

Abstract
Idiopathic pneumonia syndrome (IPS) is a common, often fatal, complication following hematopoietic stem cell transplantation (HSCT) characterized by severe pneumonitis and interstitial fibrosis. Fully reconstituted syngeneic bone marrow transplant (BMT) mice infected with murine gammaherpesvirus (γHV-68) develop IL-17-driven pneumonitis and fibrosis which mimics clinical manifestations of IPS. We found CD103+ and CD11b+ dendritic cells (DCs) are selectively deficient for the Notch ligand, DLL4, following BMT and CD4+ T-cells isolated from lungs and spleens of infected BMT mice display Notch signaling defects. Mice transplanted with CD4-Cre-driven dominant-negative Notch transcriptional regulator Mastermind Like (CCD mice) bone marrow displayed elevated IL-17 and TGFβ in the lungs, a further expansion of Th17 cells, and developed more fibrosis than WT-BMT mice. Culture of BMT lung leukocytes with recombinant Notch ligand, DLL4, restored Notch signaling and decreased production of IL-17. Adoptive transfer of CD11c+ DCs could restore Th1 and limit Th17 in WT-BMT but not CCD-BMT mice indicating a specific DC / CD4+ T-cell Notch interaction modulates IL-17 production following reconstitution in syngeneic BMT mice. Given recent clinical observations showing that patients with pulmonary complications post-transplant harbor occult herpesvirus infections, these data provide mechanistic insight and suggest potential therapies for these devastating conditions.
Introduction
Hematopoietic stem cell transplantation (HSCT) is an efficacious curative therapy for a number of malignant and auto-immune diseases 1, 2. Unfortunately, severe and often fatal lung complications such as idiopathic pneumonia syndrome (IPS) and bronchiolitis obliterans syndrome (BOS) are common following HSCT affecting between 10–15% of recipients 3, 4. Aberrant production of pro-inflammatory cytokines such as IL-6 and IL-17A are thought to play important roles in the progression of IPS and BOS 5–7; however, the cellular mechanisms that underlie the production of these cytokines remain unclear.
The Notch pathway is an evolutionarily conserved juxtacrine inter-cellular signaling pathway important in a variety of biological processes such as cell division and differentiation, organ development, and lymphocyte maturation / activation. Four Notch receptors (Notch1-4) are expressed by mammalian cells and can be activated by contact with a second cell expressing one of five Notch ligands (DLL1, 3 and 4 and Jagged 1 or 2) 8, 9. Interactions with these cognate ligands result in an initial cleavage of the Notch receptor in the plasma membrane by ADAM proteases 10. The receptor subsequently undergoes endocytosis where it undergoes a second cleavage by the intramembrane protease γ-secretase releasing an active Notch intracellular domain (NICD). The NICD transits to the nucleus where it acts as a scaffold for transcription machinery including p300 and master mind like (MAML) 11. The formation of this transcription complex de-represses the transcriptional repressor CBP / RBPj-κ turning on expression of Notch specific target genes such as the Hairy Enhancer of Split (Hes) family of proteins 12.
Engagement of Notch receptors on CD4+ T-cells by ligand expressing antigen presenting cells (APCs) has been shown to influence T-cell polarization into the various activated subsets (e.g. Th1, Th2, Th17). This process is dependent on a variety of factors including antigen and cytokine exposure and can be either activating or inhibitory. Expression of the Notch ligands DLL4 or Jagged 1 by APCs was shown to preferentially result in naive T-cells skewing towards either Th1 or Th2 respectively 13. Exposure of naive T-cells to plate-bound or APC-expressed DLL4 was shown to drive Th17 differentiation when the cells were grown in the context of the Th17 skewing cytokines, IL-6 and TGF-β 14. Further the IL-17 promoter was demonstrated to be a direct transcriptional target for the NICD complex 14, 15.
In contrast, several studies have shown that Notch ligand expression by APCs can attenuate T-cell cytokine expression. Expression of Jagged 1 by APCs pulsed with the dust mite antigen Der P1 was found to expand populations of T-reg cells and induce a tolerogenic phenotype upon antigen re-exposure 16. Pulsing of bone marrow-derived dendritic cells (BMDC) with BCG expressing recombinant Der P2 antigen increased expression of the Notch ligand DLL4 and adoptive transfer of these BMDCs decreased Th17 differentiation and lessened allergic airway inflammation in an experimental mouse model of asthma 17. DLL4 was also shown to be increased following Respiratory Syncytial Virus (RSV) infection 18. Treatment of RSV infected mice with αDLL4 antibody therapy exacerbated airway inflammation and increased IL-17 responses 18, 19. More recently, T-cells cultured with Jagged1 were found to be significantly inhibited in their ability to produce IL-17 when co-cultured with the Th17 skewing cytokines IL-6 and TGFβ 20, 21. Additionally, T-cells isolated from patients with systemic lupus erythematosus were found to be deficient in Notch 1 signaling and produced elevated levels of IL-17a 22. Thus, the ultimate role of Notch signaling during T-cell activation is highly context dependent, highlighting the need to study this pathway in detail in specific disease states.
We have previously characterized a mouse model of pneumonitis and pulmonary fibrosis following γHV-68 infection in syngeneic bone marrow transplanted (BMT) mice that strongly resembles clinical manifestations of idiopathic pneumonia syndrome (IPS) and the fibrotic properties of bronchiolitis obliterans syndrome (BOS) in humans that have undergone HSCT. In normal, non-transplanted mice, γHV-68 clearance from the lungs happens within 10 days from the onset of i.n. infection via a strong polarization of CD4+ T-cells to a Th1 phenotype secreting large amounts of IFNγ, which has been shown to limit the replication and spread of γHV-68 23–25. BMT mice however, show an increased polarization of CD4+ T-cells to a Th17 phenotype following γHV-68 challenge, secreting IL-17 instead of IFNγ resulting in a severe pneumonitis and pulmonary fibrosis, which is evident by 3 weeks post infection 26–28. Neutralization of IL-17 by administration of αIL-17 antibody therapy, or genetically ablating expression of IL-17 from donor hematopoietic cells completely abrogated disease 27. It was further demonstrated that adoptive transfer of non-BMT CD11c+ APCs could reverse polarization of CD4+ T-cells back to a Th1 phenotype and alleviate the fibrosis in BMT mice infected with γHV-68, demonstrating a role for an APC – T-cell interaction in this altered immune response 27.
Given the important role of Notch signaling in T-cell activation, we asked whether Notch signaling was involved in regulating cytokine production from T-cells following γHV-68 challenge after BMT. T-cells isolated from the lungs and spleens of BMT mice had significant decreases in the expression of Notch associated transcription factors following γHV-68 infection. Further, BMT mice transplanted with marrow specifically ablated for canonical Notch signaling only in CD4 and CD8 T-cells exhibited increased numbers of lung infiltrating Th17 cells, higher levels of pathogenic IL-17 and TGF-β production and developed more exuberant pulmonary fibrosis indicating that T-cell Notch signaling is an important regulator of pro-fibrotic cytokines following BMT and γ-herpesvirus infection.
Materials and Methods
Cell lines, virus strains, and mouse lines
Murine γ-herpesvirus (γHV-68 strain WUMS VR- 1465) was purchased from ATCC (Manassas, VA) and was propagated and titrated on NIH 3T12 fibroblast monolayers also purchased from ATCC. T-cell Notch deficient mice (CCD mice) expressing a floxed dominant negative copy of the Notch transcriptional activator MAML in combination with a Cre recombinase gene under control of the CD4 promoter were first described in 29 and were generously provided to us by Dr. Ivan Maillard, University of Michigan. Mice were maintained by mating of homozygous (flox/flox) to each other and were genotyped for the presence of the floxed DNMAML and CD4 Cre genes before use. Control C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, Maine). All mice were used between 6 and 8 weeks of age and were housed in specific pathogen free conditions. Experiments were approved by the University of Michigan Institutional Animal Committee on Use and Care. Syngeneic bone marrow transplantation has been previously described in 26. In short, C57BL/6J mice were lethally irradiated with 13 Gy (2 split doses of 6.5 Gy spaced 3 h apart) from a 137Cs radiation source. Mice were subsequently i.v. injected with 5x106 bone marrow cells isolated from either genetically identical C57BL/6J mice or congenic CCD mice. BMT mice were given acidified water (pH 3.3) for 3 weeks and then switched to plain water for an additional 2 weeks. After a 5-week reconstitution period, BMT or control mice were infected i.n. with 5x104 PFU γHV-68.
Preparation of single cell suspensions from lung tissue and flow cytometry
Collagenase digestion of mouse lung tissue was previously described in 30. In short, mice were euthanized by CO2 asphyxiation after which perfusion was carried out by injecting 3–5 mL of phosphate buffered saline (PBS) through the right ventricle of the heart. Lungs were resected, minced with scissors, and digested in complete DMEM containing 15 mg/mL collagenase A (Roche, Indianapolis, IN), and 2500 units of DNase I (Sigma-Aldrich, St. Louis, MO) for 30 m at 37°C. Digested tissue was disrupted through a 10 mL syringe, filtered through a 100 μM pore size nytex screen and centrifuged through a 20 % Percoll solution in serum free media. 10x106 cells were then stained with appropriately diluted fluorophore-conjugated antibodies for 30 m. For intracellular cytokine staining (ICS) cells were diluted to 1x106/mL and stimulated for 4 h with PMA (10 ng / mL), ionomycin (10 μM), and Golgi-stop reagent (BD Bioscience, San Jose, CA). Antibodies used in this study are as follows: anti-CD45, CD11b, CD4, IL-17A, MHCII (I-Ab), CD103, SiglecF (BD Bioscience, San Jose, CA), CD11c, IFNγ (eBioscience San Diego, CA), DLL1, DLL4, Jagged1, Jagged2, and CD64 (Biolegend, San Diego, CA).
Preparation of RNA and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from either whole lung tissue, or purified cells with TRIzol reagent (ThermoFisher, Waltham, MA) following the manufacturer’s directions. 100 ng of RNA per sample was analyzed using Taqman RNA to CT real-time master-mix (ThermoFisher, Waltham, MA) on an ABI Step-one plus thermocycler (ThermoFisher, Waltham, MA). Primers used in this study are detailed in Table 1.
Table I.
Primers and probes used for qRT-PCR
| Gene Name | Primer Sequence | |
|---|---|---|
| IL-17A | Forward | 5′-CCGCAATGAAGACCCTGATAG-3′ |
| Reverse | 5′-GCTTTCCCTCCGCATTGA-3′ | |
| Probe | 5′-GGGAAGCTCAGTGCCGCCAG-3′ | |
| Hes1 | Forward | 5′-GGCCTCTGAGCACAGAAAGT-3′ |
| Reverse | 5′-GCCGGGAGCTATCTTTCTTA-3′ | |
| Probe | 5′-TTGCCCTTCGCCTCTTCTCCA-3′ | |
| Hes5 | Forward | 5′GCAGCATAGAGCAGCTGAAG-3′ |
| Reverse | 5′-GAAGGCTTTGCTGTGTTTCA-3′ | |
| Probe | 5′-CGCGAACTCCTGCTCCAGCA-3′ | |
| Hey1 | Forward | 5′-TGCAGGAGGGAAAGGTTATT-3′ |
| Reverse | 5′-GAGGCATCGAGTCCTTCAAT-3′ | |
| Probe | 5′-AATGCCTGGCCGAAGTTGCC-3′ | |
| Notch1 | Forward | 5′-CCAGGAAACAACTGCAAGAA-3′ |
| Reverse | 5′-AGTACTGACCCGTCCACTCC-3′ | |
| Probe | 5′-CCTACAATTGCCGCTGCCCA-3′ | |
| Notch2 | Forward | 5′-TGAGAGCTTCAGCTGCTTGT-3′ |
| Reverse | 5′-AGTGTTGTGGCAGACACCAT-3′ | |
| Probe | 5′-TTTACCTTGCCAGCCAGGAGCA-3′ | |
| DLL1 | Forward | 5′-CTGGCCAGGTACCTTCTCTC-3′ |
| Reverse | 5′-CCATTCTTCTCCCACAGTGA-3′ | |
| Probe | 5′-CCGCCTGACCACACAGAGGC-3′ | |
| DLL4 | Forward | 5′-CGTCGTCAGGGACAAGAATA-3′ |
| Reverse | 5′-GAGTTTCCTGGCGAAGTCTC-3′ | |
| Probe | 5′-CCTGGCCGGGAACCTTCTCA-3′ | |
| Jagged1 | Forward | 5′-GACTCATCAGCCCTGTCTCA-3′ |
| Reverse | 5′-AGCATGCTCAGCAATTTCAC-3′ | |
| Probe | 5′-CCCGAGTAGCCCTCTGGGCA-3′ | |
| Jagged2 | Forward | 5′-CAATCCCTGTGTGAATGGAG-3′ |
| Reverse | 5′-TGGTGTTATGTGTGCAGGTG-3′ | |
| Probe | 5′-TTCTCCTGCATCTGCCGGGA-3′ |
Preparation of lung tissue sections for histology
Formalin fixed paraffin embedded (FFPE) lung sections were prepared as follows. Mice were euthanized by CO2 asphyxiation after which 3–5 mL of PBS was perfused through the right ventricle of the heart. Lungs were infused with 1 mL 10% formalin solution through the trachea and fixed for 24 h after which lungs were dehydrated in 70% ethanol, embedded in paraffin and sectioned to a 10 μM thickness. Lung sections were subsequently stained with H&E or Masson’s Trichrome stain to highlight collagen fibers.
Quantitation of collagen by hydroxyproline assay
Collagen content was assessed in whole lung homogenates using the hydroxyproline assay as described in 30.
Cytokine quantitation
Percoll-purified lung leukocytes from collagenase digested lung tissue or magnetically isolated CD4+ T-cells (3x106 per sample) were cultured for 24 h with or without additional skewing cytokines. Clarified supernatants were subsequently analyzed for the indicated cytokines using Duo-set ELISA assays (R & D systems, Minneapolis, MN). Recombinant murine DLL4 (R&D systems, Minneapolis, MN) was plate bound to tissue culture dishes at a concentration of 50 nM in PBS for 3 h at 37°C. Plates were washed 3 X with sterile PBS before addition of purified leukocytes in complete media.
Statistical analysis
Statistical analysis was done using Graph Pad Prism v6.01 (Graph Pad Software Inc. La Jolla, CA). Figures that compare only two means were analyzed by student’s T-test. Analysis of variance (ANOVA) in conjunction with Tukey’s multiple comparisons post-test was used in experiments where three or more means were analyzed together. All experiments were conducted in, at least, duplicate. N-values and other pertinent statistical information for representative experiments are given in individual figure legends.
Results
Lung DCs display reduced expression of DLL4 in virally-infected BMT mice
Notch ligand expression on CD11c+ APCs has been implicated in modulating T-cell activation and cytokine production during a variety of disease states and immunological maladies 31–35. To determine whether levels of Notch ligands were altered on APCs following BMT and viral infection, total CD11c+ cells were isolated by magnetic bead separation from either control or BMT mouse groups and infected ex-vivo at a multiplicity of infection of 1.0 for 24 h with γHV-68. Total RNA was isolated and expression levels of four Notch ligands (DLL1, DLL4, Jagged1, and Jagged2) were analyzed via qRT-PCR. Expression of DLL4 was decreased in both uninfected BMT cells and in infected non-BMT cells in comparison to control non-BMT CD11c+ APCs, and an additive downregulation of DLL4 was noted in infected BMT APCs in comparison to control cells (~5-fold downregulation in comparison to control non-BMT cells, Figure 1A). DLL1 was modestly, although not significantly, down-regulated in uninfected BMT cells in comparison to WT control cells, however, no change was observed in cells isolated from infected mice (Figure 1A). Levels of Jagged2 were down-regulated in infected BMT CD11c+ cells in comparison to WT control cells, however, no change in Jagged 2 expression was noted in comparison to WT control cells (Figure 1A). No change in Jagged1 expression was observed in any of the groups (Figure 1A). To analyze expression of Notch ligands on CD11c+ cells in vivo, we infected groups of control or BMT mice i.n. with γHV-68. 7 dpi, flow cytometry was performed on collagenase digested lung cells to assess APC expression of Notch ligands in vivo. Cells were first gated as CD45+, CD11c+, MHCII+ before gating on expression of either DLL1, DLL4, Jagged1, or Jagged2. In agreement with the ex-vivo data, significantly fewer DLL4 positive CD11c+ cells were detected in the lungs of BMT mice at 7 dpi in comparison to non-BMT mice (Figure 1B and S1). Numbers of CD11c+ APCs expressing DLL1, Jagged1 or Jagged2 were not altered in γHV-68-infected BMT mice in vivo (Figure S1).
Figure 1. Loss of Notch ligand expression in lung associated APCs following BMT.

(A) Total lung CD11c+ cells were magnetically isolated (n = 4 mice per group) and infected ex-vivo with γHV-68. 24 hpi, indicated transcript expression level was analyzed via qRT-PCR. (B) Flow cytometric analysis of DLL4 expressing APCs. Cells were first gated on CD45+, CD11C+, and MHCII+ before gating on DLL4. Expression is compared to a flow minus one (FMO) control. (C and D) DLL4 expression in CD11c+ subsets were assessed by flow cytometry. Cells were first gated on CD45+, CD11c+, MHCII+, CD64− and then subdivided as either CD103+ or CD11b+. Graphed are absolute number or percentage of both CD103+ or CD11b+ DCs. Mean values are graphed (+/− SEM), statistical significance was calculated using ANOVA (panel A) or student’s T-test (panel c), * = p < 0.05, ** = p < 0.01, *** = p < 0.001, n.s. = not significant. (E and F) Representative flow plots showing percentages of DLL4+ cells from either CD103+ or CD11b+ DCs.
We further analyzed expression of DLL4 on two distinct subsets of CD11c+ DCs, i.e. CD103+ and CD11b+ DCs, by flow cytometry. In agreement with the decreased expression of DLL4 seen in the total CD11c+ DC compartment, the absolute number as well as the percentage of DLL4+ CD103+ DCs present in the lungs of BMT mice was significantly decreased at 3 dpi (Figure 1C). Percentage of CD11b+ DCs was similarly decreased in BMT mice at 3 dpi, however, there was not a statistically significant decrease in the absolute number (Figure 1C). Similarly, at 7 dpi both absolute number and percentage of CD103+ DCs was decreased in BMT mice, no change in either absolute number or percentage of CD11b+ DCs was observed at 7 dpi (Figure 1D). Representative flow plots (7 dpi) are shown for CD103+ DCs in Figure 1E and for CD11b+ DCs in Figure 1F, the full flow cytometry gating strategy is shown in Figure S2.
We next isolated CD4+ T-cells from the spleen as a source of naive T cells from uninfected WT or BMT mice (0 dpi) or from the lungs of γHV-68-infected mice (7 dpi) and analyzed expression of the Notch target genes Hes1, Hes5, and Hey1 as well as the Notch1 and Notch2 receptors by qRT-PCR. Interestingly, in the absence of infection, expression of both Notch1 and Notch2 as well as all three Notch target genes were decreased in T-cells isolated from the spleens of BMT mice (Figure 2). At 7 dpi, the expression of Notch1 and Hes1 were significantly lower in T-cells isolated from the lungs, however expression of Hes5 and Hey1 were not significantly different between BMT and WT mice and an increase in the expression of Notch2 was also noted (Figure 2).
Figure 2. CD4+ T-cells isolated from BMT mice are deficient in Notch signaling.
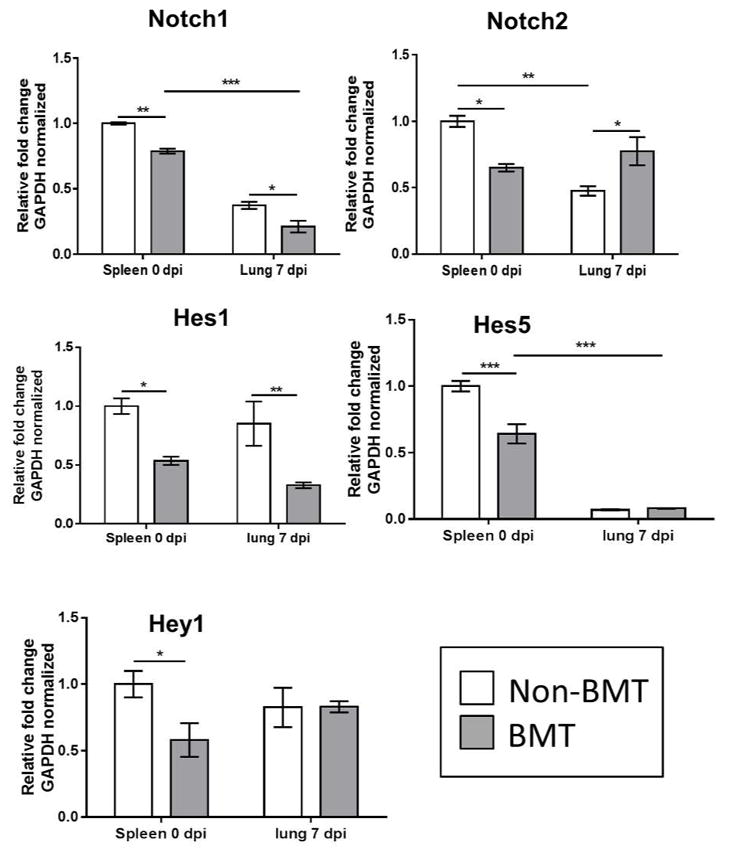
RNA was isolated from CD4+ T-cells purified from either the spleens of naive non-BMT or BMT mice (0 dpi) or the lungs at 7 dpi (n = 3 – 4 mice per group) and analyzed via qRT-PCR for expression of the indicated transcript. Statistical significance was calculated by ANOVA * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Abrogation of T-cell Notch signaling enhances Th17 differentiation and exacerbates γHV- 68-induced pulmonary fibrosis following BMT
To assess whether T-cell specific Notch signaling influenced Th17 differentiation during γHV-68 infection following syngeneic BMT, we utilized a genetic approach wherein C57BL/6J mice harboring a Cre recombinase under the control of the CD4 promoter were crossed with mice carrying a floxed dominant negative (DN) version of the Notch transcriptional regulator MAML 29. These CD4-Cre-DNMAML (CCD) mice retain a normal repertoire of T-cells but are functionally ablated for all canonical Notch signaling in both CD4+ and CD8+ T-cells. Syngeneic BMTs were performed on lethally irradiated WT mice using either WT or CCD mice as donors. Following this, both transplanted and non-transplanted control groups were infected i.n. with γHV-68. Lungs were harvested at 7 dpi, collagenase digested, and analyzed by intra-cellular cytokine staining and flow cytometry for expression of either IFNγ or IL-17 to assess Th1 and Th17 populations respectively. Following γHV-68 challenge, CCD mice lacking T-cell Notch signaling had a significantly greater number of IL-17 producing CD4+ cells in the lungs in comparison to non-transplanted control or WT BMT mice (Figure 3A and 3B). Additionally, CCD mice had significantly fewer IFN-γ producing CD4+ T-cells than non-transplanted control mice (Figure 3A and 3C). Because the DNMAML transgene is expressed by both CD4+ and CD8+ T-cells during the double positive stage of lymphocyte maturation, we also analyzed CD8+ lymphocyte populations for expression of both IL-17 and IFNγ. In contrast to CD4+ T-cells, there was no difference in CD8+ IFNγ expression between γHV-68-infected WT-BMT and CCD-BMT mouse groups (Figure 3A and 3D). Additionally, IL-17 was only detected in a small population of CD8+ T-cells in BMT mice (< 1%) and there was no difference between γHV-68 infected BMT and CCD-BMT mice (Data not shown).
Figure 3. Loss of T-cell Notch signaling increases production of IL-17 from CD4+ lymphocytes following BMT.

Control or BMT mice (n = 4 – 5 mice per group) were infected with γHV-68 for 7 d. Lungs were harvested and collagenase digested. Following PMA and Ionomycin stimulation, cells were analyzed by flow cytometry for either IL-17 or IFNγ producing CD4+ or CD8+ T-cells. (A) representative flow-plots showing percentages of Th17, Th1, or IFNγ+ CD8+ cells present in the lungs of control, BMT or CCD-BMT mice at 7 dpi. (B, C, and D) Quantification of data represented in (A). (E and F) Leukocytes from collagenase digested lungs were purified by centrifugation through Percoll. Cells were cultured overnight in complete media, without additional stimulation, and clarified supernatants were analyzed by ELISA. (G) CD4+ T-cells were magnetically purified from spleens of either WT, BMT or CCD-BMT mice (n = 3 – 4 mice per group), 1x106 cells were cultured in the presence of the indicated skewing condition for 24 h after which supernatants were harvested and analyzed for the indicated cytokine by ELISA (Unstim = no additives, CD3 = 1 μg/mL αCD3 and αCD28, Th17 = αCD3, αCD28, with 2 ng/mL TGFβ and 20 ng/mL IL-6). Statistical significance was calculated by ANOVA * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001.
To further assess the levels of cytokines produced by control and BMT mice after γHV- 68 challenge, cells from collagenase digested lungs were purified by centrifugation through Percoll at 7 or 21 dpi resulting in a 60 – 80% enrichment for CD45+ hematopoietic cells as assayed by flow cytometry (Figure S2 Panel 1). Cells were plated for 24 h in complete media (3x106 per sample) and amounts of the pro-fibrotic cytokines IL-17 and TGF-β were measured by ELISA. In agreement with the greater numbers of Th17 cells recruited to the lungs of CCD-transplanted mice; cells isolated from the lungs of CCD-BMT mice produced more IL-17 and TGF-β than non-transplanted control or WT BMT mice at both 7 and 21 dpi (Figure 3E and 3F). Additionally, CD4+ T-cells were isolated from the spleens of non-infected control, WT-BMT, or CCD-BMT mice and stimulated either with αCD3 / αCD28 alone or with αCD3 / αCD28 in conjunction with the Th17 skewing cytokines IL-6 and TGF-β. CD4+ T-cells isolated from CCD-BMT mice produced large quantities of IL-17 even in the absence of Th17 skewing cytokines and IL-17 production was further amplified in the presence of IL-6 and TGF-β (Figure 3G).
Loss of T-cell Notch signaling exacerbates γHV-68-induced pulmonary fibrosis following BMT
To address whether the increased levels of IL-17 produced by CCD mice in response to γHV-68 exacerbated the development of pulmonary fibrosis, H&E and Trichrome staining of FFPE lung sections were examined microscopically for the presence of inflammation and fibrotic foci at 21 dpi. Masson’s Trichrome staining highlights collagen rich fibrotic areas with a blue color. Non-transplanted control mice infected with γHV-68 had no indication of inflammation or pulmonary fibrosis at 21 dpi (Figure 4A and 4B) while infected BMT mice developed patchy fibrosis as evidenced by areas of interstitial thickening, inflammatory infiltrate, loss of alveolar structure, and deposition of excess collagen (Figure 4C and 4D). CCD-BMT mice however, developed a much more severe pneumonitis and fibrosis in response to γHV-68 with large areas of the lung showing loss of alveolar architecture, increased amounts of inflammatory infiltrate, and further increased collagen deposition (Figure 4E and 4F). Interestingly, non-BMT CCD mice infected with γHV-68 for 21 d did not develop pulmonary fibrosis (Figure S3A and S3B). Cytokine analysis from non-BMT WT or CCD mice revealed that, although non-BMT CCD mice produced less IFNγ at 7 dpi, levels of IL-17 were equivalent between the two groups (Figure S3D and S3E). Lung collagen content was directly quantified from either WT, BMT, or CCD-BMT mice at 21 dpi by hydroxyproline assay. In agreement with the increase in collagen noted by Masson’s Trichrome staining, CCD-BMT mice had more collagen deposition than either WT or WT-BMT mice at 21 dpi (Figure 4G). We further assayed lungs for amounts of infectious virus present at 7 dpi and noted a significant increase in viral load in the lungs of CCD-BMT mice in comparison to either WT or WT-BMT mice (Figure 4H). To assess if CCD-BMT mice had a defect in viral clearance we isolated total RNA from collagenase digested lung cells at either 7 or 21 dpi and performed qRT-PCR for two viral transcripts (vDNA polymerase and Glycoprotein B). Levels of both transcripts followed similar trends with transplanted mouse groups exhibiting increased expression at 7 dpi in comparison to non-BMT mice (Figure 4I and 4J). At 21 dpi, expression of both viral transcripts was significantly lower in all groups indicating viral clearance was preserved in both BMT and CCD-BMT mice (Figure 4I and 4J).
Figure 4. Disruption of T-cell Notch signaling exacerbates γHV-68-induced pulmonary fibrosis following BMT but does not alter viral clearance.

(A through F) Control or BMT mice were infected for 21 d with γHV-68. FFPE sections were stained with H&E (A: Untransplanted control, C: WT-BMT, E: CCD-BMT 40x magnification) or Masson’s trichrome (B: WT, D: WT-BMT, F: CCD-BMT 100x magnification). (G) Quantification of total lung collagen as measured by hydroxyproline assay at 21 dpi (n = 7 – 12 mice per group). (H) Quantification of lung associated infectious virus as measured by plaque assay (n = 4 – 5 mice per group).) (I and J) Total RNA was isolated from collagenase digested lung cells and analyzed via qRT-PCR for the indicated viral transcript (n = 4 – 5 mice per group). Mean values are graphed (+/− SEM), statistical significance was calculated using ANOVA * = p < 0.05, ** = p < 0.01.
Adoptive transfer of WT CD11c+ cells into CCD-BMT mice fails to restore T-cell Notch signaling and does not rescue fibrotic lung pathology
We previously reported that adoptive transfer of WT CD11c+ cells into BMT mice could rescue fibrotic pathology by decreasing production of IL-17 from CD4+ T-cells 27. To address the role of APC / T-cell Notch signaling in this system, we adoptively transferred magnetically sorted CD11c+ cells isolated from the lungs of γHV-68-infected WT mice into either WT-BMT or CCD-BMT mouse groups one day prior to infection with γHV-68 (1x106 CD11c+ cells transferred i.v.). As previously reported, adoptive transfer of CD11c+ APCs completely rescued fibrotic pathology in WT-BMT mice (Figure 5 A and 5B). Interestingly, collagen rich fibrotic nodes were still evident in the lungs of adoptively transferred T-cell Notch deficient CCD-BMT mice at 21 dpi (Figure 5C and 5D). We next assessed if the adoptive transfer of CD11c+ cells could restore numbers of DLL4+ cells to either the lungs or draining lymph nodes of BMT mice following γHV-68 infection. Interestingly, numbers of DLL4+ CD103+ DCs were increased in the lungs by 7 dpi following adoptive transfer (Figure 5E). In contrast, numbers of CD11b+ DCs remain unchanged in the lungs and numbers of both DLL4+ CD103+ and CD11b+ DCs remained low in the draining lymph nodes of both BMT and adoptively transferred BMT mice. To directly address the effects of CD11c+ cell transfer on T-cell Notch signaling, CD4+ T-cells were isolated from the lungs of either WT, BMT, or CCD-BMT mice at 7 dpi with or without CD11c+ APC adoptive transfer. Non-transferred WT-BMT CD4+ cells expressed significantly lower amounts of the Notch target Hes1 in comparison to WT CD4+ cells (Figure 5F). Adoptive transfer of CD11c+ cells significantly increased Hes1 expression in WT-BMT CD4+ cells, but failed to increase Hes1 expression in CCD-BMT CD4+ cells (Figure 5F). Lower numbers of Th17 cells were detected by flow cytometry in the lungs of WT-BMT mice that had received CD11c+ cellular adoptive transfer, however, CCD-BMT mice still had significantly elevated numbers of lung infiltrating Th17 cells after transfer of CD11c+ cells (Figure 5G). To address whether the adoptive transfer of CD11c+ cells had an ameliorative effect on the development of pulmonary fibrosis we analyzed lung collagen content at 21 dpi by hydroxyproline assay. In agreement with the histological analysis, WT BMT mice that had received adoptive transfer of CD11c+ cells had significantly less collagen deposition than non-transferred BMT mice (Figure 5H). In contrast, CCD-BMT mice that received CD11c+ cell transfer did not show a decrease in collagen deposition in comparison to non-transferred CCD-BMT mice and collagen levels in these mice remained significantly elevated in comparison to non-BMT mice (Figure 5H).
Figure 5. CCD-BMT mice are resistant to CD11c+ adoptive transfer.

FFPE sections were prepared from γHV-68 infected animals at 21 dpi and stained with Masson’s trichrome to highlight collagen. (A and C) BMT or CCDBMT mice, respectively without adoptive transfer of WT CD11c+ cells. (B and D) BMT or CCD-BMT mice with adoptive transfer of WT CD11c+ cells. All images are shown at 100x magnification. (E) Flow cytometric analysis of DLL4+, CD103+ or CD11b+ DCs from either lung or lung draining lymph node (dLN) at 7 dpi. (F) Indicated mouse groups were infected with γHV-68 for 7d. CD4+ T-cells were magnetically isolated from single cell suspension of collagenase digested lung tissue and analyzed by qRT-PCR for Hes1 transcript (n = 4–5 mice per group). (G) Flow cytometric analysis of lung infiltrating IL-17+, CD4+ T-cells (Th17 cells) taken from collagenase digested lungs at 7 dpi. Cells were stimulated for 4 h with PMA and Ionomycin before subsequent intracellular cytokine staining (n = 12 mice per group). (H) Mice were infected with γHV-68. 21 dpi lungs were harvested and analyzed for collagen content by hydroxyproline assay (n = 7–10 mice per group). Statistical significance was calculated by ANOVA * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, n.s. = not significant. Statistical comparisons which are labeled directly over the bars in panel G are in comparison to non-BMT mice.
In vitro culturing of lung leukocytes with recombinant DLL4 restores Notch signaling and decreases production of IL-17
We next attempted to restore Notch signaling by culturing purified lung leukocytes isolated from WT or BMT mice with plate-bound recombinant DLL4 (rDLL4). In agreement with in vivo observations, lung leukocytes cultured for 24 h in the absence of rDLL4 expressed lower levels of the Notch target gene Hes1 (Figure 6A). Addition of rDLL4 increased expression of Hes1 in BMT leukocytes, which correlated with a concomitant decrease in expression of IL-17 transcript (Figure 6B). We next quantified amounts of IL-17 produced from either WT or BMT leukocytes cultured in the presence or absence of rDLL4 by ELISA. In agreement with the level of IL-17 transcript measured by qRT-PCR, lung leukocytes from BMT mice cultured with rDLL4 secreted less IL-17 (Figure 6B and 6C). As a control, leukocytes isolated from the lungs of T-cell Notch signaling-deficient CCD-BMT mice were resistant to the effects of rDLL4 and produced increased amounts of IL-17 in comparison to rDLL4 treated WT-BMT lung leukocytes (Figure 6C).
Figure 6. Ex vivo treatment of purified lung leukocytes with recombinant DLL4 restores Notch signaling and decreases IL-17 production.

(A and B) Lung leukocytes were isolated from WT or BMT mice at 7 dpi and cultured with or without 50nM recombinant DLL4 for 24 h. Total RNA was analyzed by qRT-PCR for the indicated transcript. (C) Lung leukocytes were isolated from either WT, BMT or CCD-BMT mice at 7 dpi, incubated for 24 h with or without 50 nM DLL4. Supernatants were harvested and analyzed for IL-17 by ELISA. Statistical significance was calculated by ANOVA * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001 (n = 4 – 5 mice per group).,
Discussion
The impact of Notch signaling during the process of T-cell activation is complex and can range from activating to inhibitory depending on a variety of factors including: which specific Notch receptors and ligands are engaged, the type of T-cells and APCs involved (naive vs. previously activated or memory T-cells) and the co-stimulatory and cytokine environment experienced by the T-cell during activation 36, 37. Here we present evidence that the loss of CD4+ T-cell Notch signaling during γ-herpesvirus infection following BMT promotes the differentiation of Th17 cells and facilitates the development of IL-17-driven pneumonitis and pulmonary fibrosis that resembles features of human IPS and BOS post-transplant.
In contrast to some previously reported studies which suggested that activation of the Notch signaling pathway in T-cells preferentially enhanced IL-17 production 14, 15, we found that CD4+ T-cells isolated from the lungs of BMT mice exhibited an increased capacity to produce IL-17, however, production of IL-17 correlated with a decrease in Hes1 and Notch1 expression indicating a suppressive effect of Notch signaling on IL-17 production following BMT. These outcomes are likely due to loss of Notch 1 as Notch 2 expression does recover by 7 dpi. Furthermore, although not apparent in the normalized expression values shown in Figure 2, Notch2 was expressed at a much lower level than Notch1 under basal conditions. Several previous studies have shown that expression of Hes1 can suppress T-cell cytokine production and proliferation. Administration of activating α-Notch1 or α-Notch 2 antibodies increased expression of Hes1 and decreased phosphorylation of AKT leading to decreased secretion of IL-2 36, 38. Likewise, genetic ablation of Notch 1 and Notch 2, specifically in T-cells, led to increased expression of RoRγT and Th17 differentiation 39. Furthermore, a specific jagged1- Notch1 interaction was identified in T-cells that suppressed production of IL-17 produced due to TGF-β and IL-6 stimulation 20. Thus, it is plausible that the decreased expression of Notch1 / Hes1 by CD4+ T-cells subsequently increased production of IL-17 following BMT.
Interestingly, DLL4 expression was downregulated on BMT CD11c+ APCs in the absence of infection (Figure 1A). Likewise, Notch1 and Hes1 expression were decreased in naive T-cells isolated from the spleens of non-infected BMT mice (Figure 2, 0 dpi) indicating that the processes of radio-conditioning or immune reconstitution following conditioning may facilitate the generation of a pro IL-17 / Th17 environment. However, non-BMT CCD mice infected with γHV-68 do not develop IPS like pneumonitis or fibrosis even though they have significantly higher viral loads at 7 dpi (Figure S3A, S3B and S3C). While IFNγ levels were reduced in non-BMT CCD mice following γHV-68 infection, IL-17 levels were not elevated (Figure S3C and S3D). Furthermore, uninfected BMT mice do not develop fibrotic pathology by 21 dpi 26. Thus, it is likely that the conditioning regimen followed by an infectious stimulus is necessary to drive full elaboration of the Th17 response. In this regard, we have previously shown that lung epithelial cells are potent producers of TGFβ post-BMT, further, CD11c+ cells isolated from γHV-68 infected BMT mice secrete pro-Th17 cytokines such as IL-6, IL-23, and TGFβ which could help promote robust Th17 responses 27, 40.
We observed decreased expression of the Notch ligand DLL4 on CD11c+ APCs following syngeneic BMT, but this result is in contrast to literature related to allo-transplantation and GVHD. DLL4 expression has most recently been attributed to a population of hyper-inflammatory plasmacytoid like DCs (i-DCs) that are associated with acute GVHD following allogeneic BMT 31, 33, 34. These DLL4-hi i-DCs express CD11b and B220 and drive robust proliferation of allo-reactive T-cells that produce high levels of IFNγ and IL-17 34. Interestingly, blockade of DLL4 using α-DLL4 therapy, or genetic ablation of T-cell Notch signaling, was shown to lessen IFNγ and IL-17 production during acute GVHD improving disease outcomes 29, 34. However, adoptive transfer of allo-reactive CD4+ T-cells that had been cultured specifically with, Flt3-matured, DLL4-hi DCs were unable to mediate experimental GVHD even though they produced high levels of both IFNγ and IL-17 31. Thus, the ultimate role of DLL4 governing GVHD remains unclear and is dependent on the model system.
Given the previous data supporting an activating role of T-cell Notch signaling on Th17 differentiation during allo-BMT we were surprised to find that infiltration of DLL4-low CD11c+ APCs drives IL-17 responses and fibrosis during IPS-like pulmonary complications associated with viral infections following syngeneic BMT. One explanation for the apparent disparate functions of DLL4 in driving T-cell activation following BMT is that the subsets of DCs that are important for driving these immune reactions may be context-dependent, or may be completely different between models. The i-DCs observed during allo-reactive GVHD are thought to be host-derived and spike in number very early (3 days) after transplant and reduce in numbers by 7 days post-BMT 34. Our model uses fully immune-reconstituted BMT mice (5 weeks post-BMT at time of infection) and presumably these i-DCs would be low in number in our model.
Further, dendritic cells are highly heterogeneous with many specialized subsets. For instance, the lung contains populations of DCs that are either CD11b+ CD103- or vice versa while the major population in the intestinal lamina propria are CD11b+ CD103+ DCs 41–43. Both lung and intestinal CD11b+ DCs promote Th17 responses, however, CD103+ CD11b− DCs are thought to promote exclusively Th1 responses 41, 44, 45. Interestingly, we found that expression of DLL4 was preferentially reduced on the CD103+ CD11b− DC subset in the lungs of syngeneic BMT mice following γ-herpesvirus infection. It is plausible that the lack of DLL4 on CD103+ DCs is partially responsible for the decreases inTh1 differentiation and IFNγ production observed in BMT mice in response to γHV-68 and that in the absence of a proper Th1 response a Th17 response is generated instead.
We previously published that adoptive transfer of CD11c+ cells from non-BMT mice rescued fibrotic pathology 27. We now show adoptive transfer of these DLL4-hi DCs preferentially increased numbers of DLL4+ CD103+ DCs in the lungs following infection leading to a restoration of Hes1 expression in CD4+ T-cells and decreased Th17 differentiation. We further demonstrated that T-cell Notch-deficient CCD-BMT mice were resistant to this effect and maintained Th17 differentiation and low CD4+ Hes1 expression following adoptive transfer of CD11c+ cells suggesting a specific interaction between Notch receptor expressing CD4+ T-cells and DLL4 expressing CD103+ DCs in the lung (Fig. 6). These data support a crucial role for DC-T cell Notch signaling in regulating IL-17 levels post-BMT; however, we do acknowledge that adoptive transfer of DCs into the CCD BMT mice has some impact on lowering Th17 cells and increasing numbers of Th1 cells (Fig. 5F and data not shown). This could reflect effects on radio-resistant host T cells which would have been Notch sensitive or may suggest that there are additional Notch-independent effects of adoptively transferred DCs to limit Th17, and promote Th1, induction. Additionally, baseline secretion of low levels of IL-17 was observed in non-BMT infected mice (Figure 6C). Several other lymphocyte cell types, e.g. γδ T-cells, ILC3s, and iNKT cells, are known to secrete IL-17 and could account for these Notch independent secretion effects. However, given that the adoptive transfer of CD11c+ cells restored Notch signaling specifically in CD4+ T-cells and this correlated with decreased numbers of lung Th17 cells, we conclude that Notch signaling specifically acts to suppress IL-17 responses from CD4+ sources following BMT and viral infection.
While human IPS is defined as being non-infectious, recent studies using more sensitive PCR techniques have demonstrated that over 50% of IPS cases are characterized by a previously undetected herpesvirus infection, HHV6 46–48. These clinical data suggest that IPS could be subsequent to an occult infectious insult. This is consistent with our data showing clearance of lytic virus by 10 dpi, but development of severe pneumonitis and fibrosis by 21 dpi in the syngeneic BMT mice. Additionally, in a study analyzing 738 HSCT patients at the University of Michigan, both HHV6 and EBV infections within the first 100 days following transplant were strongly correlated with the development of IPS, with associated hazard ratios of 5.9 and 10 respectively (X. Zhou, unpublished observation). Thus, while IPS can certainly manifest because of allo-immunity following HSCT 6, we strongly feel that infection with herpesviruses is an additional stimulus to the development of pulmonary pathology following HSCT.
In summary, our results have demonstrated a selective loss of DLL4 that occurs on lung DCs at 5 weeks post-BMT, when immune reconstitution is complete. Furthermore, there is basal loss of Notch 1 and Notch 2 receptors on T cells post-BMT at this same time point. In the syngeneic BMT setting, this defect in Notch signaling promoted a robust Th17 response which worsened pneumonitis and fibrosis. We have previously shown the ability of IL-17 to promote lung mesenchymal cell activation (both proliferation and extracellular matrix deposition) 27. This pathologic Th17 response in the syngeneic setting is largely restored by adoptive transfer of primed DCs from non-transplanted mice, suggesting that in the setting of autologous HSCT complicated by herpesvirus infection, that cellular adoptive therapy or strategies to activate Notch signaling on T cells may have merit. Given the realization that pulmonary sequela of HSCT are often complicated by occult herpesvirus infection, this may provide some insight into the etiology of these complications. Further work will be needed to identify strategies and time frames to promote anti-viral response while limiting GHVD in allogeneic transplants and fibrotic outcomes in both autologous and allogeneic transplant settings.
Supplementary Material
Acknowledgments
We thank Dr. Ivan Maillard for provision of the CCD mice, helpful discussions and critical reading of the manuscript. This work was supported by NIH grants HL127805, HL115618 and AI117229 to BBM. SJG and XZ were also supported by NIH T32 HL007749 and 2UL1TR000433.
Footnotes
Author contributions: SJG: Designed and performed experiments, analyzed and interpreted data, wrote the manuscript. XFZ: Designed and performed experiments, edited the manuscript. MNF: performed experiments. CW: performed experiments. BBM: Designed experiments, analyzed and interpreted data, edited the manuscript.
References
- 1.Bouchlaka MN, Redelman D, Murphy WJ. Immunotherapy following hematopoietic stem cell transplantation: potential for synergistic effects. Immunotherapy. 2010;2(3):399–418. doi: 10.2217/imt.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hugle T, Daikeler T. Stem cell transplantation for autoimmune diseases. Haematologica. 2010;95(2):185–188. doi: 10.3324/haematol.2009.017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afessa B, Peters SG. Major complications following hematopoietic stem cell transplantation. Semin Respir Crit Care Med. 2006;27(3):297–309. doi: 10.1055/s-2006-945530. [DOI] [PubMed] [Google Scholar]
- 4.Yoshihara S, Yanik G, Cooke KR, Mineishi S. Bronchiolitis obliterans syndrome (BOS), bronchiolitis obliterans organizing pneumonia (BOOP), and other late-onset noninfectious pulmonary complications following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(7):749–759. doi: 10.1016/j.bbmt.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Varelias A, Gartlan KH, Kreijveld E, Olver SD, Lor M, Kuns RD, et al. Lung parenchyma-derived IL-6 promotes IL-17A-dependent acute lung injury after allogeneic stem cell transplantation. Blood. 2015;125(15):2435–2444. doi: 10.1182/blood-2014-07-590232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mauermann N, Burian J, von Garnier C, Dirnhofer S, Germano D, Schuett C, et al. Interferon-gamma regulates idiopathic pneumonia syndrome, a Th17+CD4+ T-cell-mediated graft-versus-host disease. Am J Respir Crit Care Med. 2008;178(4):379–388. doi: 10.1164/rccm.200711-1648OC. [DOI] [PubMed] [Google Scholar]
- 7.Vittal R, Fan L, Greenspan DS, Mickler EA, Gopalakrishnan B, Gu H, et al. IL-17 induces type V collagen overexpression and EMT via TGF-beta-dependent pathways in obliterative bronchiolitis. Am J Physiol Lung Cell Mol Physiol. 2013;304(6):L401–414. doi: 10.1152/ajplung.00080.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32(1):14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu Rev Immunol. 2005;23:945–974. doi: 10.1146/annurev.immunol.23.021704.115747. [DOI] [PubMed] [Google Scholar]
- 10.LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem. 2003;278(36):34427–34437. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- 11.Han J, Allalunis-Turner J, Hendzel MJ. Characterization and comparison of protein complexes initiated by the intracellular domain of individual Notch paralogs. Biochem Biophys Res Commun. 2011;407(3):479–485. doi: 10.1016/j.bbrc.2011.03.042. [DOI] [PubMed] [Google Scholar]
- 12.Amsen D, Antov A, Flavell RA. The different faces of Notch in T-helper-cell differentiation. Nat Rev Immunol. 2009;9(2):116–124. doi: 10.1038/nri2488. [DOI] [PubMed] [Google Scholar]
- 13.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117(4):515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol. 2009;182(12):7381–7388. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keerthivasan S, Suleiman R, Lawlor R, Roderick J, Bates T, Minter L, et al. Notch signaling regulates mouse and human Th17 differentiation. J Immunol. 2011;187(2):692–701. doi: 10.4049/jimmunol.1003658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoyne GF, Le Roux I, Corsin-Jimenez M, Tan K, Dunne J, Forsyth LM, et al. Serrate1- induced notch signalling regulates the decision between immunity and tolerance made by peripheral CD4(+) T cells. Int Immunol. 2000;12(2):177–185. doi: 10.1093/intimm/12.2.177. [DOI] [PubMed] [Google Scholar]
- 17.Qu SY, Ou-Yang HF, He YL, Li ZK, Shi JR, Song LQ, et al. Der p2 recombinant bacille Calmette-Guerin priming of bone marrow-derived dendritic cells suppresses Der p2- induced T helper17 function in a mouse model of asthma. Respirology. 2014;19(1):122– 131. doi: 10.1111/resp.12198. [DOI] [PubMed] [Google Scholar]
- 18.Schaller MA, Neupane R, Rudd BD, Kunkel SL, Kallal LE, Lincoln P, et al. Notch ligand Delta-like 4 regulates disease pathogenesis during respiratory viral infections by modulating Th2 cytokines. J Exp Med. 2007;204(12):2925–2934. doi: 10.1084/jem.20070661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ting HA, Schaller MA, de Almeida Nagata DE, Rasky AJ, Maillard IP, Lukacs NW. Notch Ligand Delta-like 4 Promotes Regulatory T Cell Identity in Pulmonary Viral Infection. J Immunol. 2017;198(4):1492–1502. doi: 10.4049/jimmunol.1601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Xing F, Ye S, Xiao J, Di J, Zeng S, et al. Jagged-1 signaling suppresses the IL- 6 and TGF-beta treatment-induced Th17 cell differentiation via the reduction of RORgammat/IL-17A/IL-17F/IL-23a/IL-12rb1. Sci Rep. 2015;5:8234. doi: 10.1038/srep08234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You P, Xing F, Mao C, Chen Z, Zhang H, Wang Y, et al. Jagged-1-HES-1 signaling inhibits the differentiation of TH17 cells via ROR gammat. J Biol Regul Homeost Agents. 2013;27(1):79–93. [PubMed] [Google Scholar]
- 22.Rauen T, Grammatikos AP, Hedrich CM, Floege J, Tenbrock K, Ohl K, et al. cAMP-responsive element modulator alpha (CREMalpha) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE) J Biol Chem. 2012;287(51):42525–42532. doi: 10.1074/jbc.M112.425371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarawar SR, Cardin RD, Brooks JW, Mehrpooya M, Tripp RA, Doherty PC. Cytokine production in the immune response to murine gammaherpesvirus 68. J Virol. 1996;70(5):3264–3268. doi: 10.1128/jvi.70.5.3264-3268.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarawar SR, Lee BJ, Giannoni F. Cytokines and costimulatory molecules in the immune response to murine gammaherpesvirus-68. Viral Immunol. 2004;17(1):3–11. doi: 10.1089/088282404322875412. [DOI] [PubMed] [Google Scholar]
- 25.Sparks-Thissen RL, Braaten DC, Hildner K, Murphy TL, Murphy KM, Virgin HWt. CD4 T cell control of acute and latent murine gammaherpesvirus infection requires IFNgamma. Virology. 2005;338(2):201–208. doi: 10.1016/j.virol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Coomes SM, Farmen S, Wilke CA, Laouar Y, Moore BB. Severe gammaherpesvirus-induced pneumonitis and fibrosis in syngeneic bone marrow transplant mice is related to effects of transforming growth factor-beta. Am J Pathol. 2011;179(5):2382–2396. doi: 10.1016/j.ajpath.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X, Loomis-King H, Gurczynski SJ, Wilke CA, Konopka KE, Ptaschinski C, et al. Bone marrow transplantation alters lung antigen-presenting cells to promote TH17 response and the development of pneumonitis and fibrosis following gammaherpesvirus infection. Mucosal Immunol. 2016;9(3):610–620. doi: 10.1038/mi.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gurczynski SJ, Procario MC, O’Dwyer DN, Wilke CA, Moore BB. Loss of CCR2 Signaling Alters Leukocyte Recruitment and Exacerbates gamma-Herpesvirus Induced Pneumonitis and Fibrosis Following Bone Marrow Transplantation. Am J Physiol Lung Cell Mol Physiol. 2016 doi: 10.1152/ajplung.00193.2016. ajplung 00193 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandy AR, Chung J, Toubai T, Shan GT, Tran IT, Friedman A, et al. T cell-specific notch inhibition blocks graft-versus-host disease by inducing a hyporesponsive program in alloreactive CD4+ and CD8+ T cells. J Immunol. 2013;190(11):5818–5828. doi: 10.4049/jimmunol.1203452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore BB, Paine R, Christensen PJ, Moore TA, Sitterding S, Ngan R, et al. Protection from Pulmonary Fibrosis in the Absence of CCR2 Signaling. The Journal of Immunology. 2001;167(8):4368–4377. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- 31.Mochizuki K, Meng L, Mochizuki I, Tong Q, He S, Liu Y, et al. Programming of donor T cells using allogeneic delta-like ligand 4-positive dendritic cells to reduce GVHD in mice. Blood. 2016;127(25):3270–3280. doi: 10.1182/blood-2015-05-644476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaller MA, Allen RM, Kimura S, Day CL, Kunkel SL. Systemic Expression of Notch Ligand Delta-Like 4 during Mycobacterial Infection Alters the T Cell Immune Response. Front Immunol. 2016;7:527. doi: 10.3389/fimmu.2016.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng L, Bai Z, He S, Mochizuki K, Liu Y, Purushe J, et al. The Notch Ligand DLL4 Defines a Capability of Human Dendritic Cells in Regulating Th1 and Th17 Differentiation. J Immunol. 2016;196(3):1070–1080. doi: 10.4049/jimmunol.1501310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mochizuki K, Xie F, He S, Tong Q, Liu Y, Mochizuki I, et al. Delta-like ligand 4 identifies a previously uncharacterized population of inflammatory dendritic cells that plays important roles in eliciting allogeneic T cell responses in mice. J Immunol. 2013;190(7):3772– 3782. doi: 10.4049/jimmunol.1202820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang HM, Hsiao G, Fan CK, Lin CL, Leu SJ, Chiang BL, et al. Notch ligand delta-like 4- pretreated dendritic cells alleviate allergic airway responses by enhancing IL-10 production. PLoS One. 2013;8(5):e63613. doi: 10.1371/journal.pone.0063613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eagar TN, Tang Q, Wolfe M, He Y, Pear WS, Bluestone JA. Notch 1 signaling regulates peripheral T cell activation. Immunity. 2004;20(4):407–415. doi: 10.1016/s1074-7613(04)00081-0. [DOI] [PubMed] [Google Scholar]
- 37.Laky K, Evans S, Perez-Diez A, Fowlkes BJ. Notch signaling regulates antigen sensitivity of naive CD4+ T cells by tuning co-stimulation. Immunity. 2015;42(1):80–94. doi: 10.1016/j.immuni.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian C, Yu Y, Jia Y, Zhu L, Zhang Y. HES1 activation suppresses proliferation of leukemia cells in acute myeloid leukemia. Ann Hematol. 2015;94(9):1477–1483. doi: 10.1007/s00277-015-2413-0. [DOI] [PubMed] [Google Scholar]
- 39.Coutaz M, Hurrell BP, Auderset F, Wang H, Siegert S, Eberl G, et al. Notch regulates Th17 differentiation and controls trafficking of IL-17 and metabolic regulators within Th17 cells in a context-dependent manner. Sci Rep. 2016;6:39117. doi: 10.1038/srep39117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domingo-Gonzalez R, Wilke CA, Huang SK, Laouar Y, Brown JP, Freeman CM, et al. Transforming growth factor-beta induces microRNA-29b to promote murine alveolar macrophage dysfunction after bone marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L86–95. doi: 10.1152/ajplung.00283.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38(5):970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14(9):937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med. 2011;208(9):1789–1797. doi: 10.1084/jem.20110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang J, Huang HI, Benzatti FP, Karlsson AB, Zhang JJ, Youssef N, et al. Inflammatory Th1 and Th17 in the Intestine Are Each Driven by Functionally Specialized Dendritic Cells with Distinct Requirements for MyD88. Cell Rep. 2016;17(5):1330–1343. doi: 10.1016/j.celrep.2016.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez-Lopez M, Iborra S, Conde-Garrosa R, Sancho D. Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur J Immunol. 2015;45(1):119–129. doi: 10.1002/eji.201444651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neurohr C, Huppmann P, Leuchte H, Schwaiblmair M, Bittmann I, Jaeger G, et al. Human herpesvirus 6 in bronchalveolar lavage fluid after lung transplantation: a risk factor for bronchiolitis obliterans syndrome? Am J Transplant. 2005;5(12):2982–2991. doi: 10.1111/j.1600-6143.2005.01103.x. [DOI] [PubMed] [Google Scholar]
- 47.Versluys AB, Rossen JW, van Ewijk B, Schuurman R, Bierings MB, Boelens JJ. Strong association between respiratory viral infection early after hematopoietic stem cell transplantation and the development of life-threatening acute and chronic alloimmune lung syndromes. Biol Blood Marrow Transplant. 2010;16(6):782–791. doi: 10.1016/j.bbmt.2009.12.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seo S, Renaud C, Kuypers JM, Chiu CY, Huang ML, Samayoa E, et al. Idiopathic pneumonia syndrome after hematopoietic cell transplantation: evidence of occult infectious etiologies. Blood. 2015;125(24):3789–3797. doi: 10.1182/blood-2014-12-617035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.