

Abstract
The rickettsial pathogen Ehrlichia chaffeensis causes a tick-borne disease, human monocytic ehrlichiosis. Mutations within certain genomic locations of the pathogen aid in understanding the pathogenesis and in developing attenuated vaccines. Our previous studies demonstrated that mutations in different genomic sites in E. chaffeensis caused variable impacts on their growth and attenuation in vertebrate and tick hosts. Here, we assessed the effect of three mutations on transcriptional changes using RNA deep-sequencing technology. RNA sequencing aided in detecting 66–80% of the transcripts of wildtype and mutant E. chaffeensis. Mutation in an antiporter gene (ECH_0379) causing attenuated growth in vertebrate hosts resulted in the down regulation of many transcribed genes. Similarly, a mutation downstream to the ECH_0490 coding sequence resulted in minimal impact on the pathogen’s in vivo growth, but caused major changes in its transcriptome. This mutation caused enhanced expression of several host stress response genes. Even though the ECH_0660 gene mutation caused the pathogen’s rapid clearance in vertebrate hosts and aids in generating a protective response, there was minimal impact on the transcriptome. The transcriptomic data offer novel insights about the impact of mutations on global gene expression and how they may contribute to the pathogen’s resistance and/or clearance from the host.
Introduction
Ehrlichia chaffeensis is a tick-transmitted intracellular bacterial pathogen causing human monocytic ehrlichiosis (HME) and it also infects dogs, deer, goats, and coyotes1–4. Mutations at certain genomic locations, leading to gene expression changes, impact the pathogen’s ability to cause infection and persistence in a host5,6. The genome of E. chaffeensis may have evolved within a host cell environment leading to the development of mechanisms to undermine the host immune response7. Pathogenesis-associated E. chaffeensis genes are likely highly active in a host microenvironment and consistent with this hypothesis, differential gene expression in response to host cell defense is known to occur8. Progress has been made towards identifying genes crucial for Ehrlichia survival in a host cell environment9–11. However, to date only a few abundantly expressed genes are identified as associated with pathogenesis. Defining the genes involved in pathogenesis and virulence, and documenting their differential expression may aid in the discovery of novel proteins valuable as targets for therapeutic interventions and vaccine development for HME.
Genetically mutated intracellular pathogens are important resources for studying microbial pathogenesis, and also aid in the efforts of vaccine development12,13. Our previous study demonstrated the feasibility of transposon-based mutations in E. chaffeensis5,6. We also found that some insertion mutations resulting in transcriptional inactivation of membrane protein genes cause attenuation of the growth of the pathogen in vertebrate hosts. Insertions within the coding regions of ECH_0379 and ECH_0660 genes offered varying levels of protection against infection in a vertebrate host14. In this study, we hypothesized that the mutations’ specific genomic locations may impact global gene expression and contribute to the pathogen’s altered survival, infection progression, and replication in a host cell environment. To test this hypothesis, we assessed the impact of three mutations, reported earlier by Cheng et al.5, on global gene transcription. We selected two mutants with mutations within the coding regions of the ECH_0660 gene encoding for a phage like protein (ECH_0660) and the ECH_0379 gene encoding for an anti-porter protein (ECH_0379). Insertion mutation in ECH_0660 gene is located at the nucleotide position 213 of the 555 base long open reading frame. Similarly, mutation in ECH_0379 gene is located at the nucleotide position 682 of the 1056 base long open reading frame. The third insertion mutant strain, ECH_0490, has the insertion mutation 166 nucleotides downstream from the stop codon of ECH_0490 gene.
High throughput RNA sequencing (RNA seq) technologies have proven to be reliable and robust tools for determining global transcriptome activity in obligate intracellular bacteria12,15–17. Comparative genomic studies identified several classes of virulence factors involved in secretion and trafficking of molecules between the pathogen and host cells and modulation of the host immune response18–20. However, studies focused on Ehrlichia gene expression have been limited mostly to outer membrane proteins genes, Type IV Secretion System (T4SS) genes, tandem repeat protein (TRP) genes, and ankyrin repeat genes (Anks)9,19,21–23. Among them, genes encoding for T4SS proteins and p28-OMP proteins have been found to be critical for pathogenicity9,24.
The obligate intracellular nature of E. chaffeensis poses a challenge in obtaining cell-free Ehrlichia from host cells25. Technical constraints in isolating Ehrlichia RNA from highly abundant host RNA remains an impediment in profiling of pathogen transcripts26. To overcome this limitation, we used an effective cell lysis strategy followed by density gradient centrifugation. Further, we enriched Ehrlichia RNA by efficiently removing polyadenylated RNA (poly(A) RNA) and eukaryotic and prokaryotic ribosomal RNAs from host and bacteria RNA mixtures. Sequencing of the enriched RNA aided in the detection of transcripts for 66–80% of the annotated E. chaffeensis genes as per the annotated genome: GenBank #CP000236.1. Comparison of transcript levels from wildtype and mutant strains revealed the highest degree of modulation in immunogenic and secretory protein genes, particularly in the mutant strains of ECH_0490 and ECH_0379, while minimal changes were observed in the ECH_0660 mutant strain.
Results
Isolation and purification of cell-free E. chaffeensis from host cells
The major challenge of undertaking transcriptome studies of intracellular pathogens is the difficulty in isolating host-cell free bacteria and subsequently recovering high-quality bacterial RNA. Rickettsial organisms, including E. chaffeensis, constitute only a very small fraction of isolated total RNA27,28. Because of the presence of highly abundant host cell RNA, recovery of bacterial RNA is a challenge for executing RNA seq analysis experiments. In this study, we first purified the host cell-free bacteria from infected host cells (canine macrophage cell line, DH82) by employing an efficient cell lysis method, coupled with density gradient centrifugation protocols. Host cell lysis was performed to efficiently rupture the host cells without causing a major damage to the bacteria. E. chaffeensis organisms are about 0.5 to 1 µm in diameter. Therefore, infected host cell lysate was filtered through 2 µm membrane to remove most of the host cell debris. A high-speed Renografin density gradient centrifugation of the resulting E. chaffeensis cell suspension aided in pelleting bacteria while host cell debris remained at the top layer of the solution. After total RNA isolation and DNase treatment, Bioanalyzer analysis revealed that despite the prior fractionation of host cell-free bacteria, the host 28 S and 18 S RNA remained at high concentrations in the recovered RNA. Bacterial mRNA enrichment was carried out by depleting the host poly(A) RNA and eukaryotic ribosomal RNA using a bacterial RNA enrichment protocol, resulting in nearly undetectable levels of host 28 S and 18 S RNA (Supplementary Figures; Fig. S1 and Fig. S2). The absence of contaminating E. chaffeensis genomic DNA in the purified RNA samples was confirmed by real-time quantitative PCR using E. chaffeensis 16 S rRNA gene primers27. We also confirmed the absence of DNA sequences in the RNA seq raw data by aligning 20 randomly selected E. chaffeensis intergenic non-coding DNA sequences (data not shown).
Ubiquitous transcription of genes in E. chaffeensis mutants
Illumina HiSeq. 4000 RNA seq of E. chaffeensis wildtype and mutants generated between 75–130 million reads. The transcriptome data were deposited in the NCBI Bio-Project ID:PRJNA428837 and SRA accession:SRP128532 (https://www.ncbi.nlm.nih.gov/sra/SRP128532). Despite efficient depletion of host ribosomal RNA, only a fraction (less than19%) of reads were mapped to E. chaffeensis genomes. Mapping of reads (10 reads minimum/gene) identified about 66–80% of the genes being expressed from the Ehrlichia genome as per the annotated genome (GenBank # CP000236.1); the transcriptome of wildtype organisms (n = 3) contained transcripts for about 920 genes of the total of 1158 genes, and similarly 888, 895, and 768 gene transcripts (n = 3) were identified in mutant organisms ECH_0660, ECH_0379, and ECH_0490, respectively (Table 1). Table S1 lists total numbers of genes, and the expression value of the genes identified in the wildtype and all three mutant organisms. The replicate RNA seq data of wildtype (R² = 0.9) (Fig. 1A) and mutants ECH_0379 (R² = 0.93), (Fig. 1B), ECH_0490 (R² = 0.68) (Fig. 1C), and ECH_0660 (R² = 0.89) (Fig. 1D) showed a high degree of expression correlation. The scatter plot expression data of wildtype vs. ECH_0379 (R² = 0.18) (Fig. 1E) and wildtype vs. ECH_0490 (R² = 0.38) (Fig. 1F) showed a negative correlation. Notably, the expression plot of wildtype vs. ECH_0660 showed a positive correlation (R² = 0.96) (Fig. 1G). Only transcripts with reads per kilobase transcriptome per million mapped reads (RPKM) ≥ 1 were considered for differential expression analysis.
Table 1.
Number of E. chaffeensis genes identified in three replicates of wildtype and its mutants.
| No. of genes identified (>3 RPKM, 10 reads minimum) | ||||
|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | Avg (std dev) | |
| Wildtype | 888 | 900 | 973 | 920 (46) |
| ECH_0379 | 920 | 882 | 883 | 895 (21) |
| ECH_0490 | 841 | 670 | 793 | 768 (88) |
| ECH_0660 | 780 | 917 | 969 | 888 (97) |
Figure 1.

Scatter plot of RNA seq expression analysis. Scatter plots of transcript expression data for replicates of E. chaffeensis wildtype (A) and mutants ECH_0379 (B), ECH_0490 (C), and ECH_066 (D) showing a high degree of correlation. Scatter plots of transcript expression data for wildtype vs. mutants: (E) wildtype vs. ECH_0379, (F) wildtype vs. ECH_0490, and (G) wildtype vs. ECH_0660. Transcripts with ≥ 1 FPKM and minimum of 10 mapped reads were used. The plot is on a log-transformed scale.
Global transcriptome of E. chaffeensis
Distribution of the transcripts in wildtype E. chaffeensis (Fig. 2) included 481 transcripts represented by less than five transcripts, followed by hypothetical protein transcripts (178) representing 19% of transcriptome, and 127 ribosomal protein gene transcripts (14%). Transcripts of major outer membrane proteins (22 transcripts) represent the next most abundant group. Conserved domain protein transcripts encoded from 14 genes are associated with NADH dehydrogenase I complex. Other highly expressed genes included molecular chaperones, ATP synthase, putative membrane protein, cytochrome c oxidase, GTP-binding protein, putative lipoprotein, translation elongation factor, ABC transporter, and DNA polymerases; all of which represented 0.5–1.7% of the transcriptome. Table S2 lists the top 100 highly expressed genes in transcriptome of wildtype E. chaffeensis.
Figure 2.

Distribution of the identified transcripts in wildtype E. chaffeensis. The inlaid numbers represent the percentage of transcripts detected in the RNA seq data (n = 3). The number of identified transcripts associated with each gene category is shown in the brackets. The minimum transcripts representation for each gene category was set to 5. The total number of genes identified was 920.
ECH_0379 mutation caused transcriptional down-regulation of many genes involved in antiporter activity, phage proteins, and those involved in transport and transcription function
Differential gene expression (DGE) was determined by comparing the RPKM expression values of mutants and wildtype (Fig. S3). Fold changes were considered significant with a p-value < 0.05, False Discovery Rate (FDR) ≤ 0.001, and consistency of expression values between replicates. The change in gene expression was not significant between wildtype and mutants for housekeeping genes. Based on these criteria, 41 genes were identified as predominantly downregulated and two genes were upregulated in the ECH_0379 gene mutant compared to wildtype (Table 2). The most prominent genes that showed a significant decrease in the transcription levels were those encoding for antiporter proteins, ABC transporters, and ATP-dependent Clp protease (ECH_0367). Four antiporter protein genes: monovalent cation/proton antiporter (ECH_0466), Na(+)/H(+) antiporter subunit C (mrpC) (ECH_0469), potassium uptake protein TrkH (ECH_1093), and nitrogen regulation protein NtrY (ECH_0299) showed a significant decline in the transcript levels. In addition, transcripts for two membrane transporters: cation ABC transporter permease protein transcript of the gene ECH_0517 and another ABC transporter permease protein transcript of the gene ECH_0972 were downregulated. Three genes coding for phage-like proteins {phage prohead protease (ECH_0032), phage portal protein (ECH_0033), and phage major capsid protein (ECH_0830)} were also downregulated in the mutant strain. Transcripts for 6 genes involved in transcription, namely DNA replication and repair protein RecF (ECH_0076), formamidopyrimidine-DNA glycosylase (ECH_0602), dimethyladenosine transferase (ECH_0648), GTP-binding protein EngA (ECH_0504), leucyl-tRNA synthetase (ECH_0794), and endonuclease III (ECH_0857) were also downregulated in this mutant strain. The enzymes of metabolic processes such as glutamate cysteine ligase (GCL) (ECH_0125), DNA/pantothenate metabolism flavoprotein (PMF) (ECH_0374), ATPase, AGF1 (ECH_0392), uroporphyrinogen III synthase (UPGS) (ECH_0480), diaminopimelate decarboxylase (DAPDC) (ECH_0485), biotin-acetyl-CoA-carboxylase ligase (BACL) (ECH_0848), and argininosuccinate lyase (ASL) (ECH_0937) are also down-regulated. Transcripts for 8 hypothetical protein genes; ECH_0021, ECH_0161, ECH_0264, ECH_0289, ECH_0725, ECH_0879, ECH_0913, and ECH_1053 were also among the downregulated genes in this mutant.
Table 2.
E. chaffeensis genes differentially transcribed in ECH_0379 compared to wildtype.
| Gene ID | Wildtype gene expression (RPKM) | ECH_0379 gene expression (RPKM) | Fold change (ECH_0379/Wildtype) FDR ≤ 0.001, p-value <0.05 | Gene name |
|---|---|---|---|---|
| Down regulated genes | ||||
| ECH_0021 | 391 | 211 | −1.88 | conserved hypothetical protein |
| ECH_0032 | 82 | 26 | −3.2 | phage prohead protease, HK97 family |
| ECH_0033 | 41 | 20 | −1.53 | phage portal protein, HK97 family |
| ECH_0076 | 287 | 59 | −5 | putative DNA replication and repair protein RecF |
| ECH_0125 | 386 | 185 | −2.08 | glutamate-cysteine ligase |
| ECH_0161 | 81 | 42 | −1.92 | hypothetical protein |
| ECH_0188 | 586 | 121 | −5 | putative surface protein |
| ECH_0264 | 814 | 194 | −4.16 | conserved hypothetical protein |
| ECH_0289 | 102 | 52 | −1.96 | hypothetical protein |
| ECH_0299 | 1432 | 442 | −1.81 | putative nitrogen regulation protein NtrY |
| ECH_0367 | 3407 | 1784 | −1.92 | ATP-dependent Clp protease, ATP-binding subunit ClpB |
| ECH_0374 | 411 | 157 | −2.63 | DNA/pantothenate metabolism flavoprotein family protein |
| ECH_0392 | 845 | 159 | −5.55 | ATPase, AFG1 family |
| ECH_0466 | 432 | 252 | −1.72 | monovalent cation/proton antiporter |
| ECH_0469 | 137 | 52 | −5.55 | Na(+)/H(+) antiporter subunit C |
| ECH_0473 | 793 | 306 | −5.55 | aromatic-rich protein family |
| ECH_0480 | 319 | 92 | −3.22 | uroporphyrinogen-III synthase |
| ECH_0485 | 537 | 172 | −3.14 | diaminopimelate decarboxylase |
| ECH_0504 | 859 | 288 | −3.03 | GTP-binding protein EngA |
| ECH_0517 | 503 | 52 | −10 | putative cation ABC transporter, permease protein |
| ECH_0523 | 1525 | 159 | −10 | conserved domain protein |
| ECH_0541 | 251 | 124 | −2 | 5-formyltetrahydrofolate cyclo-ligase family protein |
| ECH_0602 | 84 | 24 | −3.57 | formamidopyrimidine-DNA glycosylase |
| ECH_0648 | 399 | 138 | −2.94 | dimethyladenosine transferase |
| ECH_0725 | 648 | 280 | −2.32 | conserved hypothetical protein |
| ECH_0756 | 815 | 153 | −5.55 | divalent ion tolerance protein CutA1 |
| ECH_0789 | 1154 | 363 | −3.22 | cytochrome c-type biogenesis protein CcmE |
| ECH_0794 | 1593 | 306 | −5.26 | leucyl-tRNA synthetase |
| ECH_0830 | 397 | 123 | −3.22 | phage major capsid protein, HK97 family |
| ECH_0848 | 1015 | 253 | −4 | biotin—acetyl-CoA-carboxylase ligase |
| ECH_0857 | 638 | 311 | −2.04 | endonuclease III |
| ECH_0864 | 455 | 246 | −1.85 | conserved domain protein |
| ECH_0879 | 520 | 153 | −3.44 | hypothetical protein |
| ECH_0913 | 570 | 114 | −5 | conserved hypothetical protein |
| ECH_0937 | 521 | 284 | −1.85 | argininosuccinate lyase |
| ECH_0972 | 524 | 285 | −1.85 | ABC transporter, permease protein |
| ECH_0998 | 722 | 332 | −2.17 | ubiquinone/menaquinone biosynthesis methlytransferase UbiE |
| ECH_1053 | 541 | 248 | −2.22 | conserved hypothetical protein |
| ECH_1063 | 201 | 106 | −1.92 | modification methylase, HemK family |
| ECH_1081 | 310 | 78 | −4 | SURF1 family protein |
| ECH_1084 | 684 | 364 | −1.88 | AraM protein |
| ECH_1093 | 973 | 320 | −2.32 | putative potassium uptake protein TrkH |
| ECH_1101 | 1143 | 190 | −6.25 | prolipoprotein diacylglyceryl transferase |
| Up regulated genes | ||||
| ECH_0684 | 1765 | 3651 | 2.06 | ankyrin repeat protein |
| ECH_0495 | 942 | 1492 | 1.58 | type IV secretion system protein VirB4 |
Differential transcriptional regulation of T4SS and p-28 OMP gene cluster genes in mutant ECH_0490
In the ECH_0490 mutant strain, 37 genes were significantly downregulated and 17 genes were up-regulated (Table 3). Four of the downregulated genes belonged to the T4SS are ECH_0494 (VirB3), ECH_0496 (VirB6), ECH_0498 (VirB6), and ECH_0499 (VirB6); and a type I secretion membrane fusion protein (T1SS_HlyD) (ECH_0970). Molecular chaperone genes, such as a cold shock protein (CSP) (ECH_0298) and ATP-dependent Clp protease, and a ATP-binding subunit ClpA (ClpA) were also downregulated. The transport proteins including the protein export membrane protein (SecF) (ECH_0095), preprotein translocase (SecY) (ECH_0428), potassium uptake protein (TrkH) (ECH_1093), and nitrogen regulation protein (NtrY) (ECH_0299) were also among the downregulated genes. Metabolic enzymes involved in biosynthetic processes, {tetrahydropyridine-2-carboxylate N-succinyltransferasem (dapD) (ECH_0058), quinone oxidoreductase (ECH_0385), metalloendopeptidase, (MEP) (ECH_0644), peptide deformylase (PDF) (ECH_0939), serine/threonine phosphatase (PSP) (ECH_0964), pyrophosphatase (PPi) (ECH_1014), and orotate phosphoribosyltransferase (OPRTase) (ECH_1108)}, were also down-regulated. Transcription- and translation-related genes, such as elongation factors (EF-Tu) (ECH_0515), aminoacyl-tRNA synthetases (IARS) (ECH_0538), DNA-binding protein (HU) (ECH_0804), 3′-5′ exonuclease domain (ECH_1011), and DNA-binding response regulator (ECH_1012), were also downregulated.
Table 3.
E. chaffeensis genes differentially transcribed in ECH_0490 compared to wildtype.
| Gene ID | Wildtype gene expression (RPKM) | ECH_0490 gene expression (RPKM) | Fold change (ECH_0490/wildtype) FDR ≤ 0.001, p-value <0.05 | Gene name |
|---|---|---|---|---|
| Down regulated genes | ||||
| ECH_0058 | 1902 | 1006 | −1.88 | 2,3,4,5-tetrahydropyridine-2-carboxylate N-succinyltransferase |
| ECH_0085 | 1119 | 523 | −2.17 | ABC transporter, ATP-binding protein |
| ECH_0095 | 1921 | 990 | −1.96 | protein-export membrane protein SecF |
| ECH_0264 | 814 | 310 | −5.55 | conserved hypothetical protein |
| ECH_0298 | 8295 | 3870 | −2.17 | cold shock protein, CSD family; |
| ECH_0299 | 719 | 314 | −2.17 | putative nitrogen regulation protein NtrY |
| ECH_0300 | 557 | 283 | −2 | putative ribonuclease D |
| ECH_0385 | 1659 | 663 | −2.5 | quinone oxidoreductase |
| ECH_0428 | 979 | 425 | −2.32 | preprotein translocase, SecY subunit |
| ECH_0470 | 1220 | 598 | −2 | ribonuclease, Rne/Rng family |
| ECH_0475 | 977 | 444 | −2.22 | signal recognition particle protein |
| ECH_0483 | 158 | 77 | −2.04 | primosomal protein N |
| ECH_0494 | 2326 | 1034 | −2.17 | type IV secretion system protein VirB3 |
| ECH_0496 | 1059 | 435 | −2.43 | type IV secretion system protein VirB6 |
| ECH_0498 | 1154 | 490 | −2.38 | type IV secretion system protein,VirB6 family |
| ECH_0499 | 1129 | 558 | −2 | type IV secretion system protein,VirB6 family |
| ECH_0515 | 1968 | 910 | −2.17 | translation elongation factor Ts |
| ECH_0525 | 1055 | 427 | −2.5 | conserved domain protein |
| ECH_0538 | 729 | 355 | −2.08 | isoleucyl-tRNA synthetase |
| ECH_0567 | 626 | 177 | −3.57 | ATP-dependent Clp protease, ATP-binding subunit ClpA |
| ECH_0585 | 475 | 229 | −2.08 | conserved domain protein |
| ECH_0644 | 1902 | 764 | −2.5 | putative metalloendopeptidase, glycoprotease family |
| ECH_0700 | 2670 | 1073 | −2.5 | hypothetical protein |
| ECH_0804 | 3113 | 1292 | −2.43 | DNA-binding protein HU |
| ECH_0820 | 409 | 167 | −2.5 | conserved hypothetical protein |
| ECH_0840 | 935 | 296 | −3.22 | 2-polyprenylphenol 6-hydroxylase |
| ECH_0939 | 752 | 276 | −2.77 | putative polypeptide deformylase |
| ECH_0953 | 2914 | 1480 | −2 | ribosomal protein L7/L12 |
| ECH_0964 | 1281 | 557 | −2.32 | serine/threonine phosphoprotein phosphatase |
| ECH_0970 | 474 | 247 | −1.92 | type I secretion membrane fusion protein, HlyD family |
| ECH_1011 | 2253 | 1104 | −2.04 | 3′-5′ exonuclease family protein |
| ECH_1012 | 3353 | 1605 | −2.08 | DNA-binding response regulator |
| ECH_1014 | 1661 | 536 | −3.12 | inorganic pyrophosphatase |
| ECH_1093 | 973 | 416 | −2.38 | putative potassium uptake protein TrkH |
| ECH_1108 | 1903 | 938 | −2.04 | orotate phosphoribosyltransferase |
| ECH_1139 | 545 | 285 | −1.92 | major outer membrane protein OMP-1D |
| Up-regulated genes | ||||
| ECH_0009 | 7047 | 16828 | 2.38 | putative membrane protein |
| ECH_0039 | 316 | 931 | 2.94 | 120 kDa immunodominant surface protein |
| ECH_0166 | 42488 | 96364 | 2.26 | conserved hypothetical protein |
| ECH_0167 | 718 | 2654 | 3.70 | tryptophanyl-tRNA synthetase |
| ECH_0169 | 161 | 397 | 2.46 | riboflavin biosynthesis protein RibD |
| ECH_0230 | 991 | 4109 | 4.15 | putative membrane protein |
| ECH_0251 | 1042 | 2185 | 2.1 | hypothetical protein |
| ECH_0303 | 1018 | 2856 | 2.80 | BolA family protein |
| ECH_0367 | 849 | 1274 | 2.49 | ATP-dependent Clp protease, ATP-binding subunit ClpB |
| ECH_0450 | 1261 | 3710 | 2.94 | conserved hypothetical protein |
| ECH_0531 | 1363 | 11788 | 8.65 | hypothetical protein |
| ECH_0630 | 732 | 1688 | 2.30 | FeS cluster assembly scaffold IscU |
| ECH_0655 | 1840 | 2763 | 2.03 | RNA polymerase sigma-32 factor |
| ECH_0753 | 1932 | 4153 | 2.15 | conserved hypothetical protein |
| ECH_0818 | 374 | 1222 | 3.26 | major facilitator family transporter |
| ECH_0878 | 217 | 1126 | 5.17 | hypothetical protein |
| ECH_1121 | 1578 | 3132 | 3.1 | major outer membrane protein Omp-1N |
| ECH_1136 | 698 | 8270 | 2.37 | major outer membrane protein OMP-1B |
| ECH_1143 | 3957 | 8359 | 2.24 | major outer membrane protein P28 |
| ECH_1146 | 190 | 1100 | 6.73 | major outer membrane protein P28-2 |
Upregulated protein genes in this mutant included 7 that belonged to the transmembrane protein category. Of these, four belonged to the p-28 OMP gene cluster {ECH_1143 (OMP-p28), ECH_1146 (OMP-p28-2), ECH_1136 (OMP-1B), and ECH_1121 (OMP-1N)}. In addition, two putative membrane protein genes (ECH_0009, ECH_0230) and an immunodominant surface protein gene (ECH_0039) were upregulated. Transcripts for the heat shock proteins ATP-dependent Clp protease, ClpA (ECH_0567) and ATP-binding chaperon, ClpB (ECH_0367), and the stress response-associated RNA polymerase sigma factor (RpoH) (ECH_0655) were also upregulated. Transcripts for two genes coding for iron sulfur proteins {BolA family protein (ECH_0303) and FeS cluster assembly scaffold (IscU) (ECH_0630)} were similarly up-regulated. We observed differential expression of six hypothetical protein genes, which included ECH_0166, ECH_0251, ECH_0450, ECH_0531, ECH_0753, and ECH_0878.
Mutation in ECH_0660 gene led to minimal transcriptional alterations
While we observed drastic gene expression changes in both ECH_0379 and ECH_0490 mutants, ECH_0660 mutant transcriptome showed minimal variations compared to wildtype; we observed only five genes as notably differentially expressed in this mutant (Table 4). The genes included nitrogen regulation protein (NtrY) (ECH_0299) and the ABC transporter permease protein (ECH_0972) as down-regulated genes, whereas the heme exporter protein CcmA (ECH_0295) and chaperonin (ECH_0364) were upregulated. We also identified several commonly differentially-expressed genes in ECH_0379 and ECH_0490 (Table 5). The ribonuclease D (ECH_0300) and potassium uptake protein (ECH_1093) were commonly down regulated in ECH_0379 and ECH_0490. T4SS protein VirB4 gene was down-regulated in ECH_0490 mutant, whereas this gene was up-regulated in ECH_0379 mutant. Contrary to this, ClpB was down-regulated in ECH_0379 mutant and upregulated in ECH_0490 mutant.
Table 4.
E. chaffeensis genes differentially transcribed in ECH_0660 compared to wildtype.
| Gene ID | Wildtype gene expression (RPKM) | ECH_0660 gene expression (RPKM) | Fold change (ECH_0660/Wildtype) FDR ≤ 0.001, p-value <0.05 | Gene name |
|---|---|---|---|---|
| Down regulated genes | ||||
| ECH_0299 | 1432 | 720 | −2 | putative nitrogen regulation protein NtrY |
| ECH_0972 | 524 | 309 | −1.69 | ABC transporter, permease protein |
| Up regulated genes | ||||
| ECH_0295 | 336 | 631 | 1.87 | putative heme exporter protein CcmA |
| ECH_0364 | 6801 | 12150 | 1.78 | chaperonin, 10 kDa |
| ECH_1147 | 1982 | 4756 | 2.39 | conserved hypothetical protein |
Table 5.
E. chaffeensis common differentially transcribed genes in mutants.
| Gene ID | Wildtype gene expression (RPKM) | Mutant gene expression (RPKM) | Fold change FDR ≤ 0.001, p-value < 0.05 | Gene name | mutants |
|---|---|---|---|---|---|
| Down regulated genes | |||||
| ECH_0299 | 1432 | 442 | −1.81 | putative nitrogen regulation protein NtrY | ECH_0379 ECH_0490 |
| ECH_0264 | 814 | 310 | −2.63 | conserved hypothetical protein | ECH_0379 ECH_0490 |
| ECH_0300 | 557 | 283 | −2 | putative ribonuclease D | ECH_0379 ECH_0490 |
| ECH_0864 | 279 | 193 | −1.44 | conserved domain protein | ECH_0379 ECH_0490 |
| ECH_1093 | 972 | 416 | −1.81 | putative potassium uptake protein TrkH | ECH_0379 ECH_0490 |
| ECH_0495 | 833 | 517 | −1.63 | type IV secretion system protein VirB4 | ECH_0490 |
| ECH_0367 | 3407 | 1783 | −1.92 | ATP-dependent Clp protease, ATP-binding subunit ClpB | ECH_0379 |
| ECH_0745 | 712 | 437 | −1.63 | conserved domain protein | ECH_0379 |
| Up regulated | |||||
| ECH_0495 | 942 | 1492 | 1.58 | type IV secretion system protein VirB4 | ECH_0379 |
| ECH_0367 | 849 | 1275 | 2.49 | ATP-dependent Clp protease, ATP-binding subunit ClpB | ECH_0490 |
| ECH_0745 | 547 | 920 | 1.68 | conserved domain protein | ECH_0490 |
Validation of RNA seq data by quantitative real-time reverse transcription PCR
Quantitative real-time quantitative reverse transcriptase-PCR (qRT-PCR) analysis was carried out on thirteen randomly selected genes identified as differentially transcribed according to the RNA seq data. To generate qRT-PCR data, we first normalized RNA samples to a constitutively expressed E. chaffeensis gene coding for the16S RNA as previously described in Cheng et al.6. The primers and genes selected for the qRT-PCR analysis are listed in Table S3. Transcript abundance for 7 down-regulated genes in ECH_379 mutant, including ECH_0466 and mrpC, ClpB, ECH_0033, NtrY, TrkH, and ECH_0972 were validated (Fig. 3A). Similarly, 6 upregulated genes from ECH_0490 mutant strain, including four transcripts belonging to an OMP gene cluster (OMP-p28, OMP-1B, OMP-1N, OMP-p28-2) and one each from ClpB and RpoH genes were verified by qRT-PCR (Fig. 3B). Likewise, the down-regulation of transcripts for the ECH_0299 and ECH_0972 genes were confirmed in ECH_0660 mutant by qRT-PCR (Fig. 3C).
Figure 3.
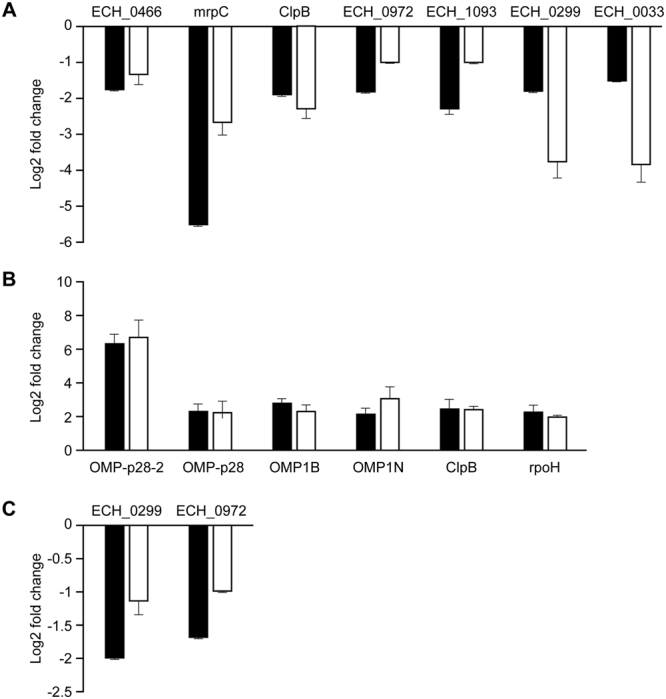
Verification of transcriptional variations observed in RNA seq data by qRT-PCR. Transcriptional fold changes in wildtype vs. ECH_0379 mutant (A), ECH_0490 mutant (B), or ECH_0660 mutant (C) were presented from the qRT-PCR data. Black bars represents RNA seq data and white bars represents qRT-PCR data.
Discussion
Isolation of cell-free bacterial RNA from highly abundant host RNA is the first challenge in transcriptional profiling of intracellular pathogens25,28,29. Rickettsiales require culturing in host cells and then need to be purified before extracting RNA for transcriptome evaluation experiments. To document the impact of three transposon mutations on E. chaffeensis transcription, we first developed a method for isolation and purification of host cell-free E. chaffeensis organisms, from which we isolated RNA and then subjected to next generation sequencing (NGS) analysis. To isolate cell-free E. chaffeensis, we started with an efficient host cell lysis protocol, and then filtration of whole cell lysate, followed by a renografin density gradient centrifugation. The second challenge was to obtain host cell-free RNA for transcriptome profiling. Previous studies report that bacterial RNA enrichment methods result in the enrichment of bacterial RNA reads only 3–10%29,30. Isolation of host cell-free bacteria and the bacterial RNA purification steps implemented in our study allowed a greater enrichment of E. chaffeensis RNA. In our current studies, we were able to enrich the bacterial RNA, which helped in generating up to 19% high mapping RNA reads. Notably, deep RNA sequencing analysis aided in mapping 80% of E. chaffeensis genes expressed in infected macrophage host cells.
Among the highly expressed genes, the p28-OMP multigene cluster was dominant in the transcriptome. The E. chaffeensis p28-OMP multigene locus contains 22 tandemly arranged genes coding for the bacterial immunodominant proteins31–33. The presence of all 22 transcripts in the RNA seq data suggest that the gene cluster is among the most abundantly expressed genes. These observations are consistent with our previous proteomic study where we reported the p28-OMP genes’ expression abundance33. NADH dehydrogenase I complex genes were also highly expressed in E. chaffeensis. NADH dehydrogenase counters the phagosomal NOX2 response to inhibit host cell apoptosis34. T4SS effector proteins in some pathogenic bacteria are considered as important in manipulating a host gene expression to undermine the host immune response35,36. The contributions of T4SS effectors in pathogenicity are already reported for rickettsiales, including for A. marginale, A. phagocytophilum, E. canis, and E. chaffeensis37–39. The RNA seq analysis identified several transcripts encoding for T4SS proteins, including VirB3, B4, B6, B8, B9, B10, and B11. Chaperone protein genes DnaK, DnaJ, GroE, and ClpB were also highly expressed in both wildtype and mutant strains. The presence of such proteins involved in cell homeostasis and the oxidative stress response is reported in other rickettsiales39–41, suggesting that their gene products are also critical for the E. chaffeensis stress response if the pathogen proteome is similarly altered as per the transcriptome reported in the current study. Indeed, our recent study suggests that the stress response proteins are important for E. chaffeensis11. Other highly expressed protein genes included those encoding for house-keeping ribosomal proteins involved in protein synthesis, putative membrane proteins, ABC transporter, and lipoprotein; all of which are likely important for the pathogen’s protein synthesis, transport, trafficking, and effector secretion into the host cells. ATP synthase subunit, cytochrome c oxidase, DNA polymerases, GTP-binding protein and translation elongation factors involved energy metabolism, cell division, and transcriptional regulation were also among the highly expressed genes in both wildtype and mutant organisms. The extent of transcriptome coverage is higher than the previously reported for E. chaffeensis in ISE6 and AAE2 tick cells8. This is substantial for both the enhanced detection of intracellular pathogen transcripts and also because of the abundance of gene expressions observed. Higher coverage of the transcriptome likely resulted from deep sequencing of the RNAs by next-generation sequencing compared to microarray analysis8. This global set of highly expressed genes may represent products involved in pathogenicity, replication and survival of E. chaffeensis in host cell environment42,43. Four transcripts that code for ankyrin repeat proteins, which are shown to mediate protein-protein interactions44, were also identified in the transcriptome. Notably, the transcriptome from the wildtype and mutant organisms contained 216 transcripts that code for hypothetical proteins with unknown function. As these were within the core transcriptome, we anticipate that they represent an important set of transcribed genes for E. chaffeensis replication.
Transcription from large numbers of genes in ECH_0379 mutant was found to be reduced compared to wildtype. Genes representing antiporters, ABC transporters, chaperons, metabolic enzymes, and transcription regulators are among the down-regulated genes (Table 2). We predict that the mutation in the anti-porter protein gene caused a metabolic depression. Antiporter and transport proteins play an important role in the transport of ions and solutes across the cell membranes of bacteria45. Antiporters are integral membrane proteins that perform secondary transport of Na+ and/or K+ for H+ across a phospholipid membrane5. The E. chaffeensis genome contains several genes having homology to antiporter proteins or their subunits, suggesting that they are needed for the pathogen’s intraphagosomal replication and survival in a host. In particular, antiporters aid bacteria in maintaining pH, salt, and temperature conditions46. We observed a significant decline in transcription of antiporter genes such as monovalent cation/H + antiporter subunit C (ECH_0469) and ECH_0466. Disrupting the antiporter function or preventing their expression may affect the pathogen’s growth in vivo. Indeed, mutation in the ECH_0379 gene resulted in the attenuated growth of the organism in both an incidental host (dog) and in the reservoir host (white-tailed deer)5,6. ABC transporters also are involved in uptake of ions and amino acids and may play an important role in a pathogen’s ability to infect and survive in a host cell environment47. The ECH_0379 mutant had low levels of transcriptional activity of the genes ECH_0517 and ECH_0972 encoding for ABC transporters, which function at different stages in the pathogenesis of infection47,48. These proteins promote the survival of pathogens in the host microenvironments49. The mutation possibly interferes with transport mechanisms, thereby affecting its ability to infect and survive in host cells5,6. The mutation may have also caused alterations to the transcriptions of genes involved in physiological responses, such as regulating the pathogen’s metabolic activities. We also found down-regulation of several transcripts encoding for metabolic enzymes: glutamate–cysteine ligase, DNA/pantothenate metabolism flavoprotein family protein, ATPase, uroporphyrinogen-III synthase, diaminopimelate decarboxylase, biotin–acetyl-CoA-carboxylase ligase, and argininosuccinate lyase. In general, a pathogen’s survival in an intracellular environment depends on its ability to derive nutrients from the host cell50. Pathogenic bacteria use metabolic pathways and virulence-associated factors that undermine the host immune system so that they can derive nutrients from their host cells51. It is possible that the downregulation of the transcripts from the aforementioned genes in the ECH_0379 mutant hampers the bacterial metabolic response and its capacity to derive nutrients from the host. The mutation also caused decreased expression of genes encoding DNA replication and repair protein, formamidopyrimidine-DNA glycosylase, dimethyladenosine transferase, and leucyl-tRNA synthetase. This may have also contributed to defects in pathogen’s intracellular growth and survival. Our prior studies suggest that despite the mutant’s attenuated growth, it failed to offer complete protection against wildtype infection challenge14. If the changes in the transcriptome correlate with changes in the proteome, variations in the mutant organisms’ protein expression relative to the wildtype E. chaffeensis may result in an altered host response, thus making the host less effective in initiating a protective host response when exposed to the mutant organisms14.
Pathogenic bacteria produce T4SS effectors to weaken the host cell gene expression and contributes to bacterial virulence52,53. RNA seq data suggested declined expressions of various T4SS component protein gene transcripts in ECH_0490 mutant. We also observed decreased transcription of chaperone proteins and several genes involved in the transcription and translational machinery, and exonuclease and DNA-binding regulator gene transcripts in the ECH_0490 mutant strain. On the contrary, ClpB (a major stress response heat shock protein) and RpoH (stress response RNA polymerase transcriptional subunit) showed increased transcription in the mutant.
Chaperone proteins play a key role in protein disaggregation and in aiding the pathogen to overcome the likely host cell-induced stress54. ClpB reactivates aggregated proteins accumulating under stress conditions and it was abundantly expressed during replication stage of E. chaffeensis54,55. Preventing or reducing protein aggregation and the associated protein inactivation during the bacterial growth within a host cell may benefit the pathogen in enhancing its survival11. The RNA polymerase transcription regulator, RpoH, is also important for the pathogen’s continued growth as it aids in promoting the expression of stress response proteins10. Consistent with the prediction, increased expression of ClpB and RpoH was observed in the current study for ECH_0490 mutant. The enhanced expression from these two important genes likely enables the mutant to grow similarly to wildtype E. chaffeensis in vertebrate and tick hosts, as reported in our previous studies5,6. Outer membrane proteins perform a variety of functions such as invasion, transport, immune response, and adhesion that are vital to the survival of Ehrlichia species, including E. chaffeensis and E. ruminantium in a host32,56–59. The ECH_0490 mutant had increased abundance of OMPs compared to wildtype organisms. We found seven transmembrane genes coding for immunodominant P28/OMP family of proteins (OMP_p28, OMP_p28-2, OMP-1B, and OMP-1N) and membrane proteins (ECH_0039, ECH_0009, and ECH_0230) to be upregulated. Significant changes in the abundance of the outer membrane proteins may be associated with overall changes in the membrane architecture, thereby altering the pathogen’s susceptibility to host defense. The transcriptional changes noted in the ECH_0490 mutant may not have had any negative impact on the pathogen, as the mutant grows similar to the wildtype pathogen both in white-tailed deer (the reservoir host) and in dogs (an incidental host), and in its tick host, Amblyomma americanum5,6. Transcriptional activity assessment of the genes ECH_0490 (lipoic acid synthetase) and ECH_0492 (putative phosphate ABC transporter), both of which are located up and down stream to the transposon insertion mutation, respectively, suggested that the mutation has no effect on these genes’ transcription (Fig. S4). The diverse changes in the transcriptome of the mutant, while having no impact near the mutation site, suggest that the mutation impacted global gene expression and yet did not adversely affect the pathogen’s survival in vertebrate and tick hosts5,6.
The most notable observation was the apparent minimal variation in the transcriptome of the ECH_0660 mutant compared to the wildtype E. chaffeensis. Importantly, mutation within ECH_0660 gene causes severe growth defects in vivo in vertebrate hosts5,6. Further, infection with this mutant also initiates a strong host response and confers protection against wildtype pathogen infection challenge14,60. In the current study, we observed only minor changes in the gene expression in this mutant compared to wildtype. The minor changes in gene expression included genes encoding for putative nitrogen regulation protein, ABC transporter, heme exporter protein and GroES, but the variations were significantly less compared to numerous changes described in the previous two mutants. Together, these data suggest that the mutation in ECH_0660 gene led to fewer transcriptional alterations. Assuming that the proteomes of the wild type and mutant strains of E. chaffeensis are similarly altered as the transcriptomes, then ECH_0660 mutant proteome may be very similar to the wildtype bacterium. The greater degree of similarity between this mutant and the wildtype may enable the vertebrate hosts to recognize this mutant as closer to wildtype organism, thus inducing a stronger host response that mimics wildtype infection14,60. The replication defect reported earlier with this mutant may have resulted due to the loss of gene expression from fewer genes such as ECH_0659 and ECH_0660, while maintaining most of the transcriptome similar to the wildtype.
Conclusions
RNA deep sequencing studies in intracellular bacteria are still a major challenge. The RNA seq data reported here provide the first snapshot of comparative transcriptomics of E. chaffeensis. Sequencing of enriched bacterial RNA from wildtype and mutant strains yielded a high coverage of genes. A mutation in the ORF of ECH_0379 gene caused drastic down-regulation of genes leading to metabolic depression, which may have contributed to the mutant’s attenuation in vertebrate hosts. While a mutation downstream to the protein coding sequence of ECH_0490 gene induced global changes in gene expression, up regulation of stress response regulatory genes may have helped the mutant survive in the vertebrate hosts and tick hosts. A mutation within ECH_0660 gene coding sequence resulted in few transcriptional changes, thus keeping the integrity of its transcriptome similar to wildtype. While the transcriptome data are suggestive of protein expression variations, additional experimental validation from protein analysis studies is necessary to confirm the results. Together, this study offers the first detailed description of transcriptome data for E. chaffeensis, suggesting that variations observed in the pathogen’s ability to survive in a host and the host’s ability to induce protection against the pathogen may be the result of global changes in the gene expression, which in turn may impact changes in the pathogen’s proteome.
Materials and Methods
In vitro cultivation and cell-free E. chaffeensis recovery
E. chaffeensis Arkansas isolate wildtype and the mutants were grown in the canine macrophage cell line, DH8258,61. Isolation and purification of cell-free E. chaffeensis wildtype and its mutants were carried out as outlined in Fig. S5. Briefly, the bacterial infection rate in DH82 cells was assessed with Diff-Quik staining. After 72 h of infection when the infection reached to about 80–90%, the culture from four T-150 confluent flasks was harvested and centrifuged at 500 × g for 5 min. Cellular pellets were resuspended in 1 × phosphate buffered saline (PBS) containing protease inhibitors (Roche, Indianapolis, IN) and cells were homogenized on ice by passing through, 15–20 strokes with a 23 g needle in a 10 mL syringe. Efficiency of homogenization, 80–90% lysis, was checked under light microscope. Whole cell lysate was centrifuged at 500 × g for 5 min at 4 °C. The resulting supernatant containing cell-free Ehrlichia organisms was filtered through a 2 µm sterile membrane filter (Millipore, Billerica, MA). Cell-free Ehrlichia from filtrates were pelleted by centrifuging at 15,000 × g for 15 min and the pellet was suspended in PBS and then layered onto 30% diatrizoate meglumine and sodium solution (Renografin) MD-76R (Mallinckrodt Inc, St. Louis, MO). The suspension was centrifuged for 1 h at 100,000 × g at 4 °C in a S50-ST swinging bucket rotor (Beckman, Indianapolis, IN). The pellet of cell-free Ehrlichia were washed at 15,000 × g for 15 min and used for experiments.
Bacterial mRNA enrichment and sequencing
Figure S6 outlines the workflow for bacteria mRNA enrichment and cDNA library preparation and RNA sequencing. Briefly, RNA form wildtype and mutants were isolated from purified cell-free Ehrlichia using TRIzol Reagent (Sigma-Aldrich, St. Louis, MO). RNA samples were then treated with DNase I (Invitrogen, Carlsbad, CA) and bacterial RNA was enriched by removing host 18 S rRNA, 28 S rRNA, and polyadenylated mRNA using MICROBEnrich Kit (Ambion, Foster City, CA). The quantity and integrity of bacterial RNA before and after enrichment was assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The Ribo-Zero Magnetic Kit was used to isolate mRNA from total RNA samples and then fragmented into short fragments as per the manufacturer’s protocols (Epicentre, Madison, WI). Subsequently, cDNA was synthesized using the mRNA fragments as templates. Libraries of cDNAs for wildtype and mutants were prepared using the TruSeq RNA Sample Prep Kit (Illumina, Ingolstadt, Germany). Sample libraries were quantified using Agilent 2100 Bioanaylzer and library quality was assessed by Real-Time PCR (ABI StepOnePlus) prior to subjecting the samples to sequencing on Illumina HiSeqTM 4000 (Beijing Genomics Institute (BGI), Philadelphia, PA).
Bioinformatics analysis
The original image data were transferred into raw sequence data via base calling. Raw reads were subjected to quality assessment to determine whether the raw reads were qualified for mapping (Fig. S5). The bases with low quality (<20) were excluded from the analysis. Raw reads were then filtered to remove adapter sequences and low quality reads, then clean reads were aligned to the E. chaffeensis Arkansas strain complete genome as per the first annotated GenBank # CP000236.1 using SOAPaligner/SOAP262. We opted to use this accession number because our prior publications, and similarly other investigators, widely used it for referring to gene names and numbers listed in it. Not more than five mismatches were allowed in the alignment, which is a standard cut off used for the alignment analysis. The alignment data were used to calculate distribution of reads on reference genes and determine the gene coverage. Alignment results were assessed for quality check and then proceed with analysis of DGE. The gene expression level was calculated using RPKM method of normalizing for total read length and the number of sequencing reads63. We used p-value < 0.05, False Discovery Rate (FDR) ≤ 0.001, and the absolute value of Log2 Ratio ≥ 1 as the threshold to judge the significance difference in gene expression. The FDR uses accurate p-values as a measure of control in multiple sample comparison of RNA seq data. Corrections for false positive and false negative errors were performed using the method described by Benjamini and Yekutieli64.
Quantitative real-time reverse transcription PCR
SYBR green detection-based quantitative real-time reverse transcription PCR (qRT-PCR assays were performed to validate the gene expression changes observed in the RNA seq data analysis. Wildtype, ECH_0379, ECH_0490, and ECH_0660 mutants’ RNAs used in generating the RNA seq data were also used to determine transcript levels by performing quantitative RT-PCR by SYBR Green assays using a SuperScript® III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen, Carlsbad, CA). RNA was reverse transcribed from all the replicates using SuperScript III and then quantitative-PCRs were performed in a 25 μL reaction containing 0.5 μM each of forward and reverse primers. Thermal cycler conditions were; 94 °C for 15 sec, 60 °C for 30 sec, and 74 °C for 15 sec for 40 cycles. Thirteen randomly selected differentially transcribed genes were used in validation experiments using StepOnePlus™ Real-Time PCR instrument (Applied Biosystems, Foster City, CA) and the data were analyzed by StepOne Software v2.3. E. chaffeensis 16 S rRNA was quantitated by real-time RT-PCR as described in27 and used for normalization of RNA concentrations among different RNA batches, prior to performing the validation experiments. For qRT-PCR data, the delta-delta Ct (ΔΔCt) calculation was employed to calculate relative change in the expression and fold change was obtained by averaging the replicate values of gene expression and the standard error. Semi-quantitative one-step RT-PCR (Life Technologies, Carlsbad, CA) targeting to E. chaffeensis genes ECH_0490 and ECH_0492 near the transposon mutation downstream to ECH_0490 gene was performed with 30 cycles of amplification using the gene specific primers as described in a previous study6. Briefly, RNA from wildtype and ECH_0490 mutant were used as the templates for RT-PCR. One tube without reverse transcriptase or template RNA was used as negative control. One tube with DNA as the template was used as positive control. Thermal cycler conditions were as follows: 50 °C for 1 h for reverse transcription step then followed by 35 cycles of 94 °C for 30 sec, 55 °C for 30 sec, and 72 °C for 30 sec; finally a 2-min 72 °C extension step was part of the reaction.
Electronic supplementary material
Acknowledgements
This work was supported by the PHS grant # AI070908 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA. This manuscript is contribution number 18-271-J from the Kansas Agricultural Experiment Station. Publication of this article was supported in part by the Kansas State University Open Access Publishing Fund. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We thank Dr. Huitao Liu and Dr.Ying Wang for their technical help in purification of E. chaffeensis and RT-PCR. We thank Don Harbidge, CVM Molecular Biology Core, for technical help in qRT-PCR. We thank Ms. Mal Rooks Hoover for her technical help in preparing the manuscript figures. We also thank Dr. Jodi McGill for critical reading of the manuscript.
Author Contributions
R.G. conceived and directed the research design and C.K. designed research plan, executed and performed the experiments, and data analysis. C.K. and R.G. prepared the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-24471-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gayle A, Ringdahl E. Tick-borne diseases. Am Fam Physician. 2001;64:461–466. [PubMed] [Google Scholar]
- 2.Dawson JE. Human ehrlichiosis in the United States. Curr Clin Top Infect Dis. 1996;16:164–171. [PubMed] [Google Scholar]
- 3.Dawson JE, Ewing SA. Susceptibility of dogs to infection with Ehrlichia chaffeensis, causative agent of human ehrlichiosis. Am J Vet Res. 1992;53:1322–1327. [PubMed] [Google Scholar]
- 4.Dumler JS, Bakken JS. Ehrlichial diseases of humans: emerging tick-borne infections. Clin Infect Dis. 1995;20:1102–1110. doi: 10.1093/clinids/20.5.1102. [DOI] [PubMed] [Google Scholar]
- 5.Cheng C, et al. Targeted and random mutagenesis of Ehrlichia chaffeensis for the identification of genes required for in vivo infection. PLoS Pathog. 2013;9:e1003171. doi: 10.1371/journal.ppat.1003171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng C, Nair AD, Jaworski D, Ganta RR. Mutations in Ehrlichia chaffeensis causing polar effects in gene expression and differential host specificities. PLoS One. 2015;10:e0132657. doi: 10.1371/journal.pone.0132657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moumène A, Meyer DF. Ehrlichia’s molecular tricks to manipulate their host cells. Microbes Infect. 2016;18:172–179. doi: 10.1016/j.micinf.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Kuriakose JA, Miyashiro S, Luo T, Zhu B, McBride JW. Ehrlichia chaffeensis transcriptome in mammalian and arthropod hosts reveals differential gene expression and post transcriptional regulation. PLoS One. 2011;6:e24136. doi: 10.1371/journal.pone.0024136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unver A, Rikihisa Y, Stich RW, Ohashi N, Felek S. The omp-1 major outer membrane multigene family of Ehrlichia chaffeensis is differentially expressed in canine and tick hosts. Infect Immun. 2002;70:4701–4704. doi: 10.1128/IAI.70.8.4701-4704.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Von Ohlen T, Cheng C, Faburay B, Ganta RR. Transcription of Ehrlichia chaffeensis genes is accomplished by RNA polymerase holoenzyme containing either sigma 32 or sigma 70. PLoS One. 2013;8:e81780. doi: 10.1371/journal.pone.0081780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuczynska-Wisnik D., Cheng, C., Ganta, R.R. & Zolkiewski, M. Protein aggregation in Ehrlichia chaffeensis during infection of mammalian cells. FEMS Microbiol Lett. 364 (2017). [DOI] [PMC free article] [PubMed]
- 12.Hammac GK, Pierlé SA, Cheng X, Scoles GA, Brayton KA. Global transcriptional analysis reveals surface remodeling of Anaplasma marginale in the tick vector. Parasit Vectors. 2014;7:193. doi: 10.1186/1756-3305-7-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McClure EE, et al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol. 2017;15:544–558. doi: 10.1038/nrmicro.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair AD, et al. Attenuated mutants of Ehrlichia chaffeensis induce protection against wild-type infection challenge in the reservoir host and in an incidental host. Infect. Immun. 2015;83:2827–2835. doi: 10.1128/IAI.00487-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierle SA, Dark MJ, Dahmen D, Palmer GH, Brayton KA. Comparative genomics and transcriptomics of trait-gene association. BMC Genomics. 2012;13:669. doi: 10.1186/1471-2164-13-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 2010;38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin J, Zhu W, Passalacqua KD, Bergman N, Borodovsky M. Bacillus anthracis genome organization in light of whole transcriptome sequencing. BMC Bioinforma. 2010;11:S10. doi: 10.1186/1471-2105-11-S3-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunning Hotopp JC, et al. Comparative Genomics of Emerging Human Ehrlichiosis Agents. PLoS Genet. 2006;2:e21. doi: 10.1371/journal.pgen.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins NE, et al. The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc Natl Acad Sci USA. 2005;102:838–843. doi: 10.1073/pnas.0406633102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frutos R, et al. Comparative genomic analysis of three strains of Ehrlichia ruminantium reveals an active process of genome size plasticity. J Bacteriol. 2006;188:2533–2542. doi: 10.1128/JB.188.7.2533-2542.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo T, McBride JW. Ehrlichia chaffeensis TRP32 interacts with host cell targets that influence intracellular survival. Infect Immun. 2012;80:2297–306. doi: 10.1128/IAI.00154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakeel A, den Dulk-Ras A, Hooykaas PJ, McBride JW. Ehrlichia chaffeensis tandem repeat proteins and ank200 are type 1 secretion system substrates related to the repeats-in-toxin exoprotein family. Front Cell Infect Microbiol. 2011;1:22. doi: 10.3389/fcimb.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu B, et al. Nuclear translocated Ehrlichia chaffeensis ankyrin protein interacts with a specific adenine-rich motif of host promoter and intronic Alu elements. Infect Immun. 2009;77:4243–4255. doi: 10.1128/IAI.00376-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noroy C, Meyer DF. Comparative genomics of the zoonotic pathogen Ehrlichia chaffeensis reveals candidate type IV effectors and putative host cell targets. Front Cell Infect Microbiol. 2017;7:120. doi: 10.3389/fcimb.2017.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012;10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 26.Kumar N, et al. Efficient Enrichment of Bacterial mRNA from Host-Bacteria Total RNA Samples. Sci Rep. 2016;6:34850. doi: 10.1038/srep34850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sirigireddy KR, Ganta RR. Multiplex detection of Ehrlichia and Anaplasma species pathogens in peripheral blood by real-time reverse transcriptase-polymerase chain reaction. J Mol Diagn. 2005;7:308–316. doi: 10.1016/S1525-1578(10)60559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westermann AJ, Barquist L, Vogel J. Resolving host-pathogen interactions by dual RNA-seq. PLoS Pathog. 2017;13:e1006033. doi: 10.1371/journal.ppat.1006033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeder CL, et al. Identification and characterization of novel small RNAs in Rickettsia prowazekii. Front Microbiol. 2016;7:859. doi: 10.3389/fmicb.2016.00859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder CL, et al. Bacterial small RNAs in the Genus Rickettsia. BMC Genomics. 2015;16:1075. doi: 10.1186/s12864-015-2293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohashi N, Zhi N, Zhang Y, Rikihisa Y. Immunodominant major outer membrane proteins of Ehrlichia chaffeensis are encoded by a polymorphic multigene family. Infect Immun. 1998;66:132–139. doi: 10.1128/iai.66.1.132-139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy GR, et al. Molecular characterization of a 28 kDa surface antigen gene family of the tribe Ehrlichia. Biochem Biophys Res Commun. 1998;247:636–643. doi: 10.1006/bbrc.1998.8844. [DOI] [PubMed] [Google Scholar]
- 33.Yu XJ, McBride JW, Zhang X, Walker DH. Characterization of the complete transcriptionally active Ehrlichia chaffeensis 28 kDa outer membrane protein multigene family. Gene. 2000;248:59–68. doi: 10.1016/S0378-1119(00)00147-5. [DOI] [PubMed] [Google Scholar]
- 34.Lin M, Rikihisa Y. Degradation of p22phox and inhibition of superoxide generation by Ehrlichia chaffeensis in human monocytes. Cell Microbiol. 2007;9:861–874. doi: 10.1111/j.1462-5822.2006.00835.x. [DOI] [PubMed] [Google Scholar]
- 35.Green, E. R. & Mecsas, J. Bacterial secretion systems: An Overview. Microbiol Spectr4 (2016) [DOI] [PMC free article] [PubMed]
- 36.Rapisarda C, Fronzes R. Secretion systems used by bacteria to subvert host functions. Curr Issues Mol Biol. 2017;25:1–42. doi: 10.21775/cimb.025.001. [DOI] [PubMed] [Google Scholar]
- 37.Rikihisa Y, Lin M, Niu H, Cheng Z. Type IV secretion system of Anaplasma phagocytophilum and Ehrlichia chaffeensis. Ann N Y Acad Sci. 2009;1166:106–111. doi: 10.1111/j.1749-6632.2009.04527.x. [DOI] [PubMed] [Google Scholar]
- 38.Lopez JE, et al. Immunogenicity of Anaplasma marginale type IV secretion system proteins in a protective outer membrane vaccine. Infect Immun. 2007;75:2333–2342. doi: 10.1128/IAI.00061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felek S, Huang H, Rikihisa Y. Sequence and expression analysis of virB9 of the type IV secretion system of Ehrlichia canis strains in ticks, dogs, and cultured cells. Infect Immun. 2003;71:6063–6067. doi: 10.1128/IAI.71.10.6063-6067.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sexton JA, Vogel JP. Type IVB secretion by intracellular pathogens. Traffic. 2002;3:178–185. doi: 10.1034/j.1600-0854.2002.030303.x. [DOI] [PubMed] [Google Scholar]
- 41.Ohashi N, Zhi N, Lin Q, Rikihisa Y. Characterization and transcriptional analysis of gene clusters for a type IV secretion machinery in human granulocytic and monocytic ehrlichiosis agents. Infect. Immun. 2002;70:2128–2138. doi: 10.1128/IAI.70.4.2128-2138.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruneau L, et al. Understanding Anaplasmataceae pathogenesis using “Omics” approaches. Front Cell Infect Microbiol. 2014;4:86. doi: 10.3389/fcimb.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin M, Kikuchi T, Brewer HM, Norbeck AD, Rikihisa Y. Global proteomic analysis of two tickborne emerging zoonotic agents: anaplasma phagocytophilum and ehrlichia chaffeensis. Front Microbiol. 2011;2:24. doi: 10.3389/fmicb.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–1448. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patiño-Ruiz M, Ganea C, Fendler K, Călinescu O. Competition is the basis of the transport mechanism of the NhaB Na+/H+ exchanger from Klebsiella pneumoniae. PLoS One. 2017;12:e0182293. doi: 10.1371/journal.pone.0182293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krulwich TA, Hicks DB, Ito M. Cation/proton antiporter complements of bacteria: why so large and diverse? Mol Microbiol. 2009;74:257–260. doi: 10.1111/j.1365-2958.2009.06842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy TF, Brauer AL, Johnson A, Kirkham C. ATP-binding cassette (ABC) transporters of the human respiratory tract pathogen, Moraxella catarrhalis: role in virulence. PLoS One. 2016;11:e0158689. doi: 10.1371/journal.pone.0158689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tjale MA, Pretorius A, Josemans A, Kleef MV, Liebenberg J. Transcriptomic analysis of Ehrlichia ruminantium during the developmental stages in bovine and tick cell culture. Ticks Tick Borne Dis. 2018;9:126–134. doi: 10.1016/j.ttbdis.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Perez Vidakovics ML, Riesbeck K. Virulence mechanisms of Moraxella in the pathogenesis of infection. Curr Opin Infect Dis. 2009;22:279–285. doi: 10.1097/QCO.0b013e3283298e4e. [DOI] [PubMed] [Google Scholar]
- 50.Eisenreich W, Heesemann J, Rudel T, Goebel W. Metabolic host responses to infection by intracellular bacterial pathogens. Front Cell Infect Microbiol. 2013;3:24. doi: 10.3389/fcimb.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olive AJ, Sassetti CM. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol. 2016;14:221–234. doi: 10.1038/nrmicro.2016.12. [DOI] [PubMed] [Google Scholar]
- 52.Sinclair SH, Rennoll-Bankert KE, Dumler JS. Effector bottleneck: microbial reprogramming of parasitized host cell transcription by epigenetic remodeling of chromatin structure. Front Genet. 2014;5:274. doi: 10.3389/fgene.2014.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gillespie JJ, et al. Phylogenomics reveals a diverse Rickettsiales type IV secretion system. Infect Immun. 2010;78:1809–1823. doi: 10.1128/IAI.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang T, et al. Aggregate-reactivation activity of the molecular chaperone ClpB from Ehrlichia chaffeensis. PLoS One. 2013;8:e62454. doi: 10.1371/journal.pone.0062454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zolkiewski M, Zhang T, Nagy M. Aggregate reactivation mediated by the Hsp100 chaperones. Arch Biochem Biophys. 2012;520:1–6. doi: 10.1016/j.abb.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumagai Y, Huang H, Rikihisa Y. Expression and porin activity of P28 and OMP-1F during intracellular Ehrlichia chaffeensis development. J Bacteriol. 2006;190:3597–605. doi: 10.1128/JB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcelino I, et al. Comparative proteomic profiling of Ehrlichia ruminantium pathogenic strain and its high-passaged attenuated strain reveals virulence and attenuation-associated proteins. PLoS One. 2015;10:e0145328. doi: 10.1371/journal.pone.0145328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singu, V. et al. Unique macrophage and tick cell-specific protein expression from the p28/p30 Omp multigene locus in Ehrlichia species. Cell Microbiol. 8, 1475–1487 (2006). [DOI] [PubMed]
- 59.Seo GM, Cheng C, Tomich J, Ganta RR. Total, membrane, and immunogenic proteomes of macrophage- and tick cell-derived Ehrlichia chaffeensis evaluated by LC-MS/MS and MALDI-TOF methods. Infect Immun. 2008;76:4823–4832. doi: 10.1128/IAI.00484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGill JL, et al. Vaccination with an attenuated mutant of Ehrlichia chaffeensis induces pathogen-specific CD4 + T cell immunity and protection from tick-transmitted wild-type challenge in the canine host. PLoS One. 2016;11:e0148229. doi: 10.1371/journal.pone.0148229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng, C. & Ganta, R. R. Laboratory maintenance of Ehrlichia chaffeensis and Ehrlichia canis and recovery of organisms for molecular biology and proteomics studies. Curr Protoc Microbiol3. Chapter 3A (2008) [DOI] [PMC free article] [PubMed]
- 62.Li R, et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 63.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Med. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 64.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics. 2001;29:1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.