

Abstract
Historically, much of our understanding of actin filaments, microtubules and intermediate filaments has come from the study of fixed cells and tissues. But the cytoskeleton is inherently dynamic, and so developing the means to image it in living cells has proved crucial. Advances in confocal microscopy and fluorescent protein technologies have allowed us to dynamically image the cytoskeleton at high resolution and so learn much more about its cellular functions. However, most of this work has been performed in cultured cells, and a critical next step is to understand how the cytoskeleton functions in the context of an intact organism. We, and others, have developed methods to image the cytoskeleton in living vertebrate embryos. Here we describe an approach to image the cytoskeleton in embryos of the frog, Xenopus laevis, using mRNA to express fluorescently-tagged cytoskeletal probes and confocal microscopy to visualize their dynamic behaviour.
Keywords: live imaging, embryos, Xenopus laevis, microtubules, actin
1. Introduction
The eukaryotic cytoskeleton has been intensively studied for over a century and yet much of our knowledge of actin filaments, microtubules, and intermediate filaments and their subcellular distribution has come from the imaging of fixed samples. However, the cytoskeleton is inherently dynamic and so the ability to image it in living cells is vital to developing a complete understanding of its functions and mechanics. In the last 15 years advances in live confocal imaging and, in particular, the development of fluorescent protein tags such as eGFP, mRFP and mCherry [1–6], have permitted major leaps forward in the study of the cytoskeleton. The majority of this work has been performed in cultured cells, which has proved to be a very powerful system for investigating the dynamics of the eukaryotic cytoskeleton. However, to build on these findings, the next step is to understand how a cell’s cytoskeleton functions within the context of an intact, living organism. To date this has been tackled predominantly by using invertebrate rather than vertebrate embryos. However, we, and others, have had increasing success imaging cytoskeletal dynamics in living vertebrate embryos [7–9] (see Figure 1). In this review, we highlight approaches to image the cytoskeleton in living embryos of the frog, Xenopus laevis.
Figure 1. Live imaging of cytoskeletal probes in Xenopus laevis embryos.
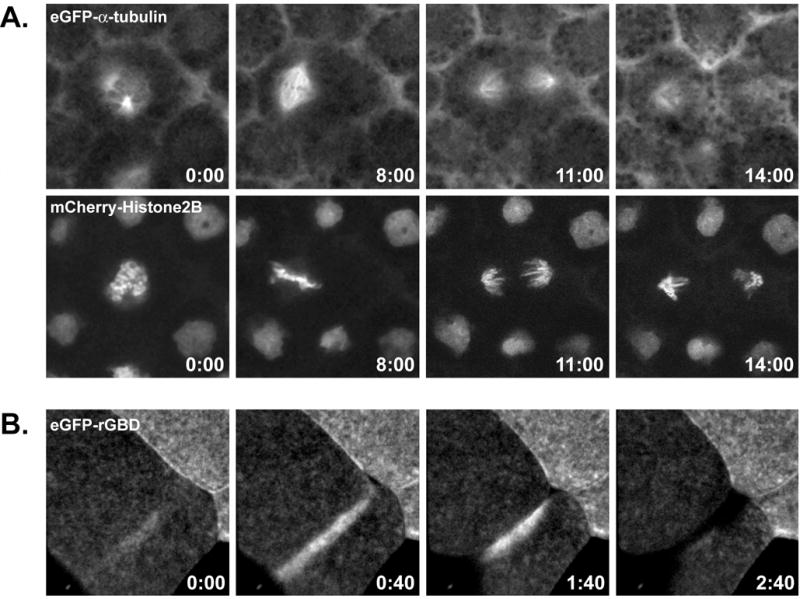
(A). A mitotic spindle imaged in an early Xenopus laevis embryo using eGFP-a-tubulin (top) to highlight microtubules and mCherry-Histone2B (bottom) to show chromosomes. (B). The activation of Rho GTPase at the site of the cytokinetic furrow is shown using eGFP-rGBD, a probe that binds specifically to active Rho. In both examples, stills were taken from timelapse movies and timestamps represent time in minutes and seconds. See Note 1 for more a more detailed description of these and other cytoskeletal probes.
Xenopus laevis embryos are an ideal system for these studies as they are large and therefore relatively easy to microinject and manipulate. Furthermore, since these embryos develop externally and at room temperature they are highly amenable to live imaging. The one disadvantage of Xenopus laevis embryos for live imaging is their opacity, which makes imaging of deep tissues more challenging; we therefore concentrate on the most superficial cells of the early embryo, where the cytoskeleton can be readily imaged using live confocal microscopy.
Traditionally, imaging of actin filaments or microtubules in cultured cells was accomplished by microinjection of actin or tubulin protein labelled with small fluorescent molecules [10]. More recently, the development of fusion constructs comprised of fluorescent proteins (e.g. eGFP or mCherry) fused to actin or tubulin has permitted generation of stable cell lines expressing these proteins by transfection with the relevant DNA [11]. However, we have found that injection of fluorescent actin or tubulin protein into embryos can be less than satisfactory, for two reasons. First, the small volumes required (5 nl or less) means that extremely high protein concentrations are required.
This, in turn, leads to clogging of the microinjection needle. Second, even if clogging is avoided, it is often difficult to obtain sufficient protein for adequate labelling in these very large cells. Conversely, microinjection of plasmid DNA encoding either fluorescent tubulin or fluorescent actin, which overcomes these problems, has its own attendant difficulties. First, it takes some time before appreciable levels of the probe accumulate, in part because transcription is very limited before the midblastula transition. Second, expression from microinjected plasmids tends to be highly mosaic, with some cells expressing robustly, and others not at all. We therefore recommend that probes for cytoskeleton proteins be expressed by microinjection of capped RNA into Xenopus embryos. The mRNA is less viscous than the protein but, unlike DNA, expresses rapidly and relatively uniformly.
2. Materials
2.1 In vitro transcription of capped RNA for cytoskeletal probes
Plasmid DNA (containing cytoskeletal probe construct).
Restriction enzyme for linearizing plasmid DNA.
20% SDS (RNase-free).
20mg/ml Proteinase K (Ambion).
Phenol-chloroform-isoamyl alcohol mixture (25:24:1; pH 7.9).
Ethanol (100% and 70%).
mMESSAGE mMACHINE® capped RNA transcription kit (Ambion).
Nuclease-free water (Ambion).
RNAseZap® (Ambion).
Chloroform.
Phenol (pH 6.6).
Isopropanol.
Pipettors wiped down with RNase Zap and nuclease-free pipette tips.
1.5 ml eppendorf tubes (nuclease-free).
Microcentrifuges (at room temperature and 4°C).
Gel electrophoresis apparatus.
Spectrophotometer.
Water bath that can be set to 37°C or 50°C.
−80°C freezer.
Speed Vac (optional).
2.2 Xenopus laevis ovulation and in vitro fertilization
Human chorionic gonadotrophin (HCG, MP Biomedicals), 1unit/μl in dH2O, filter sterilized and stored at −80°C.
Benzocaine (Ethyl 4-aminobenzoate; Sigma-Aldrich), make 10% solution in 95% ethanol and store at 4°C.
Marc’s Modified Ringer’s solution (MMR). For 1X: 100 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH7.4. Make a 10X solution and dilute to make 1X and 0.1X MMR.
Testes prep media: for 5ml: 4 ml 1X MMR, 1 ml Foetal Bovine Serum (heat inactivated), 250 μg Gentamicin, 250 Units Penicillin, 250 μg Streptomycin.
L-Cysteine (Sigma-Aldrich): 2% solution in 1X MMR, pH 7.8, made fresh on day of fertilization.
Small tank with lid for anesthetizing frog in benzocaine.
Forceps and surgical scissors.
Small Petri dishes: 35 mm diameter.
Deep Petri dishes: 60 mm (diameter) × 20 mm (depth).
50 ml falcon tubes.
3 ml transfer pipettes.
1 ml sterile syringes.
27 G 1/2 sterile needles.
Cooled incubator (set at 17°C).
2.3 Microinjection of Xenopus laevis embryos
Capped RNA (see section 3.2).
Nuclease-free water (Ambion).
0.1X MMR (see section 2.2).
Injection buffer: 5% Ficoll in 0.1X MMR, rotate overnight to dissolve.
RNaseZap® (Ambion).
Mineral oil (Sigma-Aldrich).
10 μl Drummond glass capillaries (Fisher Scientific).
Micropipette puller.
Dissecting microscope.
Microinjector, micromanipulator and N2 gas tank.
Micrometer.
Sharp forceps.
Pipettors wiped down with RNase Zap and nuclease-free pipette tips.
Parafilm.
Injection dish: 35 mm diameter Petri dish, Spectra Mesh macroporous filter (Spectrum Labs) cut to fit in bottom of Petri dish, use modelling clay to hold the mesh in place on floor of Petri dish.
35 mm diameter Petri dishes.
3 ml transfer pipettes.
2.4 Mounting Xenopus laevis embryos for live imaging
Dow Corning® high vacuum grease.
Plain glass microscope slides.
Glass cover slips: 22 × 22 mm, thickness no. 1.
10 ml syringe.
Parafilm.
P200 pipette tips.
Hair-loop: form a small loop with a single strand of hair and place into the tip of a Pasteur pipette. Melt a small amount of paraffin and use this to hold the hair-loop in place.
3 ml transfer pipettes.
Dissecting microscope.
2.5 Live imaging embryos using confocal microscopy
Confocal microscope.
Mounted embryos (see section 3.4).
3. Methods
3.1 In vitro transcription of capped RNA for cytoskeletal probes
While it is possible to microinject fluorescently-labelled cytoskeletal proteins into the Xenopus embryo, we have had by far the most success with microinjection of capped RNA encoding fluorescently-tagged cytoskeletal probes (see Note 1 for a detailed description of these probes). Here we describe our method for the in vitro transcription of probes from a vector containing an SP6 or T7 promoter (we use pCS2+-based vectors).
3.1.1 Linearizing the DNA plasmid
Prepare a mini-prep of plasmid DNA containing the fluorescently-tagged cytoskeletal probe (cloned downstream of an SP6 or T7 promoter). Note that the cleaner your template DNA is, the greater the yield of the transcription reaction should be.
-
Linearize the plasmid using a restriction enzyme which cuts downstream of the cytoskeletal probe:
- 5 μg Plasmid DNA
- 10 μl appropriate 10X Restriction Enzyme Buffer
- 1 μl BSA solution
- 8 μl (80 units) Restriction Enzyme
- make up to total volume of 100 μl with ddH2O
Note that it is necessary to linearize the plasmid because RNA polymerases are extremely processive, so circular template DNA will lead to a mess of longer-than-expected transcription products.
-
Incubate reaction at 37°C for 1 hour, then add 2 μl (20 units) Restriction Enzyme and incubate for an additional hour at 37°C.
From this point on use RNase-free reagents (see Note 2)
Add 2.5 μl 20% SDS and 0.5 μl Proteinase K to digest reaction and incubate at 50°C for 30 mins.
- Purify the linearized DNA using a phenol/chloroform extraction as follows:
- Dilute DNA digest reaction with 100 μl nuclease-free H2O.
- Add 200 μl Phenol-chloroform-isoamyl alcohol mixture (25:24:1; pH 7.9), vortex vigorously, spin in a room temperature microcentrifuge at full speed for 1 min.
- Remove and retain aqueous (top) layer in a fresh tube.
- Back extract organic layer by adding 200 μl nuclease-free H2O, vortex vigorously, spin at full speed for 1 min.
- Remove aqueous layer and combine with previous aqueous layer.
Ethanol precipitate aqueous layers with 40 μl 5M NH4OAc and 800 μl 100% ethanol. Incubate at –20°C for 1 hour to overnight.
Pellet precipitated linearized DNA by spinning at full speed in a microcentrifuge at room temperature for 15 mins. Then remove ethanol, wash with 800 μl 70% ethanol, spin at full speed for 5 mins, remove ethanol and dry pellet in a speed vac or leave to air dry.
Resuspend the linearized DNA pellet in 10 μl nuclease-free H2O. Quantify a small volume of the DNA (0.5-1 μl) by gel electrophoresis or spectrophotometer. Note that it is important to confirm that your plasmid has been linearized into a single band by gel electrophoresis before proceeding.
3.1.2 Capped RNA synthesis
We use the Ambion mMESSAGE mMACHINE® kit (Ambion, Inc; Applied Biosystems) to synthesize capped RNA, as follows:
- Set up the transcription reaction at room temp, adding the components in the order listed below:
- Nuclease-free H2O (to make total volume of 20 μl)
- 1 μg linear DNA
- 2 μl 10X mMESSAGE mMACHINE® Reaction Buffer (vortex before use)
- 10 μl 2X mMESSAGE mMACHINE® NTP/CAP (vortex before use)
- 2 μl mMESSAGE mMACHINE® Enzyme Mix (SP6 or T7 depending on vector)
Incubate at 37°C for 2-3 hours.
To terminate reaction add 115 μl nuclease-free H2O and 15 μl NH4OAc mMESSAGE mMACHINE® Stop Solution.
- Phenol/Chloroform extract the capped RNA, as follows:
- Add 75 μl chloroform and 75 μl phenol (pH 6.6) to the RNA reaction, vortex vigorously and spin at full speed in a microcentrifuge for 1 min.
- Transfer aqueous phase to a fresh tube.
- Back extract the organic layer by adding 50 μl nuclease-free H2O, vortex vigorously, spin at full speed for 1 min and combine this aqueous layer with the previous.
- Extract the combined aqueous layers with 200 μl chloroform, vortex vigorously, spin at full speed for 1 min and transfer aqueous layer to a fresh tube.
Precipitate RNA by adding 200 μl isopropanol and incubating at −80°C for 1 hour to overnight.
Pellet RNA by spinning at maximum speed in a microcentrifuge at 4°C for 15 mins.
Remove isopropanol – take care as the RNA pellet is clean and therefore quite mobile at this point. Wash pellet with 200 μl 70% ethanol and then spin at full speed at 4°C for 5 mins.
Carefully remove all ethanol and allow pellet to air-dry.
Resuspend RNA pellet in 10 μl nuclease-free H2O (to prevent loss of pellet, add H2O and leave for about 10 mins before mixing by gentle pipetting).
Quantify by spectrophotometry and aliquot into small samples (we routinely make 1 μl aliquots at 1 μg/μl concentration). Store capped RNA at –80°C.
3.2. Xenopus laevis ovulation and in vitro fertilization
Here we briefly describe the methods used to induce ovulation in female frogs, collect eggs and in vitro fertilize. More detailed information on frog husbandry can be found in Peng and Kay [12] and suggestions on how to increase egg quality can be found in Note 3. A further important consideration is whether to use pigmented or albino frogs; this will depend on the type of experiment being carried out, as is described in Note 4.
3.2.1 Inducing ovulation
Prime female frogs 4-7 days before use by injection of 50 units of human chorionic gonadotrophin (HCG) into the dorsal lymph sac using a 1 ml syringe with a 27 G 1/2 needle.
18 hours before use, inject frogs with an additional 500-800 units of HCG.
Following injection place frogs in individual tanks of water and store at 17°C until use (see Note 3). About 12-18 hours after injection with HCG most female frogs will begin to ovulate independently and eggs will be clearly visible in the tank.
3.2.2 Isolating testes
For in vitro fertilization testes are harvested from male frogs as follows:
To anesthetize frog prepare a solution of 0.02% benzocaine (Ethyl 4-aminobenzoate) in water in a small tank with lid.
Place frog into benzocaine solution and leave for 10 mins.
Remove frog from tank and check for unconsciousness by firmly squeezing the leg (the frog should be completely limp and there should be no reaction to this).
Make incisions to expose the lower belly of frog by first cutting through the outer skin and then the inner muscle layers.
The testes are attached to the yellow fat bodies and are white in color and kidney shaped. Carefully remove the testes using surgical scissors and forceps.
Blot testes on a paper towel and rinse twice in 1X MMR and then place in testes prep media. Store in a 35 mm Petri dish at 4°C and use within 7 days.
Wrap the frog carcass in your gloves and freeze at –20°C before disposal.
3.2.3 In vitro fertilization
About 18 hours following injection with HCG, female frogs should be producing eggs (which will be visible in the tank) and will be ready to “squeeze”, whereby frogs are gently massaged to lay fresh eggs. If no eggs have been laid, the frog can be injected with a further 100-200 units of HCG.
Fill a 60 × 20 mm Petri dish with 1X MMR.
Pick up ovulating frog and hold in hands with head covered by the palm of hand and legs held apart by fingers. A damp paper towel can be placed over the frogs head, as covering her eyes will help keep the frog calm.
Hold frog over the Petri dish of 1X MMR and massage her lower back until she begins to deposit eggs into the dish.
Once there are a few hundred eggs in the dish (or the frog stops laying), pipette out most of the MMR until there is just a shallow covering of liquid over the eggs (about 1/10 of the volume of the dish).
Cut a small piece of testes using forceps and add to the eggs. Macerate the testes using forceps and mix with the eggs. Try to have the testes come in direct contact with as many of the eggs as possible.
Let eggs stand in testes solution for 30 secs and then gently fill the Petri dish with dH2O (so that 1X MMR is diluted approximately 10-fold). Leave at room temperature for 30 mins. Note that fertilized eggs will rotate within 30 mins so that the animal hemisphere is facing upwards.
To dejelly the fertilized eggs, add 2% cysteine (in 1X MMR, pH 7.8), transfer to a 50 ml falcon tube and gently swirl for 2-5 mins until the jelly coat is removed (visualized by the embryos settling against each other with no gaps).
Rinse the embryos 5 times with ~20 ml 1X MMR and 5 times with ~20 ml 0.1X MMR.
Remove any dead or unfertilized eggs and store the embryos in 0.1X MMR at 17°C. The embryos will undergo their first cleavage about 1.5 hours after fertilization.
Microinjection of Xenopus laevis embryos
We microinject Xenopus embryos at the 1-, 2-, 4- or 8-cell stage using a PLI-100 picoinjector (Harvard Apparatus; see Note 5). Here we describe our most routine microinjection scenario where both cells of the 2-cell stage embryo are microinjected with capped RNA encoding a cytoskeletal probe (see Section 3.1). For information on microinjecting at alternative stages or with alternative reagents (e.g. morpholino) see Note 6. When injecting RNA, we use measures to limit RNases by cleaning injection areas with RNaseZAP® (Ambion), using RNase-free reagents and by wearing gloves at all times.
3.3.1 Needle calibration
Needle calibration can be performed while waiting for fertilized embryos to begin their first cleavage, which occurs about 1.5 hours after fertilization (at 17°C).
Pull needles using 10 μl glass capillaries and a micropipette puller.
Using dissecting microscope and sharp forceps, cut off the end of the needle to produce a point approximately 5-10 μm in diameter.
Connect needle to microinjector and secure in micromanipulator (see Note 5).
Fill needle with approximately 1 μl nuclease-free water (we do this by pipetting a drop of water onto parafilm and front-loading the needle using the “fill” function, but the needle can also be filled by back-loading prior to connecting to the microinjector, see Note 5).
Place a drop of mineral oil onto the micrometer and place under dissecting microscope.
Carefully lower the needle into oil so that the point sits just above the micrometer scale.
Dispense one injection volume and measure the diameter of the hanging water drop against the micrometer scale.
Adjust microinjector controls (pressure/time) to give a drop size that corresponds to 5 nl (e.g. on a micrometer with 0.1 mm graduations, the diameter of a 5 nl drop is 2.5 graduations).
Dispense a few more drops to ensure that the 5 nl volume is reproducibly dispensed.
3.3.2 Microinjection of capped RNA
We inject a variety of concentrations of RNA depending on the probe being used. When using a new probe it is a good idea to inject several concentrations and ultimately use the lowest concentration that gives good expression levels. High concentrations of RNA are toxic to the embryo, and a good upper limit is generally a needle concentration of 1 mg/ml. Occasionally, for weakly expressed probes, we have used concentrations higher than this (up to 2 mg/ml), however this should be avoided if at all possible. In order to help hold the embryos in place during injection we place a plastic mesh (Spectra Mesh, Spectrum Labs) in the bottom of a Petri dish and secure it with modelling clay (see Section 2.3), these “injection dishes” can be reused many times.
As the time approaches 1.5 hours since fertilization, check embryos to see if they are beginning to undergo their first cleavage. If they are, proceed to (2) if not wait a few more minutes and check again. Once the embryos start to divide, it will take around 30 mins for the next division to take place, resulting in a 4-cell embryo.
Empty water out of needle and fill with the RNA to be injected (keep RNA on ice until ready to fill needle). It is possible to mix different RNAs together and inject them simultaneously.
Pour injection buffer (5% Ficoll in 0.1X MMR) into the injection dish (see above) and pipette 15-20 embryos into dish (embryos should be selected for good health and even cleavage plane).
Place dish under dissecting microscope and lower needle into injection buffer.
To inject embryos, slowly lower needle so that it touches the cortex of a cell and then further lower needle until it pierces cell membrane and dispense one volume into cell. Once the volume has been dispensed gently raise needle until the tip is out of the cell.
Repeat with the second cell of embryo and then with all other embryos by keeping the needle centered and moving the dish.
When all are injected, pipette the embryos into 0.1X MMR to rinse and then into fresh 0.1X MMR.
Return the embryos to 17°C incubator.
1-2 h after injection, check embryos and remove any dead or dying ones and refresh 0.1X MMR.
3.4 Mounting Xenopus laevis embryos for live imaging
Here we describe a simple method for mounting Xenopus embryos using a vacuum grease ring, although mounting embryos in specially made metal shims or in glass-bottomed culture dishes also works well. For imaging the cytoskeleton during cell division, we generally mount very early embryos (up to mid-blastula transition, approximately 7 hours post fertilization at 17°C), but for imaging the mitotic spindle we use embryos at a slightly later stage (stages 10-11, approximately 18-22 hours after fertilization at 17°C) when cells are smaller and spindles are easier to see.
Fill a 10 ml syringe with vacuum grease by squeezing grease into the back of the syringe.
Place a P200 pipette tip into the end of the syringe and secure with parafilm. Cut off the tip of the pipette to make a wider hole (see Figure 2).
Use the grease-filled syringe to pipe a grease circle onto a plain glass slide. Build up the walls of the circle by piping round 1-2 more times (see Figure 2).
Pipe 4 dots of grease around the circle (these will help support the cover-slip).
Use a transfer pipette to put 1 or 2 healthy, microinjected embryos into the grease circle.
Top up with more 0.1X MMR, so that the meniscus is level with the top of the grease circle.
The embryos will automatically sit with their animal hemisphere upright and so need little manipulation. If necessary, a hair-loop (see Section 2.4) can be used to gently push the embryos towards the center of the circle.
Place a glass coverslip over the embryos and press gently onto the grease ring. The coverslip should make contact with the embryos so that they are pressed slightly against the glass and are held securely in place. Remove any excess liquid.
Figure 2. Mounting Xenopus laevis embryos using vacuum grease.
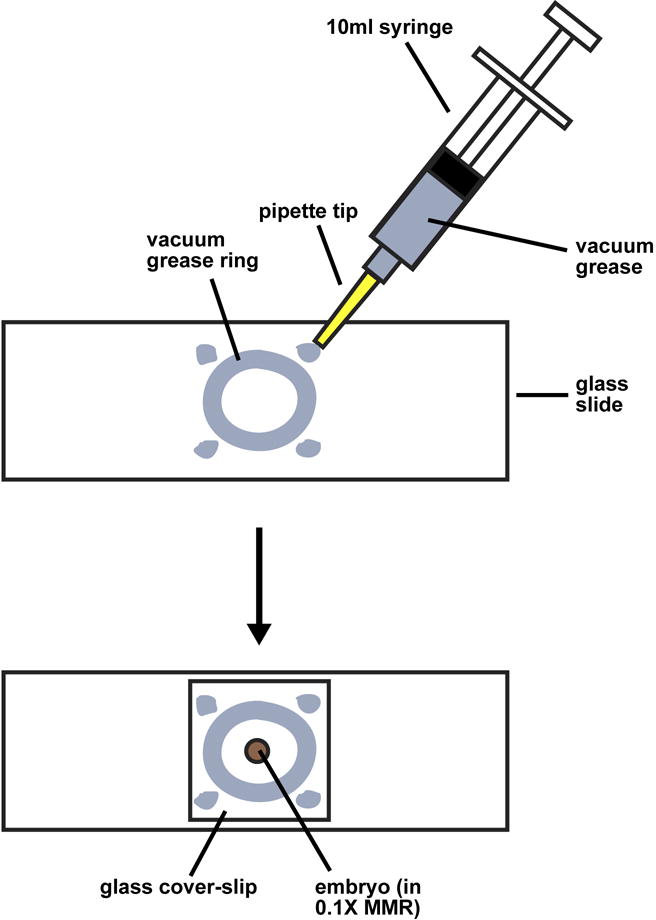
We mount embryos for live imaging by making a vacuum grease ring on glass slides. To “pipe” the grease we use a syringe with a pipette tip held at the end with parafilm. A ring of grease is then piped onto a glass slide and 4 supporting spots of grease are added as shown. The embryo, in 0.1X MMR, is then pipetted into the grease ring and a glass coverslip is placed on top. The coverslip should be gently pushed down so that it is just touching the embryo.
3.5 Live imaging embryos using confocal microscopy
Since different confocal microscope systems will vary, here we provide general pointers in order to help the reader get started with live imaging.
Place slide containing the embryos onto microscope stage and move appropriate objective into place – we generally use 25x or 63x oil objectives for very early embryos (16-cell stage up to mid-blastula transition) and a 63x oil objective for later stage embryos (stage 10 upwards).
Using bright-field or epifluorescent illumination, carefully adjust the focus until you see the most superficial cells of the embryo. It is important to be gentle when focusing, as it is possible to squash the embryos by pushing the coverslip too close to the slide.
Once the embryo is in focus, turn off the bright-field/epifluorescence and switch to confocal scanning. For live imaging it is important to set the laser power as low as possible, to prevent photobleaching and possible phototoxicity. We usually scan using a format of 512 × 512 pixels for live imaging, as this provides a good compromise between scanning speed and image resolution.
Once a good area of the embryo has been selected, and zoomed if necessary, set the gain and offset for each channel. The gain and offset should be adjusted to levels high enough to see all that is required but where there are few pixels at their maximum intensity and where a minimum of background noise is seen. These levels can be adjusted by eye or by using a look-up-table (LUT) such as the glow-over/glow-under LUT found on Leica microscopes.
For two-color live imaging it is usually best to set the confocal to scan the two channels simultaneously. Simultaneous scanning will save time, allowing faster time points and is especially important if you are imaging a highly dynamic process where changes may occur between sequential scans of the two channels. However, when scanning two channels simultaneously it is important to set the levels of gain, offset and laser power for each channel very carefully to ensure that bleed-through from one channel to another does not occur. If bleed-through still occurs, it may be necessary sacrifice some scan time and collect sequentially.
If you wish to collect a z-series over time, you must next set your upper and lower z limits and the number of sections you wish to collect. Note that if you are imaging a highly dynamic process you should limit the number of z-sections taken – when we image mitotic spindles we generally find that imaging a single plane works well and allows us to collect very frequent time points; in contrast imaging the cytokinetic furrow requires multiple z-sections (usually 13 z-sections, approximately 1 μm apart).
Next set the appropriate time interval and number of time points to be collected. Again this will vary according to how dynamic the process being imaged is. For mitotic spindles we find a time interval of 6 seconds collected 200 times works well, while for imaging cytokinesis we use a time interval of 20 seconds, and collect this 50-100 times.
The appearance of images can be greatly improved by using a scan averaging algorithm such as a Kalman accumulation. This will slow down scanning time, so should be limited to the lowest number of averages needed to improve the image quality.
Once all the settings have been finalized, start the time series. It is advisable to stay by the microscope and watch the image during scanning so that manual adjustments to the focus can be made if required.
Footnotes
As noted above, most imaging of the actin and microtubule cytoskeletons has been done using fluorescent actin or tubulin protein. This approach has many virtues, but it also has certain drawbacks. It is becoming increasingly clear that fluorescent actin does not label all pools of F-actin equally (for discussion see Burkel et al., 2007 [13]). Further, when used at high levels, both fluorescent actin and fluorescent tubulin tend to suppress the assembly of their respective polymers. An alternative approach is to use fluorescent proteins fused to isolated domains of proteins that bind to F-actin or microtubules. This typically does not prevent assembly of the polymer in question. In addition, the background is typically lower, because unlike fluorescent tubulin or actin, which represent both the assembled and unassembled forms of their respective polymers, fluorescent polymer-binding proteins accumulate specifically on the assembled form. However, herein lies their major disadvantage: too much of such probes can inhibit disassembly. Nonetheless, we have had considerable success with eGFP, mRFP or mCherry fused to the calponin homology (F-actin binding) domain of utrophin [9, 13], which permits very bright labelling of F-actin without obvious stabilization of actin filaments (see Figure 1). We have also had success with using 3XGFP (three copies of eGFP fused together) fused to the microtubule-binding domain of ensconsin, a probe originally developed by the Bulinski lab [14]. mRNA encoding this probe, however, must be carefully titered for microinjection to find a range that labels microtubules well but does not cause microtubule stabilization. Using a similar rationale, we and others have developed fluorescent probes based on effector protein domains that bind to the active forms of the Rho GTPases [15–17]. The Rho GTPases are key regulators of the cytoskeleton but one of the major problems with using the simple fluorescent protein fusions with Rho proteins is the failure of these fusions to target properly [18]. Instead, by using effector domain based probes, it is possible to visualize Rho or Cdc42 activation in Xenopus oocytes, eggs, and embryos [17, 19–21] (see Figure 1). While these probes avoid the targeting problems seen with simple Rho GTPase fusions they must still be tittered carefully to avoid inhibiting the proteins they report on.
It is vital when working with RNA to keep your samples free of any RNases; there are a number of precautions that must be taken to prevent RNases from contaminating your work. First, always wear gloves when handling samples as our fingers are a rich source of RNases; it is also good practice to change your gloves regularly during the experiment. Second, use pipette tips and eppendorf tubes that are guaranteed RNase-free. We keep all plasticware for use with RNA separate from the rest of the lab to limit contamination and we also have a set of pipettors that are exclusively used for RNA work. Third, keep all work surfaces clean; we use the RNase decontamination solution, RNaseZap® (Ambion), to wipe down lab benches and to clean pipettors. Fourth, use RNase-free solutions: these can be made either by treating with diethylpyrocarbonate (DEPC) to inactivate RNases or by purchasing guaranteed RNase-free solutions from companies such as Ambion. We tend to favour the latter, as we’ve found these reagents to be highly reliable. However, if you wish to DEPC treat solutions, add 0.1% DEPC to the solution in a fume-hood, leave overnight at room temperature and then autoclave for 15 mins; Tris solutions will reduce the activity of DEPC, so either more DEPC (e.g. 1%) should be added or solutions should be made up with molecular biology grade powdered reagents in DEPC-treated water.
Acquiring high quality eggs is crucial for these live imaging studies, and there are a number of considerations that will help to obtain them. First, we usually prime two female frogs for each experiment to maximize our chances of having an ovulating frog that produces good eggs. Second, frogs should be cycled so that they have at least a four-month break between ovulations; an even longer recovery period may help improve egg quality. Third, we have found that incubating the frogs at 17°C after their second injection of HCG increases the chances of ovulation and may improve the quality of eggs laid.
Wild-type Xenopus laevis lay eggs with a pigmented animal hemisphere; while the eggs laid by albino frogs lack this pigment. We have found albino embryos to be far superior for imaging mitotic spindles, as the pigment in wild-type embryos obscures confocal visualization of the fluorescent spindles. However, in other experiments the presence of pigment can be helpful as it is much easier to assess the health of these embryos and they are simpler to microinject.
There are many choices of microinjectors available, ranging from the basic through to the sophisticated, however the major requirement is that they are able to accurately dispense specific volumes. Some microinjectors possess a “fill” function, which allows the user to frontload the needle; this can be useful as very small volumes can be drawn into the needle – an advantage when using a precious RNA prep. The second major piece of equipment required for microinjection is a micromanipulator, which holds the glass capillary needle and allows the user to accurately move the needle up and down to inject the embryo. The micromanipulator must hold the needle firmly at an angle of approximately 45°; a micromanipulator that is fixed to a microscope and can’t be angled is not suitable for Xenopus injections [22].
As well as being injected at the 2-cell stage with capped RNA, Xenopus embryos can also be microinjected at alternative stages and with alternative reagents. 1-cell and 4-cell embryos can be microinjected with an injection volume of 5 nl just as described for 2-cell stage embryos. Individual cells of 8-cell embryos can also be easily microinjected but a slightly finer needle that dispenses an injection volume of 2.5 nl should be used. In order to track injected cells (and their progeny) as the embryo develops, a cell dye such as fluorescently-labelled dextran can be included in the injection. It is possible to microinject Xenopus embryos with fluorescently-labelled proteins using the methods described here, although protein preparations should be centrifuged (we use an air-driven ultracentrifuge for this) prior to injection to remove debris that would clog the microinjection needle. Anti-sense morpholino oligos are an effective means of gene knockdown in Xenopus embryos [23] and can be microinjected using the methods described here, except that the morpholino should be heated prior to injection for 5 mins at 65°C and kept at 37°C until ready to inject. A good starting concentration for morpholino injection is a needle concentration of 1 mM (in nuclease-free water), however we have used concentrations of up to 5 mM and down to 0.25 mM.
References
- 1.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 2.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 3.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 4.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 6.Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 7.Megason SG, Fraser SE. Digitizing life at the level of the cell: high-performance laser-scanning microscopy and image analysis for in toto imaging of development. Mech Dev. 2003;120:1407–1420. doi: 10.1016/j.mod.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Kieserman EK, Glotzer M, Wallingford JB. Developmental regulation of central spindle assembly and cytokinesis during vertebrate embryogenesis. Curr Biol. 2008;18:116–123. doi: 10.1016/j.cub.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 9.Woolner S, O’Brien LL, Wiese C, Bement WM. Myosin-10 and actin filaments are essential for mitotic spindle function. J Cell Biol. 2008;182:77–88. doi: 10.1083/jcb.200804062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mikhailov A, Gundersen GG. Relationship between microtubule dynamics and lamellipodium formation revealed by direct imaging of microtubules in cells treated with nocodazole or taxol. Cell Motil Cytoskeleton. 1998;41:325–340. doi: 10.1002/(SICI)1097-0169(1998)41:4<325::AID-CM5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 11.Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng HB, Kay BK. Xenopus laevis: Practical uses in cell and molecular biology. Vol. 36. Academic Press; 1991. [PubMed] [Google Scholar]
- 13.Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faire K, Waterman-Storer CM, Gruber D, Masson D, Salmon ED, Bulinski JC. E-MAP-115 (ensconsin) associates dynamically with microtubules in vivo and is not a physiological modulator of microtubule dynamics. J Cell Sci. 1999;112(Pt 23):4243–4255. doi: 10.1242/jcs.112.23.4243. [DOI] [PubMed] [Google Scholar]
- 15.Pertz O, Hahn KM. Designing biosensors for Rho family proteins–deciphering the dynamics of Rho family GTPase activation in living cells. J Cell Sci. 2004;117:1313–1318. doi: 10.1242/jcs.01117. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Li Z, Sacks DB. E-cadherin-mediated cell-cell attachment activates Cdc42. J Biol Chem. 2000;275:36999–37005. doi: 10.1074/jbc.M003430200. [DOI] [PubMed] [Google Scholar]
- 17.Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168:429–439. doi: 10.1083/jcb.200411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localization in cells and tissues. Exp Cell Res. 2004;295:300–314. doi: 10.1016/j.yexcr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Sokac AM, Co C, Taunton J, Bement W. Cdc42-dependent actin polymerization during compensatory endocytosis in Xenopus eggs. Nat Cell Biol. 2003;5:727–732. doi: 10.1038/ncb1025. [DOI] [PubMed] [Google Scholar]
- 20.Ma C, Benink HA, Cheng D, Montplaisir V, Wang L, Xi Y, Zheng PP, Bement WM, Liu XJ. Cdc42 activation couples spindle positioning to first polar body formation in oocyte maturation. Curr Biol. 2006;16:214–220. doi: 10.1016/j.cub.2005.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bement WM, Benink HA, von Dassow G. A microtubule-dependent zone of active RhoA during cleavage plane specification. J Cell Biol. 2005;170:91–101. doi: 10.1083/jcb.200501131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sive HL, Grainger RM, Harland RM. Early Development of Xenopus laevis: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 1998. [Google Scholar]
- 23.Heasman J. Morpholino oligos: making sense of antisense? Dev Biol. 2002;243:209–214. doi: 10.1006/dbio.2001.0565. [DOI] [PubMed] [Google Scholar]