

Abstract
Genome-scale metabolic network reconstruction offers a means to leverage the value of the exponentially growing genomics data and integrate it with other biological knowledge in a structured format. Constraint-based modeling (CBM) enables both the qualitative and quantitative analyses of the reconstructed networks. The rapid advancements in these areas can benefit both the industrial production of microbial food cultures and their application in food processing. CBM provides several avenues for improving our mechanistic understanding of physiology and genotype–phenotype relationships. This is essential for the rational improvement of industrial strains, which can further be facilitated through various model-guided strain design approaches. CBM of microbial communities offers a valuable tool for the rational design of defined food cultures, where it can catalyze hypothesis generation and provide unintuitive rationales for the development of enhanced community phenotypes and, consequently, novel or improved food products. In the industrial-scale production of microorganisms for food cultures, CBM may enable a knowledge-driven bioprocess optimization by rationally identifying strategies for growth and stability improvement. Through these applications, we believe that CBM can become a powerful tool for guiding the areas of strain development, culture development and process optimization in the production of food cultures. Nevertheless, in order to make the correct choice of the modeling framework for a particular application and to interpret model predictions in a biologically meaningful manner, one should be aware of the current limitations of CBM.
Keywords: constraint-based modeling, food fermentation, metabolic networks, microbial community modeling, microbial food cultures, strain development
Introduction
Microorganisms have been used in making food products for millennia [1]. Besides their primary role in food preservation, by virtue of their fermentative metabolism, they contribute to improving food safety and organoleptic properties and can provide additional health-promoting effects [2]. It is estimated that fermented food represents one-third of the human diet [3], which allows it to secure, together with probiotic products, a global market value of over €100 billion [4]. Although food fermentations commonly relied on the spontaneous metabolic activities of the indigenous microbiota in the food matrix, modern fermentation-based food processing mostly employs commercial starter and secondary cultures that are produced in ton quantities per annum in industrial-scale bioreactors [5–8]. These cultures typically comprise a simple or complex consortium of live microorganisms (bacteria, yeast and/or mold) that is formulated to obtain the desired characteristics in the final food product. Both the industrial production of microbial food cultures and their application in food processing are attractive areas for applying emerging molecular and systems-based approaches constantly being exploited in other fields of industrial biotechnology.
Genome-scale reconstruction of metabolic networks combined with constraint-based modeling (CBM) is an increasingly popular approach in microbial systems biology. Although the reconstruction is primarily based on genomics data, it is iteratively refined and gap-filled using biochemical, genetic and/or physiological knowledge on the organism of interest [9,10]. Converting the network reconstruction into a mathematical model, commonly referred to as a genome-scale model (GEM), enables its qualitative and quantitative analyses via an array of constraint-based methods, such as flux balance analysis [11–14]. This facilitates further refinement of the network reconstruction and provides valuable insights into genotype–phenotype relationships. Over the past three decades, CBM has found various biotechnological and medical applications ranging from the basic analysis of biological network properties to identifying and simulating complex microbe–microbe and host–microbe interactions [15–20].
Although different mathematical modeling approaches have been adopted in various areas of food biotechnology [21–23], the full potential of CBM remains largely unexplored in this field. Reconstruction and analysis of strain-specific GEMs can be used to comprehensively assess the metabolic potential of industrial strains used in food processing and generate mechanistic understanding of different physiological and biotechnological traits. Understanding interactions in microbial consortia and simulating community phenotype are also of great value in designing culture blends for food fermentations and predicting the characteristics of the final food product. In the commercial production of food cultures, CBM may enable a knowledge-driven process optimization by rationally identifying strategies for growth and stability improvement.
Here, we explore different opportunities through which CBM can guide strain development, culture development, and bioprocess optimization in the production of microbial food cultures (Figure 1), with focus on lactic acid bacteria (LAB) as a fundamental group of organisms used in the food industry. Challenges facing the systematic implementation of CBM in this field are also discussed.
Figure 1. Potential applications of constraint-based modeling in microbial food biotechnology.

CBM offers different possibilities for guiding strain development, culture development and bioprocess optimization in the industrial production of microbial food cultures.
Mechanistic understanding of microbial physiology
A GEM constitutes a structured organismal knowledgebase [24] that can be consulted for both simple and sophisticated queries — ranging from assessing the putative presence of a certain pathway to the detailed metabolic characterization of an organism in multiple conditions (Figure 2). Besides, new insights frequently originate from identifying inconsistencies between model output and experimental data, which may indicate missing or incorrect metabolic knowledge. The iterative cycles of model curation and experimental validation enable the generation of a refined model, together with new knowledge on unknown pathways, unannotated or misannotated genes and promiscuous enzymes [25,26]. CBM methods allow the calculation of optimal flux distributions under different environmental conditions, enabling the rapid simulation of multiple phenotypic states. By imposing appropriate constraints [12,27] and selecting biologically relevant cellular objectives [28], it is thereby feasible to perform a systematic metabolic characterization of an organism both quantitatively and qualitatively [14].
Figure 2. Genome-scale models provide a platform for the mechanistic understanding of physiology.
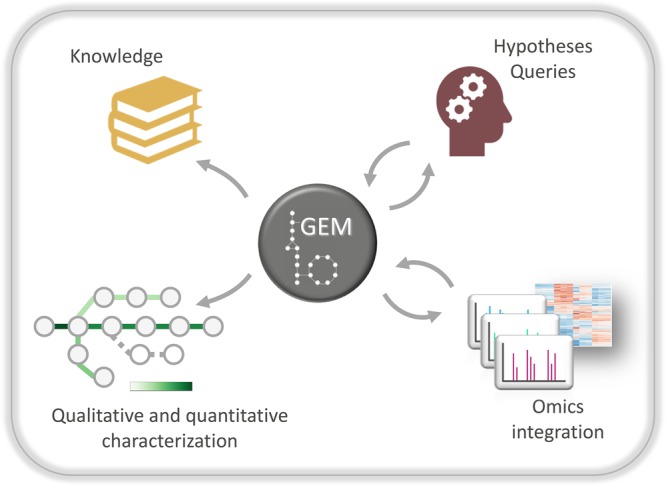
The comprehensive information contained within a GEM allows relevant biochemical knowledge to be easily extracted, ad hoc queries answered, hypotheses tested in silico through CBM and generated for subsequent in vivo verification. Moreover, integration of omics data can improve model predictions and vice versa GEMs can improve omics data analysis, while applying CBM allows a global qualitative and quantitative microbial metabolic characterization within a given environment.
The reconstruction of GEMs for several LAB used in food fermentations, e.g. Lactococcus lactis [29,30], Lactobacillus plantarum [31], Streptococcus thermophilus [32], Lactobacillus casei [33], Oenococcus oeni [34] and Leuconostoc mesenteroides [35], provides examples on how CBM can guide biological discoveries and improve our understanding of industrially relevant traits, such as carbohydrate utilization, amino acid auxotrophies, nutrient requirements or metabolite secretion. Constraint-based analysis of an L. plantarum GEM revealed how the catabolism of branched-chain and aromatic amino acids indirectly contributes to ATP generation and how the secretion of other amino acids, such as aspartate and arginine, provides a sink for the generated nitrogen [31,36]. The use of CBM and elementary mode analysis also facilitated the understanding of the adaptive growth strategy of L. plantarum, usually grown in a nutritionally rich milk environment, to a poor carbon source [37]. In L. lactis, CBM suggested that the formation of some flavor compounds is energetically favorable, hence positively affecting growth, and provided a platform for exploring flavor formation under different conditions [29]. The use of CBM also provided insights into amino acid metabolism in L. lactis and S. thermophilus and identified a unique pathway for the production of acetaldehyde, which contributes to yoghurt flavor, in the latter [30,32].
Many inherent biological constraints are not represented by reaction stoichiometry or thermodynamics alone. However, global phenotype measurements using high-throughput omics technologies provide a reflection of many of these biological constraints and can be integrated into GEMs using various approaches [38–42]. These approaches rely primarily on the detailed gene–protein–reaction mapping represented in GEMs for introducing additional constraints into the network (Figure 3). The integration of high-throughput data into GEMs can, therefore, increase the accuracy of model predictions by shrinking the solution space [43]. This enables the systematic computation of condition-specific metabolic states of the cells. The benefit of integrating omics data with GEMs is not merely unidirectional, as the latter can provide context for omics data analysis and facilitate their interpretation [38,44], which improves the chances of identifying regulatory mechanisms and possible metabolic bottlenecks [45,46]. Recently, the integrative analysis of transcriptomics data within a CBM framework in Leuconostoc mesenteroides (used in various food fermentations) contributed to understanding the metabolic behavior and regulatory mechanisms underlying the higher growth rate of the organism on sucrose than on glucose and generated new insights related to the influence of redox metabolism on cellular phenotype [35].
Figure 3. A depiction of gene–protein–reaction association typically represented in genome-scale models.

The detailed gene–protein–reaction mapping in genome-scale models offers a structured platform for the incorporation of different types of omics data, using various computational approaches.
CBM can also be integrated with comparative genomics for the identification of genetic determinants underlying strain-specific phenotypic differences [47]. This can be useful in the analysis of genetic variations arising from mutagenesis or laboratory evolution [48].
Thus, CBM provides several avenues for the systematic increase in physiological understanding, an implicit requirement for the advancement of rational strain and process development [49–51].
In silico strain design
In the food industry, microbial strains with novel or improved functional, technological and/or safety aspects are in continuous demand, either for developing new products or improving the quality of existing ones. Additionally, there is a growing interest in the use of LAB for the in situ production and delivery of functional food ingredients, such as low-calorie sweeteners, vitamins, amino acids and bioactive peptides [52–55], as well as heterologous proteins, e.g. vaccines [56]. This may require extensive strain improvement before reaching reasonable titers of the desired product.
Using GEMs as a predictive framework for identifying non-intuitive metabolic engineering strategies for strain improvement is one of the most common applications of CBM [15,16,49,57–59]. This is enabled via a variety of computational algorithms dedicated to model-based strain design (reviewed in refs [59–61]), several of which aim at finding genetic perturbations that couple the secretion of a product of interest to cellular growth, thereby forcing product formation as a requirement for increased fitness. In L. lactis, two CBM frameworks were used to find gene deletion targets for enhancing the production of diacetyl, which imparts a butter-like flavor to dairy products [30]. In silico predictions identified a combination of gene deletions that would result in 150% increase in the production of the direct diacetyl precursor (2-acetolactate), while maintaining reasonable growth. An updated version of the model was supplemented with reactions representing recombinant protein expression to identify gene targets for increasing the specific expression of heterologous proteins [62]. This is particularly relevant for using L. lactis as oral vehicles for delivering recombinant protein vaccines. Two of the in silico designs were experimentally evaluated and provided qualitative evidence on the validity of the model predictions. These approaches can easily be extrapolated to other industrially relevant traits.
CBM can also be used to systematically assess the ability to produce a range of native and non-native metabolites [63,64], which can be applied to fully catalog the potential of food microorganisms as in vivo cell factories.
A major bottleneck in food biotechnology, however, is the tight requirements of regulatory agencies currently imposed in many countries, especially in Europe [65], and the negative perception by consumers of genetically modified microorganisms. This renders the use of recombinant DNA technologies mostly inapplicable and shifts the focus of strain improvement programs toward classical methods, such as random mutagenesis and screening, dominant selection and directed evolution [66]. To address this issue, a computational algorithm was recently developed that relies on CBM and cheminformatics tools to identify metabolite analogs that can be used to manipulate cellular phenotypes without the use of recombinant DNA technologies [67]. Metabolite analogs are compounds that mimic the structure of native metabolites and, therefore, interfere with their metabolism, which makes them suitable for selection of variants with altered phenotypes. Despite that metabolite analogs are already being used in classical strain improvement schemes [66], the new algorithm extends their applicability by identifying non-intuitive metabolite targets and retrieving corresponding chemical analogs that can provide the selection pressure to redirect the metabolic fluxes towards a product of interest [67].
As the legislations regarding genetically modified organisms are constantly being revised [7], it could be possible in the near future to apply recent genome-editing technologies for implementing metabolic engineering and synthetic biology strategies to manipulate microbial strains used in the food industry [68] and hence take full advantage of CBM in this area.
Microbial community design
Food cultures are commonly based on a microbial community rather than a single strain [69]. The overall function of the culture, which dictates the characteristics of the food product, is largely modulated by the metabolic interactions of the community members. Discerning the role of individual strains within a community and their intertwined metabolic dependencies is a complex experimental task [70,71], making the design of defined microbial communities with predictable and controllable properties a challenge. Typically, commercial food cultures are developed through empirical approaches, e.g. evaluating the performance of a limited number of relevant strain combinations, which cannot practically cover all possible designs nor do they benefit from the wealth of information embedded in microbial genomes.
CBM offers a rational approach to the design of food cultures through the mechanistic understanding of microbial community characteristics and simulating community composition and phenotype [71–73]. CBM techniques developed for modeling individual organisms can be extended for community-level modeling [74]. Depending on the complexity of the community and the scope of application, different frameworks can be adopted for building and analyzing community metabolic models, such as mixed-bag modeling, species compartmentalization or multi-species dynamic modeling [72,75–77].
CBM of microbial communities allows phenotype predictions at both the overall community and individual-strain levels. This allows the identification of strain-specific roles that are an intrinsic part of many food fermentations, as exemplified in the mutualistic relationship between S. thermophilus and Lactobacillus delbrueckii subsp. bulgaricus in yoghurt production [78]. Identifying the phenotype of individual strains in a community context, in turn, enables the definition of functional strain groups comprising strains with similar capabilities within the community [79]. This could be particularly useful for model-guided simplification of otherwise complex communities (Figure 4).
Figure 4. Rational microbial community design.

Through community metabolic modeling, the phenotypes of conceived community designs can be predicted in silico. This, for instance, allows the rational modulation of existing communities, through addition, replacement or removal of individual strains for the generation of communities with improved or novel functionality. Likewise, model-guided simplification of complex communities allows the design of communities with similar functionality but lower complexity and improved practicality.
A major benefit of microbial community models lies within the ability to provide insights into potential metabolic interactions between community members [14,18,80,81], even in large communities [82]. A commensal interaction in milk between the probiotic strain Lactobacillus rhamnosus GG and the dairy bacterium S. thermophilus was predicted through CBM, where the latter supplied six different metabolites for the enhanced growth of the probiotic bacterium [83]. A community metabolic model could also predict amino acid cross-feeding between yeast and LAB, which was demonstrated in grape juice [84]. Moreover, community-level models can be used to elucidate the interactions among gut microbiota as well as their interactions with diet and their host [19,85–92], a highly relevant application for developing next-generation probiotic cultures [93,94]. Other examples of predicting metabolic interactions through CBM of microbial communities are ample [14,77,95,96].
The ability of CBM to predict metabolic interdependencies provides valuable knowledge for the modulation of community structure and function [97] and makes it possible to systematically evaluate any community designs conceivable (Figure 4). Furthermore, the effect of altering the nutrient environment (i.e. food type) can be easily modeled, enabling beneficial nutrient or even strain adjustments. By tuning model optimality criteria, at the overall community or strain-specific level, towards for instance increased production of flavor compounds [98] or texturizing polysaccharides [99], new insights into advantageous community designs for improving food products can be gained.
Rational optimization of cellular yield and stability
Industrial-scale production of microbial strains for food cultures employs advanced technologies and requires multidisciplinary knowledge on microbiology, physiology and process engineering [7,8]. Maximizing biomass yield is necessary for improving cost effectiveness [66], while process parameters should ensure stability (i.e. viability and metabolic activity) of the produced organism [6,7]. Bioprocess modeling commonly relies on unstructured models to calculate growth rates [100] and process optimization is often based on empirical approaches using either classical (one-factor-at-a-time) or statistical designs [101–103]. Implementing CBM in this context makes it possible to incorporate detailed metabolic knowledge in the process.
CBM can be used to check the consistency of complex fermentation data and analyze them in the context of the metabolic network [50]. This facilitates data interpretation and helps identifying missing end products, as was previously demonstrated in L. plantarum [31,36].
CBM is inherently suitable for calculating theoretical biomass and fermentation–product yields under different environmental conditions. This, potentiated by the mechanistic insights, CBM provides into microbial physiology (vide supra), provides a rational means for growth medium optimization, and several examples with industrial relevance already exist [32,104–107]. Industrial-scale fermentations, however, often employ complex undefined growth media [101], which may limit the usability of CBM, as complex media can be difficult to thoroughly describe, and hence less amenable to rational improvement. Even so, CBM retains the potential of identifying growth-boosting nutrients, e.g. through sensitivity analysis [31], which can be used as supplements to complex media. Alternatively, shifting towards pure substrates to ensure reproducibility, facilitate downstream processing or comply with stricter pharmaceutical standards (e.g. for live biotherapeutics), may allow a more comprehensive application of CBM in medium optimization.
The stability of food cultures is a crucial aspect; cultures have to retain viability and activity throughout their shelf life [108], despite encountering various stressors during production and downstream processing. This can be influenced by medium composition, e.g. carbon source, and the levels of certain intracellular metabolites during fermentation were shown to affect the viability of LAB during bioprocessing [109,110]. By incorporating relevant phenotypic data into CBM, metabolic flux states correlating with viability and activity can be accurately calculated [111]. Subsequent CBM can elucidate relevant medium formulations that result in such desirable metabolic states. Though not using CBM, a similar transcriptomics-based approach was used for correlating phenotype signatures and fermentation conditions in L. plantarum [112]. Additionally, correlating flux states with stability may identify candidate biomarkers that can serve as readily measurable indicators of stability [113,114], a much-needed approach for the assessment of food cultures [108].
A large-scale bioreactor constitutes a heterogeneous environment that in turn can cause heterogeneity and suboptimal growth within the microbial population [115,116]. Extended GEMs, particularly dynamic resource allocation models, offer a framework for the prediction of cellular behavior in fluctuating environments [117,118], such as areas of local heterogeneity within the bioreactor. GEMs can also be integrated with existing bioreactor and bioprocess models in a multi-scale CBM framework, which may help reveal non-trivial dependencies across different processes [119].
Challenges and future perspectives
CBM of microbial metabolism can provide quantitative answers to various biological questions and guide the generation of testable hypotheses that can benefit both the production of microbial food cultures and their application in food processing. Nevertheless, several challenges that hinder the full exploitation of CBM in microbial food biotechnology exist. Although extensive efforts are undertaken to facilitate the automated reconstruction [120–128] and refinement [26,129–134] of draft metabolic networks and to develop software tools for the analysis of the resulting models (reviewed in ref. [135]), the generation of high-quality, strain-specific models still requires a considerable amount of effort and knowledge. For industrial strains, this is further complicated by the lack of high-quality phenotypic data [16,49]. Such data are essential for the curation and validation of GEMs, especially in the case of LAB where gene function loss is frequently encountered [21]. Knowledge on strain-specific biomass composition may also be necessary to obtain reliable predictions in some applications, such as culture medium optimization. Advances in methods for the systematic generation of omics data and their incorporation into draft reconstruction pipelines may significantly aid in the automated generation of high-quality GEMs in the near future, which allows for more advanced analyses and provides more reliable predictions.
Other limitations inherent to traditional GEMs, which rely primarily on reaction stoichiometry and directionality as constraints, such as the absence of kinetic and regulatory information, remain also challenging. Incorporating such information in LAB GEMs for instance is essential for predicting homolactic fermentation [30,31,36]. Yet, extended CBM frameworks which account for resource allocation across cellular processes may partially overcome these limitations through the introduction of cellular capacity constraints [136–140].
Additional challenges arise when modeling food fermentations due to the heterogeneous nature of the food matrix, the complexity of some food-fermenting communities and the intricate mechanisms underlying the development of some food characteristics. For instance, during cheese ripening, the role of undefined secondary flora and the detailed biochemical changes that are necessary for flavor development can be challenging to model and directly correlate with sensory attributes [141]. In these scenarios, reducing the level of complexity by identifying key players together with the constant advancements in modeling complex microbial communities may prove helpful.
While constantly being addressed by researchers, awareness of these limitations may be a key to the successful choice of the CBM framework that fits an intended application as well as in the proper interpretation of model predictions in a biologically meaningful manner.
Abbreviations
- CBM
constraint-based modeling
- GEM
genome-scale model
- LAB
lactic acid bacteria
Competing Interests
M.H.R. and A.A.Z. are employed by Chr. Hansen A/S, a global supplier of food cultures and enzymes. The authors' views presented in this manuscript, however, are solely based on scientific grounds and do not reflect the commercial interests of their employer.
References
- 1.Salque M., Bogucki P.I., Pyzel J., Sobkowiak-Tabaka I., Grygiel R., Szmyt M. et al. (2012) Earliest evidence for cheese making in the sixth millennium BC in Northern Europe. Nature 493, 522–525 10.1038/nature11698 [DOI] [PubMed] [Google Scholar]
- 2.Leroy F. and De Vuyst L. (2004) Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci. Technol. 15, 67–78 10.1016/j.tifs.2003.09.004 [DOI] [Google Scholar]
- 3.Campbell-Platt G. (1994) Fermented foods — a world perspective. Food Res. Int. 27, 253–257 10.1016/0963-9969(94)90093-0 [DOI] [Google Scholar]
- 4.de Vos W.M. (2011) Systems solutions by lactic acid bacteria: from paradigms to practice. Microb. Cell Fact. 10(Suppl 1), S2 10.1186/1475-2859-10-S1-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill D., Sugrue I., Arendt E., Hill C., Stanton C. and Ross R.P. (2017) Recent advances in microbial fermentation for dairy and health. F1000Research 6, 751 10.12688/f1000research.10896.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandine W.E. (1996) Commercial production of dairy starter cultures In Dairy Starter Cultures (Cogan T.M. and Accolas J.P., eds), pp. 191–206, VCH Publishers, New York [Google Scholar]
- 7.Høier E., Janzen T., Rattray F., Sørensen K., Børsting M.W., Brockmann E. et al. (2010) The production, application and action of lactic cheese starter cultures In Technology of Cheesemaking (Law B.A. and Tamime A.Y., eds), pp. 166–192, Wiley-Blackwell, Chichester [Google Scholar]
- 8.Taskila S. (2017) Industrial production of starter cultures In Starter Cultures in Food Production (Speranza B., Bevilacqua A., Corbo M.R. and Sinigaglia M., eds), pp. 79–100, John Wiley & Sons, Ltd, Chichester [Google Scholar]
- 9.Feist A.M., Herrgard M.J., Thiele I., Reed J.L. and Palsson B.O. (2009) Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7, 129–143 10.1038/nrmicro1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiele I. and Palsson B.Ø (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121 10.1038/nprot.2009.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orth J. D., Thiele I. and Palsson B. Ø. (2010) What is flux balance analysis? Nat. Biotechnol. 28, 245–248 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price N.D., Reed J.L. and Palsson B.Ø (2004) Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2, 886–897 10.1038/nrmicro1023 [DOI] [PubMed] [Google Scholar]
- 13.Milne C.B., Kim P.-J., Eddy J.A. and Price N.D. (2009) Accomplishments in genome-scale in silico modeling for industrial and medical biotechnology. Biotechnol. J. 4, 1653–1670 10.1002/biot.200900234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis N.E., Nagarajan H. and Palsson B.O. (2012) Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305 10.1038/nrmicro2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCloskey D., Palsson B.O. and Feist A.M. (2013) Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 9, 661 10.1038/msb.2013.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim W.J., Kim H.U. and Lee S.Y. (2017) Current state and applications of microbial genome-scale metabolic models. Curr. Opin. Syst. Biol. 2, 10–18 10.1016/j.coisb.2017.03.001 [DOI] [Google Scholar]
- 17.O'Brien E.J., Monk J.M. and Palsson B.O. (2015) Using genome-scale models to predict biological capabilities. Cell 161, 971–987 10.1016/j.cell.2015.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuniga C., Zaramela L. and Zengler K. (2017) Elucidation of complexity and prediction of interactions in microbial communities. Microb. Biotechnol. 10, 1500–1522 10.1111/1751-7915.12855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoaie S. and Nielsen J. (2014) Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 5, 86 10.3389/fgene.2014.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinken A. and Thiele I. (2015) Systems biology of host-microbe metabolomics. Wiley Interdiscip. Rev.: Syst. Biol. Med. 7, 195–219 10.1002/wsbm.1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teusink B. and Smid E.J. (2006) Modelling strategies for the industrial exploitation of lactic acid bacteria. Nat. Rev. Microbiol. 4, 46–56 10.1038/nrmicro1319 [DOI] [PubMed] [Google Scholar]
- 22.Teusink B., Bachmann H. and Molenaar D. (2011) Systems biology of lactic acid bacteria: a critical review. Microb. Cell Fact. 10(Suppl 1), S11 10.1186/1475-2859-10-S1-S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Branco dos Santos F., de Vos W.M. and Teusink, B. (2013) Towards metagenome-scale models for industrial applications—the case of lactic acid bacteria. Curr. Opin. Biotechnol. 24, 200–206 10.1016/j.copbio.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 24.Palsson B.Ø. (2006) Systems Biology: Properties of Reconstructed Networks, Cambridge University Press, New York [Google Scholar]
- 25.Szappanos B., Kovacs K., Szamecz B., Honti F., Costanzo M., Baryshnikova A. et al. (2011) An integrated approach to characterize genetic interaction networks in yeast metabolism. Nat. Genet. 43, 656–662 10.1038/ng.846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan S. and Reed J.L. (2018) Advances in gap-filling genome-scale metabolic models and model-driven experiments lead to novel metabolic discoveries. Curr. Opin. Biotechnol. 51, 103–108 10.1016/j.copbio.2017.12.012 [DOI] [PubMed] [Google Scholar]
- 27.Bordbar A., Monk J.M., King Z.A. and Palsson B.O. (2014) Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120 10.1038/nrg3643 [DOI] [PubMed] [Google Scholar]
- 28.Schuetz R., Kuepfer L. and Sauer U. (2007) Systematic evaluation of objective functions forpredicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119 10.1038/msb4100162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flahaut N.A., Wiersma A., van de Bunt B., Martens D.E., Schaap P.J., Sijtsma L. et al. (2013) Genome-scale metabolic model for Lactococcus lactis MG1363 and its application to the analysis of flavor formation. Appl. Microbiol. Biotechnol. 97, 8729–8739 10.1007/s00253-013-5140-2 [DOI] [PubMed] [Google Scholar]
- 30.Oliveira A.P., Nielsen J. and Förster J. (2005) Modeling Lactococcus lactis using a genome-scale flux model BMC Microbiol. 5, 39 10.1186/1471-2180-5-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teusink B., Wiersma A., Molenaar D., Francke C., de Vos W.M., Siezen R.J. et al. (2006) Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J. Biol. Chem. 281, 40041–40048 10.1074/jbc.M606263200 [DOI] [PubMed] [Google Scholar]
- 32.Pastink M.I., Teusink B., Hols P., Visser S., de Vos W.M. and Hugenholtz J. (2009) Genome-scale model of Streptococcus thermophilus LMG18311 for metabolic comparison of lactic acid bacteria. Appl. Environ. Microbiol. 75, 3627–3633 10.1128/AEM.00138-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinay-Lara E., Hamilton J.J., Stahl B., Broadbent J.R., Reed J.L. and Steele J.L. (2014) Genome-scale reconstruction of metabolic networks of Lactobacillus casei ATCC 334 and 12A. PLoS ONE 9, e110785 10.1371/journal.pone.0110785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendoza S., Cañón P., Contreras A., Ribbeck M. and Agosin E. (2017) Genome-scale reconstruction of the metabolic network in Oenococcus oeni to assess wine malolactic fermentation. Front. Microbiol. 8, 534 10.3389/fmicb.2017.00534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koduru L., Kim Y., Bang J., Lakshmanan M., Han N.S. and Lee D.-Y. (2017) Genome-scale modeling and transcriptome analysis of Leuconostoc mesenteroides unravel the redox governed metabolic states in obligate heterofermentative lactic acid bacteria. Sci. Rep. 7, 15721 10.1038/s41598-017-16026-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goffin P., van de Bunt B., Giovane M., Leveau J.H., Hoppener-Ogawa S., Teusink B. et al. (2010) Understanding the physiology of Lactobacillus plantarum at zero growth. Mol. Syst. Biol. 6, 413 10.1038/msb.2010.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teusink B., Wiersma A., Jacobs L., Notebaart R.A. and Smid E.J. (2009) Understanding the adaptive growth strategy of Lactobacillus plantarum by in silico optimisation. PLoS Comput. Biol. 5, e1000410 10.1371/journal.pcbi.1000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyduke D.R., Lewis N.E. and Palsson B.O. (2013) Analysis of omics data with genome-scale models of metabolism. Mol. Biosyst. 9, 167–174 10.1039/C2MB25453K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joyce A.R. and Palsson B.O. (2006) The model organism as a system: integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol. 7, 198–210 10.1038/nrm1857 [DOI] [PubMed] [Google Scholar]
- 40.Blazier A.S. and Papin J.A. (2012) Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3, 299 10.3389/fphys.2012.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunk E., George K.W., Alonso-Gutierrez J., Thompson M., Baidoo E., Wang G. et al. (2016) Characterizing strain variation in engineered E. coli using a multi-omics-based workflow. Cell Syst. 2, 335–346 10.1016/j.cels.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aurich M.K., Paglia G., Rolfsson O., Hrafnsdottir S., Magnusdottir M., Stefaniak M.M. et al. (2015) Prediction of intracellular metabolic states from extracellular metabolomic data. Metabolomics 11, 603–619 10.1007/s11306-014-0721-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M., Rai N., Zorraquino V. and Tagkopoulos I. (2016) Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 7, 13090 10.1038/ncomms13090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santos F., Spinler J.K., Saulnier D.M., Molenaar D., Teusink B., de Vos W.M. et al. (2011) Functional identification in Lactobacillus reuteri of a PocR-like transcription factor regulating glycerol utilization and vitamin B12 synthesis. Microb. Cell Fact. 10, 55 10.1186/1475-2859-10-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patil K.R. and Nielsen J. (2005) Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl Acad. Sci. U.S.A. 102, 2685–2689 10.1073/pnas.0406811102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L., Agren R., Bordel S. and Nielsen J. (2010) Use of genome-scale metabolic models for understanding microbial physiology. FEBS Lett. 584, 2556–2564 10.1016/j.febslet.2010.04.052 [DOI] [PubMed] [Google Scholar]
- 47.Monk J. and Bosi E. (2018) Integration of comparative genomics with genome-scale metabolic modeling to investigate strain-specific phenotypical differences. Methods Mol. Biol. 1716, 151–175 10.1007/978-1-4939-7528-0_7 [DOI] [PubMed] [Google Scholar]
- 48.Cardoso J.G., Andersen M.R., Herrgard M.J. and Sonnenschein N. (2015) Analysis of genetic variation and potential applications in genome-scale metabolic modeling. Front. Bioeng. Biotechnol. 3, 13 10.3389/fbioe.2015.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopes H. and Rocha I. (2017) Genome-scale modeling of yeast: chronology, applications and critical perspectives. FEMS Yeast Res. 17 10.1093/femsyr/fox050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahadevan R., Burgard A.P., Famili I., Dien S. and Schilling C.H. (2005) Applications of metabolic modeling to drive bioprocess development for the production of value-added chemicals. Biotechnol. Bioprocess Eng. 10, 408–417 10.1007/BF02989823 [DOI] [Google Scholar]
- 51.Mienda B.S. (2017) Genome-scale metabolic models as platforms for strain design and biological discovery. J. Biomol. Struct. Dyn. 35, 1863–1873 10.1080/07391102.2016.1197153 [DOI] [PubMed] [Google Scholar]
- 52.Hugenholtz J. (2008) The lactic acid bacterium as a cell factory for food ingredient production. Int. Dairy J. 18, 466–475 10.1016/j.idairyj.2007.11.015 [DOI] [Google Scholar]
- 53.Waters D.M., Mauch A., Coffey A., Arendt E.K. and Zannini E. (2015) Lactic acid bacteria as a cell factory for the delivery of functional biomolecules and ingredients in cereal-based beverages: a review. Crit. Rev. Food Sci. Nutr. 55, 503–520 10.1080/10408398.2012.660251 [DOI] [PubMed] [Google Scholar]
- 54.Thakur K., Tomar S.K. and De S. (2016) Lactic acid bacteria as a cell factory for riboflavin production. Microb. Biotechnol. 9, 441–451 10.1111/1751-7915.12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown L., Pingitore E.V., Mozzi F., Saavedra L., Villegas J.M. and Hebert E.M. (2017) Lactic acid bacteria as cell factories for the generation of bioactive peptides. Protein Pept. Lett. 24, 146–155 10.2174/0929866524666161123111333 [DOI] [PubMed] [Google Scholar]
- 56.Miyoshi A., Bermúdez-Humarán L.G., de Azevedo M.S.P., Langella P. and Azevedo V. (2010) Lactic acid bacteria as live vectors: heterologous protein production and delivery systems In Biotechnology of Lactic Acid Bacteria: Novel Applications (Mozzi F., Raya R.R. and Vignolo G.M., eds), Wiley-Blackwell, Chichester [Google Scholar]
- 57.King Z.A., Lloyd C.J., Feist A.M. and Palsson B.O. (2015) Next-generation genome-scale models for metabolic engineering. Curr. Opin. Biotechnol. 35, 23–29 10.1016/j.copbio.2014.12.016 [DOI] [PubMed] [Google Scholar]
- 58.Maia P., Rocha M. and Rocha I. (2016) In silico constraint-based strain optimization methods: the quest for optimal cell factories. Microbiol. Mol. Biol. Rev. 80, 45–67 10.1128/MMBR.00014-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klamt S., Hädicke O. and von Kamp A. (2014) Stoichiometric and constraint-based analysis of biochemical reaction networks In Large-Scale Networks in Engineering and Life Sciences (Benner P., Findeisen R., Flockerzi D., Reichl U. and Sundmacher K., eds), pp. 263–316, Springer International Publishing, Cham [Google Scholar]
- 60.Cvijovic M., Bordel S. and Nielsen J. (2011) Mathematical models of cell factories: moving towards the core of industrial biotechnology. Microb. Biotechnol. 4, 572–584 10.1111/j.1751-7915.2010.00233.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Machado D. and Herrgård M.J. (2015) Co-evolution of strain design methods based on flux balance and elementary mode analysis. Metab. Eng. Commun. 2, 85–92 10.1016/j.meteno.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oddone G.M., Mills D.A. and Block D.E. (2009) A dynamic, genome-scale flux model of Lactococcus lactis to increase specific recombinant protein expression. Metab. Eng. 11, 367–381 10.1016/j.ymben.2009.07.007 [DOI] [PubMed] [Google Scholar]
- 63.Campodonico M.A., Sukumara S., Feist A.M. and Herrgård M.J. (2018) Computational methods to assess the production potential of bio-based chemicals In Synthetic Metabolic Pathways: Methods and Protocols (Jensen M.K. and Keasling J.D., eds), pp. 97–116, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 64.Zhang X., Tervo C.J. and Reed J.L. (2016) Metabolic assessment of E. coli as a biofactory for commercial products. Metab. Eng. 35, 64–74 10.1016/j.ymben.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 65.Renault P. (2010) Genetically modified lactic acid bacteria In Biotechnology of Lactic Acid Bacteria: Novel Applications (Mozzi F., Raya R.R. and Vignolo G.M., eds), pp. 361–381, Wiley-Blackwell, Chichester [Google Scholar]
- 66.Derkx P.M., Janzen T., Sorensen K.I., Christensen J.E., Stuer-Lauridsen B. and Johansen E. (2014) The art of strain improvement of industrial lactic acid bacteria without the use of recombinant DNA technology. Microb. Cell Fact. 13(Suppl 13), S5 10.1186/1475-2859-13-S1-S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cardoso J.G.R., Zeidan A.A., Jensen K., Sonnenschein N., Neves A.R. and Herrgård M.J. (2018) MARSI: metabolite analogues for rational strain improvement. Bioinformatics bty108 10.1093/bioinformatics/bty108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gaj T., Sirk S.J., Shui S.L. and Liu J. (2016) Genome-editing technologies: principles and applications. Cold Spring Harb. Perspect. Biol. 8, a023754 10.1101/cshperspect.a023754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cogan T.M. and Accolas J.P. (eds) (1996) Dairy Starter Cultures, VCH Publishers, New York [Google Scholar]
- 70.Ponomarova O. and Patil K.R. (2015) Metabolic interactions in microbial communities: untangling the Gordian knot. Curr. Opin. Microbiol. 27, 37–44 10.1016/j.mib.2015.06.014 [DOI] [PubMed] [Google Scholar]
- 71.Tan J., Zuniga C. and Zengler K. (2015) Unraveling interactions in microbial communities — from co-cultures to microbiomes. J. Microbiol. 53, 295–305 10.1007/s12275-015-5060-1 [DOI] [PubMed] [Google Scholar]
- 72.Biggs M.B., Medlock G.L., Kolling G.L. and Papin J.A. (2015) Metabolic network modeling of microbial communities. Wiley Interdiscip. Rev.: Syst. Biol. Med. 7, 317–334 10.1002/wsbm.1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blasche S., Kim Y., Oliveira A.P. and Patil K.R. (2017) Model microbial communities for ecosystems biology. Curr. Opin. Syst. Biol. 6, 51–57 10.1016/j.coisb.2017.09.002 [DOI] [Google Scholar]
- 74.Gottstein W., Olivier B.G., Bruggeman F.J. and Teusink B. (2016) Constraint-based stoichiometric modelling from single organisms to microbial communities. J. R. Soc. Interface 13, 20160627 10.1098/rsif.2016.0627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henry C.S., Bernstein H.C., Weisenhorn P., Taylor R.C., Lee J.-Y., Zucker J. et al. (2016) Microbial community metabolic modeling: a community data-driven network reconstruction. J. Cell Physiol. 231, 2339–2345 10.1002/jcp.25428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bosi E., Bacci G., Mengoni A. and Fondi M. (2017) Perspectives and challenges in microbial communities metabolic modeling. Front. Genet. 8, 88 10.3389/fgene.2017.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faria J.P., Khazaei T., Edirisinghe J.N., Weisenhorn P., Seaver S. M.D., Conrad N. et al. (2017) Constructing and analyzing metabolic flux models of microbial communities In Hydrocarbon and Lipid Microbiology Protocols: Genetic, Genomic and System Analyses of Communities (McGenity T.J., Timmis K.N. and Nogales B., eds), pp. 247–273, Springer, Berlin, Heidelberg [Google Scholar]
- 78.Herve-Jimenez L., Guillouard I., Guedon E., Boudebbouze S., Hols P., Monnet V. et al. (2009) Postgenomic analysis of Streptococcus thermophilus cocultivated in milk with Lactobacillus delbrueckii subsp. bulgaricus: involvement of nitrogen, purine, and iron metabolism. Appl. Environ. Microbiol. 75, 2062–2073 10.1128/AEM.01984-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanemaaijer M., Roling W.F., Olivier B.G., Khandelwal R.A., Teusink B. and Bruggeman F.J. (2015) Systems modeling approaches for microbial community studies: from metagenomics to inference of the community structure. Front. Microbiol. 6, 213 10.3389/fmicb.2015.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li C., Lim K.M., Chng K.R. and Nagarajan N. (2016) Predicting microbial interactions through computational approaches. Methods 102, 12–19 10.1016/j.ymeth.2016.02.019 [DOI] [PubMed] [Google Scholar]
- 81.Reed J.L. (2017) Genome-scale metabolic modeling and its application to microbial communities In The Chemistry of Microbiomes: Proceedings of a Seminar Series, The National Academies Press, Washington (DC) PMID: 28806041 [PubMed] [Google Scholar]
- 82.Zelezniak A., Andrejev S., Ponomarova O., Mende D.R., Bork P. and Patil K.R. (2015) Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl Acad. Sci. U.S.A. 112, 6449–6454 10.1073/pnas.1421834112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kort R., Westerik N., Mariela Serrano L., Douillard F.P., Gottstein W., Mukisa I.M. et al. (2015) A novel consortium of Lactobacillus rhamnosus and Streptococcus thermophilus for increased access to functional fermented foods. Microb. Cell Fact. 14, 195 10.1186/s12934-015-0370-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ponomarova O., Gabrielli N., Sevin D.C., Mulleder M., Zirngibl K., Bulyha K. et al. (2017) Yeast creates a niche for symbiotic lactic acid bacteria through nitrogen overflow. Cell Syst. 5, 345–357.e6 10.1016/j.cels.2017.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Magnúsdóttir S., Heinken A., Kutt L., Ravcheev D.A., Bauer E., Noronha A. et al. (2017) Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 10.1038/nbt.3703 [DOI] [PubMed] [Google Scholar]
- 86.Shoaie S., Ghaffari P., Kovatcheva-Datchary P., Mardinoglu A., Sen P., Pujos-Guillot E. et al. (2015) Quantifying diet-Induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331 10.1016/j.cmet.2015.07.001 [DOI] [PubMed] [Google Scholar]
- 87.Shoaie S., Karlsson F., Mardinoglu A., Nookaew I., Bordel S. and Nielsen J. (2013) Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 3, Article number: 2532 10.1038/srep02532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.El-Semman I.E. Karlsson F.H., Shoaie S., Nookaew I., Soliman T.H. and Nielsen J. (2014) Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2-32 and Faecalibacterium prausnitzii A2-165 and their interaction. BMC Syst. Biol. 8, 41 10.1186/1752-0509-8-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinken A., Sahoo S., Fleming R.M. and Thiele I. (2013) Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 4, 28–40 10.4161/gmic.22370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sahoo S. and Thiele I. (2013) Predicting the impact of diet and enzymopathies on human small intestinal epithelial cells. Hum. Mol. Genet. 22, 2705–2722 10.1093/hmg/ddt119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levy R. and Borenstein E. (2013) Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc. Natl Acad. Sci. U.S.A. 110, 12804–12809 10.1073/pnas.1300926110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Magnúsdóttir S. and Thiele I. (2018) Modeling metabolism of the human gut microbiome. Curr. Opin. Biotechnol. 51, 90–96 10.1016/j.copbio.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 93.O'Toole P.W., Marchesi J.R. and Hill C. (2017) Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat. Microbiol. 2, 17057 10.1038/nmicrobiol.2017.57 [DOI] [PubMed] [Google Scholar]
- 94.Sheth R.U., Cabral V., Chen S.P. and Wang H.H. (2016) Manipulating bacterial communities by in situ microbiome engineering. Trends Genet. 32, 189–200 10.1016/j.tig.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hanemaaijer M., Olivier B.G., Roling W.F., Bruggeman F.J. and Teusink B. (2017) Model-based quantification of metabolic interactions from dynamic microbial-community data. PLoS ONE 12, e0173183 10.1371/journal.pone.0173183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stolyar S., Van Dien S., Hillesland K.L., Pinel N., Lie T.J., Leigh J.A. et al. (2007) Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 3, 92 10.1038/msb4100131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lindemann S.R., Bernstein H.C., Song H.-S., Fredrickson J.K., Fields M.W., Shou W. et al. (2016) Engineering microbial consortia for controllable outputs. ISME J. 10, 2077–2084 10.1038/ismej.2016.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smid E.J. and Kleerebezem M. (2014) Production of aroma compounds in lactic fermentations. Annu. Rev. Food Sci. Technol. 5, 313–326 10.1146/annurev-food-030713-092339 [DOI] [PubMed] [Google Scholar]
- 99.Zeidan A.A., Poulsen V.K., Janzen T., Buldo P., Derkx P.M.F., Øregaard G. et al. (2017) Polysaccharide production by lactic acid bacteria: from genes to industrial applications. FEMS Microbiol. Rev. 41, S168–S200 10.1093/femsre/fux017 [DOI] [PubMed] [Google Scholar]
- 100.Villadsen J., Nielsen J. and Lidén G. (2011) Bioreaction Engineering Principles, Springer, New York [Google Scholar]
- 101.Stanbury P.F., Whitaker A. and Hall S.J. (2013) Principles of Fermentation Technology, Butterworth Heinemann, Oxford [Google Scholar]
- 102.Singh V., Haque S., Niwas R., Srivastava A., Pasupuleti M. and Tripathi C.K. (2016) Strategies for fermentation medium optimization: an in-depth review. Front. Microbiol. 7, 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parekh S., Vinci V.A. and Strobel R.J. (2000) Improvement of microbial strains and fermentation processes. Appl. Microbiol. Biotechnol. 54, 287–301 10.1007/s002530000403 [DOI] [PubMed] [Google Scholar]
- 104.Song H., Kim T.Y., Choi B.-K., Choi S.J., Nielsen L.K., Chang H.N. et al. (2008) Development of chemically defined medium for Mannheimia succiniciproducens based on its genome sequence. Appl. Microbiol. Biotechnol. 79, 263–272 10.1007/s00253-008-1425-2 [DOI] [PubMed] [Google Scholar]
- 105.Santos B.., Olivier F., Boele B.G., Smessaert J., De Rop V., Krumpochova P. et al. (2017) Probing the genome-scale metabolic landscape of Bordetella pertussis, the causative agent of whooping cough. Appl. Environ. Microbiol. 83, e01528-17 10.1128/AEM.01528-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feist A.M., Scholten J.C., Palsson B.O., Brockman F.J. and Ideker T. (2006) Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol. Syst. Biol. 2, 2006.0004 10.1038/msb4100046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Imielinski M., Belta C., Rubin H. and Halasz A. (2006) Systematic analysis of conservation relations in Escherichia coli genome-scale metabolic network reveals novel growth media. Biophys. J. 90, 2659–2672 10.1529/biophysj.105.069278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lacroix C. and Yildirim S. (2007) Fermentation technologies for the production of probiotics with high viability and functionality. Curr. Opin. Biotechnol. 18, 176–183 10.1016/j.copbio.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 109.Carvalho A.S., Silva J., Ho P., Teixeira P., Malcata F.X. and Gibbs P. (2004) Effects of various sugars added to growth and drying media upon thermotolerance and survival throughout storage of freeze-dried Lactobacillus delbrueckii ssp. bulgaricus. . Biotechnol. Prog. 20, 248–254 10.1021/bp034165y [DOI] [PubMed] [Google Scholar]
- 110.Wisselink H.W., Weusthuis R.A., Eggink G., Hugenholtz J. and Grobben G.J. (2002) Mannitol production by lactic acid bacteria: a review. Int. Dairy J. 12, 151–161 10.1016/S0958-6946(01)00153-4 [DOI] [Google Scholar]
- 111.Bordbar A., Yurkovich J.T., Paglia G., Rolfsson O., Sigurjonsson O.E. and Palsson B.O. (2017) Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 7, 46249 10.1038/srep46249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bron P.A., Wels M., Bongers R.S., van de Veen H.v.B., Wiersma A., Overmars L. et al. (2012) Transcriptomes reveal genetic signatures underlying physiological variations imposed by different fermentation conditions in Lactobacillus plantarum. PLoS ONE 7, e38720 10.1371/journal.pone.0038720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bordbar A. and Palsson B.O. (2012) Using the reconstructed genome-scale human metabolic network to study physiology and pathology. J. Intern. Med. 271, 131–141 10.1111/j.1365-2796.2011.02494.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jerby L. and Ruppin E. (2012) Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 18, 5572–5584 10.1158/1078-0432.CCR-12-1856 [DOI] [PubMed] [Google Scholar]
- 115.Delvigne F. and Goffin P. (2014) Microbial heterogeneity affects bioprocess robustness: dynamic single-cell analysis contributes to understanding of microbial populations. Biotechnol. J. 9, 61–72 10.1002/biot.201300119 [DOI] [PubMed] [Google Scholar]
- 116.van Heerden J.H., Wortel M.T., Bruggeman F.J., Heijnen J.J., Bollen Y.J., Planque R. et al. (2014) Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343, 1245114 10.1126/science.1245114 [DOI] [PubMed] [Google Scholar]
- 117.Reimers A.-M., Lindhorst H. and Waldherr S. (2017) A protocol for generating and exchanging (genome-scale) metabolic resource allocation models. Metabolites 7, 47 10.3390/metabo7030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lindhorst H., Lucia S., Findeisen R. and Waldherr S. (2017) Modeling metabolic networks including gene expression and uncertainties. arXiv preprint, arXiv:1609.08961 [Google Scholar]
- 119.Zhuang K.H. and Herrgard M.J. (2015) Multi-scale exploration of the technical, economic, and environmental dimensions of bio-based chemical production. Metab. Eng. 31, 1–12 10.1016/j.ymben.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 120.Agren R., Liu L., Shoaie S., Vongsangnak W., Nookaew I. and Nielsen J. (2013) The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 9, e1002980 10.1371/journal.pcbi.1002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Henry C.S., DeJongh M., Best A.A., Frybarger P.M., Linsay B. and Stevens R.L. (2010) High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 10.1038/nbt.1672 [DOI] [PubMed] [Google Scholar]
- 122.Swainston N., Smallbone K., Mendes P., Kell D. and Paton N. (2011) The suBliMinaL toolbox: automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 8, 186 10.1515/jib-2011-186 [DOI] [PubMed] [Google Scholar]
- 123.Dias O., Rocha M., Ferreira E.C. and Rocha I. (2015) Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 43, 3899–3910 10.1093/nar/gkv294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cuevas D.A., Edirisinghe J., Henry C.S., Overbeek R., O'Connell T.G. and Edwards R.A. (2016) From DNA to FBA: how to build your own genome-scale metabolic model. Front. Microbiol. 7, 907 10.3389/fmicb.2016.00907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thorleifsson S.G. and Thiele I. (2011) Rbionet: A COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 27, 2009–2010 10.1093/bioinformatics/btr308 [DOI] [PubMed] [Google Scholar]
- 126.Büchel F., Rodriguez N., Swainston N., Wrzodek C., Czauderna T., Keller R. et al. (2013) Path2models: large-scale generation of computational models from biochemical pathway maps. BMC Syst. Biol. 7, 116 10.1186/1752-0509-7-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamilton J.J. and Reed J.L. (2014) Software platforms to facilitate reconstructing genome-scale metabolic networks. Environ. Microbiol. 16, 49–59 10.1111/1462-2920.12312 [DOI] [PubMed] [Google Scholar]
- 128.Machado D., Andrejev S., Tramontano M. and Patil K. R. (2018) Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. bioRxiv, 223198 10.1101/223198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar V.S., Dasika M.S. and Maranas C.D. (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinf. 8, 212 10.1186/1471-2105-8-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reed J.L., Patel T.R., Chen K.H., Joyce A.R., Applebee M.K., Herring C.D. et al. (2006) Systems approach to refining genome annotation. Proc. Natl Acad. Sci. U.S.A. 103, 17480–17484 10.1073/pnas.0603364103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Orth J.D. and Palsson B.O. (2010) Systematizing the generation of missing metabolic knowledge. Biotechnol. Bioeng. 107, 403–412 10.1002/bit.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thiele I., Vlassis N. and Fleming R.M. (2014) Fastgapfill: efficient gap filling in metabolic networks. Bioinformatics 30, 2529–2531 10.1093/bioinformatics/btu321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Krumholz E.W. and Libourel I.G. (2015) Sequence-based network completion reveals the integrality of missing reactions in metabolic networks. J. Biol. Chem. 290, 19197–19207 10.1074/jbc.M114.634121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Santos S. and Rocha I. (2016) Estimation of biomass composition from genomic and transcriptomic information. J. Integr. Bioinform. 13, 285 10.1515/jib-2016-285 [DOI] [PubMed] [Google Scholar]
- 135.Dandekar T., Fieselmann A., Majeed S. and Ahmed Z. (2014) Software applications toward quantitative metabolic flux analysis and modeling. Brief. Bioinform. 15, 91–107 10.1093/bib/bbs065 [DOI] [PubMed] [Google Scholar]
- 136.O'Brien E.J. and Palsson B.O. (2015) Computing the functional proteome: recent progress and future prospects for genome-scale models. Curr. Opin. Biotechnol. 34, 125–134 10.1016/j.copbio.2014.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goelzer A. and Fromion V. (2017) Resource allocation in living organisms. Biochem. Soc. Trans. 45, 945–952 10.1042/BST20160436 [DOI] [PubMed] [Google Scholar]
- 138.Nilsson A., Nielsen J. and Palsson B.O. (2017) Metabolic models of protein allocation call for the kinetome. Cell Syst. 5, 538–541 10.1016/j.cels.2017.11.013 [DOI] [PubMed] [Google Scholar]
- 139.Bachmann H., Bruggeman F.J., Molenaar D., dos Santos F.B. and Teusink B. (2016) Public goods and metabolic strategies. Curr. Opin. Microbiol. 31, 109–115 10.1016/j.mib.2016.03.007 [DOI] [PubMed] [Google Scholar]
- 140.Sanchez B.J., Zhang C., Nilsson A., Lahtvee P.J., Kerkhoven E.J. and Nielsen J. (2017) Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 13, 935 10.15252/msb.20167411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Law B.A. and Tamime A.Y. (eds) (2010) Technology of Cheesemaking, Wiley-Blackwell, Chichester [Google Scholar]