

Abstract
H19 is an imprinted long non-coding RNA abundantly expressed in embryonic liver and repressed after birth. Here we show that H19 serves as a lipid sensor by synergizing with RNA-binding protein PTBP1 to modulate hepatic metabolic homeostasis. H19RNA interacts with PTBP1 to facilitate its association with SREBP1c mRNA and protein, leading to increased stability and nuclear transcriptional activity. H19 and PTBP1 are upregulated by fatty acids in hepatocytes and in diet-induced fatty liver, which further augments lipid accumulation. Ectopic expression of H19 induces steatosis and pushes the liver into a “pseudo fed” state in response to fasting by promoting SREBP1c protein cleavage and nuclear translocation. Deletion of H19 or knockdown of PTBP1 abolishes high-fat and high-sucrose (HFHS) diet-induced steatosis. Our study unveils a H19/PTBP1/SREBP1 feedforward amplifying signaling pathway to exacerbate the development of fatty liver.
Keywords: Fatty liver, lipid metabolism, non-coding RNA, RNA binding protein
Nonalcoholic fatty liver disease (NAFLD), one of the most common causes of chronic liver disease, encompasses a histological spectrum of liver diseases ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which can progress to cirrhosis and liver cancer (1, 2). Steatosis is defined as hepatic accumulation of triglyceride (TG) above 5% of liver weight and represents the hallmark of NAFLD (3). Beyond NASH, steatosis may progress to cirrhosis and ultimately hepatocellular carcinoma (4). Increased TG deposition in the liver results from an imbalance of input, output, oxidation, and de novo synthesis of hepatic free fatty acids (FFA) (5) (6), which is controlled by genes in pathways of lipid uptake, secretion, oxidation, and synthesis. The induction of lipogenesis is mainly controlled by sterol regulatory element-binding protein 1c (SREBP1c), which is an endoplasmic reticulum (ER) membrane-bound protein that functions as a transcription factor in the liver (7). In sterol-depleted cells, the membrane-bound precursor is cleaved to generate a soluble NH2-terminal fragment that translocates to the nucleus to activate the hepatic lipogenic program. Hyper-activation of SREBP1 causes TG accumulation and leads to hepatic steatosis (8).
Long non-coding RNAs (lncRNAs) are defined as transcripts that are above 200 nucleotides in length and without proof of protein coding potential. In the past decade, lncRNAs have come to the forefront of research interest in many fields. Recent studies have shown that lncRNAs are involved in the regulation of various cellular processes including lipid metabolism, microbial susceptibility, nuclear reprogramming, neural differentiation, and epigenetics (9-11).
Paternally imprinted and maternally expressed H19 is one of the first identified lncRNAs. The physiological and pathological effect of H19 on mRNA stability, tumor metastasis, and osteoblast differentiation have been reported (12, 13). Of particular interest, expression of H19RNA was high in fetal liver but diminished significantly in adult liver. Intriguingly, such expression is upregulated in human chronic liver diseases (14), implicating an important regulatory role in hepatic function and metabolism.
Polypyrimidine tract-binding protein 1 (PTBP1, also known as PTB, or hnRNP I) is an RNA binding protein that regulates mRNA stability and pre-mRNA splicing (15). It was also implicated in directing cell reprogramming (16) and brain tumor growth (17). The finding that PTBP1 controls genes in insulin biosynthesis (18) suggests a potential function of PTBP1 in metabolic diseases.
In this study, we unravel a H19/PTBP1 metabolic circuit that dictates SREBP1c expression and function in steatosis. When hepatic lipid homeostasis is disrupted, fatty acids induce the expression of H19RNA and PTBP1 and enhance their interaction. This facilitates PTBP1 protein interaction with Srebp1c mRNA and protein, further stimulating the lipogenic program. Consequently, overexpression of H19 results in hepatic metabolic reprograming and exacerbated diet-induced fatty liver. Our study provides a new lncRNA-mediated regulatory module to control steatosis.
Materials and Methods
Mouse models include maternal H19-deleted mice (Mat H19-/-) with paternal H19-deleted mice (Pat H19-/-) as wild type controls, H19 overexpressed and Ptbp1 knockdown mice. Basic procedures to analyze animal metabolic phenotypes and serum parameters (19), RNA pull-down, and RIP were described previously (20). Primary hepatocytes isolation and culture, in vitro virus transduction and plasmid transfection, qPCR, Western blot (WB), liver histology, hepatic lipid extraction and blood chemistry, immunocytochemistry, and confocal images were described previously (21-25). Primer sequences were provided in Supporting Table 1. Other potential H19 binding proteins from MassSpec analysis were provided in Supporting Table 2. The coded human liver specimens were obtained through the Liver Tissue Cell Distribution System (LTCDS) (Minneapolis, Minnesota), which was funded by NIH Contract # HSN276201200017C (see Supporting Table 3). Because we did not ascertain individual identities associated with the samples, the Institutional Review Board for Human Research at the University of Connecticut determined that the project was not research involving human subjects.
Please see Supplemental Information for detailed experimental models and conditions.
Statistical Analysis
Data are shown as the mean ± standard error of the mean (SEM) and are representative of at least three independent experiments. Statistical analysis was carried out using Student's t test between two groups and one way ANOVA among multiple groups. P<0.05 was considered statistically significant. The sample variation within each group was estimated to ensure that the variance is similar. All experiments were randomized by simple randomization based on a single sequence of random assignments.
Results
H19 interacts with PTBP1 to enhance PTBP1-binding to SREBP1 mRNA and protein
lncRNAs often regulate target genes through interactions with RNA binding proteins. To understand the metabolic function of H19, the first step was to identify H19-interacting proteins. We cloned and synthesized biotin-labeled sense (S) and antisense (AS) full length (F) and 5′-end (5′) of H19RNA (Fig. 1A left). We conducted RNA-pull down assay by incubating biotin-labeled full length H19 S or AS (negative control) RNA with Hepa1 cell extracts. A band in the sense lane (red arrow) was excised, digested, and subjected to mass spectrometry (LC-MS) to identify proteins that bound to the H19RNA (Fig. 1A). (Fig. 1A right; Supporting Fig. S2A). The band likely contained multiple proteins of similar sizes. Indeed, a total of 48 candidates were identified (Supporting Table 2). From the 15 proteins that received the highest scores (Supporting Fig. S2A bottom), six were selected for further validation based on the availability of antibodies. PTBP1 was the only protein validated as a binding partner of H19, and the interaction of PTBP1 with the 5′end of H19 was stronger than full length H19, as demonstrated by RNA pull down (Fig. 1B left-top). In contrast, the sphingosine-1-phosphate lyase 1 (SGPL1) antibody failed to detect SGPL1 binding to PTBP1, which was used as a negative control. The results with other five proteins are presented in Supporting Fig. S2B.
Figure 1. H19 interacts with PTBP1 to enhance SREPB1 nuclear translocation and H19/PTBP1 increases SREBP1 stability and transactivation.
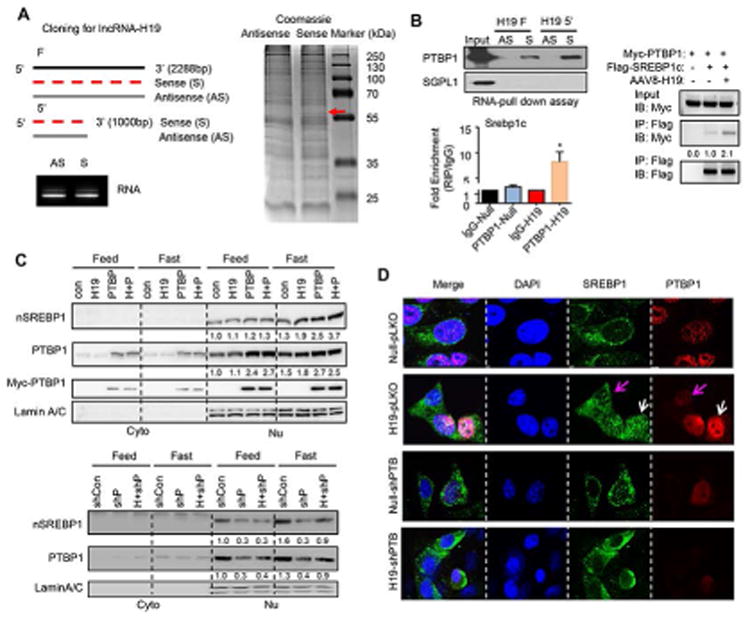
(A) Left-top: Diagram showing cloning strategy of full length (F) H19 (2288 bp) or 5′-end of H19 (5′) (1000 bp). Left-bottom: Synthesis of biotin-labeled antisense (AS) and sense (S) H19RNA by in vitro transcription. Right: RNA-pull down assay with biotin-labeled sense or antisense (negative control) full length H19RNA in Hepa1 cells. After pull-down, proteins were subjected to SDS-PAGE and stained by coomassie blue. Red arrow indicates the unique band observed in the sense lane. The band was cut, digested, and subjected to Mass spectrometry.
(B) Left-top: RNA-pull down with Western blot to determine the specific interaction of sense H19RNA with the PTBP1 protein, but not with the SGPL1 protein (negative control). Left-bottom: RNA IP to determine the interaction between Srebp1c mRNA and PTBP1 protein using anti-PTBP1 or IgG (negative control) in Hepa1 cells in the presence or absence of H19RNA overexpression. Right: Co-IP followed by Western blot to determine the effect of H19 on PTBP1 and SREBP1c protein interaction. Hepa1 cells were transduced with AAV-Null or AAV-H19 for 24 hr followed by transfection with Myc-PTBP1 or Flag-SREBP1c plasmid for 24 hr. Cells were then fasted in serum free medium for 24 hr. Total proteins were collected for Co-IP with the anti-Flag antibody, and WB analysis was performed with anti-Myc-tag or anti-Flag antibody.
(C) Top: Western blot analysis to determine the effects of H19 and PTBP1 on nSREBP1 protein expression. Hepa1 cells were transduced with AAV8-Null or AAV8-H19 virus for 24hr, and then transfected with Myc-PTBP1 for 24hr. For fasting conditions, the cells were treated for 24hr with serum free medium, followed by fractionation of cytoplasm and nucleus proteins. Anti-SREBP1, anti-PTBP1 and anti-Myc antibodies were used to detect SREBP1 and PTBP1 proteins, respectively. Bottom: Western blot analysis to determine the effect of knockdown of PTBP1 on nSREBP1 protein expression. Hepa1 cells were transduced with Lenti-Con or Lenti-shPTBP1 virus for 72 hr, followed by AAV8-Null or AAV8-H19 for 72 hr. Anti-PTBP1 and anti-SREBP1 antibodies were used to detect nSREBP1 and PRBP1 proteins, respectively. shCon, shcontrol; shP, shPTBP1; H+shP, AAV8-H19 + shPTBP1.
(D) Confocal images of immunocytochemistry analysis of SREBP1 and PTBP1 protein subcellular localization. Hepa1 cells were transduced with Lenti-pLKO or Lenti-shPTBP1 virus and selected with 1 μg puromycin for 7 days to generate stable cell lines. The cells were then transduced with AAV8-Null or AAV8-H19 for 48hr and fasted for 24hr. SREBP1 and PTBP1 proteins were detected using respective antibodies. Pink arrow: cells with low PTBP1 expression; white arrow: cells with high PTBP1 expression.
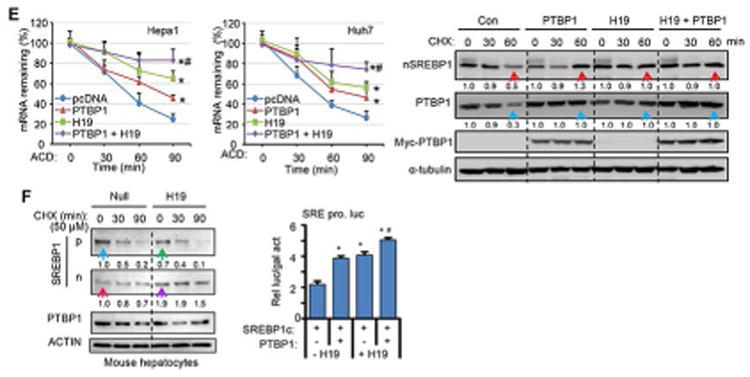
(E) Top: Hepa1 (left) and Huh7 (right) cells transfected with pcDNA3 or H19 with or without PTBP1 were treated with actinomycin D (5 μg/ml). RNA was extracted at different time points (0, 30, 60 mins) and Srebp1c mRNA was analyzed by qPCR and normalized to β-actin. Data are shown in mean ± SEM from triplicate assays. *p<0.01 vs pcDNA; #p<0.001 vs H19 or PTBP1 alone. Bottom: Western blot analysis of protein expression in Hepa1 cells treated with CHX. H19 and PTBP1 were expressed alone or in combination using the same conditions in Fig. 1E. Red arrow: nSREBP1 increased in PTBP1 or H19 vs Con. Blue arrow: PTBP1 increased in PTBP1 or H19 vs Con.
(F) Top: Western blot analysis of SREBP1 protein expression in primary hepatocytes treated with CHX. Primary hepatocytes were isolated from WT mice, cultured overnight, and transduced with AAV8-Null or AAV8-H19 the next day for 24 hr. The cells were then fasted for 24 hr before the treatment with CHX. Bottom: Transient transfection assay. The SRE promoter luciferase reporter was transfected with SREBP1c expression plasmid in the presence or absence of PTBP1 or H19 co-expression in Hepa1 cells. The luc activity was normalized to β-gal activity. *p<0.01 vs lane 1; #p<0.01 lane 2 or lane 3.
See also Fig. S2-S3.
Because we are interested in the function of H19 in crosstalk with PTBP1 in lipid metabolism, we used online bioinformatics analysis tools (RBPmap, StarBase v2.0) to predict PTBP1 targets in lipid metabolic pathways. Srebp1c mRNA was predicted to contain multiple PTBP1 binding sites within its CDS and 3′UTR (Supporting Fig. S3A-3B). RNA IP (RIP) assay was carried out using an anti-PTBP1 antibody or mouse lgG with Hepa1 cell lysates, which demonstrated that Srebp1c mRNA, but not farnesoid X receptor (Fxr), fatty acid synthase (Fasn), peroxisome proliferator-activated receptor gamma (Pparγ), or acetyl-CoA carboxylase (Acc2) mRNAs, was moderately enriched by PTBP1 vs control lgG antibody (Supporting Fig. S3C). However, we observed a significant interaction of PTBP1 with Srebp1c mRNA in the presence of H19 (Fig. 1B left-bottom). Interestingly, Co-IP and WB also revealed a physical interaction between PTBP1 and SREBP1 proteins, and the interaction was enhanced by H19 (Fig. 1B right; Supporting Fig. S3D).
H19 induces SREBP1 protein nuclear translocation in a PTBP1-dependent fashion under fasting conditions
To better understand the regulation of SREBP1 protein by H19 and PTBP1, we overexpressed H19 and PTBP1 alone or in combination in Hepa1 cells and collected both cytosolic and nuclear proteins under either feeding or fasting (24 hr) conditions (Fig. 1C up). H19 or PTBP1 slightly increased the endogenous nuclear SREBP1 (nSREBP1) protein under feeding conditions. Upon fasting, the induction of SREBP1 protein by H19 or PTBP1 was detectable to a much greater extent, which was further augmented by co-expression of both genes (Fig. 1C top; Supporting Fig. S3E). We also examined the effect of PTBP1 knockdown on SREBP1 expression (Fig. 1C bottom).
ShPTBP1 decreased nSREBP1 protein in both fed and fasted cells, and this effect was partially rescued by H19 in fasted cells. Furthermore, changes in sub-cellular localization of SREBP1 protein were evaluated under fasting conditions using confocal microscopy (Fig. 1D). In Null cells (row 1), the SREBP1 protein was observed in cytoplasm and around the nuclear membrane. In H19 cells (row 2), more nuclear SREBP1 proteins were detected in cells that expressed higher levels of PTBP1 protein (white vs purple). Knockdown of PTBP1 in the absence of H19 did not alter SREBP1 subcellular distribution (row 3), although it impeded SREBP1 nuclear translocation mediated by H19 (row 4).
H19 and PTBP1 increase SREBP1 protein cleavage, stability, and transcriptional activity
In Hepa1 and Huh7 cells treated with the transcriptional inhibitor actinomycin D (ACD) to block RNA synthesis, overexpression of H19 and PTBP1 decreased Srebp1c mRNA degradation (Fig. 1E left). When Hepa1 cells were treated with cycloheximide (CHX) to inhibit protein synthesis, an increased SREBP1 protein stability by PTBP1 and H19 was observed (Fig. 1E right). Interestingly, H19 also increased PTBP1 protein half-life. We next treated primary hepatocytes with CHX to examine the effect of H19 on SREBP1 protein expression (Fig. 1F left). It was interesting to note that the basal precursor SREBP1 (pSREBP1) protein was decreased by H19 (p, green arrow vs blue arrow) concomitantly with increased nSREBP1 protein (n, purple arrow vs red arrow), suggesting increased protein cleavage. In addition, using a promoter reporter containing a SRE response element, we observed enhanced SREBP1c transcriptional activity by overexpression of PTBP1 and H19 (Fig. 1F right).
H19 enhances fatty acid-induced lipid accumulation in hepatocytes
The important role of H19 in PTBP1-mediated SREBP1c expression promoted us to determine the impact of H19RNA on neutral lipid accumulation in vitro in hepatocytes. Transient overexpression of H19 using AAV8-H19 virus markedly augmented oleic acid (OA)-induced lipid accumulation in Hepa1 and Huh7 cells (Fig. 2A) as well as in mouse primary hepatocytes (Fig. 2B left). Intriguingly, H19RNA was highly induced by FAs, including OA, palmitic acid (PA), butyric acid (BA), and stearic acid (SA) (Fig. 2B right; Supporting Fig. S4A). In addition, H19 enhanced lipid staining in Hepa1 and Huh7 cells treated with PA and SA (Supporting Fig. S4B). The results suggest that H19 may function as a fatty acid sensor to modulate lipogenesis.
Figure 2. H19 increases fatty acid-mediated lipid accumulation in hepatocytes.

(A) Confocal images of Nile red staining and quantification of neutral lipids in Hepa1 and Huh7 cells. Cells were transduced with AAV8-Null or AAV8-H19 in the presence or absence of oleic acid (OA). *P<0.05 vs BSA Null; #P<0.05 vs OA Null.
(B) Left: Oil red O staining of neutral lipid in mouse primary hepatocytes. Right: qPCR of H19RNA in Hepa1 and primary hepatocytes treated with various fatty acids.
OA, oleic acid 200 μM; POA, palmitoleic acid 200 μM; LLA, linoleic acid 200 μM; ADA, arachidonic acid 10 μM; PA, palmitic acid 200 μM; LA, lauric acid 100 μM; BA, butyric acid 5 mM; MA, myristic acid 100 μM; SA, stearic acid 200 μM. Cells were treated for 16 hr. *P<0.05 vs control.
(C) Western blot analysis of protein expression in Hepa1 cells treated with OA in the presence or absence of H19RNA. Cells were transduced with AAV8-Null or AAV8-H19 virus for 72 hr and then treated with BSA or oleic acid (OA, 200 μM) for 16 hr. Whole cell lysates were collected and examined with the indicated antibodies.
(D) Confocal images of Nile red staining and quantifications of neutral lipids in Hepa1 (histogram). Cells were transduced with AAV8-Null or AAV8-H19 for 24 hr and then transfected with Myc-PTBP1 plasmid overnight, followed by the treatment of BSA or oleic acid (OA, 200 μM) for 16 hr. *P<0.05 vs BSA Con; ¥P<0.05 vs OA Con; #P<0.05 vs OA H19 or PTBP1.
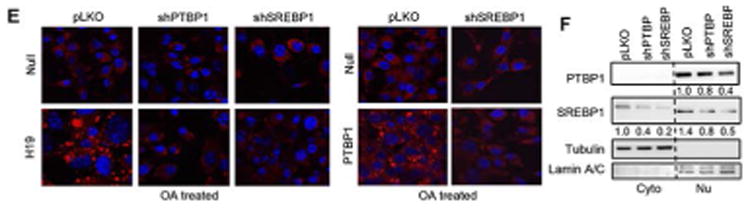
(E) Confocal images of Nile red staining. Left: Hepa1 cells stably overexpressed pLKO, pLKO-shPTBP1 or pLKO-shSREBP1 were transduced with AAV8-Null or AAV8-H19 (left) or Lenti-Null or Lenti-PTBP1 virus (right) for 48 hr and fasted for another 24 hr, followed by the treatment of BSA or oleic acid (OA, 200 μM) for 16 hr.
(F) Western blot analysis of PTBP1 and SREBP1 proteins in cytosolic and nuclear fractions in Hepa1 cells stably overexpressed pLKO, pLKO-shPTBP1 or pLKO-shSREBP1.
See also Fig. S5.
It was noticed that transient expression of H19RNA alone had less effect on cellular lipid levels (Fig. 2A-2B), although it induced SREBP1 protein (Fig. 2C; Supporting Fig. S4C). SREBP1c target gene FASN, which is a critical enzyme to catalyze fatty acid synthesis, was not correspondingly induced by H19 alone; however its expression was stimulated by OA and further enhanced by H19 in OA treated cells. Thus, the modest impact of H19 on lipid accumulation could be attributed to the delayed activation of FASN.
Similar to H19, transient overexpression of PTBP1 promoted OA-mediated lipid synthesis and enhanced the lipogenic effect of H19 (Fig. 2D), which could be attributed to the enhanced binding of H19 to PTBP1 by OA treatment (Supporting Fig. S4D). Knockdown of PTBP1 or SREBP1 significantly diminished H19-induced lipid accumulation (Fig. 2E left), and knockdown of SREBP1 also impeded PTBP1-induced lipid accumulation (Fig. 2E right). Interestingly, shSREBP1 also diminished PTBP1 protein (Fig. 2F), suggesting a potential feedforward regulation of PTBP1 expression by SREBP1. Therefore, the interaction of PTBP1 and H19 facilitates SREBP1c-mediated de novo lipogenesis.
Prolonged H19 expression facilitates the development of hepatic Steatosis
We next investigated the in vivo effect of H19 by using two experimental approaches: First to overexpress H19 alone for a prolonged period of time (presented in Fig. 3) and second to overexpress H19 for a short time in the presence of a HFHS diet (presented in Fig. 4).
Figure 3. Hepatic overexpression of H19 induces steatosis.
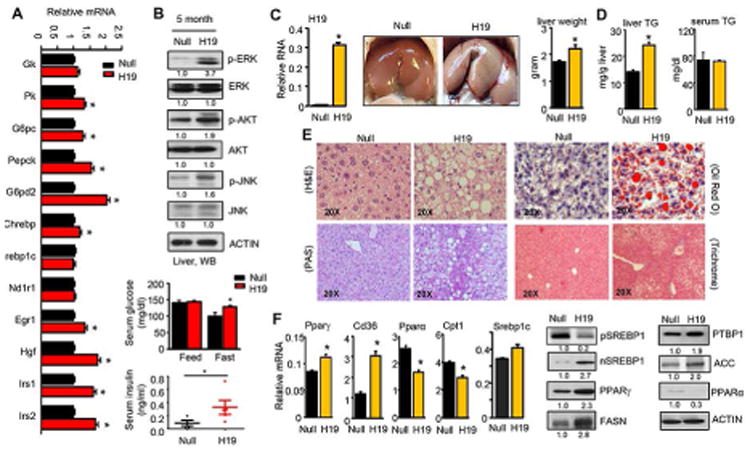
(A) qPCR of genes involved in lipid and glucose metabolism. Mice were transduced with AAV8-null or AAV8-H19 for 5 months (n=5/group). Data are shown as mean ± SEM. *P<0.05 vs Null control.
(B) Top: Western blot analysis of hepatic protein expression. Bottom: Blood glucose levels in mice under feeding or fasting (overnight) conditions and insulin levels under fasting conditions. The same mice as in A.
(C) Left: qPCR of hepatic H19RNA. Middle: Gross liver appearance. Right: Average liver weight. Mice were transduced with AAV8-Null or AAV8-H19 for 10 months (n=5/group). Data are shown as mean ± SEM, *P<0.05 H19 vs Null.
(D) Hepatic triglyceride (TG) content and serum TG levels. The same mice as in C.
(E) Representative images of H&E, Oil red O, PAS and Trichrome Masson staining of liver sections. The same mice as in C.
(F) qPCR and Western blot analysis of hepatic expression of genes involved in lipid metabolism. The same mice as in C. Each band represents pooled sample (equal amount of protein) from 5 individual mice.
See also Fig. S5A-S5D.
Figure 4. HFHS diet-induced steatosis is diminished by H19- and PTBP1-deficiency.
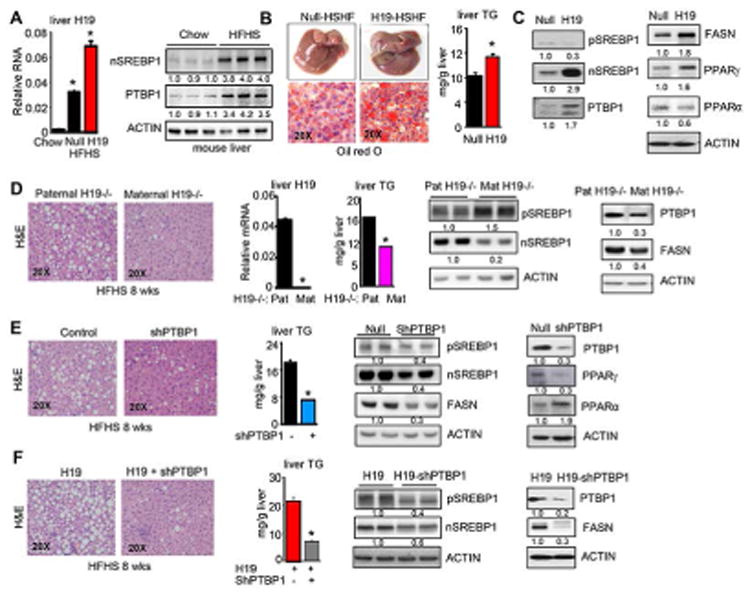
(A) Left: qPCR of hepatic H19RNA in mouse livers. Right: WB analysis of nSREBP1 and PTBP1 protein in mouse livers.
(B) Left: Gross liver appearance and images of oil red O staining of liver sections. Right: Liver TG content. Mice were transduced with AAV8-Null or AAV8-H19 for 1 week, followed by HFHS feeding for 5 weeks. Data are shown as mean ± SEM (n=5/group). *P<0.05 H19 vs Null.
(C) Western blot of hepatic protein expression. The same mice as in A.
(D) Left: H&E staining of liver sections in wild-type control Pat H19-/- and Mat H19-/- mice. Middle: qPCR of H19RNA and hepatic TG content. Right: Western blot of hepatic protein expression. Mice were fed with a HFHS diet for 8 weeks. Data are shown as mean ± SEM (n=5/group). *P<0.05 vs NC control.
(E) Left: H&E staining of liver sections in control and PTBP1 knockdown (shPTBP1) mice. Middle: Hepatic TG content. Right: Western blot analysis of hepatic protein expression. Mice were transduced with lenti-Null and lenti-shPTBP1 for 1 week, followed by HFHS feeding for 8 weeks. Data are shown as mean ± SEM (n=5/group). *P<0.05 vs control.
(F) Left: H&E staining of liver sections in H19 and H19-shPTBP1 mice. Middle: Hepatic TG content. Right: Western blot analysis of hepatic protein expression. Mice were transduced with AAV8-H19/lenti-Null and AAV8-Null/lenti-shPTBP1 for 1 week, followed by HFHS feeding for 8 weeks. Data are shown as mean ± SEM (n=5/group). *P<0.05 vs H19 group.
See also Fig. S6A-S6C and Fig. S7A-S7C.
We overexpressed H19 in mouse liver via tail vein injection of AAV8-H19 (thereafter referred to as H19 mice) or AAV8-Null (scramble control, Null mice) driven by using a specific promoter thyroxine binding globulin (TBG) for hepatic-directed delivery. H19 expression for five months resulted in global changes in hepatic gene expression in glucose and lipid metabolism (Fig. 3A). Several signaling pathways were activated, including extracellular signal-regulated kinase (ERK) (26), phosphoinositol 3-kinase (PI3K) AKT/protein kinase B (PKB) (27), and c-Jun N-terminal kinase (JNK) (28). Specifically, the levels of p-ERK, p-AKT, and p-JNK were induced to various extents in H19 mouse livers (Fig. 3B top). In addition, H19 mice exhibited elevated fasting glucose levels and insulin levels (Fig. 3B bottom), suggesting a dysfunction in response to fasting as a result of insulin resistance. The results were in agreement with the report that mice with activation of ERK in the livers showed fasting hyperglycemia (26). Despite these noticeable metabolic alterations, hepatic and serum TG levels remained unaltered (not shown).
Hepatic H19 expression remained constantly expressed (∼15 fold) in mice ten months after AAV8-H19 transduction (Fig. 3C left). The H19 liver appeared pale in color as compared to the Null liver (middle), which was associated with significantly increased liver weight (right) and TG content (Fig. 3D). Interestingly, serum TG levels did not differ between H19 and Null mice. Severe steatosis (both macrovesicular and microvasicular steatosis) was visualized by H&E and oil red O staining in H19 mice (Fig. 3E). Glycogen content was moderately increased in H19 mice compared to Null mice (PAS staining), and no hepatic fibrosis was observed in H19 or Null mice (Trichrome Masson staining).
We next analyzed representative genes in pathways coordinating lipid homeostasis, including lipid uptake, oxidation and synthesis (19). The mRNA expression of Pparγ and its target Cd36 was elevated whilst Pparα and its target Cpt1 were downregulated (Fig. 3F left), suggesting increased lipid uptake and decreased oxidation. Consistently, the nuclear protein expression of SREBP1, a key de novo lipogenic transcription factor, and its targets PPARγ and FASN, as well as PTBP1 and ACC proteins, were coordinately elevated, whereas PPARα protein was downregulated (Fig. 3F right).
Srebp1c mRNA showed no significant changes in H19 vs Null liver, whereas the elevated nSREBP1 protein was correlated with the decreased pSREBP1 protein, suggesting increased cleavage. On the other hand, the expression of key genes in VLDL secretion (Mtp, Apob) did not change in H19 vs Null liver (Supporting Fig. S5A), which was consistent with the unaltered serum TG levels (Fig. 3D right). We also analyzed the expression of genes in lipoprotein metabolism (Supporting Fig. S5B) and observed unique change patterns. Because the body weight of H19 and Null mice was similar (Supporting Fig. S5C), the global changes in hepatic metabolic genes were likely due to a direct effect of H19 in the liver. Consistent with the severe steatosis, increased insulin resistance and glucose intolerance were developed in H19 vs Null mice (Supporting Fig. S5D).
Overexpression of H19 exaggerates HFHS-induced steatosis
Although it took more than 5 months for H19 to spontaneously induce fatty liver, based on the results that H19 enhanced FA-induced lipid accumulation in hepatocytes (Fig. 2), we hypothesized that H19 may facilitate the development of steatosis induced by a HFHS diet (29). We transduced mice with AAV8-H19 or Null control for one week followed by HFHS feeding for five weeks. H19RNA, nSREBP1 and PTBP1 proteins were markedly induced in mice fed a HFHS diet compared to chow diet (Fig. 4A). As predicted, overexpression of H19 exaggerated hepatic steatosis by the HFHS diet, as indicated by the increased neutral lipid staining and elevated liver TG levels (Fig. 4B). Importantly, nSREBP1 protein levels were strongly elevated in H19 mice, which was accompanied by the induction of PTBP1 protein, decreased PPARα and increased PPARγ proteins (Fig. 4C). No significant changes in body weight, white fat, brown fat, and serum TG levels were observed between Null-HFHS and H19-HFHS mice (Supporting Fig. S6A). No significant differences in PAS staining and Trichrome Masson staining were observed between the two groups (Supporting Fig. S6B). qPCR revealed common as well as differential gene expression profiles in Null-HFHS and H19-HFHS mice (Supporting Fig. S6C) compared with Null and H19 mice (Supporting Fig. S5A-5C), suggesting the influence of HFHS diet on H19-mediated steatosis. Overall, the results showed that H19 exacerbated hepatic lipid phenotype induced by a HFHS diet.
Deficiency in H19 and PTBP1 prevents HFHS-induced steatosis
To further establish the endogenous effect of H19 in lipogenesis, Mat H19-/- and its wild-type control Pat H19-/- (Supporting Fig. S6D) were challenged with a HFHS diet for two months, a feeding period chosen to ensure that steatosis was well developed in Pat H19-/- mice. Lipid droplets became evident in Pat H19-/- mice but were largely prevented from developing in Mat H19-/- mice (Fig. 4D). In agreement with this observation, hepatic TG content, as well as the levels of nSREBP1, PTBP1, and FASN proteins, were concurrently lower in Mat H19-/- vs Pat H19-/- mice. pSREBP1 protein, on the other hand, was higher in Mat H19-/- vs Pat H19-/- mice, suggesting that the diminished nSREBP1 was due to decreased cleavage.
We next determined the effect of PTBP1 knockdown in HFHS-induced steatosis. We used lenti-shPTBP1 virus to transduce WT mice for one week followed by a HFHS feeding for two months. PTBP1 knockdown markedly diminished hepatic lipid accumulation and TG content, which was accompanied by the downregulation of pSREBP1, nSREBP1, FASN, PTBP1, and PPARγ proteins, and increased PPARα protein, as compared to the control mice (Fig. 4E).
We further conducted rescue experiments by knockdown of PTBP1 in H19 mice. We observed markedly diminished steatosis in H19-shPTBP1 compared with H19 mice (Fig. 4F). pSREBP1, nSREBP1, PTBP1, and FASN proteins were coordinately decreased. shPTBP1 did not alter glycogen storage (PAS staining) (Supporting Fig. S7A) or H19RNA levels (Supporting Fig. S7B), as compared with the control. It was interesting to note that Srebp1c mRNA was not altered by shPTBP1 in control or H19 mice, but was increased in Mat H19-/- vs Pat H19-/- mice (Supporting Fig. S7C). We speculate that H19 functions as a guide RNA scaffold to recruit PTBP1 to Srebp1c mRNA, and loss of H19 likely causes Srebp1c release, resulting in increased levels. Taken together, the results suggest that H19 induces fatty liver via a PTBP1-dependent mechanism.
H19 increases nuclear localization of SREBP1 protein in response to fasting
We examined hepatic nSREBP1 and PTBP1 proteins in cytoplasm and nucleus isolated from Null or H19 mice fed with a HFHS diet for 5 wks under feeding and fasting conditions (Fig. 5A). In agreement with the decreased Srebp1c mRNA upon fasting (Supporting Fig. S7D), the nuclear SREBP1 protein was also reduced by fasting vs feeding in Null (-) mice (blue arrow vs green arrow). In H19 (+) mice, nSREBP1 protein was highly upregulated under feeding conditions, which was further elevated by fasting (purple arrow vs red arrow). Under feeding conditions, PTBP1 protein was mostly observed in the nuclear fractions compared to the cytosolic fractions. Upon fasting, the levels of PTBP1 proteins were decreased in the cytoplasm but slightly increased in the nucleus, suggesting an increased nuclear translocation.
Figure 5. H19 promotes PTBP1 subcellular translocation and PTBP1/SREBP1c interaction.
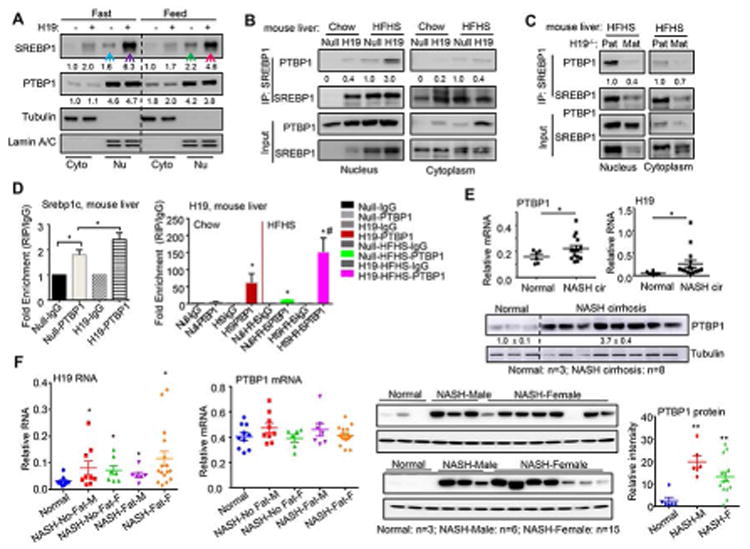
(A) Western blot analysis of cytoplasmic (Cyto) and nuclear (Nu) proteins isolated from Null and H19 mouse livers fed with a HFHS diet under feeding and fasting conditions. Loading controls: Tubulin (Cyto) and Lamin A/C (Nu) proteins.
(B-C) Co-IP followed by Western blot to determine the in vivo endogenous PTBP1 and SREBP1c protein interaction. Nuclear and cytoplasmic extracts were isolated from mouse livers of Null and H19 mice fed a chow or HFHS diet (B), or Pat-H19-/- (wild-type con) and Mat-H19-/- mice fed a HFHS diet (C). Co-IP was performed using SREBP1 antibody and WB analysis (IB) was performed using PTBP1, and SREBP1 antibodies. Input: The same amount of protein from each group was loaded.
(D) Left: RNA IP to determine the in vivo interaction between Srebp1c mRNA and PTBP1 protein using anti-PTBP1 or IgG (negative control) in livers of Null (lanes 1-2) or H19 (lanes 3-4) mice. Right: RNA IP to detect the association of H19RNA with endogenous PTBP1 protein in mouse livers from Null or H19 mice fed a chow or HFHS diet. Data are shown as mean ± SEM. *P<0.05 red vs black; #P<0.05 pink vs green.
(E) Top: qPCR of PTBP1 mRNA and H19RNA in human normal livers (n=6) and NASH cirrhotic livers (n=17). Each dot represents an individual sample. Data are shown as mean ± SEM. *P<0.05 vs normal. Bottom: Western blot analysis of PTBP1 protein in human normal livers (n=3) and NASH cirrhotic livers (n=8). Each line represents an individual sample.
(F) Left and middle: qPCR of H19RNA and PTBP1 mRNA in human normal livers (n=10), NASH-No-Fat livers (male, M: n=9; female, F: n=7), and NASH-Fat livers (M: n=6; F: n=15). Each dot represents an individual sample. Data are shown as mean ± SEM. *P<0.05 vs normal. Right: Western blot analysis of PTBP1 protein in human normal livers (n=6), NASH-Fat livers (male: n=6; female: n=15). The samples were analyzed in two gels. Each line represents an individual sample. **P<0.01 vs normal.
See also Fig. S7D.
The in vivo SREBP1 and PTBP1 protein interaction in the nucleus and the cytoplasm of mouse liver was assessed. Co-IP assays revealed a stronger interaction in chow-H19 vs chow-Null liver (Fig. 5B left), and the interaction was enhanced by HFHS diet. A weaker interaction but with a similar pattern was observed in the cytoplasm (Fig. 5B right). The interaction, however, was largely diminished in H19-/- mice (Fig. 5C). RNA IP revealed a significant enrichment of PTBP1 protein to Srebp1c mRNA in Null liver, which was further augmented in H19 liver (Fig. 5D left). In addition, the association of H19RNA with endogenous PTBP1 protein in mouse liver was markedly enhanced by the HFHS diet (Fig. 5D right). Hepatic H19RNA maintained at constant high levels 1 year after H19 overexpression (Supporting Fig. S7E). In contrast, H19RNA expression in muscle and adipose tissues did not alter in H19 vs control mice (Supporting Fig. S7F).
Importantly, the expression of PTBP1 mRNA and protein, as well as H19RNA, was highly induced in human NASH cirrhotic livers vs normal livers (Fig. 5E). We also analyzed H19RNA and PTBP1 expression in human NASH-No-Fat and NASH-Fat livers from both male (M) and female (F) patients to determine gender difference. H19RNA was similarly elevated in male and female NASH-No-Fat specimens (Fig. 5F left). It was more highly induced in NASH-Fat-F vs NASH-Fat-M, which did not reach statistical significance between the two groups. To our surprise, PTBP1 mRNA was not induced in NASH-No-Fat and NASH-Fat livers vs normal livers (Fig. 5F middle). Nonetheless, PTBP1 protein was strikingly elevated in both male and female NASH patients (Fig. 5F right). Taken together, these results demonstrate the clinical significance of both genes in regulating the development of human chronic liver diseases.
H19 disrupts metabolomics profiling in response to fasting
Metabolomics serves as a powerful approach to identify different molecules produced in cells and offers new insight into the cellular metabolic pathways regulated by H19. Untargeted metabolomics profiling (GC-TOF mass spectrometer) was used to elucidate changes in liver metabolites in Null and H19 overexpressed mice by 10 months under both fasting and feeding conditions. The data sets were subjected to non-supervised multivariate data analysis to evaluate the presence of a metabolomic pattern that could discriminate between Null and H19 mice subjected to fasting.
Correlation matrix and cluster analysis revealed both positively and negatively correlated metabolites (Supporting Fig. S8A). The differential abundance of metabolites in response to fasting vs feeding in Null and H19 mice was further visualized by Heatmap (Supporting Fig. S8B; Fig. 6A). It was noted that the levels of majority of metabolites were either increased or decreased in response to fasting in Null mice vs Null-feed, and such responses were blunted to a large extent in H19 mice. The PCA (principal component analysis) (Supporting Fig. S8C) and PLS-DA (Fig. 6B) scores plots showed an almost complete separation between Null-fast with Null-feed and those overexpressed with H19. Due to the lost response to fasting, metabolites altered in H19-fast vs Null-fast were largely enriched in glucose metabolic pathways, including glycolysis and gluconeogenesis (Fig. 6C; Supporting Fig. S9A). In contrast, other metabolites including fatty acids and amino acids remained at low levels in H19-fast vs Null-fast mice, whereas cholesterol levels remained unchanged in H19 vs Null mice (Supporting Fig. S9B-9F). Among all the metabolites, glucose-1-phosphate (G1P) and glucose-6-phosphate (G6P) were most highly enriched in H19-fast vs. Null-fast mice, as shown in the Volcano plot (Fig. 6D), whereas the concentrations of N-methylalalanine and methionine were markedly reduced (Fig. 6E). Overall, the results suggest that hepatic overexpression of H19 has a substantial impact on the stress-response capacity of liver, which results in a wide-ranging disruption of metabolic pathways.
Figure 6. H19 expression disrupts stress-induced hepatic metabolomics profiles.
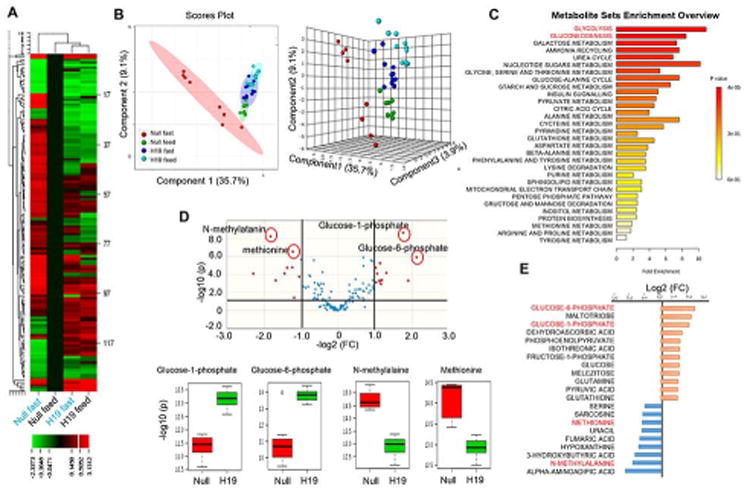
(A) Heatmap showing differential abundance of metabolites in response to fasting vs feeding in Null and H19 mice. Liver tissues were used for untargeted metabolomics profiling (GC-TOF mass spectrometer). Four groups include: Null-fast (n=8), Null-feed (n=8), H19-fast (n=12), H19-feed (n=12). Raw data were averaged in each group (each column) and normalized to Null-feed (set as 1), and log2 fold changes were used to generate Heatmap using CIMminer. Each row represents a metabolite.
(B) PLS-DA scores plot performed using two principal components corresponding to a model built using two latent variables aimed at discrimination among four groups described in A. The ovals filled with different color donate 95% confidence interval Hotelling's ellipses.
(C) Metabolite Sets Enrichment analysis to identify enriched metabolites in various pathways in H19-fast vs Null-fast mice.
(D) Volcano plot (upper) indicates the size of the biological effect (fold change, FC) vs the statistical significance of the result (statistical p-value). Each dot represents a metabolite plotted as a function of fold change (log 2 (FC), x-axis) and statistical significance (−log 10 (p-value), y-axis). The red dots represent metabolites (H19-fast vs Null-fast) with a p-value ≤0.05 and fold-change cut off of > or < 2. Red-Circled dots were selected metabolites shown in boxplots (lower).
(E) Top 21 metabolites altered in H19-fast vs Null-fast mice presented as log2 fold change (FC). Orange: increased levels; blue: decreased levels.
See also Fig. S8A-S8C and Fig. S9A-S9F.
Overexpression of H19 reprograms hepatic lipid species
Lipidomics analysis provides an unbiased approach to further define the altered hepatic lipid species by H19. In general, a majority of individual TG lipid species were found to be highly upregulated in H19 mice compared with Null mice under feeding conditions and were further elevated by fasting (Fig. 7A-7C; Supporting Fig. S10A-10C). In contrast, there were no differences in levels of absolute and relative TG abundance between fasting and feeding conditions in Null mice. Among other lipid species, diacylglycerol DG (34:1), DG (36:2), DG (38:3), DG (38:5), and cholesterol ester CE (22:6) exhibited similar changes to TG species (Supporting Fig. S11A-11B). Interestingly, several phosphatidyl choline (PC) species, including PC (38:4) A and PC (38:5) A, were only elevated in H19-Fast mice (Supporting Fig. S11C). On the contrary, FA (18:2), FA (18:3), FA (20:4), and FA (22:6) were induced by fasting in Null but not in H19 mice (Supporting Fig. S11D). In addition, ceramide species 38:1 were similarly elevated in H19-Feed and H19-Fast mice (Supporting Fig. S11E).
Figure 7. Lipidomics analysis revealed H19-induced triglyceride (TG) species.
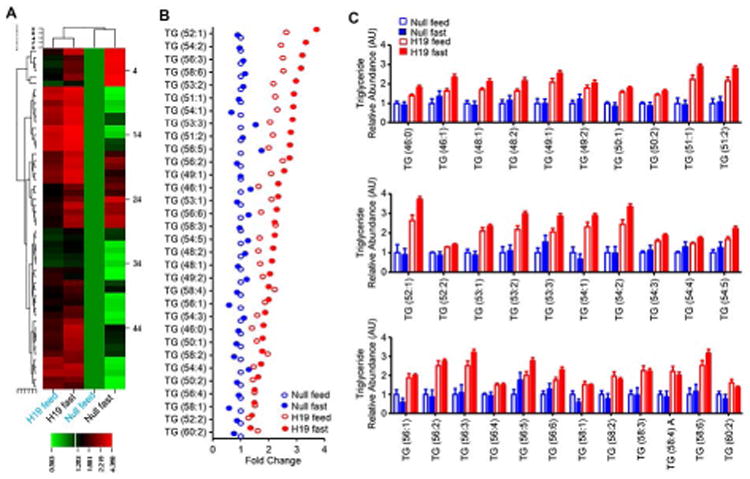
(A) Heatmap showing differential abundance of liver TG species in response to fasting vs feeding in Null and H19 overexpressed mice, as revealed by lipidomics (LC-MS) analysis. Four groups include: Null-fast (n=8), Null-feed (n=8), H19-fast (n=12), H19-feed (n=12).
(B) TG lipid species are expressed as fold change of H19 relative to Null mice according to the greatest fold difference (top to bottom).
(C) Representative TG species in Null and H19 mice under feeding and fasting conditions.
See also Fig. S10A-S10C and Fig. S11A-S11E.
Discussion
H19 has been documented as a tumor suppressor or oncogene in various tissues or cells (12, 13, 30), but its function in the liver remains to be defined. In this study, we report a novel role of H19 in modulating hepatic lipid metabolism. The key findings include: 1) H19 acts as an interacting partner of PTBP1, and their association is enhanced by nutrient status (fatty acid and HFHS diet); 2) H19 facilitates PTBP1 binding to Srebp1c mRNA in cytoplasm, resulting in increased stability; 3) H19 promotes PTBP1 binding to SREBP1 protein in nucleus, which in turn augments Srebp1c transcriptional activity; 4) H19 induces pSREBP1 protein cleavage and nSREBP1 nuclear translocation via a PTBP1-dependent fashion; 5) The expression of H19 and PTBP1 is upregulated by HFHS diet, which further elevates SREBP1 and stimulates the PTBP1 and SREBP1 interaction; and 6) H19 reprograms hepatic fasting metabolic response into a “pseudo fed” state by inducing nuclear accumulation of SREBP1 protein, which exacerbates HFHS-induced fatty liver. Both H19 and PTBP1 are markedly induced in human cirrhotic livers, demonstrating their clinical significance in the regulation of chronic liver diseases.
Several lncRNAs have been reported to be associated with PTBP1. Pnky and TUNA can recruit PTBP1 to regulate neuronal differentiation of neural stem cells (NSCs) (31) and embryonic stem cells (ESCs) (32). MEG3 binds to PTBP1 to control small heterodimer partner (SHP) mRNA stability and cholestatic liver injury (20). Algorithms to predict mRNA targets of PTBP1 across the genome using genomic sequences have not been developed yet; therefore the prediction of PTBP1 targeted mRNAs is limited to specific genes of interest. In this study, we focused on genes in lipid metabolic pathways and identified Srebp1c as a downstream gene regulated by PTBP1.
Based on the results obtained from in vitro and in vivo models in this study, PTBP1-mediated elevation of SREBP1 protein likely results from increased stability, because knockdown of PTBP1 decreases both pSREBP1 and nSREBP1 in Null and H19-overexpresed mice. On the other hand, H19 increases SREBP1 at least in part by inducing SREBP1 protein cleavage, as evidenced by decreased pSREBP1 and increased nSREBP1 in H19 mice, but increased pSREBP1 and decreased nSREBP1 in H19-/- mice. MassSpec analysis did not identify SREBP1 as a potential H19 interacting partner; therefore H19 may regulate SREBP1 cleavage through an unknown intermediate protein. Interestingly, PTBP1 is essential for H19-induced SREBP1 nuclear translocation under fasting conditions. Additional evidence supporting PTBP1-dependent regulation of SREBP1 expression by H19 includes: 1) overexpression of H19 increases both SREBP1 and PTBP1 protein; 2) H19-deficiency decreases both SREBP1 and PTBP1 protein; 3) knockdown of PTBP1 decreases SREBP1 without altering H19 levels; and 4) knockdown of PTBP1 in H19 overexpressed mice decreases SREBP1.
It is noted that Srebp1c mRNA is not markedly elevated in H19 mice, suggesting that H19 does not likely regulate Srebp1c gene transcription. However, overexpression of H19 and PTBP1 in vitro in cell models increases Srebp1c mRNA stability. Because PTBP1 is mostly expressed in the nucleus, its effect in cytoplasm may be moderate in vivo in mouse liver. The effect of PTBP1 could also be masked by other transcription factors and signaling events controlling Srebp1c expression.
In addition to the changed metabolic and lipogenic program, other pathways are also altered in H19 mice, including lipid uptake and oxidation, and lipoprotein metabolism. Interestingly, H19 pushes the liver into a “fed” like state, as supported by hyperglycemia and hyperinsulinemia, increased nuclear SREBP1c protein despite decreased Srebp1c mRNA in the fasted state, and activation of the insulin-signaling pathway in liver. Overall, a global change of lipid metabolic homeostasis by H19 facilitated the development of fatty liver.
New lncRNAs associated with human liver diseases are constantly identified (33). Recent studies have demonstrated that H19RNA was elevated in human liver fibrosis and cirrhosis (14), as well as in human primary sclerosing cholangitis (PSC) and primary biliary cirrhosis (PBC) patients (34). The present study provides further evidence for the induction of H19 and PTBP1 in human NASH fatty liver. These results may have important clinical implications, suggesting that reducing the expression or blocking the function of H19 and PTBP1 could provide novel treatment strategies for chronic liver diseases.
In summary, the interplay between H19RNA and PTBP1 protein functions as an essential component of the hepatic metabolic network (Fig. 8). Intriguingly, FAs stimulate H19 and PTBP1 expression, which in turn augments the lipogenic effect of FAs by enhancing SREBP1c expression and transactivation, forming a robust feedforward regulatory loop. The importance of such a mechanism becomes more prominent under stress or disease conditions. In addition to modulating lipid homeostasis, upregulation of H19 also exerts a major influence on glucose metabolism. Because glucose homeostasis and lipid metabolism are closely intertwined, future work will continue to identify additional targets regulated by the H19/PTBP1 circuit in glucose metabolic pathways that are associated with the development of liver metabolic disorders.
Figure 8. A H19/PTBP1 feedforward metabolic circuit to reprogram hepatic response to fasting via modulating SREBP1c expression.
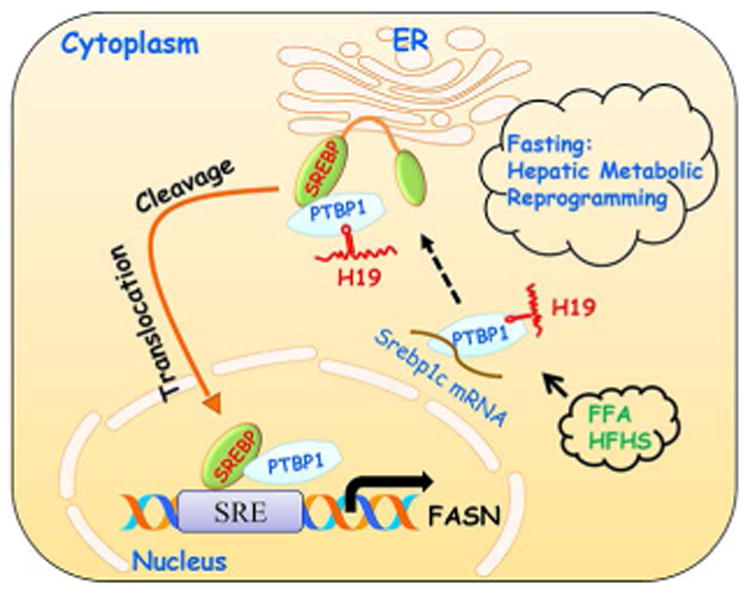
, H19 functions as a lipid sensor and its expression is induced by fatty acids and HFHS diet, which in turn enhances H19 association with the PTBP1 protein. The interaction of H19RNA and PTBP1 protein results in dual effects: it facilitates PTBP1 binding to Srebp1c mRNA in the cytoplasm, leading to increased Srebp1c mRNA stability, and it stimulates PTBP1 binding to SREBP1c protein in the nucleus, augmenting SREBP1c transcriptional activity. In addition, H19 induces pSREBP1c protein cleavage; it also promotes nSREBP1c nuclear translocation in PTBP1-dependent fashion under fasting conditions. Therefore, H19 reprograms hepatic response to fasting and pushes the liver into a “fed” like state. The H19/PTBP1/SREBP1 metabolic circuit generates a feedforward amplifying signal to exacerbate the development of fatty liver by the HFHS diet. Question mark: To be identified H19 interacting protein.
Supplementary Material
Acknowledgments
We sincerely thank the research community: Drs. Douglas Black (UCLA) and Jiuyong Xie (University of Manitoba, Canada) for PTBP1 antibodies and plasmids; Linheng Li (Stowers Institue) and Xiaobo Zhong (UConn) for the H19-/- mice; TuKiet T. Lam at Yale Keck Proteomics Center for Mass Spectrometry (LC-MS) analysis; West Coast Metabolomics Center, UC Davis for Metabolomics and Lipidomics analysis (Grant: NIDDK 1R01DK104656-01); and Liver Tissue Liver Tissue Cell Distribution System (# HSN276201200017C) for the human liver specimens.
Grant Support: L.W. is supported by NIH R01DK104656, R01DK080440, R01ES025909, R21AA022482, R21AA024935, VA Merit Award 1I01BX002634, National Natural Scientific Foundation of China (Grant No. 81572443) and P30 DK34989 (Yale Liver Center).
Footnotes
Conflict of Interest: There are none to declare for all authors.
Author contributions: C.L., Z.Y., J.W., L.Z., S.L., D.S. designed and performed experiments. M.L. analyzed the data.
C.L. drafted the manuscript. L.W. supervised the work, obtained funding, and wrote the manuscript.
Authors names in bold designate shared co-first authorship.
Supplemental Information: Supplemental Information for this article includes three tables, eleven figures, and detailed experimental procedures.
References
- 1.Pacana T, Sanyal AJ. Vitamin E and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2012;15:641–648. doi: 10.1097/MCO.0b013e328357f747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerrero R, Vega GL, Grundy SM, Browning JD. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology. 2009;49:791–801. doi: 10.1002/hep.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–1375. doi: 10.1053/j.gastro.2008.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 5.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2016 doi: 10.1136/gutjnl-2016-312431. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, Moore DD, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, et al. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature. 2015;524:243–246. doi: 10.1038/nature14557. [DOI] [PubMed] [Google Scholar]
- 9.Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, Chen Y, et al. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab. 2015;21:455–467. doi: 10.1016/j.cmet.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell. 2013;152:743–754. doi: 10.1016/j.cell.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durruthy-Durruthy J, Sebastiano V, Wossidlo M, Cepeda D, Cui J, Grow EJ, Davila J, et al. The primate-specific noncoding RNA HPAT5 regulates pluripotency during human preimplantation development and nuclear reprogramming. Nat Genet. 2016;48:44–52. doi: 10.1038/ng.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang WC, Fu WM, Wang YB, Sun YX, Xu LL, Wong CW, Chan KM, et al. H19 activates Wnt signaling and promotes osteoblast differentiation by functioning as a competing endogenous RNA. Sci Rep. 2016;6:20121. doi: 10.1038/srep20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giovarelli M, Bucci G, Ramos A, Bordo D, Wilusz CJ, Chen CY, Puppo M, et al. H19 long noncoding RNA controls the mRNA decay promoting function of KSRP. Proc Natl Acad Sci U S A. 2014;111:E5023–5028. doi: 10.1073/pnas.1415098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Liu C, Barbier O, Smalling R, Tsuchiya H, Lee S, Delker D, et al. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci Rep. 2016;6:20559. doi: 10.1038/srep20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keppetipola N, Sharma S, Li Q, Black DL. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol. 2012;47:360–378. doi: 10.3109/10409238.2012.691456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell. 2013;152:82–96. doi: 10.1016/j.cell.2012.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrarese R, Harsh GRt, Yadav AK, Bug E, Maticzka D, Reichardt W, Dombrowski SM, et al. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J Clin Invest. 2014;124:2861–2876. doi: 10.1172/JCI68836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knoch KP, Nath-Sain S, Petzold A, Schneider H, Beck M, Wegbrod C, Sonmez A, et al. PTBP1 is required for glucose-stimulated cap-independent translation of insulin granule proteins and Coxsackieviruses in beta cells. Mol Metab. 2014;3:518–530. doi: 10.1016/j.molmet.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabbi-Anneni I, Cooksey R, Gunda V, Liu S, Mueller A, Song G, McClain DA, et al. Overexpression of nuclear receptor SHP in adipose tissues affects diet-induced obesity and adaptive thermogenesis. Am J Physiol Endocrinol Metab. 2010;298:E961–970. doi: 10.1152/ajpendo.00655.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Yang Z, Trottier J, Barbier O, Wang L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology. 2017;65:604–615. doi: 10.1002/hep.28882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smalling RL, Delker DA, Zhang Y, Nieto N, McGuiness MS, Liu S, Friedman SL, et al. Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am J Physiol Gastrointest Liver Physiol. 2013;305:G364–374. doi: 10.1152/ajpgi.00077.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, Wang L. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr-1/SHP/EID1 network. Hepatology. 2014;60:919–930. doi: 10.1002/hep.27121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SM, Zhang Y, Tsuchiya H, Smalling R, Jetten AM, Wang L. Small heterodimer partner/neuronal PAS domain protein 2 axis regulates the oscillation of liver lipid metabolism. Hepatology. 2015;61:497–505. doi: 10.1002/hep.27437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuchiya H, da Costa KA, Lee S, Renga B, Jaeschke H, Yang Z, Orena SJ, et al. Interactions Between Nuclear Receptor SHP and FOXA1 Maintain Oscillatory Homocysteine Homeostasis in Mice. Gastroenterology. 2015;148:1012–1023 e1014. doi: 10.1053/j.gastro.2015.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Yang Z, Wu J, Wu R, Keshipeddy SK, Wright D, Wang L. High-mobility-group protein 2 regulated by microRNA-127 and small heterodimer partner modulates pluripotency of mouse embryonic stem cells and liver tumor initiating cells. Hepatology Communications. 2017:n/a–n/a. doi: 10.1002/hep4.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao P, Feng B, Li Y, He Q, Xu H. Hepatic ERK activity plays a role in energy metabolism. Mol Cell Endocrinol. 2013;375:157–166. doi: 10.1016/j.mce.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 28.Lawan A, Zhang L, Gatzke F, Min K, Jurczak MJ, Al-Mutairi M, Richter P, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol Cell Biol. 2015;35:26–40. doi: 10.1128/MCB.00503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Liu J, Saha P, Huang J, Chan L, Spiegelman B, Moore DD. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2005;2:227–238. doi: 10.1016/j.cmet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Matouk IJ, Halle D, Raveh E, Gilon M, Sorin V, Hochberg A. The role of the oncofetal H19 lncRNA in tumor metastasis: orchestrating the EMT-MET decision. Oncotarget. 2015 doi: 10.18632/oncotarget.6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos AD, Andersen RE, Liu SJ, Nowakowski TJ, Hong SJ, Gertz CC, Salinas RD, et al. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell. 2015;16:439–447. doi: 10.1016/j.stem.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, Zhang D, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell. 2014;53:1005–1019. doi: 10.1016/j.molcel.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z, Ross RA, Zhao S, Tu W, Liangpunsakul S, Wang L. LncRNA AK054921 and AK128652 are potential serum biomarkers and predictors of patient survival with alcoholic cirrhosis. Hepatology Communications. 2017;1:513–523. doi: 10.1002/hep4.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L, Pope C, et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule. Hepatology. 2017;66:1183–1196. doi: 10.1002/hep.29209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.