

SUMMARY
Cerebral malaria is a deadly complication of Plasmodium infection and involves blood brain barrier (BBB) disruption following infiltration of white blood cells. During experimental cerebral malaria (ECM), mice inoculated with Plasmodium berghei ANKA-infected red blood cells develop a fatal CM-like disease caused by CD8+ T cell-mediated pathology. We found that treatment with interleukin-15 complex (IL-15C) prevented ECM, whereas IL-2C treatment had no effect. IL-15C-expanded natural killer (NK) cells were necessary and sufficient for protection against ECM. IL-15C treatment also decreased CD8+ T cell activation in the brain and prevented BBB breakdown without influencing parasite load. IL-15C induced NK cells to express IL-10, which was required for IL-15C-mediated protection against ECM. Finally, we show that ALT-803, a modified human IL-15C, mediates similar induction of IL-10 in NK cells and protection against ECM. These data identify a regulatory role for cytokine-stimulated NK cells in the prevention of a pathogenic immune response.
In Brief
NK cells can display both pro-inflammatory and regulatory function, but their role in the pathogenesis of malaria is not fully understood. Burrack et al. demonstrate that IL-15 complex (IL-15C) therapy prevents mice from succumbing to experimental cerebral malaria (ECM). IL-15C treatment stimulates NK cells to produce IL-10, suppressing the pathogenic CD8+ T cell response during ECM.
INTRODUCTION
A successful response to infection requires controlled, coordinated efforts by multiple cells of the immune system without causing an overly robust immune response that damages the host. While populations such as CD4+ regulatory T (Treg) cells have been well characterized for their ability to restrain immune responses, other components of the immune system can also exert immunosuppressive actions with suitable stimulation. For example, natural killer (NK) cells, which are chiefly perceived as a population dedicated to promoting early inflammatory responses, can also substantially restrain CD4+ and CD8+ T cell responses through cytotoxic activity or production of immunosuppressive cytokines (Biron, 2012; Crome et al., 2013; Crouse et al., 2015; Welsh and Waggoner, 2013). However, how NK cells acquire immunosuppressive function or whether these cells can be induced therapeutically is less clear.
Cytokines strongly influence the intensity and duration of immune responses, and these effects can be magnified using cytokine complexes: cytokines combined in vitro with either specific antibodies or presenting receptors. Studies in our laboratory and others have shown that treatment with cytokine complexes can have robust effects on immune cells that enhance the normal biological activity of the cytokine due to both increased half-life and strengthened signal on target cells (Boyman et al., 2006; Hamilton et al., 2010; Rubinstein et al., 2006). For example, complexes containing interleukin 2 (IL-2) and IL-15 induce expansion of CD8+ T cells and NK cells, and treatment of mice with these complexes can protect against bacterial and viral pathogens, as well as promote the control of tumors (Epardaud et al., 2008; Hamilton et al., 2010; Verdeil et al., 2008; Votavova et al., 2014). However, less is known about whether these cytokine complexes can be used to restrain damaging immune responses and how that process might occur. Here, we tested this possibility by exploring the capacity of cytokine complex stimulation to inhibit a well-characterized model of T cell-mediated immunopathology: experimental cerebral malaria (ECM).
Cerebral malaria (CM) is a deadly complication of Plasmodium infection that kills approximately 500,000 people each year, mostly children under the age of 5 (WHO, 2016). The pathogenesis of CM remains incompletely understood but is known to involve sequestration of Plasmodium-infected erythrocytes within the microvasculature of the brain and white blood cell infiltration, leading to disruption of the blood brain barrier (BBB), edema, and hemorrhaging (Hora et al., 2016; Storm and Craig, 2014). There are no adjunctive therapies currently available for CM patients, and resistance to available malaria drugs is rising (WHO, 2016).
In ECM, C57BL/6 mice inoculated with Plasmodium berghei ANKA (PbA)-infected red blood cells die within 5–10 days post-infection (dpi) from a CM-like disease (de Oca et al., 2013). CD8+ T cells are the primary pathogenic effectors in the progression of ECM, and effector functions such as cytolysis and interferon-γ (IFN-γ) production are critical for ECM immunopathology (Belnoue et al., 2002; Nitcheu et al., 2003; Yañez et al., 1996). The role of NK cells is less clear: the capacity of NK cells to kill and produce IFN-γ might suggest that they would contribute to ECM pathology; however, findings that NK cells can restrain CD8+ T cell responses in some infectious disease settings (Welsh and Waggoner, 2013) raised the possibility that, with appropriate activation, the immunosuppressive capacity of NK cells might be harnessed to control ECM.
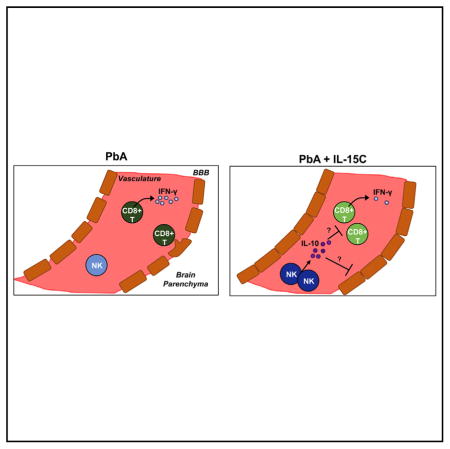
Using the rapid and ultimately lethal response to infection in ECM as a model for immunopathology, we sought to investigate whether cytokine complexes could be used to modulate the immune response and ultimately provoke an immunosuppressive state to prevent ECM. Here, we show that treatment with IL-15 complexes (IL-15C), but not IL-2C, prevented the development of ECM. NK cells were necessary and sufficient for IL-15C-mediated survival from ECM. Moreover, IL-15C treatment reduced the activation and functional response of malarial antigen-specific CD8+ T cells in the brain. IL-15C but not IL-2C induced IL-10 expression in NK cells, and NK cell-derived IL-10 rescued mice from ECM disease without affecting parasite load. Similar results were observed using a modified human IL-15C that is being evaluated in clinical trials for other conditions. These studies uncovered an important role for NK cells in the prevention of ECM and demonstrate the unique potential of IL-15C therapy to restrain a normally devastating immune response.
RESULTS
IL-15C but Not IL-2C Protects against ECM
As ECM is an immunopathologic disease, we sought to modulate immune cell activation using cytokine complex therapy. We evaluated the influence of two γc-cytokine complexes on the development of ECM following PbA infection: IL-2C (IL-2 bound to the anti-IL-2 S4B6 antibody) and IL-15C (IL-15 bound to an IL-15 receptor α-Fc fusion protein). We first treated mice prior to PbA infection (Figure S1) and found that prophylactic treatment with IL-15C but not IL-2C prevented lethal ECM in the majority of infected mice (Figure 1A). As prophylactic treatment has limited clinical relevance, we extended these studies to test whether therapeutic treatment with IL-15C was also effective. Indeed, 70% of PbA-infected mice treated therapeutically with IL-15C on days 2 and 3 pi (Figure S1) were protected from developing ECM (Figure 1B).
Figure 1. IL-15C Prevents the Development of ECM.

(A) Survival curve of mice left untreated (n = 15) or treated prophylactically with IL-2C (n = 14) on days −7, 5, and −3 or IL-15C (n = 13) on days −5 and −3 relative to PbA infection. Data are combined from three independent experiments. ****p < 0.0001 as determined by log-rank (Mantel-Cox) test.
(B) Survival curve of mice left untreated (n = 14) or treated therapeutically with IL-15C (n = 20) on days 2 and 3 after PbA infection. Data are combined from four independent experiments. ****p < 0.0001 as determined by log-rank (Mantel-Cox) test.
(C) Parasitemia analysis on day 6 pi of mice left untreated or treated prophylactically or therapeutically with IL-15C. Each symbol represents one mouse. Data are combined from three independent experiments. Each symbol represents one mouse; horizontal bar indicates the mean. *p < 0.05 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
(D) PbA-infected mice (n = 7/group) were treated therapeutically with PBS or IL-15C. Representative gadolinium-enhanced T1-weighted MRI scans (midsagittal orientation) of mice on day 6 pi. Arrowheads indicate regions of hyperintensity.
(E) Quantification of hyperintensity measured from T2-weighted MRI of the same mice as in (D). Uninfected mice (n = 5) were used as a control. Each symbol represents one mouse; data are presented as the mean ± SEM. Data are combined from two independent experiments. **p < 0.01 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
(F) PbA-infected mice (n = 5/group) were treated therapeutically with PBS or IL-15C on days 2+3 pi. Representative images of coronal slices from cerebral cortex showing FITC-albumin leakage (green) with DAPI counter-stain (blue).
(G) Quantification of cerebral FITC leakage from mice in (F). Uninfected mice (n = 4) were used as a control. Each symbol represents one mouse; data are presented as the mean ± SEM. ***p < 0.001, ****p < 0.0001 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
Related to Figure S1.
The ability of IL-15C treatment to prevent ECM might arise from enhanced clearance of infected red blood cells. However, blood parasitemia levels were not significantly altered in mice treated prophylactically or therapeutically with IL-15C compared with untreated mice (Figure 1C), similar to other studies that demonstrated no correlation between blood parasitemia levels and survival from ECM (Dende et al., 2015; Fauconnier et al., 2012; Gordon et al., 2015). Both CM and ECM are characterized by brain edema, which can be visualized in mice and humans via magnetic resonance imaging (MRI) techniques (Penet et al., 2005; Potchen et al., 2012), and survival from ECM is associated with reduced BBB breakdown in mice (Dende et al., 2015; Gordon et al., 2015). We sought to determine whether IL-15C-mediated survival from ECM was associated with reduced edema, as visualized by T2- and T1-weighted MRI’s on PbA-infected mice treated therapeutically with IL-15C or with phosphate-buffered saline (PBS) as a control. Similar to studies performed by others (Zhao et al., 2014), we found gadolinium enhancement within the olfactory bulb, ventricles, and meninges, and significantly increased hyperintensity volume in control treated PbA-infected mice compared with uninfected controls (Figures 1D and 1E). However, hyperintensity volume was no different between uninfected controls and IL-15C-treated mice (Figures 1D and 1E). As a second measure of BBB leakage, FITC-albumin was injected prior to euthanasia. In control-treated PbA-infected mice, FITC-albumin localization in the brain was detected by histology (Figure 1F) and was significantly increased compared with uninfected mice (Figure 1G). In contrast, therapeutic IL-15C treatment prevented localization of FITC-albumin in the brain (Figures 1F and 1G). Together, these data suggest that IL-15C-induced survival from ECM is associated with reduced cerebral leakage rather than inhibition of parasite replication.
IL-15C- but Not IL-2C-Treated NK Cells Prevent the Development of ECM
Both IL-2C and IL-15C engage the signaling chains CD122 and CD132 and do not bind the α chains that normally distinguish these cytokines (Boyman et al., 2006; Ma et al., 2006; Votavova et al., 2014). Both complexes have also been reported to induce robust expansion of memory phenotype CD8+ T cells and NK cells (which express high levels of CD122) and augment protection against pathogens and tumors (Hamilton et al., 2010; Kamimura et al., 2006; Rubinstein et al., 2006; Stoklasek et al., 2006; Tomala et al., 2009). Indeed, we observed similar expansion of splenic NK and CD8+ T cells following treatment with either cytokine complex (Figures 2A and 2B); moreover, similar numbers of NK cells and CD8+ T cells were observed in the spleen and brain following either cytokine complex treatment on days 3 and 6 post-PbA infection (Figures S2A and S2B). Our earlier studies showed that prophylactic IL-2C treatment enhanced control of Listeria monocytogenes (LM), which involved both CD8+ T cell and NK cell activity (Hamilton et al., 2010). We observed a similar reduction in LM burden following treatment with IL-15C (Figure 2C). Hence, survival from ECM by IL-15C treatment does not reflect some generalized attenuation of the cellular immune response. Instead, we considered that either NK cells or CD8+ T cells in IL-15C-treated animals could interfere with ECM immunopathology. As CD8+ T cells contribute to ECM in PbA-infected mice (Howland et al., 2015a), we focused on the potential role of NK cells in protecting against ECM. Depletion of NK cells by administration of anti-NK1.1 monoclonal Ab resulted in complete loss of the protective effects of IL-15C treatment (Figure 2D), demonstrating that NK1.1-expressing cells are required for IL-15C-mediated survival from ECM. To complement these studies, we tested whether IL-15C-treated NK cells were sufficient to prevent ECM. NK cells enriched from the spleens of mice treated with IL-2C or IL-15C or left untreated were transferred into untreated mice, which were subsequently infected with PbA the next day. NK cell purity, including the absence of NKT and T cells, was confirmed by flow cytometry prior to adoptive transfer (Figures S2C and S2D). While recipients of NK cells from control and IL-2C-treated mice succumbed to ECM, animals that received NK cells from IL-15C-treated animals were protected against ECM (Figure 2E). Similar protection was induced by therapeutic adoptive transfer of NK cells from IL-15C-treated mice, on day 2 after PbA infection (data not shown). Adoptive transfer of purified CD8+ T cells from IL-15C-treated mice, in contrast, did not alter the progression of ECM (Figure 2E). Together, these data indicate that IL-15C-treated NK cells are both necessary and sufficient for protection against ECM.
Figure 2. IL-15C-Treated NK Cells Prevent the Development of ECM.

(A and B) Total number of CD8+ T cells (A) and NK cells (B) in the spleens of mice left untreated (n = 13) or treated with IL-2C three times (n = 17) or IL-15C two times (n = 16). Data are combined from six independent experiments and are presented as the means ± SEM.
(C) Mice were treated with IL-2C on days −7, −5, and −3 and IL-15C on days −5 and −3 relative to LM-OVA infection. CFU of LM in the spleen on day 5 pi. Each symbol represents one mouse; horizontal bar indicates the mean. Data are combined from two independent experiments. LOD, limit of detection.
(D) Survival curve of mice (n = 9/group) left untreated or treated prophylactically with IL-2C on days −7, −5, and −3 or IL-15C on days −5 and −3 and/or treated with anti-NK1.1 Ab on days −3, −1, and +1 relative to PbA infection. Data are combined from two independent experiments.
(E) Survival curve of mice that, 1 day prior to PbA infection, received NK cells enriched from the spleens of mice left untreated or treated with IL-2C three times or IL-15C two times. Groups: PbA (n = 15), NK + PbA (n = 7), IL-2C NK + PbA (n = 10), IL-15C + PbA (n = 15). Data are combined from three independent experiments.
(A–C) *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
(D and E) ***p < 0.001, ****p < 0.0001, as determined by log-rank (Mantel-Cox) test.
Related to Figure S2.
IL-15C Treatment Does Not Substantially Diminish the Expansion or Brain Localization of PbA-Specific CD8+ T Cells
In some viral models, NK cells limit the CD8+ T cell response to antigen by mechanisms that include preventing CD4+ T cell help or antigen presenting cell function and the direct elimination of antigen-specific CD8+ T cells (Biron, 2012; Crouse et al., 2015; Welsh and Waggoner, 2013). As CD8+ T cells are pathogenic effectors in ECM, we hypothesized that IL-15C-stimulated NK cells were suppressing the CD8+ T cell response and thus preventing ECM development. To test this, we quantified the number of total and antigen-specific CD8+ T cells in the spleens and brains of PbA-infected mice that had or had not been treated with IL-15C. C57BL/6 mice generate a robust response to a CD8+ TCR epitope in the Plasmodium glideosome-associated protein 50 (GAP50) (Howland et al., 2013), so we used GAP5041-48/Db tetramers to identify antigen-specific CD8+ T cells. IL-15C treatment increased the frequency and total number of CD8+ T cells overall, resulting in an increase in the number of GAP50-specific CD8+ T cells in IL-15C-treated mice even though the frequency of GAP50-specific splenic CD8+ T cells was similar between untreated and IL-15C-treated mice at 6 dpi (Figures 3A–3C). These data indicate that IL-15C treatment does not result in deletion of CD8+ T cells in the spleens of infected mice. In the brain, the frequency and number of total CD8+ T cells was similar between untreated and IL-15C-treated mice, and while the frequency of GAP50-specific CD8+ T cells was significantly reduced in the brains of IL-15C-treated mice, the total number of these cells, although decreased, did not reach significance (Figures 3D–3F). Hence, these data argue that IL-15C treatment does not substantially reduce the magnitude of the antigen-specific CD8+ T cell response in the spleen or completely prevent the accumulation of these cells in the brain during PbA infection.
Figure 3. IL-15C Treatment Has Little Effect on T Cell Numbers in the Spleen and Brain.

The spleens and brains of untreated or prophylactically IL-15C-treated PbA-infected mice (n = 9/group) were harvested on day 6 pi for CD8+ T cell analysis. Gating strategy to identify GAP50-specific CD8+ T cells in the spleen (A) and brain (D). The frequency (B and E) and number (C and F) of total and GAP50-specific CD8+ T cells in the spleen (B and C) and brain (E and F) were quantified. Data are combined from three independent experiments and presented as the mean ± SEM (B, C, E, F). *p < 0.05, **p < 0.01, **p < 0.01, ****p < 0.0001 as determined by two-way Student’s t test.
IL-15C Treatment Results in Decreased T Cell Activation and IFN-γ Production in the Brain
Although IL-15C treatment did not significantly decrease numbers of antigen-specific CD8+ T cells in the brains of PbA-infected mice on day 6 pi, we considered that there may be a reduction in the antigen-induced activation of those cells. To test this, we utilized Nr4a1GFP reporter mice: expression of Nur77 (encoded by Nr4a1) is directly proportional to the strength of TCR stimulation, thus GFP signal intensity is a readout for TCR stimulation intensity (Moran et al., 2011). During ECM, CD8+ T cells are thought to receive TCR stimulation in the microvessels in the brain when they detect specific peptide:MHC complexes on the surface of endothelial cells and/or monocytes (Howland et al., 2013, 2015b; Swanson et al., 2016). We detected Nr4a1GFP expression in CD8+ T cells isolated from the brains (Figure 4A) but not spleens (data not shown) of PbA-infected mice at 6 dpi. In agreement with previous studies (Howland et al., 2015a), this suggests that after being initially activated in the spleen and trafficking to the brain, CD8+ T cells receive additional TCR signals that lead to pathology. Bulk and GAP50-specific CD8+ T cells isolated from the brains of IL-15C-treated mice showed significantly reduced Nr4a1GFP induction, as measured by mean fluorescence intensity (MFI) and the frequency of Nr4a1GFP+ cells, compared with CD8+ T cell populations isolated from the brains of untreated mice (Figures 4A–4C). Additionally, the number of Nr4a1GFP+ GAP50-specific CD8+ T cells was significantly reduced in the brains of IL-15C-treated mice compared with untreated mice (Figure 4D). Since CD8+ T cell-mediated effector functions such as cytolysis and IFN-γ production are critical for ECM immunopathology (Belnoue et al., 2002; Nitcheu et al., 2003; Yañez et al., 1996), we also explored the impact of IL-15C treatment on cytokine expression using IfngYFP reporter mice (Reinhardt et al., 2009). Again, we found that, compared with untreated mice, IL-15C-treated mice had significantly reduced frequencies of IfngYFP+ total and GAP50-specific CD8+ T cells and reduced IfngYFP MFI of IfngYFP+ cells, although the total number of IfngYFP+ cells was not different between the groups (Figures 4E–4H). The reduction in IfngYFP MFI with IL-15C treatment was also observed in the spleen and blood (Figures S3A–S3C). Combined, these data show that IL-15C treatment results in reduced CD8+ T cell activation and cytokine production in the brain on day 6 pi.
Figure 4. IL-15C Treatment Results in Decreased T Cell Activation in the Brain on Day 6 pi.

The brains of PbA-infected Nr4a1(Nur77)GFP (n = 9/group) or IfngYFP mice (n = 6–7/group) that were left untreated or treated with IL-15C on days −5 and −3 relative to PbA infection were harvested on day 6 pi. Infiltrating leukocytes were isolated and analyzed by flow cytometry. Representative histograms of Nr4a1GFP or IfngYFP expression in total CD8 T cells or GAP50-specific CD8+ T cells from Nr4a1GFP (A) or IfngYFP (E) mice. To combine multiple experiments, the Nr4a1GFP (B) or IfngYFP (F) MFI was normalized and graphed. The frequency (C and G) and total number (D and H) of Nr4a1GFP+ (C and D) or IfngYFP+ (G and H) CD8 T cells were quantified. Data are combined from three (A–D) or two (E–H) independent experiments and presented as the mean ± SEM (B–D, F–H). *p < 0.05, **p < 0.01 as determined by one-way ANOVA followed by Tukey’s multiple comparison test. Related to Figure S3.
IL-15C Treatment Induces IL-10 Expression by NK Cells
Despite the expectation that IL-2C and IL-15C treatments would have similar effects on NK cells, we found that IL-15C but not IL-2C provokes NK cells to develop immunoregulatory properties that control ECM. This was not due to differential survival of NK cells stimulated by IL-2C versus IL-15C, since both populations showed comparable maintenance following adoptive transfer (Figures S4A–S4C). Therefore, we investigated the differentiation state and gene expression characteristics of NK cells stimulated by IL-2C versus IL-15C treatment. Expression of activation and maturation markers (including KLRG1, CD11b, and CD27) were broadly similar on NK cells from IL-2C- and IL-15C-treated mice (Figures S4D and S4E), although we did observe some differences in expression of maturation markers that implied a higher frequency of less mature NK cells in the IL-15C-treated population (Chiossone et al., 2009). NK cells from IL-2C- and IL-15C-treated mice produced similar levels of IFN-γ following ex vivo stimulation with IL-12 and IL-18 on days 0 and 3 after PbA infection (Figure S4F). In addition, serum IFN-γ levels were not different between untreated, IL-2C-treated, and IL-15C-treated mice on day 5 pi (Figure S4G), indicating that IL-15C treatment does not hinder IFN-γ production during PbA infection.
For a more global and unbiased analysis of differences between IL-2C- and IL-15C-treated NK cells, we subjected these cells to RNA-seq analysis. As expected, there were relatively few substantial (≥2-fold) and significant changes in gene expression between the two groups (Figure 5A). Expression from two genes that encode cell surface markers CD43 and Ly-6A was found to differ between IL-2C- and IL-15C-treated NK cells, and we confirmed these findings at the protein level by flow cytometry (Figures S4H and S4I), although the potential functional significance of these changes is unclear. Strikingly, we observed strong induction of Il10 in NK cells from IL-15C- but not IL-2C-treated mice (Figure 5A). Since IL-10 expression is difficult to detect by intracellular staining, we used Il10GFP reporter mice (Vert-X mice; Madan et al., 2009) to track Il10 expression following IL-2C or IL-15C treatment. NK cells from the spleen, blood, or brain of untreated or IL-2C-treated mice had minimal to no Il10GFP expression (Figure 5B). In contrast, more than half of splenic NK cells in IL-15-treated mice upregulated Il10GFP expression (Figure 5B). IL-15C treatment also resulted in Il10GFP expression by a small subset of CD8+ T cells while very little to no Il10GFP expression was detected in CD4+ T cells from any of the groups of mice (Figures S5A and S5B). Indeed, while some Il10GFP+ cells were detected among other lymphocyte subsets, including a small number of myeloid cells (data not shown), NK cells were the major population within Il10GFP+ cells in the spleen following IL-15C treatment (Figures S5C and S5D). Similarly, of the IL-10-expressing cells in the spleen, the largest number was NK cells (Figure S5E), suggesting that NK cells are a prominent source of the cytokine following IL-15C treatment.
Figure 5. IL-15C Treatments Induce IL-10 Expression in NK Cells.

(A) WT mice (n = 3/group) were treated with IL-2C on days −7, −5, and −3 or IL-15C on days −5 and −3 relative to harvest, and splenic NK1.1+NKp46+CD3− NK cells were FACS sorted. RNA-seq analysis was performed on RNA isolated from the purified NK cell populations. Genes shown were significantly differentially expressed between the two treatments with ≥2-fold change in expression. Arrowhead indicates Il10 on the heatmap.
(B and C) Il10GFP mice were left untreated (n = 11) or treated with IL-2C (n = 8) or IL-15C (n = 11) on days −5 and −3 relative to harvest.
(B) Representative histograms of Il10GFP expression in NK1.1+NKp46+TCRβ− cells in the spleen, blood, and brain.
(C and D) The frequency (C) and total number (D) of Il10GFP+ NK cells in the spleen, blood, and brain on days 0, 3, and 6 after PbA infection were quantified. Data are combined from four independent experiments and presented as the mean ± SEM (C and D); *p < 0.05, **p < 0.01, ****p < 0.0001 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
Related to Figures S4 and S5.
IL-10 production by NK cells has been reported previously in the context of various systemic inflammatory infections (Maroof et al., 2008; Perona-Wright et al., 2009; Tarrio et al., 2014). Therefore, we next sought to investigate whether IL-10 is produced by NK cells following PbA infection. Very few NK cells in the spleen, blood, or brain were Il10GFP+ on day 3 pi (Figure 5C); however, the majority of NK cells in those tissues expressed Il10GFP on day 6 pi (Figures 5C and 5D), suggesting that the systemic inflammation during ECM induces NK cells to express IL-10 but that it is induced too late to prevent death. We then analyzed Il10GFP expression in NK cells from the spleen, blood, and brain of mice treated with IL-2C or IL-15C prior to PbA infection. The frequency and total number of Il10GFP+ NK cells in each tissue was significantly increased in the IL-15C-treated mice compared to untreated and IL-2C-treated mice on day 3 pi (Figures 5C and 5D), while by day 6 pi, the majority of NK cells in the spleen, blood, and brain from all of the groups expressed Il10GFP, although the total numbers of NK cells had declined considerably (Figure 5D). These data indicate that IL-15C treatment induces IL-10-expressing NK cells prior to infection, while PbA infection itself can induce IL-10-expressing NK cells but only at late stages (6 dpi) of this rapidly progressing disease.
Human IL-15C Induces IL-10 Expression in Mouse and Human NK Cells and Protects Mice from ECM
IL-15C therapy is effective at controlling tumor growth in mice, and its potential use in humans has been proposed (Epardaud et al., 2008; Stoklasek et al., 2006). Indeed, ALT-803, a mutant IL-15N72D/IL-15Rα sushi domain-Fc fusion complex that is structurally similar to human IL-15C, has potent immune stimulatory function in various murine models and is currently in several clinical trials for treatment of hematological malignancies and solid tumors (Basher et al., 2016; Kim et al., 2016; Mathios et al., 2016; Rosario et al., 2016; Xu et al., 2013). Hence, we sought to determine whether our findings with mouse IL-15C would be mimicked by ALT-803 treatment. Treatment with either complex resulted in a similar increase in the frequency of splenic NK cells (Figure 6A) but ALT-803 treatment resulted in greater expansion of splenic lymphocytes, and the numbers of NK cells (Figure 6B) and CD8+ T cells (Figures S6A and S6B) was increased relative to IL-15C-treated animals. ALT-803 and IL-15C treatment produced similar frequencies of KLRG1+CD11b+ NK cells (Figure S6C), although there were differences in the patterns of CD11b and CD27 co-expression (Figure S6D). ALT-803 and IL-15C treatments induced similar frequencies and numbers of IL-10-expressing NK cells (Figures 6C and 6D), and NK cells comprised the majority of Il10GFP+ cells in both IL-15C- and ALT-803-treated mice (Figures S6E and S6F). Furthermore, as for mouse IL-15C, therapeutic treatment using ALT-803 was able to prevent ECM in most PbA-infected mice (Figure 6E). Despite inducing more IL-10-producing NK cells, ALT-803 treatment was slightly less efficacious than mouse IL-15C in protecting against ECM: this may relate to ALT-803 inducing greater expansion of CD8+ T cells (Figures S6A and S6B) (which may include pathogenic cells), but suboptimal dosing cannot be excluded. These data provide pre-clinical evidence that the human immunotherapeutic ALT-803 can control ECM.
Figure 6. Human IL-15C Treatment Induces IL-10 Expression in NK Cells and Protects Mice against ECM.

Il10GFP mice were left untreated (n = 5) or treated with IL-15C (n = 4) or ALT-803 (n = 5) on days −5 and −3 relative to harvest.
(A–D) The frequency (A) and number (B) of splenic NK cells were quantified. After gating on NK cells, the frequency (C) and number (D) of Il10GFP+ cells were quantified. Data are combined from two independent experiments and presented as the mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 as determined by one-way ANOVA followed by Tukey’s multiple comparison test.
(E) Primary human NK cells (n = 10 donors) were cultured for 6 days in the presence or absence of ALT-803 or IL-15 with or without IL-21 and with or without addition of IL-12 24 hr prior to harvest. IL-10 in the supernatant was quantified. Data are combined from three independent experiments and represented as the mean ± SEM; ND, not detected; ***p < 0.001, ****p < 0.0001 as determined by one-way ANOVA followed by Tukey’s multiple comparison test, comparing ALT-803 or IL-15 culture groups.
(F) Mice (n = 10/group) were infected with PbA then treated therapeutically with PBS, IL-15C, or ALT-803 on days 2 and 3 pi. Data are combined from two independent experiments.
Related to Figure S6.
These findings prompted investigation into whether human NK cells can also produce IL-10 in response to cytokine stimulation in vitro. Previous studies showed that IL-12 and IL-21 provoked IL-10 synthesis by proliferating mouse NK cells (Tarrio et al., 2014). Similarly, we found that human NK cells made IL-10 in response to ALT-803 (or free IL-15) but only in combination with IL-21 or with IL-12 and IL-21 (Figure 6F). Unlike our in vivo mouse studies, however, IL-2 was equally competent at cooperating with IL-12 and IL-21 to promote IL-10 production (Figure S6G). Nevertheless, these data indicate that human NK cells, like their mouse counterparts, can develop the ability to produce IL-10 in response to cytokine stimulation.
NK Cell-Derived IL-10 Is Required for IL-15C-Mediated Inhibition of ECM
Although our data suggested that IL-10 production was a hallmark of IL-15-stimulated NK cells, it was unclear whether IL-10 played a direct role in their protection against ECM. To address this, we first tested whether IL-10 expression was required for IL-15C-mediated protection from ECM. WT and Il10−/− mice were infected with PbA, then treated with IL-15C therapeutically. As expected (Niikura et al., 2011), untreated WT and Il10−/− mice succumbed to ECM with similar kinetics. However, while WT mice treated with IL-15C were protected from ECM, this was not the case for IL-15C-treated Il10−/− mice (Figure 7A), suggesting that IL-10 is essential for IL-15C-mediated protection from ECM. We observed no effect of IL-10 deficiency or IL-15C treatment on blood parasitemia (Figure S7A), suggesting that these effects relate to the development of cerebral malaria rather than changes in the infection per se. Since genetic deficiency of IL-10 might impact immune populations prior to infection, we also evaluated the effect of treating PbA-infected mice with IL-10R-blocking Ab during IL-15C therapy: 70% of these mice succumbed to ECM, whereas mice given a control Ab and IL-15C all survived ECM (Figure 7B). Once again, these differences in survival rates were not reflected in blood parasitemia (Figure S7B).
Figure 7. IL-10 Expression by NK Cells Is Required for IL-15C-Mediated Survival from ECM.

(A) Survival curve of WT or Il10−/− mice infected with PbA then left untreated or treated therapeutically with IL-15C on days 2 and 3 pi. Groups: PbA (WT), n = 15; PbA + IL-15C (WT), n = 15; PbA (Il10−/−), n = 7; PbA + IL-15C (Il10−/−), n = 13.
(B) Survival curve of mice infected with PbA then left untreated or treated with IL-15C on days 2 and 3 pi along with either a control Ab or anti-IL-10R Ab on days 1, 4, and 6 pi (n = 9–10 mice/group).
(C) 4 × 106 WT CD8+ T cells and 4 × 106 Il10rb−/− CD8+ T cells were transferred into congenically disparate WT mice on day −6, treated with IL-15C on days −5 and −3, and infected with PbA on day 0. Proportion of WT or Il10rb−/− CD8+ T cells in the spleen and brain on day 6 pi. *p = 0.01 as determined by 2-way ANOVA followed by Sidak’s multiple comparison test.
(D) Survival curve of mice infected with PbA then left untreated (n = 13), treated with IL-15C therapeutically (n = 14), or given 3 × 106 NK cells isolated from IL-15C-treated WT (n = 25) or Il10−/− mice (n = 21) on day 2 pi.
(A, B, D) **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by log-rank (Mantel-Cox) test. Related to Figure S7.
IL-10 acts on many cell types to restrain immune responses (Filippi and von Herrath, 2008). To investigate whether IL-10 acts directly on CD8+ T cells during ECM, we transferred polyclonal WT and IL-10Rβ-deficient CD8+ T cells into congenically disparate WT hosts, treated the recipients with IL-15C, and infected with PbA. While similar frequencies of donor WT and IL-10Rβ-deficient CD8+ T cells were found in the spleen on day 6 pi, the IL-10Rβ-deficient CD8+ T cells were significantly more abundant in the brain (Figure 7C). However, IFN-γ production was similar between the two donor populations (Figure S7G). These data suggest that IL-10R sensitivity by CD8+ T cells can directly impair their accumulation in the brain during ECM following IL-15C treatment but does not exclude potential effects of IL-10 on additional cell types or other CD8+ T cell functions during ECM.
It was still possible that NK cell production of IL-10 was a characteristic of IL-15C treatment but that this function was not required for NK cells to control ECM. We showed earlier that NK cells from IL-15C-treated mice could transfer protection against ECM (Figure 2E), offering an opportunity to test whether NK cell-derived IL-10 was important for their protective function. NK cells from IL-15C-treated WT or Il10−/− mice were transferred into WT recipients on day 2 post-PbA infection. Strikingly, while therapeutic transfer of WT NK cells protected against ECM, most PbA-infected mice receiving Il10−/− NK cells succumbed, similar to untreated controls (Figure 7D). Importantly, IL-15C treatment of Il10−/− mice induced similar expression of activation and maturation markers (Figures S7C–S7F), and NK cell adoptive transfer had no effect on parasitemia levels (Figure S7G). Taken together, our results indicate that IL-15C induces IL-10 production by NK cells and that this function is essential for NK cell-mediated protection against ECM.
DISCUSSION
NK cells deploy the same effector functions (cytolysis and IFN-γ production) that CD8+ T cells use to induce ECM, and some studies have suggested that NK cells actively contribute to ECM (Hansen et al., 2007; Ryg-Cornejo et al., 2013), while others have suggested that NK cells play no role (Yañez et al., 1996). We found that IL-15C-treated NK cells were necessary and sufficient to mediate the effects of IL-15C therapy but that they did so without interfering with parasite load. Furthermore, we found that NK cells were the predominant population of IL-10-producing cells induced by IL-15C treatment, that both NK cells and IL-10 were required for IL-15C-mediated protection against ECM, and that the capacity of IL-15C-treated NK cells to transfer protection depended on their ability to make IL-10.
Our finding that IL-2C treatment does not protect against ECM and also fails to induce IL-10 production by NK cells led us to reveal the key role of NK cell-produced IL-10 in control of the disease: the different effects of in vivo IL-2C and IL-15C treatments occurred despite the fact that the cytokine complexes are believed to signal through the same receptor chains and both lead to expansion of NK cells and memory phenotype CD8+ T cells (Boyman et al., 2006; Votavova et al., 2014). In vitro studies show that mouse NK cells stimulated to proliferate (by MCMV infection or culture with high doses of IL-2) make IL-10 in response to IL-12 or IL-21 (Tarrio et al., 2014), and our studies suggest that either IL-2 or IL-15 can cooperate with IL-21 and IL-12 to induce human NK cells to make IL-10. Why IL-15C but not IL-2C induces IL-10 expression in mouse NK cells in vivo is unclear, but it is possible that IL-15C uniquely promotes production of cytokines (such as IL-12 or IL-21) to act on proliferating NK cells, or that this difference reflects distinct pharmacokinetics of IL-2C and IL-15C. Further studies will be required to resolve this issue. Regardless, our data indicate that both mouse and human NK cells can be induced to produce IL-10 in response to cytokine stimulation, suggesting that human NK cells may also play immunoregulatory roles and could perhaps be harnessed for control of immunopathological diseases in humans.
A host-protective role for IL-10 in malaria has been proposed previously (Hunt and Grau, 2003; Niikura et al., 2011). Indeed, CBA/J mice treated with recombinant IL-10 are protected from developing ECM 70% of the time (Kossodo et al., 1997), and ECM-resistant BALB/c mice develop ECM following treatment with IL-10R blocking Ab (Claser et al., 2017). Moreover, BALB/c mice treated with anti-IL-10R Ab have an increased number of IFN-γ-producing T cells on day 6 pi (Claser et al., 2017). This is consistent with our findings that IL-15C-treated mice had reduced IFN-γ-expressing T cells in the spleen and brain on day 6 pi. Studies investigating the effect of co-infections on malaria pathogenesis have identified that co-infection skews the immune system, resulting in altered disease pathogenesis and immunity (Onkoba et al., 2016; Rénia and Potter, 2006). Mice co-infected with PbA and the non-lethal PbXAT strain or previously infected with the filarial parasite Litomosoides sigmodontis were protected from ECM, and this protection was lost in Il10−/− mice (Niikura et al., 2011; Specht et al., 2010). As a large proportion of NK cells from IL-15C-treated mice express IL-10, we hypothesized that IL-10 would be critical for IL-15C-mediated protection from ECM. Indeed, Il10−/− mice treated therapeutically with IL-15C and WT mice treated therapeutically with IL-15C and an IL-10R blocking Ab were not protected from ECM, indicating that IL-10 is required for IL-15C-mediated protection from ECM. Critically, we found that NK cells isolated from IL-15C-treated Il10−/− mice do not protect mice from ECM, indicating that NK cell-derived IL-10 is required for IL-15C-mediated protection from ECM.
IL-10-producing NK cells have been identified following other systemic infections, including MCMV, LM, Leishmania donovi, and Toxoplasma gondii, in a pathway that involves IL-12 (Maroof et al., 2008; Perona-Wright et al., 2009; Tarrio et al., 2014). The impact of IL-10-producing NK cells on disease progression varies with the infection. During experimental visceral leishmaniasis and LM infection, IL-10-producing NK cells inhibited resistance to infection, resulting in increased microbial burdens (Clark et al., 2016; Maroof et al., 2008). In contrast, during MCMV infection, IL-10-expressing NK cells protected mice from CD8+ T cell-mediated death (Lee et al., 2009). Our studies showed that IL-15C treatment did not diminish the magnitude of the PbA-specific CD8+ T cell response but did cause significantly reduced activation and IFN-γ production by these cells in the brain and spleen, suggesting a more nuanced effect.
IL-10 produced by IL-15C-treated NK cells could suppress T cells directly during ECM, either in the spleen during initial antigen encounter or in the brain vasculature as the CD8+ T cells traffic to and accumulate in that organ. As we did not detect IL-10 in the serum of IL-15C-treated PbA-infected mice (data not shown), we favor the hypothesis that IL-10 is acting locally in tissues such as the brain rather than systemically. We saw only a modest decrease in GAP50-specific CD8+ T cells in the brains of IL-15C-treated mice after ECM; but, in direct competition studies, we found a significantly increased proportion of IL-10Rβ-deficient CD8+ T cells accumulate in the brain compared with WT CD8+ T cells, suggesting that CD8+ T cells are directly inhibited by IL-10. These experiments did not reveal increased IFN-γ production by IL-10Rβ-deficient CD8+ T cells, raising the possibility that IL-10 could be directly inhibiting pathogenic functions beyond IFN-γ. Additionally, IL-10 could inhibit T cell activation indirectly by suppressing dendritic cell (DC) activation in the spleen early after infection. IL-10 decreases the expression of MHC and costimulatory molecules on antigen-presenting cells (Filippi and von Herrath, 2008). Moreover, NK cell-derived IL-10 has been shown to suppress IL-12 production from DCs in vitro (Perona-Wright et al., 2009). Since PbA-specific CD8+ T cells are primed in the spleen by CD8α+ DCs (Lundie et al., 2008), reduced DC activation could contribute to the diminished CD8+ T cell activation seen on day 6 pi. Endothelial cells in the brain vasculature could also be affected by IL-10 production from IL-15C-stimulated NK cells. Indeed, IL-10 signaling suppresses T cell stimulation by endothelial cells in vitro (Gleissner et al., 2007). In ECM, it has been hypothesized that endothelial cells present PbA peptides to CD8+ T cells in the vasculature of the brain, resulting in disruption of the BBB (Howland et al., 2015a; Swanson et al., 2016). Thus, the reduced edema seen in IL-15C-treated mice could be a result of decreased endothelial cell-mediated presentation of PbA antigens to CD8+ T cells. However, the complete model by which pathogenic CD8+ T cells promote ECM is still being unraveled (Huggins et al., 2017; Shaw et al., 2015). Future studies will further investigate the mechanism by which IL-15C treatment results in reduced CD8+ T cell activation in the brain and the different cell types directly affected by IL-10.
Our data show that IL-15C can promote generation of IL-10-producing NK cells and protect against lethal ECM. Similar findings were also observed using a human IL-15 “superagonist” complex (ALT-803), which is an engineered form of human mutant IL-15N72D/human IL-15Rα currently being tested in clinical trials for safety and efficacy in cancer therapies (Floros and Tarhini, 2015). Human NK cells have been shown to produce IL-10 (Park et al., 2011), and peripheral blood NK cells from patients with chronic viremia after HCV infection produced IL-10 following cell-mediated triggering (De Maria et al., 2007).
Our finding that IL-15C treatment can ameliorate pathogenic CD8+ T cell responses during PbA infection while also being able to promote bacterial control during LM infection suggests that appropriate use of such complexes may permit both enhancement and restraint of the immune response, with NK cells serving as important regulators in this process. This flexibility holds promise for the use of IL-15C in adjunctive therapies for human diseases, potentially including CM.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NK1.1 (clone PK136) | BioLegend | Cat#108739; RRID: AB_2562273 |
| NKp46 (clone 29A1.4) | BioLegend | Cat#137611; RRID: AB_10915472 |
| CD3ε (clone 17A2) | BioLegend | Cat#100217; RRID: AB_1595597 |
| TCRβ (clone H57-597) | eBioscience | Cat#47-5961-82; RRID: AB_1272173 |
| CD4 (clone RM4-5) | BD Biosciences | Cat#557956; RRID: AB_396956 |
| CD8α (clone 53-6.7) | BD Biosciences | Cat#563332; RRID: AB_2721167 |
| CD44 (clone IM7) | BioLegend | Cat#103044; RRID: AB_2650923 |
| Ly-6A (clone D7) | eBioscience | Cat#12-5981-81; RRID: AB_466085 |
| CD43 (clone 1B11) | BioLegend | Cat#121217; RRID: AB_528812 |
| CD11b (clone M1/70) | BD Biosciences | Cat#565976; RRID: AB_2721166 |
| CD27 (clone LG.7F9) | eBioscience | Cat#17-0271-81; RRID: AB_469369 |
| KLRG1 (clone 2F1) | TONBO Biosciences | Cat#20-5893; RRID: AB_2621607 |
| IFN-γ (clone XMG1.2) | TONBO Biosciences | Cat#60-7311; RRID: AB_2621871 |
| CD45.2 (clone 104) | BioLegend | Cat#109813; AB_389210 |
| Ter119 | eBioscience | Cat#25-5921-82; RRID: AB_469661 |
| CD3ε (clone 500A2) | BD Biosciences | Cat#553239; RRID: AB_394728 |
| NK1.1 (clone PK136) | Bio X Cell | Cat#BE0036 |
| IL-10R (clone 1B1.3A) | Bio X Cell | Cat#BE0050 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes-OVA | Dr. Hao Shen, U of Pennsylvania | (Pope et al., 2001) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse recombinant IL-15 | eBioscience | Cat#14-8153-80 |
| Mouse recombinant IL-15Rα-Fc | R&D Systems | Cat#551-MR |
| Mouse recombinant IL-2 | eBioscience | Cat#14-8021-64 |
| Recombinant Anti-IL-2 (S4B6) | Bio X Cell | Cat#BE0043-1 |
| ALT-803 | Altor Bioscience Corp | Cat#ALT-803 |
| Glutaraldehyde | Calbiochem | Cat#354400 |
| Hoescht | Sigma-Aldrich | Cat#B2261 |
| Dihydroethidium | Sigma-Aldrich | Cat#37291 |
| Percoll | GE HealthCare | Cat#17-0891-09 |
| Streptavidin-APC | ThermoFisher | Cat#S868 |
| Streptavidin-PE | ThermoFisher | Cat#S21388 |
| Critical Commercial Assays | ||
| Mouse NK isolation kit II | Miltenyi Biotec | Cat#130-096-892 |
| Foxp3 Fixation/Permeabilization Kit | eBioscience | Cat#00-5521-00 |
| IFN-γ ELISA kit | eBioscience | Cat#50-173-21 |
| Deposited Data | ||
| RNA-seq | This paper | GEO: 109586 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Charles River Laboratories | C57BL/6NCrl |
| C57BL/6-Ly5.1 | Charles River Laboratories | B6.SJL-PtprcaPepcb/BoyCrl |
| Nr4a1GFP | Dr. Kristin Hogquist | (Moran et al., 2011) |
| IfngYFP | The Jackson Laboratory | JAX: 017581 |
| Il10GFP | The Jackson Laboratory | JAX: 014530 |
| Il10−/− | The Jackson Laboratory | JAX: 002251 |
| Il10rb−/− | The Jackson Laboratory | JAX: 005027 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc. | https://www.graphpad.com/ |
| TopHat | (Kim et al., 2013) | http://cole-trapnell-lab.github.io/projects/tophat/ |
| CuffLinks | (Trapnell et al., 2010, 2013) | http://cole-trapnell-lab.github.io/projects/cufflinks/ |
| CummRbund | (Goff et al., 2014) | http://compbio.mit.edu/cummeRbund/ |
| Other | ||
| Plasmodium berghei ANKA | Dr. Susan Pierce, NIH | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sara Hamilton Hart (hamil062@umn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All strains are on the C57BL/6 background: C57BL/6, C57BL/6-Ly5.1, Nr4a1GFP, IfngYFP, Il10GFP, Il10−/−, and Il10rb−/−. All experimental mice, both male and female, were 8–12 weeks old, were housed in specific pathogen-free conditions, and were bred following all Institutional Animal Care and Use Committee Procedures at the University of Minnesota.
Microbe strains
Plasmodium berghei ANKA, originally obtained from Dr. Susan Pierce at the NIH, was passaged in vivo, and stocks frozen in Alsever’s solution and glycerol (9:1 ratio) were stored in liquid nitrogen until use. Recombinant Listeria monocytogenes strains LM-OVA or attenuated LM-OVA (both expressing secreted OVA protein) have been described elsewhere (Pope et al., 2001). Bacteria were grown in tryptic soy broth with 50 μg/ml streptomycin to an OD600 of 0.1.
METHOD DETAILS
Cytokine complex treatment
IL-2C was formed by combining 1.0 μg carrier-free recombinant mouse IL-2 (eBioscience) with 10 μg anti-IL-2 Ab S4B6 (Bio X Cell). IL-15C was formed by combining 0.75 μg IL-15 (eBioscience) with 7 μg IL-15Rα-Fc (R&D Systems) and incubating for 20–30 min at 37°C. ALT-803 (IL-15N72D/IL-15RαSu-Fc) was generated as previously described (Han et al., 2011) and was provided by Altor BioScience Corporation; 10 μg was injected per mouse per treatment.
Plasmodium berghei ANKA infections
Freshly passaged P. berghei ANKA-infected RBCs (0.5 – 1 × 106) were injected (i.v. or i.p.) into experimental mice, as previously described (de Oca et al., 2013). A donor mouse was infected with a stock vial of PbA-infected RBCs. Once parasitemia levels reached > 2%, usually on day 6 pi, blood from the donor mouse was collected via cardiac puncture. RBCs were counted, and blood was diluted in PBS for infection of experimental mice. Alternatively, iRBCs from donor mice were cryopreserved in liquid nitrogen, and freshly thawed stocks were used as the source for infection of experimental mice.
Quantification of peripheral blood parasitemia by flow cytometry
Parasitemia was determined by flow cytometry using a modification of a previously described method (Gordon et al., 2015; Malleret et al., 2011). Blood was obtained from mouse tail veins, fixed with 1 mL 0.025% aqueous glutaradehyde solution (Calbiochem), washed with 2 mL PBS, and stained with the following: the DNA dye Hoechst 33342 (Sigma) (8 μM), the DNA and RNA dye dihydroethidium (diHEt; Sigma) (10 μg/mL), the pan C57BL/6 lymphocyte marker CD45.2 (BioLegend), and the RBC marker Ter119 (eBioscience). Cells were analyzed on a BD LSR Fortessa flow cytometer equipped with UV (325 nm), violet (407 nm), blue (488 nm), and red (633 nm) lasers. Data were analyzed using FlowJo software (Tree Star Technologies). Parasitemia was calculated as the percent of RBCs (Ter119+CD45.2− cells) that stained positive for Hoescht and diHEt.
FITC-albumin permeability assay
Mice were injected intravenously with 10 mg FITC-albumin (Sigma-Aldrich). After a one hour incubation, brains were harvested, flash frozen, and stored at −80°C. Frozen brains were homogenized and spun down. The total protein concentration of the brain lysate was quantified by BCA. Equal protein was loaded, and the tissue fluorescence intensity was quantified using 488 nm excitation and 525 nm emission on a Synergy H1 plate reader (BioTek).
Magnetic resonance imaging
Magnetic resonance images were acquired using a Bruker Avance II 7 Tesla vertical bore small animal MRI system (Bruker Biospin). Mice were anesthetized using isofluorane for scanning. For T1-weighted scans, mice were administered an intraperitoneal injection of gadolinium (100mg/kg), as initially reported (Johnson et al., 2012). T1-weighted MSME sequence with TR: 300ms, TE: 9.5ms, FOV: 4.0×2.0×2.0cm, matrix: 192×96×96 was used. For T2-weighted scans a RARE pulse sequence was used with repetition time (TR) = 1500ms, echo time (TE) = 70ms, RARE factor: 16, field of view (FOV): 3.2×1.92×1.92cm, matrix: 256×128×128. T2-weighted MRI: MSME, TR: 300ms, TE: 9.5ms, FOV: 4.0×2.0×2.0cm, matrix: 192×96×96. MRI scans were analyzed using Analyze 10.0 (Biomedical Imaging Resource, Mayo Clinic).
NK cell depletion, enrichment, and adoptive transfer
NK cells were depleted by injection of 600 μg of anti-NK1.1 (PK136) on days −3, −1 and +1 relative to PbA infection. In separate experiments, NK cells were enriched from the spleen via negative selection (Miltenyi Biotec NK cell isolation kit II). To further deplete T cells from the splenocyte suspension, we added 2 μg/ml biotinylated anti-CD3ε Ab (clone 500A2, BD Biosciences) to the Miltenyi Biotin Ab Cocktail. Following enrichment, an aliquot of cells were stained for NK1.1, NKp46, TCRβ, and CD1d tetramer to determine purity; NK cells were identified as NK1.1+NKp46+TCRβ−CD1d tetramer−. Based on the NK cell purity, 3 × 106 NK cells were injected i.v. into recipient mice in 200 μl PBS.
Anti-IL-10R Ab treatment
Mice were treated with 200 μg of anti-IL-10R Ab (clone 1B1.3A, Bio X Cell) or a control rat IgG1 Ab (clone HRPN, Bio X Cell) on days +1, +4, and +6 and treated with IL-15C on days +2 and +3 relative to PbA infection.
Leukocyte isolation and flow cytometry
Mice were euthanized by CO2 overdose then bled by cardiac puncture to remove circulating blood cells, and spleens and brains were removed. Blood was collected in 1.5 mL tubes containing 30 μl of heparin. After the volume of blood was determined, blood samples were treated twice with ACK lysis buffer to lyse RBCs. The cells were washed and resuspended in FACS buffer consisting of PBS and 2% FBS. Spleens were mashed through 70 μm cell strainers (BD Bioscience), and RBCs were lysed using ACK lysis buffer. The cells were washed and resuspended in FACS buffer (PBS with 2% FBS). To obtain leukocytes from the brain, dissected brains were minced and digested in 1 mg/mL collagenase and incubated in an orbital shaker for 30 min at 37°C. Tissues were further homogenized using a GentleMACS Dissociator (Miltenyi Biotec). After the tissue was passed through 70-μm strainer, homogenates were placed on a 90–60%–40% discontinuous Percoll gradient and centrifuged for 18 min at 1,000 × g at 4°C. After centrifugation, the cells at the 40%–60% and 90%–60% interfaces containing leukocytes were collected and washed with 1X HBSS. The pellet was then treated with ACK lysis buffer to lyse RBCs. The cells were washed and resuspended in FACS buffer (PBS with 2% FBS). The following fluorescent dye-conjugated antibodies specific for the following cell-surface markers were used for staining: anti-NK1.1 (PK136, BioLegend), anti-NKp46 (29A1.4, BioLegend), anti-CD3ε (17A2, BioLegend), anti-TCRβ (H57-597, eBioscience), anti-CD4 (RM4-5, BD Biosciences), anti-CD8α (53-6.7, BD Biosciences), anti-CD44 (IM7, BioLegend), anti-Ly-6A (D7, eBioscience), anti-CD43 (1B11, BioLegend), anti-CD11b (M1/70, BD Biosciences), anti-CD27 (LG.7F9, eBioscience), anti-KLRG1 (2F1, TONBO Biosciences). Db-GAP50 monomer was obtained from the NIH tetramer core facility and tetramerized with streptavidin-allophycocyanin (APC, LifeTechnologies). CD1d monomer was obtained from the NIH tetramer core facility and tetramerized with streptavidin-phycoerythrin (PE, LifeTechnologies).
For analysis of IFN-γ production by NK cells, spleens were processed as described above and 4 × 106 splenocytes were plated in 24 well plates in RPMI medium containing Golgi Plug (BD Biosciences), IL-12 (10 ng/mL), and IL-18 (10 ng/mL). After a 4 h incubation at 37°C, cells were stained for surface markers as described above. Following fixation/permeabilization (Foxp3 Kit, eBioscience), cells were stained for the presence of intracellular IFN-γ (XMG1.2, TONBO Biosciences).
Samples were acquired on a BD LSRII Fortessa using BD FACSDiva (BD Bioscience), and data were analyzed with FlowJo v9 software (Tree Star Technologies).
Listeria monocytogenes infections and determination of CFU
For LM-OVA challenges, 2 × 105 CFU were injected i.v. The actual number of bacteria injected was confirmed by dilution and growth on tryptic soy broth plates containing streptomycin. On day 5 post-LM infection, the spleen was removed and placed in a 0.2% IGEPAL solution (Sigma-Aldrich). Organs were homogenized, and serial dilutions were plated onto tryptic soy broth plates containing 50 μg/ml streptomycin. Bacterial colonies were counted after plate incubation for 24 h at 37°C. The limit of detection (100 organisms) is indicated by a dashed line on graphs in the figure.
IFN-γ ELISA
On day 5 pi, blood was collected by cardiac puncture, allowed to clot for 30 minutes at room temperature, and spun at 10,000 RPM for 10 min at room temperature. The serum layer was collected and analyzed for IFN-γ via ELISA (eBioscience).
Human NK cell analysis
Peripheral blood was drawn from healthy donors with written consent. The institutional review board at the University of Minnesota approved these studies, and all research was conducted in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque Premium (GE Healthcare). CD3−CD56+ NK cells were isolated by negative selection using the EasySep Human NK Cell Isolation Kit (STEMCELL Technologies). Primary NK cells were cultured for 7 days at a concentration of 0.5×106 cells per ml in B0 media (Cichocki and Miller, 2010) with 20% heat-inactivated human AB serum. The following recombinant cytokines were added alone or in combination: ALT-803 (72 ng/mL, Altor), IL-15 (NCI), IL-2 (10 ng/mL, Peprotech), IL-21 (50 ng/mL, eBioscience), and IL-12 (1 ng/mL, R&D Systems). IL-12 was added on day 6 for overnight stimulation. Supernatants were collected before and after culture for analysis of IL-10 concentration by ELISA using the Human IL-10 Quantikine ELISA Kit (R&D Systems).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
RNA was isolated from FACS-sorted NK cells (NK1.1+NKp46+CD3−CD1d tetramer−) using a RNAeasy kit (QIAGEN). RNA was submitted to the University of Minnesota Genomics Core for HiSeq 2500 analysis (paired-end reads of 125 base pairs). The resulting data (in triplicate) was aligned to the mouse genome (mm10) using TopHat [v2.0.13; (Kim et al., 2013)], and gene expression was quantified and tested for differential expression using cufflinks [v2.2.1; (Trapnell et al., 2010, 2013)]. A heatmap of significantly differentially expressed genes where at least a 2-fold change in expression between treatments was constructed using CummeRbund [v2.16.0; (Goff et al., 2014)].
Statistical analysis
All data were analyzed on GraphPad Prism 6. Data were evaluated for statistically significant differences using a two-tailed, unpaired t test with or without Welch’s correction, a one-way analysis of variance (ANOVA) test followed by Tukey’s multiple comparison test, a two-way ANOVA followed by a Bonferroni multiple comparison test, or a log-rank (Mantel-Cox) test. A P-value < 0.05 was considered statistically significant. All differences not specifically indicated to be significant were not significant (p > 0.05).
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO 109586.
Supplementary Material
Highlights.
IL-15 complex (IL-5C) treatment protects mice from experimental cerebral malaria (ECM)
NK cell-derived IL-10 is required for IL-15C-mediated survival from ECM
IL-15C inhibits CD8+ T cell activation and cytokine production in the brain
Human NK cells also produce IL-10 after cytokine stimulation
Acknowledgments
We thank Susan Pierce, PhD for parasites and advice. We thank Kristin Hogquist, PhD, and Thomas Morrison, PhD, for advice and critical review of the manuscript. This project was supported by a Grant-in-Aid from the University of Minnesota Office of the Vice President for Research (S.E.H.) and NIH grants R21-AI100088 (S.C.J. and S.E.H.), F32-AI120312-01 (K.S.B.), R56-NS094150 (A.J.J.), and T32-AI7425-19 (M.A.H.).
Footnotes
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.immuni.2018.03.012.
AUTHOR CONTRIBUTIONS
Experiments were performed by K.S.B., M.A.H., E.T., P.D., and S.E.H. RNA-seq data were analyzed by C.M.H. and R.Y. S.A., E.K.J., and H.C.W. contributed to the development of ALT-803 and provided this reagent for the current study. G.T.H., F.C., J.S.M., and M.F. contributed to the design of the study. G.T.H. and A.J.J. provided technical guidance. F.C., J.S.M., and M.F. supervised the human NK cell experiments. S.C.J. and S.E.H. supervised the project. K.S.B., S.C.J., and S.E.H. wrote the manuscript.
DECLARATION OF INTERESTS
S.A., E.K.J., and H.C.W. are employees and stockholders of Altor Bioscience Corporation. H.C.W. holds a patent on ALT-803 (US Patent No. 8,507,222). No other competing interests were declared by the other authors.
References
- Basher F, Jeng EK, Wong H, Wu J. Cooperative therapeutic anti-tumor effect of IL-15 agonist ALT-803 and co-targeting soluble NKG2D ligand sMIC. Oncotarget. 2016;7:814–830. doi: 10.18632/oncotarget.6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Rénia L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- Biron CA. Yet another role for natural killer cells: cytotoxicity in immune regulation and viral persistence. Proc Natl Acad Sci USA. 2012;109:1814–1815. doi: 10.1073/pnas.1120528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- Cichocki F, Miller JS. In vitro development of human killer-immunoglobulin receptor-positive NK cells. Methods Mol Biol. 2010;612:15–26. doi: 10.1007/978-1-60761-362-6_2. [DOI] [PubMed] [Google Scholar]
- Clark SE, Filak HC, Guthrie BS, Schmidt RL, Jamieson A, Merkel P, Knight V, Cole CM, Raulet DH, Lenz LL. Bacterial manipulation of NK cell regulatory activity increases susceptibility to Listeria monocytogenes infection. PLoS Pathog. 2016;12:e1005708. doi: 10.1371/journal.ppat.1005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claser C, De Souza JB, Thorburn SG, Grau GE, Riley EM, Rénia L, Hafalla JCR. Host Resistance to Plasmodium-induced acute immune pathology is regulated by interleukin-10 receptor signaling. Infect Immun. 2017;85:85. doi: 10.1128/IAI.00941-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crome SQ, Lang PA, Lang KS, Ohashi PS. Natural killer cells regulate diverse T cell responses. Trends Immunol. 2013;34:342–349. doi: 10.1016/j.it.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Crouse J, Xu HC, Lang PA, Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. 2015;36:49–58. doi: 10.1016/j.it.2014.11.001. [DOI] [PubMed] [Google Scholar]
- De Maria A, Fogli M, Mazza S, Basso M, Picciotto A, Costa P, Congia S, Mingari MC, Moretta L. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol. 2007;37:445–455. doi: 10.1002/eji.200635989. [DOI] [PubMed] [Google Scholar]
- de Oca MM, Engwerda C, Haque A. Plasmodium berghei ANKA (PbA) infection of C57BL/6J mice: a model of severe malaria. Methods Mol Biol. 2013;1031:203–213. doi: 10.1007/978-1-62703-481-4_23. [DOI] [PubMed] [Google Scholar]
- Dende C, Meena J, Nagarajan P, Panda AK, Rangarajan PN, Padmanaban G. Simultaneously targeting inflammatory response and parasite sequestration in brain to treat experimental cerebral malaria. Sci Rep. 2015;5:12671. doi: 10.1038/srep12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, Hamerman JA, Goldrath AW, Turley SJ. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008;68:2972–2983. doi: 10.1158/0008-5472.CAN-08-0045. [DOI] [PubMed] [Google Scholar]
- Fauconnier M, Palomo J, Bourigault ML, Meme S, Szeremeta F, Beloeil JC, Danneels A, Charron S, Rihet P, Ryffel B, et al. IL-12Rβ2 is essential for the development of experimental cerebral malaria. J Immunol. 2012;188:1905–1914. doi: 10.4049/jimmunol.1101978. [DOI] [PubMed] [Google Scholar]
- Filippi CM, von Herrath MG. IL-10 and the resolution of infections. J Pathol. 2008;214:224–230. doi: 10.1002/path.2272. [DOI] [PubMed] [Google Scholar]
- Floros T, Tarhini AA. Anticancer cytokines: biology and clinical effects of interferon-α2, interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol. 2015;42:539–548. doi: 10.1053/j.seminoncol.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleissner CA, Zastrow A, Klingenberg R, Kluger MS, Konstandin M, Celik S, Haemmerling S, Shankar V, Giese T, Katus HA, Dengler TJ. IL-10 inhibits endothelium-dependent T cell costimulation by up-regulation of ILT3/4 in human vascular endothelial cells. Eur J Immunol. 2007;37:177–192. doi: 10.1002/eji.200636498. [DOI] [PubMed] [Google Scholar]
- Goff L, Trapnell C, Kelley D. CummeRbund: visualization and exploration of cufflinks high-throughput sequencing data. 2014 https://www.bioconductor.org/packages/3.7/bioc/vignettes/cummeRbund/inst/doc/cummeRbund-manual.pdf.
- Gordon EB, Hart GT, Tran TM, Waisberg M, Akkaya M, Kim AS, Hamilton SE, Pena M, Yazew T, Qi CF, et al. Targeting glutamine metabolism rescues mice from late-stage cerebral malaria. Proc Natl Acad Sci USA. 2015;112:13075–13080. doi: 10.1073/pnas.1516544112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SE, Schenkel JM, Akue AD, Jameson SC. IL-2 complex treatment can protect naive mice from bacterial and viral infection. J Immunol. 2010;185:6584–6590. doi: 10.4049/jimmunol.1001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, Vyas VV, Marcus WD, Chavaillaz PA, Romero CA, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine. 2011;56:804–810. doi: 10.1016/j.cyto.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DS, Bernard NJ, Nie CQ, Schofield L. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol. 2007;178:5779–5788. doi: 10.4049/jimmunol.178.9.5779. [DOI] [PubMed] [Google Scholar]
- Hora R, Kapoor P, Thind KK, Mishra PC. Cerebral malaria–clinical manifestations and pathogenesis. Metab Brain Dis. 2016;31:225–237. doi: 10.1007/s11011-015-9787-5. [DOI] [PubMed] [Google Scholar]
- Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Rénia L. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med. 2013;5:984–999. doi: 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland SW, Claser C, Poh CM, Gun SY, Rénia L. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015a;37:221–231. doi: 10.1007/s00281-015-0476-6. [DOI] [PubMed] [Google Scholar]
- Howland SW, Poh CM, Rénia L. Activated brain endothelial cells cross-present malaria antigen. PLoS Pathog. 2015b;11:e1004963. doi: 10.1371/journal.ppat.1004963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins MA, Johnson HL, Jin F, Songo NA, Hanson LM, LaFrance SJ, Butler NS, Harty JT, Johnson AJ. Perforin expression by CD8 T cells is sufficient to cause fatal brain edema during experimental cerebral malaria. Infect Immun. 2017;85:85. doi: 10.1128/IAI.00985-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- Johnson HL, Chen Y, Jin F, Hanson LM, Gamez JD, Pirko I, Johnson AJ. CD8 T cell-initiated blood-brain barrier disruption is independent of neutrophil support. J Immunol. 2012;189:1937–1945. doi: 10.4049/jimmunol.1200658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D, Sawa Y, Sato M, Agung E, Hirano T, Murakami M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. J Immunol. 2006;177:306–314. doi: 10.4049/jimmunol.177.1.306. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, Schlom J, Hodge JW. IL-15 superagonist/IL-15RαSushi-Fc fusion complex (IL-15SA/IL-15RαSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget. 2016;7:16130–16145. doi: 10.18632/oncotarget.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology. 1997;91:536–540. doi: 10.1046/j.1365-2567.1997.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med. 2009;206:2235–2251. doi: 10.1084/jem.20082387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci USA. 2008;105:14509–14514. doi: 10.1073/pnas.0806727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret B, Claser C, Ong ASM, Suwanarusk R, Sriprawat K, Howland SW, Russell B, Nosten F, Rénia L. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci Rep. 2011;1:118. doi: 10.1038/srep00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, Wong HC, Pardoll DM, Lim M. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer. 2016;138:187–194. doi: 10.1002/ijc.29686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura M, Inoue S, Kobayashi F. Role of interleukin-10 in malaria: focusing on coinfection with lethal and nonlethal murine malaria parasites. J Biomed Biotechnol. 2011;2011:383962. doi: 10.1155/2011/383962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol. 2003;170:2221–2228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- Onkoba WN, Chimbari MJ, Kamau JM, Mukaratirwa S. Differential immune responses in mice infected with the tissue-dwelling nematode Trichinella zimbabwensis. J Helminthol. 2016;90:547–554. doi: 10.1017/S0022149X15000723. [DOI] [PubMed] [Google Scholar]
- Park JY, Lee SH, Yoon SR, Park YJ, Jung H, Kim TD, Choi I. IL-15-induced IL-10 increases the cytolytic activity of human natural killer cells. Mol Cells. 2011;32:265–272. doi: 10.1007/s10059-011-1057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penet MF, Viola A, Confort-Gouny S, Le Fur Y, Duhamel G, Kober F, Ibarrola D, Izquierdo M, Coltel N, Gharib B, et al. Imaging experimental cerebral malaria in vivo: significant role of ischemic brain edema. J Neurosci. 2005;25:7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, Johnson LL, Smiley ST, Mohrs M. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe. 2009;6:503–512. doi: 10.1016/j.chom.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C, Kim SK, Marzo A, Masopust D, Williams K, Jiang J, Shen H, Lefrançois L. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J Immunol. 2001;166:3402–3409. doi: 10.4049/jimmunol.166.5.3402. [DOI] [PubMed] [Google Scholar]
- Potchen MJ, Kampondeni SD, Seydel KB, Birbeck GL, Hammond CA, Bradley WG, DeMarco JK, Glover SJ, Ugorji JO, Latourette MT, et al. Acute brain MRI findings in 120 Malawian children with cerebral malaria: new insights into an ancient disease. AJNR Am J Neuroradiol. 2012;33:1740–1746. doi: 10.3174/ajnr.A3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10:385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rénia L, Potter SM. Co-infection of malaria with HIV: an immunological perspective. Parasite Immunol. 2006;28:589–595. doi: 10.1111/j.1365-3024.2006.00903.x. [DOI] [PubMed] [Google Scholar]
- Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, Han K, Jeng EK, Rhode PR, Leong JW, et al. The IL-15-based ALT-803 complex enhances FcγRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin Cancer Res. 2016;22:596–608. doi: 10.1158/1078-0432.CCR-15-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, Sprent J. Converting IL-15 to a superagonist by binding to soluble IL-15Ralpha. Proc Natl Acad Sci USA. 2006;103:9166–9171. doi: 10.1073/pnas.0600240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryg-Cornejo V, Nie CQ, Bernard NJ, Lundie RJ, Evans KJ, Crabb BS, Schofield L, Hansen DS. NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology. 2013;218:263–271. doi: 10.1016/j.imbio.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Shaw TN, Stewart-Hutchinson PJ, Strangward P, Dandamudi DB, Coles JA, Villegas-Mendez A, Gallego-Delgado J, van Rooijen N, Zindy E, Rodriguez A, et al. Perivascular arrest of CD8+ T cells is a signature of experimental cerebral malaria. PLoS Pathog. 2015;11:e1005210. doi: 10.1371/journal.ppat.1005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht S, Ruiz DF, Dubben B, Deininger S, Hoerauf A. Filaria-induced IL-10 suppresses murine cerebral malaria. Microbes Infect. 2010;12:635–642. doi: 10.1016/j.micinf.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Stoklasek TA, Schluns KS, Lefrançois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol. 2006;177:6072–6080. doi: 10.4049/jimmunol.177.9.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm J, Craig AG. Pathogenesis of cerebral malaria–inflammation and cytoadherence. Front Cell Infect Microbiol. 2014;4:100. doi: 10.3389/fcimb.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PA, 2nd, Hart GT, Russo MV, Nayak D, Yazew T, Peña M, Khan SM, Janse CJ, Pierce SK, McGavern DB. CD8+ T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen-specific engagement of brain vasculature. PLoS Pathog. 2016;12:e1006022. doi: 10.1371/journal.ppat.1006022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrio ML, Lee SH, Fragoso MF, Sun HW, Kanno Y, O’Shea JJ, Biron CA. Proliferation conditions promote intrinsic changes in NK cells for an IL-10 response. J Immunol. 2014;193:354–363. doi: 10.4049/jimmunol.1302999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. 2009;183:4904–4912. doi: 10.4049/jimmunol.0900284. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdeil G, Marquardt K, Surh CD, Sherman LA. Adjuvants targeting innate and adaptive immunity synergize to enhance tumor immunotherapy. Proc Natl Acad Sci USA. 2008;105:16683–16688. doi: 10.1073/pnas.0805054105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votavova P, Tomala J, Kovar M. Increasing the biological activity of IL-2 and IL-15 through complexing with anti-IL-2 mAbs and IL-15Rα-Fc chimera. Immunol Lett. 2014;159:1–10. doi: 10.1016/j.imlet.2014.01.017. [DOI] [PubMed] [Google Scholar]
- Welsh RM, Waggoner SN. NK cells controlling virus-specific T cells: rheostats for acute vs. persistent infections. Virology. 2013;435:37–45. doi: 10.1016/j.virol.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Malaria Report 2016. Geneva: World Health Organization); 2016. http://apps.who.int/iris/bitstream/10665/252038/1/9789241511711-eng.pdf?ua=1. [Google Scholar]
- Xu W, Jones M, Liu B, Zhu X, Johnson CB, Edwards AC, Kong L, Jeng EK, Han K, Marcus WD, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor αSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 2013;73:3075–3086. doi: 10.1158/0008-5472.CAN-12-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol. 1996;157:1620–1624. [PubMed] [Google Scholar]
- Zhao H, Aoshi T, Kawai S, Mori Y, Konishi A, Ozkan M, Fujita Y, Haseda Y, Shimizu M, Kohyama M, et al. Olfactory plays a key role in spatiotemporal pathogenesis of cerebral malaria. Cell Host Microbe. 2014;15:551–563. doi: 10.1016/j.chom.2014.04.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.