh T. G., Hynes R. O., Freeman G. J., Kroemer G., Zitvogel L., Weissleder R., Pittet M. J., Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 44, 343–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathios D., Kim J. E., Mangraviti A., Phallen J., Park C.-K., Jackson C. M., Garzon-Muvdi T., Kim E., Theodros D., Polanczyk M., Martin A. M., Suk I., Ye X., Tyler B., Bettegowda C., Brem H., Pardoll D. M., Lim M., Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 8, 370ra180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Apetoh L., Ghiringhelli F., Tesniere A., Obeid M., Ortiz C., Criollo A., Mignot G., Maiuri M. C., Ullrich E., Saulnier P., Yang H., Amigorena S., Ryffel B., Barrat F. J., Saftig P., Levi F., Lidereau R., Nogues C., Mira J.-P., Chompret A., Joulin V., Clavel-Chapelon F., Bourhis J., André F., Delaloge S., Tursz T., Kroemer G., Zitvogel L., Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J.-L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., Zitvogel L., Kroemer G., Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Ghiringhelli F., Apetoh L., Tesniere A., Aymeric L., Ma Y., Ortiz C., Vermaelen K., Panaretakis T., Mignot G., Ullrich E., Perfettini J.-L., Schlemmer F., Tasdemir E., Uhl M., Génin P., Civas A., Ryffel B., Kanellopoulos J., Tschopp J., André F., Lidereau R., McLaughlin N. M., Haynes N. M., Smyth M. J., Kroemer G., Zitvogel L., Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Green D. R., Ferguson T., Zitvogel L., Kroemer G., Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kroemer G., Galluzzi L., Kepp O., Zitvogel L., Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Ma Y., Mattarollo S. R., Adjemian S., Yang H., Aymeric L., Hannani D., Portela Catani J. P., Duret H., Teng M. W. L., Kepp O., Wang Y., Sistigu A., Schultze J. L., Stoll G., Galluzzi L., Zitvogel L., Smyth M. J., Kroemer G., CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. 74, 436–445 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Krysko D. V., Garg A. D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P., Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Sistigu A., Yamazaki T., Vacchelli E., Chaba K., Enot D. P., Adam J., Vitale I., Goubar A., Baracco E. E., Remédios C., Fend L., Hannani D., Aymeric L., Ma Y., Niso-Santano M., Kepp O., Schultze J. L., Tüting T., Belardelli F., Bracci L., La Sorsa V., Ziccheddu G., Sestili P., Urbani F., Delorenzi M., Lacroix-Triki M., Quidville V., Conforti R., Spano J.-P., Pusztai L., Poirier-Colame V., Delaloge S., Penault-Llorca F., Ladoire S., Arnould L., Cyrta J., Dessoliers M.-C., Eggermont A., Bianchi M. E., Pittet M., Engblom C., Pfirschke C., Préville X., Uzè G., Schreiber R. D., Chow M. T., Smyth M. J., Proietti E., André F., Kroemer G., Zitvogel L., Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014). [DOI] [PubMed] [Google Scholar]
- 25.van der Sluis T. C., van Duikeren S., Huppelschoten S., Jordanova E. S., Beyranvand Nejad E., Sloots A., Boon L., Smit V. T. H. B. M., Welters M. J. P., Ossendorp F., van de Water B., Arens R., van der Burg S. H., Melief C. J. M., Vaccine-induced tumor necrosis factor–producing T cells synergize with cisplatin to promote tumor cell death. Clin. Cancer Res. 21, 781–794 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Rios-Doria J., Durham N., Wetzel L., Rothstein R., Chesebrough J., Holoweckyj N., Zhao W., Leow C. C., Hollingsworth R., Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 17, 661–670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.ClinicalTrials.gov, Safety and efficacy study of Pembrolizumab (MK-3475) in combination with chemotherapy as neoadjuvant treatment for participants with triple negative breast cancer (TNBC) (MK-3475-173/KEYNOTE 173); https://clinicaltrials.gov/ct2/show/NCT02622074.
- 28.ClinicalTrials.gov, Pembrolizumab and doxorubicin hydrochloride or anti-estrogen therapy in treating patients with triple-negative or hormone receptor-positive metastatic breast cancer; https://clinicaltrials.gov/ct2/show/NCT02648477.
- 29.ClinicalTrials.gov, A(B)VD followed by Nivolumab as frontline therapy for higher risk patients with classical Hodgkin lymphoma; https://clinicaltrials.gov/ct2/show/NCT03033914.
- 30.ClinicalTrials.gov, Study of Pembrolizumab (MK-3475) plus chemotherapy vs placebo plus chemotherapy as neoadjuvant therapy and pembrolizumab vs placebo as adjuvant therapy in participants with triple negative breast cancer (TNBC) (MK-3475-522/KEYNOTE-522); https://clinicaltrials.gov/ct2/show/NCT03036488.
- 31.Arola O. J., Saraste A., Pulkki K., Kallajoki M., Parvinen M., Voipio-Pulkki L.-M., Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 60, 1789–1792 (2000). [PubMed] [Google Scholar]
- 32.Spain L., Diem S., Larkin J., Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 44, 51–60 (2016). [DOI] [PubMed] [Google Scholar]
- 33.MacKay J. A., Chen M., McDaniel J. R., Liu W., Simnick A. J., Chilkoti A., Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 8, 993–999 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogawara K.-i., Un K., Tanaka K.-i., Higaki K., Kimura T., In vivo anti-tumor effect of PEG liposomal doxorubicin (DOX) in DOX-resistant tumor-bearing mice: Involvement of cytotoxic effect on vascular endothelial cells. J. Control. Release 133, 4–10 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Etrych T., Šubr V., Strohalm J., Šírová M., Říhová B., Ulbrich K., HPMA copolymer-doxorubicin conjugates: The effects of molecular weight and architecture on biodistribution and in vivo activity. J. Control. Release 164, 346–354 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Maksimenko A., Dosio F., Mougin J., Ferrero A., Wack S., Reddy L. H., Weyn A.-A., Lepeltier E., Bourgaux C., Stella B., Cattel L., Couvreur P., A unique squalenoylated and nonpegylated doxorubicin nanomedicine with systemic long-circulating properties and anticancer activity. Proc. Natl. Acad. Sci. U.S.A. 111, E217–E226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye D.-X., Ma Y.-Y., Zhao W., Cao H.-M., Kong J.-L., Xiong H.-M., Möhwald H., ZnO-based nanoplatforms for labeling and treatment of mouse tumors without detectable toxic side effects. ACS Nano 10, 4294–4300 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Kingwell B. A., Chapman M. J., Kontush A., Miller N. E., HDL-targeted therapies: Progress, failures and future. Nat. Rev. Drug Discov. 13, 445–464 (2014). [DOI] [PubMed] [Google Scholar]
- 39.D. Li, S. Gordon, A. Schwendeman, A. T. Remaley, Apolipoprotein mimetic peptides for stimulating cholesterol efflux, in Apolipoprotein Mimetics in Management of Human Disease, G. M. Anantharamaiah, D. Goldberg, Eds. (Springer, 2015), pp. 29–42. [Google Scholar]
- 40.Kuai R., Li D., Chen Y. E., Moon J. J., Schwendeman A., High-density lipoproteins: Nature’s multifunctional nanoparticles. ACS Nano 10, 3015–3041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schumacher T. N., Schreiber R. D., Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 42.van Rooij N., van Buuren M. M., Philips D., Velds A., Toebes M., Heemskerk B., van Dijk L. J. A., Behjati S., Hilkmann H., el Atmioui D., Nieuwland M., Stratton M. R., Kerkhoven R. M., Keşmir C., Haanen J. B., Kvistborg P., Schumacher T. N., Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizvi N. A., Hellmann M. D., Snyder A., Kvistborg P., Makarov V., Havel J. J., Lee W., Yuan J., Wong P., Ho T. S., Miller M. L., Rekhtman N., Moreira A. L., Ibrahim F., Bruggeman C., Gasmi B., Zappasodi R., Maeda Y., Sander C., Garon E. B., Merghoub T., Wolchok J. D., Schumacher T. N., Chan T. A., Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le D. T., Durham J. N., Smith K. N., Wang H., Bartlett B. R., Aulakh L. K., Lu S., Kemberling H., Wilt C., Luber B. S., Wong F., Azad N. S., Rucki A. A., Laheru D., Donehower R., Zaheer A., Fisher G. A., Crocenzi T. S., Lee J. J., Greten T. F., Duffy A. G., Ciombor K. K., Eyring A. D., Lam B. H., Joe A., Kang S. P., Holdhoff M., Danilova L., Cope L., Meyer C., Zhou S., Goldberg R. M., Armstrong D. K., Bever K. M., Fader A. N., Taube J., Housseau F., Spetzler D., Xiao N., Pardoll D. M., Papadopoulos N., Kinzler K. W., Eshleman J. R., Vogelstein B., Anders R. A., Diaz L. A. Jr., Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian C., Kuai R., Zhu Q., White P., Moon J. J., Schwendeman A., Cohen M. S., Synthetic high-density lipoprotein nanoparticles: A novel therapeutic strategy for adrenocortical carcinomas. Surgery 159, 284–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang J., Kuai R., Yuan W., Drake L., Moon J. J., Schwendeman A., Effect of size and pegylation of liposomes and peptide-based synthetic lipoproteins on tumor targeting. Nanomedicine 13, 1869–1878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rensen P. C. N., de Vrueh R. L., Kuiper J., Bijsterbosch M. K., Biessen E. A. L., van Berkel T. J. C., Recombinant lipoproteins: Lipoprotein-like lipid particles for drug targeting. Adv. Drug Deliv. Rev. 47, 251–276 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Ma Y., Adjemian S., Mattarollo S. R., Yamazaki T., Aymeric L., Yang H., Catani J. P. P., Hannani D., Duret H., Steegh K., Martins I., Schlemmer F., Michaud M., Kepp O., Sukkurwala A. Q., Menger L., Vacchelli E., Droin N., Galluzzi L., Krzysiek R., Gordon S., Taylor P. R., Van Endert P., Solary E., Smyth M. J., Zitvogel L., Kroemer G., Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Zou W., Wolchok J. D., Chen L., PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gubin M. M., Zhang X., Schuster H., Caron E., Ward J. P., Noguchi T., Ivanova Y., Hundal J., Arthur C. D., Krebber W.-J., Mulder G. E., Toebes M., Vesely M. D., Lam S. S. K., Korman A. J., Allison J. P., Freeman G. J., Sharpe A. H., Pearce E. L., Schumacher T. N., Aebersold R., Rammensee H.-G., Melief C. J. M., Mardis E. R., Gillanders W. E., Artyomov M. N., Schreiber R. D., Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., Delamarre L., Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y., Davis C., Ryan J., Janney C., Peña M. M. O., Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clin. Exp. Metastasis 30, 903–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodwin T. J., Zhou Y., Musetti S. N., Liu R., Huang L., Local and transient gene expression primes the liver to resist cancer metastasis. Sci. Transl. Med. 8, 364ra153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J. C., Tadmor A. D., Schoenberger S. P., Huber C., Türeci Ö., Sahin U., Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barter P. J., Nicholls S., Rye K.-A., Anantharamaiah G. M., Navab M., Fogelman A. M., Antiinflammatory properties of HDL. Circ. Res. 95, 764–772 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Cooney M. T., Dudina A., De Bacquer D., Wilhelmsen L., Sans S., Menotti A., De Backer G., Jousilahti P., Keil U., Thomsen T., Whincup P., Graham I. M., HDL cholesterol protects against cardiovascular disease in both genders, at all ages and at all levels of risk. Atherosclerosis 206, 611–616 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Frias M. A., Lang U., Gerber-Wicht C., James R. W., Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc. Res. 85, 118–126 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Lorusso D., Di Stefano A., Carone V., Fagotti A., Pisconti S., Scambia G., Pegylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia (‘hand-foot’ syndrome). Ann. Oncol. 18, 1159–1164 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Ishida T., Ichihara M., Wang X., Yamamoto K., Kimura J., Majima E., Kiwada H., Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 112, 15–25 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Cui L., Lin Q., Jin C. S., Jiang W., Huang H., Ding L., Muhanna N., Irish J. C., Wang F., Chen J., Zheng G., A PEGylation-free biomimetic porphyrin nanoplatform for personalized cancer theranostics. ACS Nano 9, 4484–4495 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Schwendeman A., Sviridov D. O., Yuan W., Guo Y., Morin E. E., Yuan Y., Stonik J., Freeman L., Ossoli A., Thacker S., Killion S., Pryor M., Chen Y. E., Turner S., Remaley A. T., The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J. Lipid Res. 56, 1727–1737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan Y., Wen J., Tang J., Kan Q., Ackermann R., Olsen K., Schwendeman A., Synthetic high-density lipoproteins for delivery of 10-hydroxycamptothecin. Int. J. Nanomedicine 11, 6229–6238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaao1736/DC1
fig. S1. Schematic for the synthesis of the lipid-DOX conjugate.
fig. S2. Mass spectroscopy confirmed the conjugation of DOX to PTD.
fig. S3. sHDL-DOX was lyophilized and stored for 2 months before reconstitution by adding water to the lyophilized powder.
fig. S4. Cellular uptake of DOX or sHDL-DOX by CT26 cells in vitro.
fig. S5. Confocal images of CT26 cells and their CRT staining after sHDL-DOX treatment.
fig. S6. Biodistribution of Lipo-DiR, PEG-Lipo-DiR, and sHDL-DiR in CT26 tumors in vivo.
fig. S7. Intratumoral uptake of DOX or sHDL-DOX in CT26 tumors in vivo.
fig. S8. Analysis of DCs in TDLNs after treatment of CT26 tumor-bearing mice with DOX or sHDL-DOX.
fig. S9. Antitumor immune responses in the tumor microenvironment.
fig. S10. Efficacy of sHDL-DOX + αPD-1 therapy against MCA205 fibrosarcoma.