

Abstract
Hepatocellular lipid accumulation characterizes nonalcoholic fatty liver disease (NAFLD). However, the types of lipids associated with disease progression are debated, as is the impact of their localization. Traditional lipidomics analysis using liver homogenates or plasma dilutes and averages lipid concentrations, and does not provide spatial information about lipid distribution. We aimed to characterize the distribution of specific lipid species related to NAFLD severity by performing label-free molecular analysis by mass spectrometry imaging (MSI). Fresh frozen liver biopsies from obese subjects undergoing bariatric surgery (n = 23) with various degrees of NAFLD were cryosectioned and analyzed by matrix-assisted laser desorption/ionization (MALDI)-MSI. Molecular identification was verified by tandem MS. Tissue sections were histopathologically stained, annotated according to the Kleiner classification, and coregistered with the MSI data set. Lipid pathway analysis was performed and linked to local proteome networks. Spatially resolved lipid profiles showed pronounced differences between nonsteatotic and steatotic tissues. Lipid identification and network analyses revealed phosphatidylinositols and arachidonic acid metabolism in nonsteatotic regions, whereas low–density lipoprotein (LDL) and very low–density lipoprotein (VLDL) metabolism was associated with steatotic tissue. Supervised and unsupervised discriminant analysis using lipid based classifiers outperformed simulated analysis of liver tissue homogenates in predicting steatosis severity. We conclude that lipid composition of steatotic and nonsteatotic tissue is highly distinct, implying that spatial context is important for understanding the mechanisms of lipid accumulation in NAFLD. MSI combined with principal component–linear discriminant analysis linking lipid and protein pathways represents a novel tool enabling detailed, comprehensive studies of the heterogeneity of NAFLD.
Nonalcoholic fatty liver disease (NAFLD), characterized by hepatocellular lipid accumulation, refers to liver damage ranging from simple steatosis through steatohepatitis to cirrhosis.1,2 Recent data indicate that NAFLD is the most common form of chronic liver disease, and the second leading cause of liver transplantation.3 The prevalence of NAFLD is estimated to be ∼30% in healthy versus ∼80% in obese individuals among the general Western population.1 Comorbidities are frequently observed in patients with NAFLD,4 which may exacerbate steatosis or vice versa. Hepatic steatosis is a known risk factor for postoperative complications after major hepatic surgery;5−7 however, the relevant amount and type of fat necessary to cause injury are debated.6,8−10 Despite its high prevalence, the pathogenesis of NAFLD remains unclear, making patient stratification and diagnosis difficult. Currently, liver biopsy, followed by histopathological examination, is the gold standard for the diagnosis of NAFLD and assessment of steatosis severity. Severity is determined by the Kleiner scoring system, which address 14 histological features.11 Confounding factors, such as sampling bias, factitious staining artifacts, liver tissue heterogeneity, and the variable manifestation of hepatic steatosis,12 result in subjective interpretations.13 Heterogeneity is the most complex factor to consider in that normal liver consists of zonations with multiple cell types. Inflammation and steatosis add additional complexity to liver structure and organization, which contributes to NASH/NAFLD progression. Histologically these livers samples appear similar, which renders diagnostics difficult. Altogether, improved diagnostic methods for NAFLD are urgently required to benefit treatment decisions and ultimately patient outcomes.
Currently, hepatic lipid metabolism is primarily studied using liver homogenates or serum/plasma. However, homogenization of tissue results in dilution and averaging of molecules, and plasma lipids do not exclusively reflect liver lipids. Nonetheless, these methods have been widely applied to assess the potential role of triacylglycerides (TAGs),1,14−18 phosphatidylcholines (PCs),19−22 phosphatidylethanolamines (PEs),19,21−24 phosphatidylglycerols (PGs),23 as well as free fatty acids (FFAs)17,22,24 in the development of NAFLD. Whereas the presence of these lipids and their association with NAFLD has been reported and debated, the spatial distribution of lipids in liver tissue in the context of NAFLD has been described in just a few recent studies.20,25,26 Wattacheril et al. and Hall et al. performed predominantly positive ion mode mass spectrometry imaging (MSI) of hepatic lipids and showed distinct distribution patterns of PC lipids. Furthermore, Hall et al. hypothesized a mechanism for disrupted lipid metabolism in steatosis. Whereas their analyses are primarily based on a small fraction of lipid species detectable in positive ion mode MSI, it is known that many lipid classes exhibit preferential ionization in a negative polarity. For example, phosphatidylinositols (PIs), phosphatidylserines (PSs), PEs, PGs, and FFAs are ionized more efficiently in negative ion mode.27,28 In view of this, we performed negative ion mode MSI of lipid species in human liver biopsies. The aim of this study was to identify lipids and their distribution profiles within and between tissue samples, and to integrate our findings with publicly available molecular databases to pinpoint potential biological processes directing regional lipid accumulation in NAFLD. Furthermore, mathematical tissue classifiers based on spatial vs simulated homogenate data sets were constructed and compared. Our data demonstrate the potential of MSI for more detailed NAFLD diagnosis, based on precise molecular histological classification.
Methods
Materials
A list with information on additional materials, consumables, and instrumentation is provided in Supporting Information.
Samples
Between 2006 and 2009, liver wedge biopsies were obtained from severely obese individuals with a body mass index (BMI) from 36 to 74 kg m–2 undergoing bariatric surgery at the Maastricht University Medical Centre (MUMC+). Patients with acute or chronic inflammatory diseases, degenerative diseases, reported alcohol consumption (>10 g/day), or with prescribed anti-inflammatory drugs were excluded. This study was approved by the Medical Ethics Board of MUMC+, in line with the ethical guidelines of the 1975 Declaration of Helsinki. Informed consent was obtained in writing from each individual. Patient characteristics are shown in Supporting Table 1. Human liver biopsies were categorized for severity of steatosis by a pathologist into 4 groups using the scoring system of Kleiner et al.11
Sample Preparation and MSI Data Acquisition
Fresh frozen tissue was cryosectioned at 10 μm thickness, at −18 °C on a Cryostat HM525 (Microm, Walldorf, Germany) and thaw mounted to Indium tin oxide (ITO)-coated glass slides (Delta Technologies, Loveland, CO, USA).29 The 23 liver biopsies, 1 cryosection per biopsy, were distributed onto three ITO slides in a randomized manner to minimize batch effects. MSI experiments were performed on two technical replicates.
A protocol for sample preparation that targets lipids was employed. Briefly, norharmane matrix (Merck KGaA, Darmstadt, Germany) was sublimed onto the vacuum-dried tissue with a custom-built sublimation device (IDEE, Maastricht University, The Netherlands) to enable the analysis of lipid species by MALDI-MSI. Data acquisition was performed with the Bruker RapifleX MALDI Tissuetyper system operating in reflectron mode (Bruker Daltonik GmbH, Bremen, Germany). Data were acquired in negative ion mode at the mass range 300–2000 Da with the raster pixel size of 50 μm. On average, 3400 spatially resolved spectra were recorded per tissue section with typical mass accuracy ∼50 ppm. The speed of acquisition was roughly 25 pixels/s, which resulted in an analysis time of approximately 2 min per biopsy.
High mass resolution imaging was performed on a representative tissue section using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to a reduced-pressure MALDI source30 to identify characteristic lipids that were deemed distinctive by MALDI-Time of Flight (TOF) mass spectrometer. Data was acquired with a pixel size of 40 μm and a mass resolution of 240 000 at mass-to-charge ratio (m/z) 400. Mass accuracy was typically 2 ppm or better. To further identify lipids, high-mass-resolution (240 000 at m/z 400), tandem mass spectrometry (MS/MS) was performed using higher energy collisional dissociation (HCD) with a normalized collision energy of 50 and an isolation width of 0.7 Da on dedicated samples. A 5-s injection time was used while the sample continuously moved to accumulate sufficient precursor ions and increase the fragment ion signal for low intensity lipids during tandem MS. This methodology allowed for confident lipid ID assignment, however, the total analysis time per tissue was several hours. This targeted identification approach complements the high throughput MSI of the complete cohort. The data were analyzed using Xcalibur software v.2.3.26 (Thermo Fisher Scientific, Bremen, Germany). When multiple isobaric ions were observed, manual comparison of Orbitrap ion distribution image and the MALDI-TOF data set was performed for correct correlation, and the most abundant ion was chosen for identification.
Histological Staining
The MALDI-MS images were overlaid with microscopic images of the same tissue section to facilitate alignment of the molecular distribution with the morphology of liver tissue. The MALDI matrix was removed post-MSI analysis by slide submersion in 100% ethanol to enable overlay of different imaging modalities from the same tissue. Subsequently, standard hematoxylin and eosin (H&E) staining was applied according to the supplier’s instructions (Merck KGaA, Darmstadt, Germany). Subsequently, the slides were imaged with a Microscope using the bright-field Leica DM6000B (Buffalo Grove, IL, USA) light microscope and MIRAX scanner (Zeiss, Breda, The Netherlands).
Tissue Annotation
The histologically stained tissues were blindly evaluated by an experienced liver pathologist (J.V.), including morphological tissue annotation of steatotic regions, inflammation areas, and portal sclerosis.
MSI Data Analysis
Several data analysis strategies were applied, as schematically represented in Figure 1.
Figure 1.
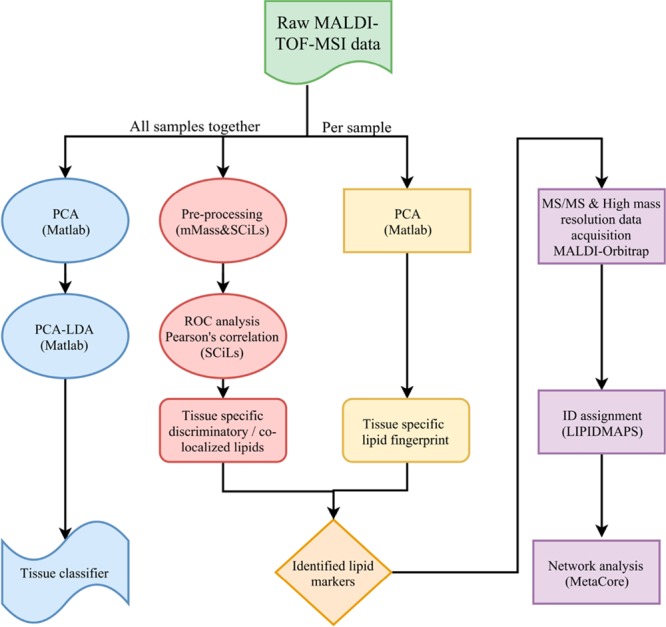
Schematic overview of the parallel data analyses performed.
MSI Data Processing
The raw MALDI-TOF data was loaded into Fleximaging software v.4.1 (Bruker Daltonik GmbH, Bremen, Germany), where the MSI image was semiautomatically overlaid (using fiducial markers) with a histological optical image of the same tissue section. This overlay was necessary to define the tissue vs background boundaries. All individually aligned samples were then imported to SCiLS software v.2016a (SCiLS, Bremen, Germany) where they were together normalized by total ion count. The overall mean spectrum from all 23 samples was extracted and exported to mMass v.5.531 where it was subjected to autoscaling, baseline subtraction, and smoothing (Gaussian algorithm with window size of 0.1 m/z and 2 cycles). Peak picking was performed on the spectrum with the peak intensity threshold relative to base peak set to 1%. Finally, the resulting peak list was imported back to SCiLS with the window size per peak adjusted to 0.13 Da yielding the optimal area under the curve (AUC) for integration.
Identification of Molecular Signature Per Region of Interest
The ion’s distribution and colocalization with an annotated tissue region were assessed by Pearson’s correlation coefficient computed using SCiLS software. In order to verify and quantify the importance and the discriminatory power of a particular m/z ion, receiver operating characteristic (ROC)32 analysis was performed.
Integration of Lipid MSI Data into Pathway Analysis
After the extraction of m/z values of interest based on their spatial distribution, MADLI-Orbitrap identification of the molecules was performed first at the sum-composition level as [M – H]− ions utilizing accurate mass measurements and further by MS/MS to identify fatty acyl compositions. The lipid nomenclature is explained in the Supporting Information.
To explore known and predicted interactions and to build a lipid–protein interaction network from our MSI data, each identified molecule was represented by a unique PubChem compound ID. Subsequently, using these identified molecules, GeneGo’s MetaCore software (Thomson Reuters, NY, USA)33 (v.6.32 build 69020) constructed the lipid–protein interaction network based on known and predicted interactions in curated human databases. Dijkstra’s34 shortest-path algorithm with the maximum number of steps in the path set to 10 was used to ensure a fully connected network per region. Briefly, this algorithm builds a network with the smallest possible number of directed one-step interactions between pairs of initial objects. Finally, the identified connections were further investigated to highlight the biomolecular mechanisms and extrapolate their biological relevance.
Multivariate Data Analysis
Principal component analysis (PCA), a linear unsupervised statistical method describing the largest variances within the data set, was performed using the ChemomeTricks toolbox for Matlab (v.R2015, The MathWorks, Natick, MA).35 PCA was first applied to discard nontissue–specific signal, second to reduce the dimensionality of the data. We assumed that the molecular differences between the steatosis groups were the main sources of variance and could thus be captured by PCA. PCA was applied pixel-by-pixel to samples individually as well as to all 23 samples at once. We performed PCA coupled to linear discriminant analysis (LDA) to capture the molecular signature of steatosis. Contrary to PCA, LDA is a supervised statistical method used to define classes within predefined groups, for example, steatosis grade 0 versus steatosis grade 1. The input variables for LDA were principle components (PCs) from prior PCA. The crucial step in PCA-LDA is the estimation of the required input variables, addressed as previously described by Mascini et al.36 Here, the number of PCs that explained 80% of the variance was the optimum. Linear combinations of the PCs that maximized the Fisher’s criterion, that is, the ratio of the between-class variance and the within-class variance, were calculated. A sample was categorized to class A if the mean discriminant score of the sample spectra was closest to the score of class A.
To address overfitting common to analyzing multidimensional data sets, we cross-validated the PCA-LDA results using the leave-one-out methodology, which is widely accepted for small sample sizes (n < 30).35,36 Additional validation of the PCA-LDA classifier was based on its classification accuracy performance test using the replicate of all 23 samples as the test set. Accuracy is expressed as percentage of correctly classified samples during the leave-one-out cross-validation.
Simulation of Tissue Homogenization Effect
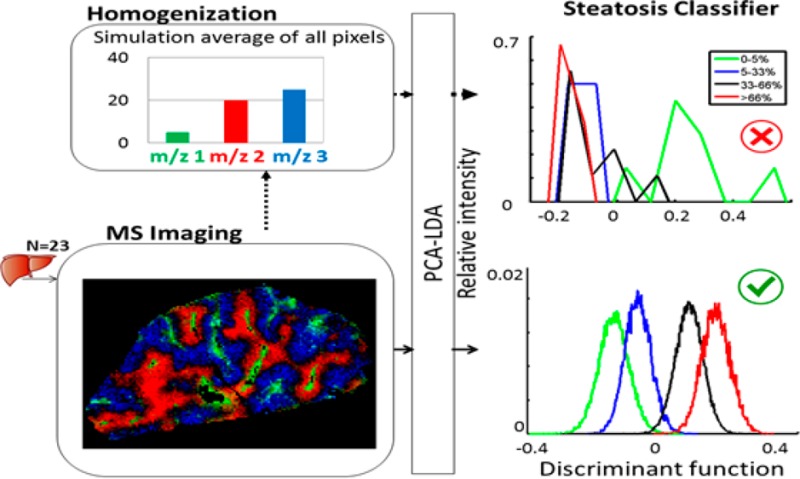
Tissue homogenization effects were simulated by averaging of all pixels (spectra) per biopsy from MALDI-TOF-MSI to one representative spectrum. The 23 average spectra were subjected to the same PCA-LDA analysis, where 80% of variance-containing PCs were used to build the 4-class LDA.
Gene Expression
We extracted gene expression data37 from 35 patient samples, of which 15 were taken from the same patients analyzed by MSI. Samples were categorized into steatosis groups using the Kleiner11 scoring system: grade 0, n = 7; grade 1, n = 9; grade 2, n = 16; grade 3, n = 3. Gene expression was analyzed in the ‘R’ statistical computing environment (version 3.3.0, https://www.R-project.org/). Gene expression levels with identical gene symbols were averaged. Box plots were created for the genes identified by the lipid–protein interaction analysis, in which the samples with corresponding MSI data are shown as markers.
Results
MSI of Tissue Samples
MSI has proven to be an exceptional tool for rapid detection, localization, and identification of thousands of molecules simultaneously and directly from complex sample surfaces.29,38,39 MSI retains sensitive spatial molecular information, typically with spatial resolution of 20–50 μm. We collected 112 539 spectra for 23 obese patients categorized into 4 groups, ranging from no steatosis (grade 0) to severe steatosis (grade 3) (Supporting Table 1). These spectra were analyzed as shown in Figure 1. Images of H&E stained tissue were coregistered with the MSI images to visualize individual molecular species in relation to tissue morphology.
Pixel-Wise PCA Analysis Robustly Identified Steatotic Tissue Regions
After initial preprocessing of the raw MALDI-TOF data, pixel-wise PCA was performed. Co-registration of PC score plots to histology images exposed particular PCA patterns corresponding to the pathological annotations. PC1 and PC2 mainly contained foreground versus background signal. PC3 delineated steatotic (positive loading, PC+3) versus nonsteatotic (negative loading, PC–3) regions (Figure 2). The corresponding loading spectrum exhibits different molecular signatures matching morphologically distinct tissue regions which indicate a heterogeneous and region-specific lipid distribution within the tissue. Next, we tested the robustness of PC3 identified as the steatotic signature from this one sample by projecting it onto the remaining 22 samples. The pixel-wise pattern resulting from the projection coarsely matches the annotated H&E staining in each of the 22 tissues (Supporting Figure 1). This suggests that the steatosis-specific signature is robust across different patients with histomorphological significant steatosis (>5%). The top 10 PCA identified ions for steatotic and nonsteatotic tissue (Supporting Table 2; full ion list in Supporting Tables 3 and 4) revealed a unique lipid composition for nonsteatotic and steatotic tissues.
Figure 2.

Molecular classification of liver tissue by PCA of the MALDI-TOF-MSI data in negative-ion mode. Projection of PCA loading (PC3) from a representative tissue section delineates nonsteatotic (top, PC–3) and steatotic (bottom, PC+3) regions. Ion images are scaled to relative intensity. These regions correspond well to histological annotations (middle). Molecular intensity-scaled loading spectra of the PC function show unique molecular mass profiles for each tissue region. Note that PC–3 is shown with absolute values for easier interpretation.
Distinct Lipid Species Distributions in Steatotic and Nonsteatotic Regions
Independent supervised data analysis was employed on the same MALDI-TOF data set to further investigate the lipid species profiles in steatosis. The steatotic and nonsteatotic tissue regions were manually annotated. We aimed to identify individual ions discriminating the two tissue types. Several ions showed high correlation coefficients with tissue type as well as high ROC values (Supporting Table 5) which indicates that their distribution was associated with steatosis. Identification and structural assignments of the discriminatory ions were performed. Combining the MALDI-Orbitrap high mass resolution data and MS/MS searches allowed for identification of over 80 lipid species (Supporting Tables 3 and 4). Among the most discriminatory ions, PG(18:1_20:4) (m/z 795.4) exhibited distinct localization in steatotic areas (Figure 3A), abundance in each pixel, and a high ROC value (AUC 0.838) (Figure 3B), demonstrating it may be a steatosis-specific lipid. Another lipid species, PG(18:2_22:6) (m/z 817,4), also exhibited a distinct distribution pattern in steatosis. In addition, its distribution was visualized in all 23 biopsies. On a pixel level, clear regional intensity of PG(18:2_22:6) was observed with localized differences in abundance (Figure 4A) corresponding to histological steatotic regions (Supporting Figure 4). However, when the overall ion intensity of PG(18:2_22:6) across the cohort was displayed (Figure 4B), the differences in ion abundance were not profound, which may be explained by the lack of localized annotation. Among the top 10 PCA discriminatory ions, PIs were identified six times in nonsteatotic tissues but not identified at all in steatotic tissues (Supporting Table 2). Indeed, correlation and ROC analysis showed a lower abundance of PI lipid species in steatotic tissue compared to nonsteatotic tissue. These observations suggest a disruption in cellular signaling and trafficking via the phosphatidylinositol cascade in steatosis.40,41 Conversely, PEs were identified as 5 of the top 10 discriminatory ions in steatotic tissues but not identified in nonsteatotic tissues (Supporting Table 2).
Figure 3.
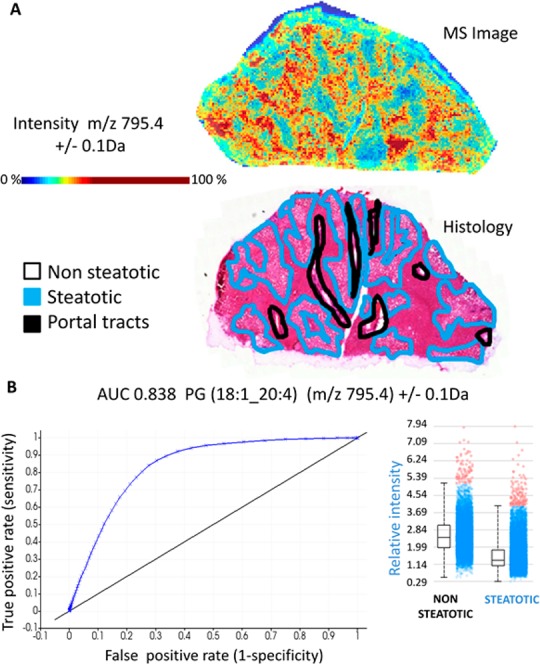
Preferential tissue distribution and high predictive value of phosphatidylglycerol (18:1_20:4) to steatotic regions. (A) MS ion image (top) of PG(18:1_20:4) (m/z 795.4) alongside the annotated histological image (bottom) of the same tissue section reveals preferential localization to steatotic areas. Color scale is in relative intensity. (B) Receiver operating characteristic (ROC) analysis of PG(18:1_20:4) shows high discriminatory power with AUC of 0.838. Inset, the relative ion intensity of PG(18:1_20:4) shows higher abundance in steatotic compared to nonsteatotic tissue regions (pixels). The horizontal line denotes the average value, the box indicates the 95% confidence interval, and the bars signify the standard deviation.
Figure 4.
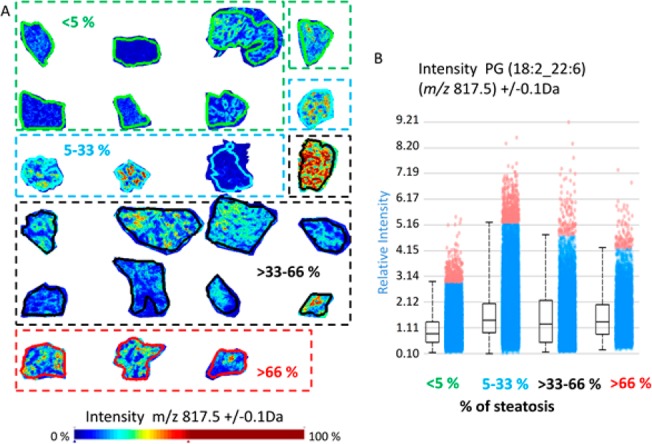
Increased PG(18:2_22:6) (m/z 817.5) abundance in regions with steatosis. (A) Relative intensity of PG(18:2_22:6) (m/z 817.5) in MS ion images shows increase in steatotic regions. (B) Box plots showing the intensity of this lipid in complete tissue regions grouped according to steatosis content: group 1 (<5% steatosis, green); group 2 (5–33% steatosis, blue); group 3 (>33–66% steatosis, black); and group 4 (>66% steatosis, red).
Network Enrichment Analyses Identify Lipid–Protein Interaction Networks Related to Inflammation Processes
Next, we performed a network analysis to determine whether the specific lipid species identified in our data set could be involved in specific metabolic pathways. The MALDI-MSI PCA identified 39 lipids enriched in nonsteatotic regions and 47 lipids in steatotic regions. Since many isobaric species may be present, multiple PubChem IDs were allowed per lipid peak (Supporting Tables 3 and 4), yielding 68 lipid IDs enriched in nonsteatotic and 95 lipid IDs enriched in steatotic regions. Despite the high number of PubChem IDs available for the analysis, only 4 (PE(16:0/20:4), PE(16:0/18:2), PS(16:0/18:1), PE(16:0/18:1)) and 3 (PG(16:0/18:0), PS(20:3/20:4), PA(18:0/20:4)) were recognized in the database for steatotic and nonsteatotic lipids, respectively. The network analysis resulted in almost independent networks for each tissue type, pointing out that the metabolic processes in these tissue types are divergent. The interaction networks consisted of 3 highly interconnected nodes (hubs): arachidonic acid (AA) and the diacylglycerol kinase (DGK) reaction (Icosa-5,8,11,14-tetraenoic acid [1-(hydroxymethyl)-2-octadecanoyloxy-ethyl] ester + ATP = 1-Stearoyl-2-arachidonoylphosphatidic acid + ADP), and phosphatidylethanolamine (PE). The network (52 nodes and 57 interactions, Supporting Table 6) revealed AA metabolism in nonsteatotic tissue, whereas low–density lipoprotein (LDL) metabolism including small, very small and large LDL were identified in steatotic regions (Figure 5). Gene expression confirmed the expression of enzymes with important functions within the inflammation pathways (Supporting Figure 5). In particular, several genes of the phospholipase group of enzymes (PLA2) and diacylglycerol kinase (DGK) family were expressed in patients with more severe steatosis.
Figure 5.

Lipid–protein interaction network determined from lipids prevalent in nonsteatotic (blue) and steatotic (green) regions.
Data-Driven PCA-LDA Classifier Discriminates 4 Steatosis Groups with Tissue-Specific Lipid Profiles
Next, we performed PCA-LDA to evaluate whether the lipid-specific signature for steatosis identified by MALDI-MSI could predict steatosis development and progression. We stratified the samples into 4 groups per steatosis grade as determined by the pathologist (Supporting Table 1), and PCA-LDA was built using 200 PCs (80% variance) from all ∼100 000 pixels/spectra. The resulting discriminant function showed the separation of the 4 steatosis grades (Figure 6), with a direct hit (4 groups) leave-one-out cross-validation accuracy of 48%. When a shift of neighboring groups was allowed (that is grade 0 ↔ 1 or 2 ↔ 3), predictive accuracy improved to 96%. Additional validation was performed using a technical replicate of all 23 biopsies as test set and confirmed the accuracy of the results. Classification performance together with confusion matrix is shown in Supporting Table 7.
Figure 6.

PCA-LDA data-driven classifier. Histogram (middle right) showing the distribution of the 4 classes along the discriminant function 1 (DF1, middle left). Intensity-scaled loading spectra (top and bottom) displaying the mass channels associated with the discriminatory power of DF1 for nonsteatotic (top) and steatotic grades (bottom). Projection of the DF score onto the training set of MS images, where each pixel is given a color based on its DF1 score. The color code indicates the 4 classes used in the PCA-LDA classifier, which corresponds to steatosis stages 0 to 4, blue to orange, respectively.
Since the spatial information was retained, the discriminant function (its score) could be projected onto each pixel and color-coded blue to red. In this way, we visualized pixel by pixel the heterogeneous lipid composition of steatotic and nonsteatotic tissue regions for the whole data set. Figure 6 shows that tissues with steatosis grade 0 or 1 were mostly color-coded blue, whereas tissues with grade 2–3 were mostly orange. Within a tissue, MSI-based PCA-LDA patterns matched the histological annotation of nonsteatotic versus steatotic regions. Interestingly, the regions denoted by histology as nonsteatotic in biopsies with grade 2 or 3 were color coded based on the MS profiles as yellow or green. This indicates an already altered metabolic profile in these regions compared with the nonsteatotic tissue region in biopsies with grade 0 or 1 which were colored blue based on their MS profile.
The effect of homogenization was simulated by averaging the spectra over all pixels within a liver biopsy and then building a PCA-LDA using the resulting 23 spectra. A histogram displaying the first discriminant function for the averaged 23 spectra PCA-LDA classifier (Supporting Figure 6) clearly showed the failure of the homogenate simulation data to discriminate the 4 steatosis groups. Results did not improve when more PCs were considered. The loss of spatial information, reflected in a reduced number of PCs, is detrimental to the performance of the classifier.
Discussion
We applied an innovative methodology to retain spatial information and evaluate local lipid profile changes in progressive stages of steatosis in NAFLD. Whereas previous studies focused predominantly on TAGs and PCs,17,20,25 we identified other lipid species and their local distribution, including specific PI, PE, PG, and PA species in steatosis. Moreover, our data are from clinically relevant human biopsies. By presenting a unique combination of high-quality structural lipid identification and evaluation of its interrelationship by systematic network analysis, we demonstrate how systematic lipid identification delineates the metabolic changes during NAFLD progression at the cellular/tissue level.
First, using pixel-wise analysis of MALDI-MSI data, we distinguished steatotic from nonsteatotic tissue. To demonstrate the strength of this approach, we performed simulated homogenate values, by averaging the spectra over all pixels within a liver biopsy, and showed that the resulting PCA-LDA classifier failed to discriminate the steatosis groups. The inability to detect subtle changes in the simulated homogenized data may be attributed to the dilution effect upon homogenization. In contrast, these subtle changes are retained when imaging data is analyzed. Localized MSI measurements are therefore likely to contribute to our understanding of the pathophysiology of lipid accumulation in NAFLD and its consequences.
Next, we showed that the steatotic lipid fingerprint identified by MALDI-MSI had high predictive accuracy, demonstrating potential use for clinical applications. Whereas the direct hit classification performed with 48% accuracy, allowing one group shift improved the accuracy to 96%. This doubling in accuracy can be explained in two ways. First, the pathological scoring system underlying the classification delineates four discrete groups, whereas steatosis severity in reality is continuous. There is poor conformity and agreement among expert pathologists concerning the assessment of steatosis severity. A recent study12 used intra class correlation coefficients (ICC) to evaluate intra- and interobserver performances and found ICC of 0.57 regarding the assessment of total steatosis (ICC of 0.7 indicates acceptable agreement). The significant jump in accuracy when we allowed a one-group shift at the extreme groups is likely to partially result from misclassification of borderline cases by the pathologist. Second, the small sample size and overfitting of the classifier could be responsible. The inherent nature of LDA causes the removal of interpatient variance within one group when building the classifier. This is further compounded when the test sample is scored, yielding misclassification because the small sample size is unable to compensate for heterogeneity within one group of patients.
Our data set uncovers potential underlying lipid metabolism and homeostasis changes in the liver that may contribute to steatosis progression. The top 10 discriminatory peaks that were identified by PCA analysis belonged to PI, PG and PS classes in nonsteatotic regions and PE, PG, and PA classes in steatotic regions. The loss of PI and increased prevalence of PE species in steatosis may suggest how lipid metabolism is dysregulated in NAFLD. Additionally, network analysis revealed diacylglycerol kinase (DGK) as a core reaction in nonsteatotic tissue (Figure 5). Interestingly, DGK is downstream of the phosphoinositide pathway, indicating its importance in nonsteatotic metabolic processes. The phosphoinositide pathway also plays a key role in liver insulin signal transduction, linking this back to the insulin resistance comorbidity of NAFLD.42 Additionally, AA-containing lipid species were enriched in nonsteatotic tissue. Both AA (as precursor for eicosanoids17,24,43) and the PI cascade are linked to inflammation, and network analysis also revealed AA metabolism to be involved. Gene expression data indicated an increase of phospholipases (PLA1a and PLA2) in patients with more severe steatosis. These enzymes have important functions within the inflammation pathways, recognizing and hydrolyzing the sn-2 acyl bond of AA-containing phospholipids thereby releasing AA and lysolipids. Therefore, this observation suggests that local elevation of AA concentrations in steatotic regions may directly contribute to the initiation of inflammatory responses at these sites. AA is also associated with insulin resistance,44 which further underscores the potential impact of AA on the development of NAFLD. Because of the tight regulation of AA levels in healthy tissues, we hypothesize that AA dysregulation in steatosis is due to aberrant hydrolysis of intracellular pools of AA-containing phospholipids by phospholipases, increasing cytoplasmic AA concentrations and stimulating the eicosanoid inflammation cascade.45 Our findings are in line with those from Hall et al., who indicated that AA could be linked to local inflammation using data obtained from mouse models of NAFLD and a validation cohort of patients with NAFLD with unknown etiology. Furthermore, our lipid analysis supports the hypothesis of Chiappini et al.22, indicating decrease of lipid synthesis such as the eicosanoid precursors leading to global reduction of phospholipids in NAFLD. Despite the reduction of AA-containing lipids and increased PLA2 gene expression in steatosis, we did not observe increases in the corresponding hydrolysis products (free AA and lysolipids) in steatotic regions. A plausible explanation for lysolipids could be increased lipid remodeling, i.e., the rapid addition of another fatty acyl to the lysolipid yielding another intact lipid moiety. With respect to AA, our network analysis also revealed low–density lipoprotein metabolism to be associated with steatosis, where the LDL receptor pathway was previously found to deliver AA for eicosanoid formation.43 Thus, the absence of free AA may be due to its immediate processing within these inflammatory pathways. Another reason for not observing the lysolipids as well as free fatty acids could be their very reactive nature and cytotoxicity. These molecules are thus cleared and processed by the cell machinery immediately upon release. Finally, detection of these low molecular mass ions is complicated by inference of matrix ions and lower sensitivity of MALDI-TOF MSI in this mass range.
Conclusion
We conclude that regions associated with a steatotic phenotype exhibit a distinct lipid composition compared to nonsteatotic tissue. In particular, steatotic regions showed a loss of AA-containing intracellular phospholipids: PI and PE species predominantly. These findings are consistent with an important role for the eicosanoid cascade in driving inflammatory responses in NAFLD, and may directly link lipid metabolism changes to inflammation-mediated clinical comorbidities. The combination of MSI and bioinformatic analyses in a systems biology approach provided molecule-based histological classifications of liver tissue. This integration strategy represents a new way to study localized lipid distribution and will contribute to our understanding of how altered lipid metabolism, homeostasis, and distributions contribute to steatosis progression in NAFLD.
Acknowledgments
This research was funded with the support of the Dutch Province of Limburg under the LINK program. The research was also supported by IOP genomics grant IGE05012A. K.S. acknowledges support from FP7 European Union Marie Curie IAPP Program, BRAINPATH. The authors thank Hang Nguyen for assistance with editing of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b05215.
Detailed subject characteristics and categorization, top ten discriminatory peaks in identified by PCA analysis, complete list of PCA identified lipid ions in nonsteatotic tissue, complete list of PCA identified lipid ions in steatotic tissue, statistical analysis of the MALDI-TOF data, interaction matrix and additional information regarding the network analysis, and performance and confusion matrix for the PCA-LDA classifier (XLSX)
Materials, lipid nomenclature, projection of one sample built PCA onto the remaining 22 samples, HCD MS/MS spectrum of the ion at m/z 847.5326, HCD MS/MS spectrum of the ion at m/z 871.5329, localized lipid accumulation and potential discriminatory power of a single lipid biomarker of steatosis, box plots showing hepatic expression of the genes from the network analysis, and PCA-LDA classifier based on averaged 23 spectra showing the homogenization effect (PDF)
Author Present Address
# G.E.: Unit Sustainable Health, Flemish Institute for Technological Research (VITO), Mol, Belgium.
The authors declare no competing financial interest.
Supplementary Material
References
- Than N. N.; Newsome P. N. Atherosclerosis 2015, 239 (1), 192–202. 10.1016/j.atherosclerosis.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Rinella M. E. JAMA 2015, 313 (22), 2263. 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- van Mierlo K. M. C.; Schaap F. G.; Dejong C. H. C.; Olde Damink S. W. M. J. Hepatol. 2016, 65 (6), 1217–1231. 10.1016/j.jhep.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Townsend S. A.; Newsome P. N. Br. Med. Bull. 2016, 119 (1), 143–156. 10.1093/bmb/ldw031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasberg S. M.; Howard T. K.; Molmenti E. P.; Hertl M. Hepatology 1994, 20 (4), 829–838. 10.1002/hep.1840200410. [DOI] [PubMed] [Google Scholar]
- McCormack L.; Petrowsky H.; Jochum W.; Furrer K.; Clavien P.-A. Ann. Surg. 2007, 245 (6), 923–930. 10.1097/01.sla.0000251747.80025.b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavien P.-A.; Petrowsky H.; DeOliveira M. L.; Graf R. N. Engl. J. Med. 2007, 356 (15), 1545–1559. 10.1056/NEJMra065156. [DOI] [PubMed] [Google Scholar]
- Nocito A.; El-Badry A. M.; Clavien P.-A. J. Hepatol. 2006, 45 (4), 494–499. 10.1016/j.jhep.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Angele M. K.; Rentsch M.; Hartl W. H.; Wittmann B.; Graeb C.; Jauch K. W.; Loehe F. Am. J. Surg. 2008, 195 (2), 214–220. 10.1016/j.amjsurg.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Yoong K.; Gunson B.; Neil D.; Mirza D.; Mayer A.; Buckels J. A.; McMaster P. Transplant. Proc. 1999, 31 (1–2), 550–551. 10.1016/S0041-1345(98)01550-4. [DOI] [PubMed] [Google Scholar]
- Kleiner D. E.; Brunt E. M.; Van Natta M.; Behling C.; Contos M. J.; Cummings O. W.; Ferrell L. D.; Liu Y.; Torbenson M. S.; Unalp-arida A.; Yeh M.; Mccullough A. J.; Sanyal A. J. Hepatology 2005, 41, 1313–1321. 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- El-Badry A. M.; Breitenstein S.; Jochum W.; Washington K.; Paradis V.; Rubbia-Brandt L.; Puhan M. A.; Slankamenac K.; Graf R.; Clavien P.-A. Ann. Surg. 2009, 250 (5), 691–697. 10.1097/SLA.0b013e3181bcd6dd. [DOI] [PubMed] [Google Scholar]
- Chalasani N.; Younossi Z.; Lavine J. E.; Diehl A. M.; Brunt E. M.; Cusi K.; Charlton M.; Sanyal A. J. American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterologyh. Gastroenterology 2012, 142 (7), 1592–1609. 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Perseghin G.; Manzoni G.; Grassi G. J. Hypertens. 2017, 35 (4), 715–717. 10.1097/HJH.0000000000001290. [DOI] [PubMed] [Google Scholar]
- Burt A.; Lackner C.; Tiniakos D. Semin. Liver Dis. 2015, 35 (3), 207–220. 10.1055/s-0035-1562942. [DOI] [PubMed] [Google Scholar]
- Byrne C. D.; Targher G. Diabetologia 2016, 59 (6), 1141–1144. 10.1007/s00125-016-3910-y. [DOI] [PubMed] [Google Scholar]
- Puri P.; Baillie R. A.; Wiest M. M.; Mirshahi F.; Choudhury J.; Cheung O.; Sargeant C.; Contos M. J.; Sanyal A. J. Hepatology 2007, 46 (4), 1081–1090. 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- Sa R.; Zhang W.; Ge J.; Wei X.; Zhou Y.; Landzberg D. R.; Wang Z.; Han X.; Chen L.; Yin H. J. Mol. Cell Biol. 2016, 8 (3), 195–206. 10.1093/jmcb/mjw016. [DOI] [PubMed] [Google Scholar]
- García-Cañaveras J. C.; Donato M. T.; Castell J. V.; Lahoz A. J. Proteome Res. 2011, 10 (10), 4825–4834. 10.1021/pr200629p. [DOI] [PubMed] [Google Scholar]
- Wattacheril J.; Seeley E. H.; Angel P.; Chen H.; Bowen B. P.; Lanciault C.; Caprioli R. M.; Abumrad N.; Flynn C. R. PLoS One 2013, 8 (2), e57165. 10.1371/journal.pone.0057165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K.; Uebanso T.; Maekawa K.; Ishikawa M.; Taguchi R.; Nammo T.; Nishimaki-Mogami T.; Udagawa H.; Fujii M.; Shibazaki Y.; Yoneyama H.; Yasuda K.; Saito Y. Sci. Rep. 2015, 5, 12466. 10.1038/srep12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappini F.; Coilly A.; Kadar H.; Gual P.; Tran A.; Desterke C.; Samuel D.; Duclos-Vallée J.-C.; Touboul D.; Bertrand-Michel J.; Brunelle A.; Guettier C.; Le Naour F. Sci. Rep. 2017, 7 (April), 46658. 10.1038/srep46658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjani K.; Lhomme M.; Sokolovska N.; Poitou C.; Aron-wisnewsky J.; Bouillot J.; Lesnik P.; Bedossa P.; Kontush A.; Clement K.; Dugail I.; Tordjman J. J. Hepatol. 2015, 62 (4), 905–912. 10.1016/j.jhep.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Ma D. W. L.; Arendt B. M.; Hillyer L. M.; Fung S. K.; McGilvray I.; Guindi M.; Allard J. P. Nutr. Diabetes 2016, 6 (7), e220 10.1038/nutd.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Bond N. J.; Ashmore T.; Sanders F.; Ament Z.; Wang X.; Murray A. J.; Bellafante E.; Virtue S.; Vidal-Puig A.; Allison M.; Davies S. E.; Koulman A.; Vacca M.; Griffin J. L. Hepatology 2017, 65 (4), 1165–1180. 10.1002/hep.28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Chu Y.; Griffin J. L. Anal. Chem. 2017, 89 (9), 5161–5170. 10.1021/acs.analchem.7b01097. [DOI] [PubMed] [Google Scholar]
- Pulfer M.; Murphy R. C. Mass Spectrom. Mass Spectrom. Rev. 2003, 22 (5), 332–364. 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- Murphy R. C.; Hankin J. A.; Barkley R. M. J. Lipid Res. 2009, 50 (Supplement), S317–S322. 10.1194/jlr.R800051-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chughtai K.; Heeren R. M. A. Chem. Rev. 2010, 110, 3237–3277. 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov M. E.; Ellis S. R.; Dilillo M.; Paine M. R. L.; Danielson W. F.; Anderson G. A.; de Graaf E. L.; Eijkel G. B.; Heeren R. M. A.; McDonnell L. A. Anal. Chem. 2017, 89 (14), 7493–7501. 10.1021/acs.analchem.7b01168. [DOI] [PubMed] [Google Scholar]
- Strohalm M.; Hassman M.; Košata B.; Kodíček M. Rapid Commun. Mass Spectrom. 2008, 22 (6), 905–908. 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- Altman D. G.Practical Statistics for Medical Research; Chapman and Hall, 1991. [Google Scholar]
- MetaCore Login|Thomson Reuters. https://portal.genego.com/ (accessed May 16, 2017).
- Dijkstra E. W. Numer. Math. 1959, 1 (1), 269–271. 10.1007/BF01386390. [DOI] [Google Scholar]
- Eijkel G. B.; Kükrer Kaletaş B.; van der Wiel I. M.; Kros J. M.; Luider T. M.; Heeren R. M. A. Surf. Interface Anal. 2009, 41, 675–685. 10.1002/sia.3088. [DOI] [Google Scholar]
- Mascini N. E.; Eijkel G. B.; ter Brugge P.; Jonkers J.; Wesseling J.; Heeren R. M. A. J. Proteome Res. 2015, 14 (2), 1069–1075. 10.1021/pr501067z. [DOI] [PubMed] [Google Scholar]
- Wolfs M. G. M.; Gruben N.; Rensen S. S.; Verdam F. J.; Greve J. W.; Driessen A.; Wijmenga C.; Buurman W. A.; Franke L.; Scheja L.; Koonen D. P. Y.; Shiri-Sverdlov R.; van Haeften T. W.; Hofker M. H.; Fu J. Nutr. Diabetes 2015, 5 (2), e146 10.1038/nutd.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balluff B.; Schöne C.; Höfler H.; Walch A. Histochem. Cell Biol. 2011, 136, 227–244. 10.1007/s00418-011-0843-x. [DOI] [PubMed] [Google Scholar]
- Chaurand P.; Schwartz S. A.; Reyzer M. L.; Caprioli R. M. Toxicol. Pathol. 2005, 33, 92–101. 10.1080/01926230590881862. [DOI] [PubMed] [Google Scholar]
- Arifin S.; Falasca M. Metabolites 2016, 6 (1), 6. 10.3390/metabo6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T. Physiol. Rev. 2013, 93 (3), 1019–1137. 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta J.; Gaikwad A. B.; Tikoo K. J. Cell. Biochem. 2010, 111 (4), 944–954. 10.1002/jcb.22783. [DOI] [PubMed] [Google Scholar]
- Habenicht A. J. R.; Salbach P.; Goerig M.; Zeh W.; Janssen-Timmen U.; Blattner C.; King W. C.; Glomset J. A. Nature 1990, 345 (6276), 634–636. 10.1038/345634a0. [DOI] [PubMed] [Google Scholar]
- Sears B.; Perry M. Lipids Health Dis. 2015, 14 (1), 121. 10.1186/s12944-015-0123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Prostaglandins, Leukotrienes Essent. Fatty Acids 1995, 53 (4), 239–254. 10.1016/0952-3278(95)90123-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.