

Abstract
To reduce attrition in drug development, it is crucial to consider the development and implementation of translational phenotypic assays as well as decipher diverse molecular mechanisms of action for new molecular entities. High-throughput fluorescence and confocal microscopes with advanced analysis software have simplified the simultaneous identification and quantification of various cellular processes through what is now referred to as high-content screening (HCS). HCS permits automated identification of modifiers of accessible and biologically relevant targets and can thus be used to detect gene interactions or identify toxic pathways of drug candidates to improve drug discovery and development processes. In this review, we summarize several HCS-compatible, biochemical, and molecular biology-driven assays, including immunohistochemistry, RNAi, reporter gene assay, CRISPR-Cas9 system, and protein-protein interactions to assess a variety of cellular processes, including proliferation, morphological changes, protein expression, localization, post-translational modifications, and protein-protein interactions. These cell-based assay methods can be applied to not only 2D cell culture but also 3D cell culture systems in a high-throughput manner.
Keywords: High content screening (HCS), drug discovery, cell-based assay, 3D cell culture system
Introduction
Drug discovery and development are crucial to finding treatments for diseases with unmet medical needs, such as cancers and neurodegenerative diseases; however, drug development is a challenging process that is expensive, time consuming, and troubled by failures. Specifically, developing a marketable drug can take 12–15 years and cost in excess of $1 billion [53, 84]. During pre-clinical development, drug candidates are identified, validated, optimized, and tested for safety and efficacy in animal models, a process which can last for ~3–6 years [84, 115]. Following this, an investigational new drug (IND) application is filed with the FDA and, if accepted, clinical trials begin. One-third of drugs fail the first clinical phase, and approximately half of drug candidates entering clinical trials fail at some point owing to unforeseen toxicity in humans [44, 65]. All of these failures had been predicted to be non-toxic in humans based on animal models prior to beginning clinical trials. Additionally, another quarter of drug candidates fail in clinical trials because the drug proves ineffective in humans [68, 70]. Thus, it is a goal to test compounds for safety and effectiveness in the early stages of the drug discovery process with in vitro testing on human cells and tissues to predict human-compound interactions prior to clinical trials. Owing to the large combinatorial space associated with drug candidates, testable outcomes, and organ systems within the human body, it is desirable to do so in a high-throughput manner.
High-throughput screening (HTS) has been widely applied to rapidly evaluate diverse compound libraries. However, maximizing throughput has not directly translated into an expected increase in new drug approvals, primarily as a result of biochemical screening assays being unable to reflect the complexity of living systems [32]. Thus, more sophisticated biological assays are needed to accurately evaluate the biological activity and potential toxicity of compound libraries.
To address this challenge, cell-based assays have been increasingly used in HTS campaigns. In vitro cell culture models can provide substantive information on various cellular responses from exposure to a compound [16]. Integrating advanced microscopy with cell-based assays is attractive because cellular events can be monitored with both spatial and temporal resolution. This approach has driven the development of high-content screening (HCS) technologies. Broadly speaking, HCS refers to any cell-based experiment that is monitored through myriad techniques, including reporter signals, morphological analysis, and phenotypic profiling, all used to measure cellular responses to a controlled stimulus. Several pioneering studies have used HCS to characterize drug efficacy [89], rapidly screen small molecules for biological activity [106], categorize subcellular localizations of target proteins [8], identify subcellular phenotypes by using RNA interference (RNAi) [18], and screen gene-deletion libraries by time-lapse imaging of live human cells [81].
These assays require sophisticated biochemical and molecular biology tools that permit quantitative observation of cell phenotypes, including characterization of cellular morphology, protein expression, localization, and post-translational modifications. Herein, we summarize several methods commonly used in phenotypic characterization of cells that are readily adaptable to high content screening purposes. Additionally, we highlight studies that have successfully integrated three-dimensional (3D) cell culture models with HTS and HCS experiments.
Immunohistochemistry-Based Assays
Immunohistochemistry (IHC) is a powerful and widespread method used to understand the distribution, localization, and extent of protein expression in various cell types and biological tissues. IHC is used to detect protein expression in a similar manner to that of western blotting and ELISA, both of which use antibodies for identification of specific proteins obtained from homogeneous cell lysates. However, unlike western blotting and ELISA, IHC can be used to detect protein expression within an intact cellular structure. Importantly, IHC is able to identify spatial patterns of protein expression. This is particularly advantageous when assessing the effects of drugs on a specific, often low fraction cell type within a heterogeneous cell population, such as within a stem cell niche or a tumor [72]. Spatial resolution prevents signal dilution, which can occur when a biomarker is partially expressed within a heterogeneous cell population (e.g., a specific type of cell within a culture of differentiating stem cells). Thus, unlike western blotting and ELISA, IHC has the ability to detect both the level of protein expression and the percentage of cells expressing target proteins. IHC can be performed on small tissue biopsies or prepared samples that can be stored in paraffin for long times, making IHC readily available for drug discovery efforts. Additionally, there are many readily available antibodies that have been developed and validated for detecting target proteins (i.e., http://www.antibodies-online.com). However, many IHC assays are time-consuming and involve area density measurements, semi-quantitative hand counts, and scoring based on “representative” microscopy fields of view of heterogeneous cells and tissues. To overcome these limitations, multiplexed and automated IHC assays have been advanced, such as for rapid quantification of protein expression. This has facilitated the use of fluorescence-based IHC assays in HCS campaigns [31] to measure multiple cellular parameters and define relationships among molecular target activities, subcellular localization, and morphologic features [31].
In addition to microscopy, flow cytometry can be used to simultaneously measure multiple events within single cells. High-end instruments use multiple lasers and detectors to measure signals from as many as 17 different fluorophores simultaneously [88]. However, it is difficult to image individual cells. For example, detailed cellular processes such as subcellular localization of proteins are unobservable. To overcome this limitation, an imaging flow cytometry method has been described that combines the precise electronic tracking of moving cells associated with flow cytometry with a high-resolution multispectral imaging system [25]. Imaging flow cytometry could be used by a clinician or researcher to obtain traditional fluorescent cytometry data (e.g., histograms and dot plots) while also analyzing cell morphology and phenotype, but the adoption of this technology has been limited to date.
RNAi Screening
RNA interference (RNAi) is a method used to silence expression of a specific gene. A complementary RNA strand of a specific gene is introduced into a cell, leading to binding to its complementary mRNA strand and breakdown of the target mRNA. The process is self-propagative and leads to destruction of all complementary RNA to the introduced strand, which results in the knockdown of a given gene’s function. Thus, the crucial components of an RNAi experiment include generating appropriate RNAi libraries and efficiently introducing the RNAi into cells primarily via transfection. RNAi technology has provided researchers with the appropriate tools needed to perform functional genomic analysis in human cells [104]. The results of large-scale RNAi screens have led to new understanding of gene function relevant in many fields, including infection, cancer, obesity, and aging [77].
RNAi screening has been accelerated through high-throughput parallel transfection, and this has enabled genome-wide RNAi screens in mammalian cell culture [86, 89, 103]. Transfections are typically performed with twodimensional (2D) monolayers of cells grown in 96- or 384-well plates through the addition of RNAi-liposome complexes using liquid-handling robotics [38]. As an alternative, reverse transfection of RNAi has been studied to perform high-throughput transfections for phenotypic analysis coupled with simple light microscopy [30, 63, 81, 101, 120]. For example, cell transfection arrays can be prepared by spotting siRNA oligonucleotides pre-mixed with transfection and matrix reagents. These spots are allowed to dry and a uniform layer of cells is subsequently seeded onto transfection arrays, which permits local transfection at each spot through a solid phase transfection into a monolayer (2D) cell culture [28, 30]. A large number of conditions can be spotted in parallel with several replicates. These arrays can be stored for subsequent use in various screening applications without the need of additional robotic handling [81]. The risk of cross-contamination from neighboring spots has been reduced by using multiwell plates for solid phase transfection [29].
Cell-Based Fluorescence Reporter Gene Assays
Unlike IHC assays, non-invasive techniques can provide better insight into the biological role of target molecules by allowing researchers to study the dynamic processes that occur in living cells in real time. One of the most commonly used cell-based assays is a reporter gene assay, which has been used for cell-based screening to identify primary signal pathway modulators [10, 20, 36]. Traditional RGAs are based on introducing reporter genes, transiently or stably transfected, into an appropriate host. Among the most commonly used reporter genes are enzymes such as β-galactosidase, chloramphenicol acyltransferase (CAT), and luciferase [3, 80]. Green fluorescent protein (GFP) has also been used widely as a reporter because multiple colored variants are available and quantification does not require cell lysis or substrate addition [100]. Additionally, GFP allows non-invasive kinetic studies to be performed in living cells [66].
GFP expression is controlled by cis-regulatory elements, recognized by a target transcription factor that can easily be monitored through fluorescence as a surrogate indicator of transcriptional activity. Regulatory elements upstream of the reporter gene might include known or putative promoters, or portions of them, or response and enhancer elements. Thousands of predicted transcription factor binding sites are known, which enables a large number of targets to be screened using RGAs [71]. In addition, hundreds of artificial promoter-reporter gene assays have been developed, many of which are commercially available. Promoter-reporter gene assays require internal controls because of potential for artifacts or off-target interference. To overcome this concern, dual promoter systems have been developed that employ two distinct reporter genes under the control of an inducible promoter of interest and a constitutive promoter used as control for a specific effect, respectively. The main concern in using RGAs for HCS is that the use of an artificially engineered assay system may not accurately reflect the endogenous cellular regulation. Endogenous gene promoters are regulated by many signaling circuits, and artificial promoters often do not account for such tight regulation.
To circumvent this limitation, bacterial artificial chromosome (BAC) reporter gene constructs have been generated leading to BAC reporter cell lines. The large capacity of BACs (up to 350 kb of genomic sequence) enables the inclusion of all required regulatory elements and ensures appropriate regulation of the gene of interest. Specifically, sequences encoding GFP can be introduced into the target locus (e.g., an exon or C-terminus of a gene) by homologous recombination (Fig. 1A). The modified BAC can then be transfected into a specific cell line [79, 82], which results in introduction of a gene associated with a GFP reporter with its endogenous regulatory elements [26, 116]. BAC reporter cell lines allow highly accurate representation of endogenous gene expression. As an example, different mouse embryonic GFP reporter stem cell lines have been constructed for specific safety evaluation (ToxTracker Assay), including apoptotic cellular stress (Btg2-GFP), DNA damage (Bscl2-GFP), oxidative stress (Srxn1-GFP), and the unfolded protein response (Ddit3-GFP) [46] (Fig. 1B). Reporter cell lines such as these can be used to identify the biological reactivity and potential carcinogenic properties of newly developed compounds in a single test and can easily be automated for high-throughput compound screening [46, 59].
Fig. 1.

Schematics of (A) introduction of GFP into a specific target locus (Exon 2) and (B) the construction of GFP reporter cell lines using BAC technology for chemical safety assessment (ToxTracker Assay).
PGK, phosphoglycerate kinase promoter; IRES, internal ribosomal entry site; NEO, neomycin resistance gene.
CRISPR-Cas9 System
CRISPR-Cas9 was originally discovered to be a bacterial “immune-like” response against bacteriophages [51, 114]. The system is composed of two essential components: the targeting RNA and Cas9 nuclease. The RNA component is composed of a CRISPR RNA (crRNA) fused to a normally trans-encoded tracrRNA, which can direct Cas9 nuclease to sequence-specifically cleave target DNA matching crRNA [69]. For the binding of Cas9 protein, the target DNA sequence must also contain the correct protospacer adjacent motif sequence (NGG) immediately following the target sequence [57].
New genome engineering technologies are beginning to be developed based on CRISPR-associated RNA-guided Cas9 (endonuclease), which enable the rapid and precise manipulation of cellular genomes [17, 70]. Whereas the utility of RNAi is limited by the inherent incompleteness of protein depletion, temporary inhibition of gene function, and unpredictable off-target effects [56], CRISPR/Cas9-mediated gene knockout provides effective methods of introducing targeted loss-of function mutations at specific sites in the genome, resulting in increasing specificity and efficiency [52]. Moreover, CRISPR/Cas9 can target promoters, enhancers, and introns. Conversely, RNAi is limited to RNA transcripts only. Recently, Shalem et al. [98] successfully performed genome-scale CRISPR-Cas9 knockout (GeCKO) screening using a sequence-specific guide RNA (sgRNA) library, resulting in targeting 18,080 genes in human cells.
In addition to enabling genome-wide knockouts using the GeCKO library, CRISPR/Cas9-mediated genome engineering is able to fuse fluorescent proteins (FP) to an endogenous gene. Recently, Yang et al. [117] reported one-step generation of an endogenous reporter allele. As shown in Fig. 2, FP could be inserted into an endogenous locus (e.g., Nanog gene) by co-transfecting a pCas-guide plasmid carrying both Cas9 and sgRNA genes and a donor plasmid carrying the gene encoding the 2A-FP reporter with homologous arms into embryonic stem (ES) cells. They designed a sgRNA targeting the stop codon of the target gene (Nanog) and a corresponding oligo to fuse the FP before the stop codon. When the fusion protein (Nanog-2AFP) was expressed in ES cells, the self-cleaving 2A peptides allowed simultaneous individual expression of Nanog and FP. Fluorescent detection of FP could quantify the endogenous expression of the target protein, Nanog (Fig. 2).
Fig. 2. One-step incorporation of fluorescent protein (FP) into the specific locus of an endogenous target gene (e.g., Nanog).
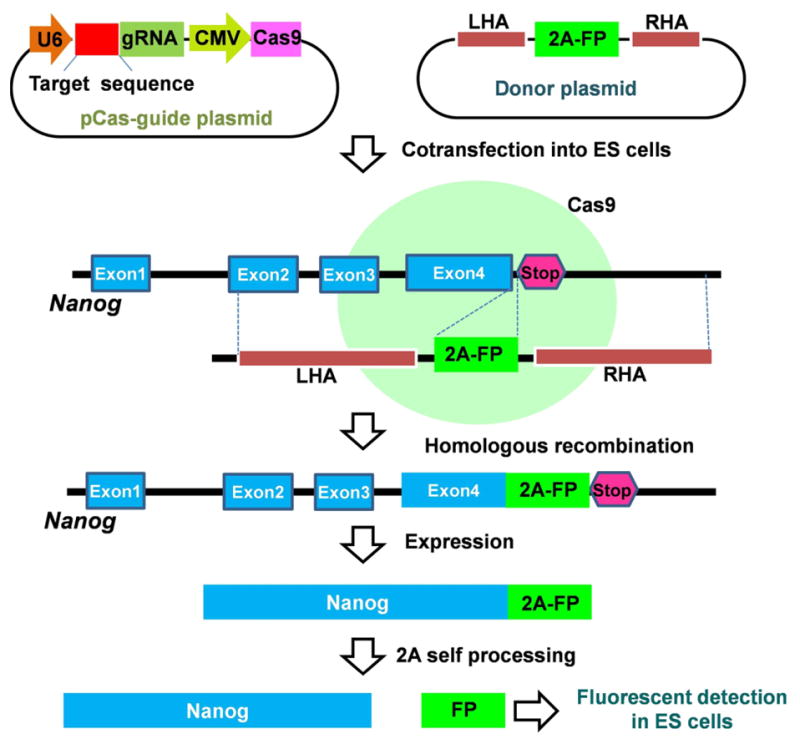
The CRISPR-Cas9 genome editing system requires the co-expression of a Cas9 protein with a guide RNA vector expressed from the human U6 polymerase III promoter. LHA, left homologous arm; RHA, right homologous arm; CMV, constitutive cytomegalovirus promoter; gRNA; guide RNA.
Cell-Based Assays for Detecting Protein-Protein Interactions
Protein–protein interactions (PPIs) are cellular processes guided by a complex spatial and temporal interplay of proteins that are essential for cell functions. PPIs are PGK, phosphoglycerate kinase promoter; IRES, internal ribosomal entry site; NEO, neomycin resistance gene. therapeutically important because abnormal PPIs are associated with various diseases, including cancer, diabetes, and viral infections [94]. Thus, modulators of PPIs are attractive drug candidates [2, 112]. Cell-based PPI assays are useful because they offer insight into a test compound’s activity within a more relevant cellular environment [55, 60]. A classical approach to analyze PPIs is the yeast two-hybrid (Y2H) system. In such a system, a gene encoding a protein of interest (Bait) is fused to the DNA-binding domain of the yeast protein GAL4, while another gene (Prey) is fused to the transcriptional activation domain (AD) [35, 43] (Fig. 3A). These two-hybrid constructs are co-transfected into a suitable yeast strain. If Bait (target protein) and Prey (proteins for screening) interact, they create a functional transcription activator by bringing the activation domain into close proximity with the DNA binding domain. This can be detected by expression of the reporter gene, but it is associated with several technical difficulties such as generation of false positives [43]. An alternative to the Y2H system, with relevance to human drug screening, is use of mammalian cells. A major advantage of the mammalian two-hybrid assay over the Y2H system is that protein-protein interactions of mammalian proteins can be studied in their endogenous environments. This permits researchers to study interactions between mammalian proteins that may not fold correctly in yeast or require post-translational modifications or external stimulations that are not present in yeast [85, 113].
Fig. 3. Schematics of cell-based assay for identifying proteinprotein interactions.
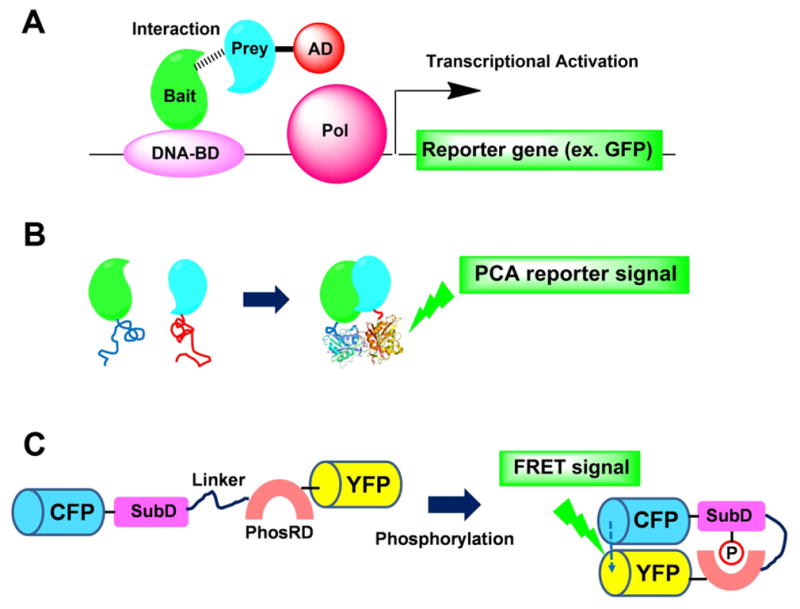
(A) Mammalian two-hybrid system. Interaction between the two test proteins (Bait and Prey), expressed as DNA binding domain (BD-Bait) and RNA polymerase activation domain (PolAD-Prey) fusion constructs, results in an increase in the expression of reporter genes over the negative controls. (B) Protein-fragment complementation assays (PCAs). Interacting proteins are fused to either of the two complementary fragments of a reporter protein (blue and red). Interaction of the two proteins brings the unfolded reporter-protein fragments into proximity, allowing them to fold into their active conformation. (C) Fluorescent indicators for protein phosphorylation in living cells. Upon phosphorylation of the substrate domain (SubD) within the fusion protein by the protein kinase, the adjacent phosphorylation recognition domain (PhosRD) binds with the phosphorylated substrate domain, which changes the efficiency of FRET between the GFP mutants. CFP, cyan fluorescent protein; YFP, yellow fluorescent protein; P in an open circle, the phosphorylated residue.
Another elegant approach to detect PPIs in mammalian cells is protein-fragment complementation assays (PCAs), which is based on protein complementation. In the PCA method, PPIs can be detected by fusing each of the proteins of interest to one of two fragments of a “reporter” protein that has been rationally dissected into two fragments using protein engineering strategies (Fig. 3B) [58, 76, 87]. When the reporter protein fragments are brought into proximity of one another through the association of the two interacting proteins of interest, they can recombine and fold into a functional reporter protein. PCAs can be created with many types of reporter proteins, including murine dihydrofolate reductase [74, 87, 92], glycinamide ribonucleotide transformylase [76], aminoglycoside kinase [76], hygromycin B kinase [76], β-lactamase [37], GFP [41], and luciferases [91]. However, because reconstitution is often irreversible, this method is not well suited to screen for compounds that disrupt protein-protein interactions after they have been formed.
Similarly, it is possible to quantify signal transduction based on protein phosphorylation within the cell [96, 97, 107]. For example, Sato et al. [97] visualized signal transduction based on protein phosphorylation in living cells by joining two different GFP variants (YFP and CFP) through a tandem fusion domain composed of a substrate domain (SubD) for the protein kinase of interest, a flexible linker sequence, and a phosphorylation recognition domain (PhosRD) that binds with the phosphorylated substrate domain (Fig. 3C) [97]. Intramolecular interaction of the substrate domain and the adjacent phosphorylation recognition domain is then dependent upon phosphorylation of the substrate domain by a protein kinase, which influences the efficiency of fluorescence resonance energy transfer (FRET) between the fluorescent proteins. This assay was originally designed to visualize protein phosphorylation by the insulin receptor but can also be adapted for other proteins (e.g., the phosphorylation of α-synuclein at Serine 129, which is closely associated with α-synuclein aggregate formation) [14, 42]. Thus, these fluorescent indicators can provide biologically significant molecular events in single living cells with high spatial and temporal resolution. In addition, the indicators provide a powerful tool for performing high-throughput, highcontent screening of pharmaceuticals that regulate second messengers, protein kinases, or nuclear receptors.
HCS in 3D Cell Culture System
The cell-based assay methods for detecting target genes and proteins have been applied to high-throughput 2D cell culture systems. However, the relevance of studying cells in traditional 2D culture environments has begun to be questioned because such an environment is not highly representative of the natural environments and niches present in vivo [45]. Alternatively, the use of artificial 3D matrices for cell encapsulation or growth of 3D organoid cultures permits screening of cells within more complex cellular microenvironments [54, 83]. Implementing 3D cell culture in HTS and HCS campaigns, however, remains challenging often due to the lack of methods available to interrogate and analyze cells within a 3D environment in a high-throughput manner.
Recently, immunofluorescence-based IHC methods were described for the detection and quantification of target proteins within microscale 3D cell cultures [33, 34]. These on-chip, in-cell immunofluorescence assays involve the binding of a primary antibody to a target protein and the subsequent binding of a secondary antibody conjugated with horseradish peroxidase (HRP), followed by addition of a signal amplification substrate (Fig. 4A). A laser scanner was then used to assess population-level protein expression in 3D cultured cells in addition to evaluation of the effects of small molecules on target protein levels in cancer cells [34], primary cell lines [64], and stem/progenitor cells [73]. High content analysis, such as single-cell protein expression, of cells within 3D systems is possible with this type of approach if coupled with more advanced imaging platforms (i.e., automated fluorescence microscopes).
Fig. 4. HCS strategies in 3D cell culture system.

(A) In-cell immunofluorescence assay of biomarkers (target proteins) in 3D cell culture on the microarray chip [34, 64]. (B) Micropillar/microwell chip platform for RNAi delivery into 3D cell culture [64]. The micropillar chip can be transferred to a microwell chip containing a target compound after incubation with lentiviruses carrying the RNAi library.
In addition to IHC methods, fluorescent reporter cell lines can be used for purposes such as detecting target proteins, promoter-specific activity, and PPI within 3D culture systems [12, 33, 108]. Stable cell lines can be prepared prior to formation of organoid or embedded culture systems and analyzed with fluorescent techniques (i.e., confocal microscopy). However, there is a notable shortage of methods that can be used to accurately measure high-content readouts of 3D systems. A combination of automated confocal microscopy with advanced analytical methods was recently described for analysis of cytotoxicity, architectural changes, and size-dependent responses of cells in 3D culture [13], but additional methods are needed to measure more complex phenomena (such as protein co-localization). Additionally, genome-wide loss-of-function screens to identify associations between common genetic variation and drug response remain challenging in 3D cell culture systems owing to inefficient gene delivery [21]. Viral gene delivery using recombinant adenoviruses has recently been described for 3D culture gene delivery into organoids [111]. A similar gene delivery approach was also described for 3D microscale cultures on a complementary micropillar/microwell chip system that is compatible with high-throughput screening methods (Fig. 4B) [64]. These advances could potentially be used for viral delivery of RNAi to mediate target gene knockdown for subsequent screening of large chemical libraries in 3D microenvironments.
Application of Cell-Based Assay Tools in Drug Discovery
Successful drug discovery requires a comprehensive understanding of the genetic variations and corresponding molecular phenotypes in relation to clinical phenotypes. However, functional annotation of the human genome is still challenging because genetic variants associated with a disease cannot easily be evaluated in a proper molecular environment. A multidisciplinary approach integrating genomic information, gene and protein expression, and biological parameters is often required. HCS approaches have begun to generate enormous amounts of raw data that can be used to understand the interactions between different cellular processes or the function of unknown genes. Since methods to systematically modify gene function (e.g., RNAi-mediated gene silencing, overexpression libraries) are now accessible [18], high-throughput RNAi screening has become a suitable investigative tool to interrogate the gene or pathway function within cell models [9, 78]. However, the majority of cell-based assays have used simplistic readouts (i.e., population-level changes) of complex biological processes. These one-dimensional assays can potentially miss subtle but essential effects or changes (i.e., morphological or gene expression alteration from treatment with a compound) that are only present in a subset of cells. To better understand how genes, signal networks, and environmental factors such as the extracellular matrix (ECM) cooperate to regulate the complex phenotypes in heterogeneous populations of cells, we have to progress toward multi-dimensional/multi-parametric assays at the single-cell level using advanced techniques associated with HCS. Although continued development of automated high-quality fluorescent and confocal microscopes and advanced analysis software (e.g., the Cellomics ArrayScan from Thermo Scientific) [16, 18, 19] is essential to achieving this goal, appropriate cell-based tools must also be developed and optimized. Importantly, these tools and assays reviewed herein and summarized in Table 1 can be used to genetically or chemically modify a cell and its environment while also simultaneously providing feedback on the consequences of each manipulation. When combined, HCS can be used to aid in drug discovery and addressing meaningful biological questions.
Table 1.
Biochemical and molecular biology tools for HCS: summary of their application, advantages, and potential pitfalls.
| Assay tools | Applications | Advantages | Potential pitfalls | Selected References |
|---|---|---|---|---|
| IHC |
|
|
|
[31] [118] [50] [95] |
| RNAi screen |
|
|
|
[11] [24] [49] [61, 62] [9] [78] |
| RGA |
|
|
|
[39] [6] [23] [27] [68] [46] [47] |
| CRISPR-Cas9 |
|
|
|
[99] [109] [7] [93] [119] |
| PPI |
|
|
|
[96] [102] [107] [75] [48] [1] [90] |
Future Directions
Initial integration of cell-based HTS assays into the drug development process resulted in relatively poor efficiency in the identification of safe and effective drug candidates. It is believed that population-averaged readouts (i.e., a result on an entire well, not individual cells) associated with cell-based assays contributed to the poor translation from benchtop to clinic [105]. Thus, a continual challenge is to acquire accurate information from cell-based assays to increase success rates of drugs in clinical trials. This is where the potential of HCS lies, in that HCS can be used to image cell populations and assess complex phenotypic outcomes of individual cells within a heterogeneous cell population. This eliminates the variability of population-based averages. Overall, these profiling methods allow for the measurement of multiple integrated parameters at the level of single cells to facilitate more complex analysis such as target prediction of a drug candidate [67, 89] or precise identification of proteins involved in biological processes [40].
HCS has been exploited in screening campaigns to identify changes in biological pathways that impact specific cellular responses to small molecule drug candidates [22, 110]. Moreover, HCS can facilitate “drug repositioning” strategies, which seek to identify new pharmacological targets for existing molecules that have already been evaluated for toxicity and pharmacokinetics. This approach would reduce costs associated with drug discovery [4, 15]. However, drug repositioning is often limited commercially owing to the lack of clear patent coverage, which often deters pharmaceutical companies from broader pursuit of this approach [5].
Recent developments in ES and induced pluripotent stem (iPS) cells have opened up new opportunities for HCS to be used in understanding phenotypic profiling and molecular mechanisms associated with diseased cell phenotypes and differentiating cells. Specifically, patient-derived iPS cells from familial cases of genetically linked diseases may represent unique sources of biological material to better understand disease progression without the need for exogenous expression systems. There remains a need for robust phenotypic assays that can be used in combination with target-based assays (described in this review), however, and continual improvements with automated fluid dispensing and miniaturized detection systems compatible with HCS campaigns. Combined with advances in biochemical and molecular biology tools, 3D cellular models, live cell imaging and data analysis, and cell-based HCS, the research community should be able to obtain important answers to key questions surrounding molecular mechanisms of diseases, resulting in accelerated drug discovery.
References
- 1.Arkin MR, Glicksman MA, Fu H, Havel JJ, Du Y. Inhibition of protein-protein interactions: non-cellular assay formats. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, et al., editors. Assay Guidance Manual. Bethesda, MD: 2004. [Google Scholar]
- 2.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 3.Arnone MI, Dmochowski IJ, Gache C. Using reporter genes to study cis-regulatory elements. Methods Cell Biol. 2004;74:621–652. doi: 10.1016/s0091-679x(04)74025-x. [DOI] [PubMed] [Google Scholar]
- 4.Aronson JK. Old drugs – new uses. Br J Clin Pharmacol. 2007;64:563–565. doi: 10.1111/j.1365-2125.2007.03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 6.Aswendt M, Adamczak J, Tennstaedt A. A review of novel optical imaging strategies of the stroke pathology and stem cell therapy in stroke. Front Cell Neurosci. 2014;8:226. doi: 10.3389/fncel.2014.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrangou R, Birmingham A, Wiemann S, Beijersbergen RL, Hornung V, Smith A. Advances in CRISPR-Cas9 genome engineering: lessons learned from RNA interference. Nucleic Acids Res. 2015;43:3407–3419. doi: 10.1093/nar/gkv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boland MV, Murphy RF. A neural network classifier capable of recognizing the patterns of all major subcellular structures in fluorescence microscope images of HeLa cells. Bioinformatics. 2001;17:1213–1223. doi: 10.1093/bioinformatics/17.12.1213. [DOI] [PubMed] [Google Scholar]
- 9.Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9:554–566. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- 10.Bronstein I, Fortin J, Stanley PE, Stewart GS, Kricka LJ. Chemiluminescent and bioluminescent reporter gene assays. Anal Biochem. 1994;219:169–181. doi: 10.1006/abio.1994.1254. [DOI] [PubMed] [Google Scholar]
- 11.Campeau E, Gobeil S. RNA interference in mammals: behind the screen. Brief Funct Genomics. 2011;10:215–226. doi: 10.1093/bfgp/elr018. [DOI] [PubMed] [Google Scholar]
- 12.Canton I, Cole DM, Kemp EH, Watson PF, Chunthapong J, Ryan AJ, et al. Development of a 3D human in vitro skin co-culture model for detecting irritants in real-time. Biotechnol Bioeng. 2010;106:794–803. doi: 10.1002/bit.22742. [DOI] [PubMed] [Google Scholar]
- 13.Celli JP, Rizvi I, Blanden AR, Massodi I, Glidden MD, Pogue BW, Hasan T. An imaging-based platform for high-content, quantitative evaluation of therapeutic response in 3D tumour models. Sci Rep. 2014;4:3751. doi: 10.1038/srep03751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 15.Chong CR, Sullivan DJ., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 16.Clemons PA. Complex phenotypic assays in high-throughput screening. Curr Opin Chem Biol. 2004;8:334–338. doi: 10.1016/j.cbpa.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Cong L, Ran FA, Cox D, Lin S, Barret to R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrad C, Erfle H, Warnat P, Daigle N, Lorch T, Ellenberg J, et al. Automatic identification of subcellular phenotypes on human cell arrays. Genome Res. 2004;14:1130–1136. doi: 10.1101/gr.2383804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conrad C, Gerlich DW. Automated microscopy for high-content RNAi screening. J Cell Biol. 2010;188:453–461. doi: 10.1083/jcb.200910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crisman TJ, Parker CN, Jenkins JL, Scheiber J, Thoma M, Kang ZB, et al. Understanding false positives in reporter gene assays: in silico chemogenomics approaches to prioritize cell-based HTS data. J Chem Inf Model. 2007;47:1319–1327. doi: 10.1021/ci6005504. [DOI] [PubMed] [Google Scholar]
- 21.Dhaliwal A, Oshita V, Segura T. Transfection in the third dimension. Integr Biol (Camb) 2013;5:1206–1216. doi: 10.1039/c3ib40086g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diamandis P, Wildenhain J, Clarke ID, Sacher AG, Graham J, Bellows DS, et al. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat Chem Biol. 2007;3:268–273. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- 23.Duryagina R, Anastassiadis K, Maitz MF, Gramm S, Schneider S, Wobus M, et al. Cellular reporter systems for high-throughput screening of interactions between bioactive matrices and human mesenchymal stromal cells. Tissue Eng Part C Methods. 2014;20:828–837. doi: 10.1089/ten.TEC.2013.0590. [DOI] [PubMed] [Google Scholar]
- 24.Echeverri CJ, Perrimon N. High-throughput RNAi screening in cultured cells: a user’s guide. Nat Rev Genet. 2006;7:373–384. doi: 10.1038/nrg1836. [DOI] [PubMed] [Google Scholar]
- 25.Edwards BS, Sklar LA. Flow cytometry: impact on early drug discovery. J Biomol Screen. 2015;20:689–707. doi: 10.1177/1087057115578273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elliott DA, Braam SR, Koutiss K, Ng ES, Jenny R, Lagerqvist EL, et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- 27.Enikolopov G, Overstreet-Wadiche L, Ge S. Viral and transgenic reporters and genetic analysis of adult neurogenesis. Cold Spring Harb Perspect Biol. 2015;7:a018804. doi: 10.1101/cshperspect.a018804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erfle H, Neumann B, Liebel U, Rogers P, Held M, Walter T, et al. Reverse transfection on cell arrays for high content screening microscopy. Nat Protoc. 2007;2:392–399. doi: 10.1038/nprot.2006.483. [DOI] [PubMed] [Google Scholar]
- 29.Erfle H, Neumann B, Rogers P, Bulkescher J, Ellenberg J, Pepperkok R. Work flow for multiplexing siRNA assays by solid-phase reverse transfection in multiwell plates. J Biomol Screen. 2008;13:575–580. doi: 10.1177/1087057108320133. [DOI] [PubMed] [Google Scholar]
- 30.Erfle H, Simpson JC, Bastiaens PI, Pepperkok R. siRNA cell arrays for high-content screening microscopy. Biotechniques. 2004;37:454–458. 460, 462. doi: 10.2144/04373RT01. [DOI] [PubMed] [Google Scholar]
- 31.Falcon BL, Stewart J, Ezell S, Hanson J, Wijsman J, Ye X, et al. High-content multiplexed tissue imaging and quantification for cancer drug discovery. Drug Discov Today. 2013;18:510–522. doi: 10.1016/j.drudis.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Feng Y, Mitchison TJ, Bender A, Young DW, Tallarico JA. Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat Rev Drug Discov. 2009;8:567–578. doi: 10.1038/nrd2876. [DOI] [PubMed] [Google Scholar]
- 33.Fernandes TG, Diogo MM, Clark DS, Dordick JS, Cabral JM. High-throughput cellular microarray platforms: applications in drug discovery, toxicology and stem cell research. Trends Biotechnol. 2009;27:342–349. doi: 10.1016/j.tibtech.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandes TG, Kwon SJ, Lee MY, Clark DS, Cabral JM, Dordick JS. On-chip, cell-based microarray immunofluorescence assay for high-throughput analysis of target proteins. Anal Chem. 2008;80:6633–6639. doi: 10.1021/ac800848j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 36.Fishman MC, Porter JA. Pharmaceuticals: a new grammar for drug discovery. Nature. 2005;437:491–493. doi: 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- 37.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 38.Ganesan AK, Ho H, Bodemann B, Petersen S, Aruri J, Koshy S, et al. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genet. 2008;4:e1000298. doi: 10.1371/journal.pgen.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gasparri F, Galvani A. Image-based high-content reporter assays: limitations and advantages. Drug Discov Today Technol. 2010;7:e1–e94. doi: 10.1016/j.ddtec.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Genovesio A, Kwon YJ, Windisch MP, Kim NY, Choi SY, Kim HC, et al. Automated genome-wide visual profiling of cellular proteins involved in HIV infection. J Biomol Screen. 2011;16:945–958. doi: 10.1177/1087057111415521. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 42.Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, et al. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci USA. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamdi A, Colas P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol Sci. 2012;33:109–118. doi: 10.1016/j.tips.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 45.Haycock JW. 3D cell culture: a review of current approaches and techniques. Methods Mol Biol. 2011;695:1–15. doi: 10.1007/978-1-60761-984-0_1. [DOI] [PubMed] [Google Scholar]
- 46.Hendriks G, Atallah M, Morolli B, Calleja F, Ras-Verloop N, Huijskens I, et al. The ToxTracker assay: novel GFP reporter systems that provide mechanistic insight into the genotoxic properties of chemicals. Toxicol Sci. 2012;125:285–298. doi: 10.1093/toxsci/kfr281. [DOI] [PubMed] [Google Scholar]
- 47.Hendriks G, van de Water B, Schoonen W, Vrieling H. Cellular-signaling pathways unveil the carcinogenic potential of chemicals. J Appl Toxicol. 2013;33:399–409. doi: 10.1002/jat.2845. [DOI] [PubMed] [Google Scholar]
- 48.Herce HD, Deng W, Helma J, Leonhardt H, Cardoso MC. Visualization and targeted disruption of protein interactions in living cells. Nat Commun. 2013;4:2660. doi: 10.1038/ncomms3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hibbitts A, Lieggi N, McCabe O, Thomas W, Barlow J, O’Brien F, Cryan SA. Screening of siRNA nanoparticles for delivery to airway epithelial cells using high-content analysis. Ther Deliv. 2011;2:987–999. doi: 10.4155/tde.11.73. [DOI] [PubMed] [Google Scholar]
- 50.Hofman F. Immunohistochemistry. Curr Protoc Immunol. 2002;Chapter 21(Unit 21.4) doi: 10.1002/0471142735.im2104s49. [DOI] [PubMed] [Google Scholar]
- 51.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 52.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hynds RE, Giangreco A. Concise review: the relevance of human stem cell-derived organoid models for epithelial translational medicine. Stem Cells. 2013;31:417–422. doi: 10.1002/stem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 56.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci USA. 1994;91:10340–10344. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karlsson HL, Gliga AR, Calleja FM, Goncalves CS, Wallinder IO, Vrieling H, et al. Mechanism-based genotoxicity screening of metal oxide nanoparticles using the ToxTracker panel of reporter cell lines. Part Fibre Toxicol. 2014;11:41. doi: 10.1186/s12989-014-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korn K, Krausz E. Cell-based high-content screening of small-molecule libraries. Curr Opin Chem Biol. 2007;11:503–510. doi: 10.1016/j.cbpa.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 61.Krausz E. High-content siRNA screening. Mol Biosyst. 2007;3:232–240. doi: 10.1039/b616187c. [DOI] [PubMed] [Google Scholar]
- 62.Krausz E, Korn K. High-content siRNA screening for target identification and validation. Expert Opin Drug Discov. 2008;3:551–564. doi: 10.1517/17460441.3.5.551. [DOI] [PubMed] [Google Scholar]
- 63.Kumar R, Conklin DS, Mittal V. High-throughput selection of effective RNAi probes for gene silencing. Genome Res. 2003;13:2333–2340. doi: 10.1101/gr.1575003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwon SJ, Lee DW, Shah DA, Ku B, Jeon SY, Solanki K, et al. High-throughput and combinatorial gene expression on a chip for metabolism-induced toxicology screening. Nat Commun. 2014;5:3739. doi: 10.1038/ncomms4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ledford H. Translational research: 4 ways to fix the clinical trial. Nature. 2011;477:526–528. doi: 10.1038/477526a. [DOI] [PubMed] [Google Scholar]
- 66.Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- 67.Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nat Methods. 2007;4:445–453. doi: 10.1038/nmeth1032. [DOI] [PubMed] [Google Scholar]
- 68.Machleidt T, Whitney P, Bi K. Multiplexing of pathway-specific beta-lactamase reporter gene assays by optical coding with Qtracker nanocrystals. J Biomol Screen. 2009;14:845–852. doi: 10.1177/1087057109335742. [DOI] [PubMed] [Google Scholar]
- 69.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meli L, Barbosa HS, Hickey AM, Gasimli L, Nierode G, Diogo MM, et al. Three-dimensional cellular microarray platform for human neural stem cell differentiation and toxicology. Stem Cell Res. 2014;13:36–47. doi: 10.1016/j.scr.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michnick SW, Ear PH, Landry C, Malleshaiah MK, Messier V. A toolkit of protein-fragment complementation assays for studying and dissecting large-scale and dynamic protein-protein interactions in living cells. Methods Enzymol. 2010;470:335–368. doi: 10.1016/S0076-6879(10)70014-8. [DOI] [PubMed] [Google Scholar]
- 75.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 76.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN. Detection of protein-protein interactions by protein fragment complementation strategies. Methods Enzymol. 2000;328:208–230. doi: 10.1016/s0076-6879(00)28399-7. [DOI] [PubMed] [Google Scholar]
- 77.Mohr SE, Perrimon N. RNAi screening: new approaches, understandings, and organisms. Wiley Interdiscip Rev RNA. 2012;3:145–158. doi: 10.1002/wrna.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mohr SE, Smith JA, Shamu CE, Neumuller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Narayanan K, Chen Q. Bacterial artificial chromosome mutagenesis using recombineering. J Biomed Biotechnol. 2011;2011:971296. doi: 10.1155/2011/971296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naylor LH. Reporter gene technology: the future looks bright. Biochem Pharmacol. 1999;58:749–757. doi: 10.1016/s0006-2952(99)00096-9. [DOI] [PubMed] [Google Scholar]
- 81.Neumann B, Held M, Liebel U, Erfle H, Rogers P, Pepperkok R, Ellenberg J. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat Methods. 2006;3:385–390. doi: 10.1038/nmeth876. [DOI] [PubMed] [Google Scholar]
- 82.Ohtsuka M, Kimura M, Tanaka M, Inoko H. Recombinant DNA technologies for construction of precisely designed transgene constructs. Curr Pharm Biotechnol. 2009;10:244–251. doi: 10.2174/138920109787315033. [DOI] [PubMed] [Google Scholar]
- 83.Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 84.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 85.Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 86.Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, Krausz E, Zerial M. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 87.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci USA. 1998;95:12141–12146. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 89.Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 90.Rao VS, Srinivas K, Sujini GN, Kumar GN. Proteinprotein interaction detection: methods and analysis. Int J Proteomics. 2014;2014:147648. doi: 10.1155/2014/147648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3:977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- 92.Remy I, Wilson IA, Michnick SW. Erythropoietin receptor activation by a ligand-induced conformation change. Science. 1999;283:990–993. doi: 10.1126/science.283.5404.990. [DOI] [PubMed] [Google Scholar]
- 93.Ren Q, Li C, Yuan P, Cai C, Zhang L, Luo GG, Wei W. A dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci Rep. 2015;5:8865. doi: 10.1038/srep08865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 95.Samsel L, Dagur PK, Raghavachari N, Seamon C, Kato GJ, McCoy JP., Jr Imaging flow cytometry for morphologic and phenotypic characterization of rare circulating endothelial cells. Cytometry B Clin Cytom. 2013;84:379–389. doi: 10.1002/cyto.b.21088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato M, Kawai Y, Umezawa Y. Genetically encoded fluorescent indicators to visualize protein phosphorylation by extracellular signal-regulated kinase in single living cells. Anal Chem. 2007;79:2570–2575. doi: 10.1021/ac062171d. [DOI] [PubMed] [Google Scholar]
- 97.Sato M, Ozawa T, Inukai K, Asano T, Umezawa Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat Biotechnol. 2002;20:287–294. doi: 10.1038/nbt0302-287. [DOI] [PubMed] [Google Scholar]
- 98.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 101.Silva JM, Mizuno H, Brady A, Lucito R, Hannon GJ. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc Natl Acad Sci USA. 2004;101:6548–6552. doi: 10.1073/pnas.0400165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 103.Sonnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434:462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 104.Sui G, Soohoo C, Affarel B, Gay F, Shi Y, Forrester WC. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA. 2002;99:5515–5520. doi: 10.1073/pnas.082117599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 106.Tanaka M, Bateman R, Rauh D, Vaisberg E, Ramachandani S, Zhang C, et al. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005;3:e128. doi: 10.1371/journal.pbio.0030128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ting AY, Kain KH, Klemke RL, Tsien RY. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci USA. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst. 2011;136:473–478. doi: 10.1039/c0an00609b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wade M. High-throughput silencing using the CRISPR-Cas9 system: a review of the benefits and challenges. J Biomol Screen. 2015;20:1027–1039. doi: 10.1177/1087057115587916. [DOI] [PubMed] [Google Scholar]
- 110.Walsh DP, Chang YT. Chemical genetics. Chem Rev. 2006;106:2476–2530. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 111.Wang N, Zhang H, Zhang BQ, Liu W, Zhang Z, Qiao M, et al. Adenovirus-mediated efficient gene transfer into cultured three-dimensional organoids. PLoS One. 2014;9:e93608. doi: 10.1371/journal.pone.0093608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 113.Weston CR, Davis RJ. Signal transduction: signaling specificity – a complex affair. Science. 2001;292:2439–2440. doi: 10.1126/science.1063279. [DOI] [PubMed] [Google Scholar]
- 114.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 115.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 116.Wink S, Hiemstra S, Huppelschoten S, Danen E, Niemeijer M, Hendriks G, et al. Quantitative high content imaging of cellular adaptive stress response pathways in toxicity for chemical safety assessment. Chem Res Toxicol. 2014;27:338–355. doi: 10.1021/tx4004038. [DOI] [PubMed] [Google Scholar]
- 117.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010;28:237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 119.Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 120.Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]