

Abstract
Kainate receptors are members of the glutamate receptor family that function by both generating ionotropic currents through an integral ion channel pore and coupling to downstream metabotropic signaling pathways. They are highly expressed in the striatum, yet their roles in regulating striatal synapses are not known. Using mice of both sexes, we demonstrate that GluK2-containing kainate receptors expressed in direct pathway spiny projection neurons (dSPNs) inhibit glutamate release at corticostriatal synapses in the dorsolateral striatum. This inhibition requires postsynaptic kainate-receptor-mediated mobilization of a retrograde endocannabinoid (eCB) signal and activation of presynaptic CB1 receptors. This pathway can be activated during repetitive 25 Hz trains of synaptic stimulation, causing short-term depression of corticostriatal synapses. This is the first study to demonstrate a role for kainate receptors in regulating eCB-mediated plasticity at the corticostriatal synapse and demonstrates an important role for these receptors in regulating basal ganglia circuits.
SIGNIFICANCE STATEMENT The GRIK2 gene, encoding the GluK2 subunit of the kainate receptor, has been linked to several neuropsychiatric and neurodevelopmental disorders including obsessive compulsive disorder (OCD). Perseverative behaviors associated with OCD are known to result from pathophysiological changes in the striatum and kainate receptor knock-out mice have striatal-dependent phenotypes. However, the role of kainate receptors in striatal synapses is not known. We demonstrate that GluK2-containing kainate receptors regulate corticostriatal synapses by mobilizing endocannabinoids from direct pathway spiny projection neurons. Synaptic activation of GluK2 receptors during trains of synaptic input causes short-term synaptic depression, demonstrating a novel role for these receptors in regulating striatal circuits.
Keywords: endocannabinoid, kainate receptor, short-term plasticity, spiny projection neuron, striatum
Introduction
Plasticity at corticostriatal synapses is regulated by endocannabinoids (eCBs), lipophilic signaling molecules that inhibit transmitter release by activating the presynaptic CB1 receptor (Gerdeman et al., 2002; Lovinger, 2007). The striatum is the input nucleus of the basal ganglia and receives convergent excitatory cortical and thalamic input. Outputs of the basal ganglia in turn reciprocally regulate cortical activity via ascending projections to thalamocortical neurons (Alexander et al., 1986). The basal ganglia play key roles in regulating voluntary movement and instrumental conditioning and alterations in corticostriatal plasticity produce changes in these striatal-dependent behaviors (Yin and Knowlton, 2006; Surmeier et al., 2014; Graybiel and Grafton, 2015). A diverse array of neurotransmitter receptors have been demonstrated to modulate corticostriatal plasticity (Kreitzer and Malenka, 2008). However, it is not known whether kainate receptors, a subtype of ionotropic glutamate receptor expressed at SPN synapses (Xu et al., 2017), regulate corticostriatal synaptic transmission.
Kainate receptors, in contrast to the structurally similar AMPA receptors, fulfill a more varied range of synaptic and extrasynaptic roles (Contractor et al., 2011). The receptors assemble as heterotetramers from combinations of the GluK1–5 subunits encoded by the Grik1-5 genes and are named for their sensitivity to kainic acid (KA) while lacking affinity for AMPA and NMDA (Dingledine, 1991). They are localized to postsynaptic densities in only a small subset of synapses, where they contribute to a component of the EPSC (Castillo et al., 1997; Fernandes et al., 2009). In most instances, they function to modulate neural circuits through roles as direct presynaptic regulators of neurotransmitter release (Contractor et al., 2001; Schmitz et al., 2001), indirect modulators through the release of additional signaling molecules (Chergui et al., 2000; Lourenço et al., 2011), or regulators of intrinsic excitability (Melyan et al., 2002).
Recently, we demonstrated that mice lacking genes encoding all five subunits of the kainate receptor, 5ko mice, have alterations in striatal physiology and elevations in perseverative behaviors such as grooming and digging (Xu et al., 2017). Perseverative behaviors are hallmarks of psychiatric diseases involving striatal dysfunction such as obsessive compulsive disorder (OCD) (Monteiro and Feng, 2016); mice with aberrant behaviors serve as models for the perseverative behavior in patients (Pauls et al., 2014; Monteiro and Feng, 2016). 5ko mice showed changes in properties of corticostriatal synaptic transmission and alterations in synapse density in SPNs (Xu et al., 2017). However, it remained unclear whether these changes were adaptive changes due to a loss of kainate receptors during development or if ongoing kainate receptor activity in the mature circuit contributes to striatal function. In the current work, we have proposed to establish whether there is an ongoing role for kainate receptors at corticostriatal synapses.
GluK2 is the primary subunit that is expressed in the principal striatal cell type, the spiny projection neuron (SPN) (Chergui et al., 2000). GluK2 knock-out mice display elevations in perseverative digging behavior (Xu et al., 2017), are hyperactive (Shaltiel et al., 2008), and genetic evidence has linked mutations in the GRIK2 gene with OCD. Family-based studies have identified single nucleotide polymorphisms (SNPs) in GRIK2 that are undertransmitted in, and significantly associated with, OCD (Delorme et al., 2004; Sampaio et al., 2011). In addition, two recent genome wide association studies identified several GRIK2 SNPs with high probability among candidate OCD susceptibility genes (Stewart et al., 2013; Mattheisen et al., 2015).
There are two distinct types of SPNs in the striatum: “direct” pathway dopamine D1 receptor-expressing SPNs (dSPNs) that project directly to the substantia nigra and “indirect” dopamine D2 receptor expressing SPNs (iSPNs) that project to the globus pallidus, which inhibits the substantia nigra (Surmeier et al., 2014). Here, we present evidence that acute activation of GluK2-containing kainate receptors inhibits glutamate release at cortical synapses onto dSPNs. This depression is mediated by postsynaptic receptors that mobilize the eCB 2-arachidonoylglycerol (2-AG). Short trains of presynaptic stimulation within the range of physiological frequencies selectively engage kainate receptor eCB mobilization, but not plasticity mediated by the group I metabotropic glutamate receptor family (Lerner and Kreitzer, 2012), demonstrating a specific role for kainate receptors in corticostriatal plasticity.
Materials and Methods
Animals.
Animals were group housed with 14 h/10 h light/dark cycle and food and water were provided ad libitum. Mice of both sexes during the third postnatal week [postnatal day 21 (P21) to P28] were used and all experiments were performed in accordance with procedures approved by the Northwestern University institutional animal care and use committee.
Generation of Grik2 conditional allele.
Standard gene-targeting techniques were used to generate mutant floxed Grik2 mice. Genomic DNA for Grik2 was initially cloned from a phage library of 129 SVJ mouse genomic DNA fragments (Stratagene) (Mulle et al., 1998). A 4.8 kb DNA fragment for the 3′ targeting arm was inserted into a shuttle plasmid containing positive and negative selection markers (Xu et al., 2009). Next, a 2.0 kb Grik2 DNA fragment containing exon 13 that encodes the reentrant p-loop of the channel was subcloned into the same plasmid between the two loxP sites in the vector upstream of the neocassette. Finally, a 3.4 kb Grik2 DNA fragment for the 5′ targeting arm was subcloned into the targeting vector upstream of the floxed exon. The targeting vector was linearized by Sal1 and transfected into R1 mouse ES cells by electroporation. Transfected ES cells were cultured in selection medium with G418 (Geneticin) and surviving clones were isolated for PCR and Southern blot analysis. Southern blot analysis was performed with 3′ probes and 5′ probes. Both probes are located outside of recombination arms to exclude clones with random integration of the targeting vector DNA. Blastocyst injection and embryo transfer were performed by the transgenic core facility at Northwestern University. Mice with germline transmission were further bred with EIIa-Cre transgenic mice (The Jackson Laboratory Stock #003724) to remove the neomycin cassette.
D1-cre;Grik2f/f mice were generated by intercrossing mice homozygous for the floxed Grik2 allele with mice expressing Cre recombinase under control of the drd1a promoter for expression in dSPNs (Gong et al., 2007) and a Cre-dependent tdTomato reporter inserted in the Gt26Sor (Rosa26) locus (Ai9) for identification of dSPNs expressing Cre (Madisen et al., 2010).
Slice procedure and electrophysiological recording.
Parasagittal slices containing the dorsal striatum (250 μm) were prepared using techniques modified from those described previously (Xu et al., 2017). Briefly, the brain was rapidly removed under ice-cold oxygenated sucrose-slicing ACSF containing the following (in mm): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 74 sucrose, 0.5 CaCl2, 4 MgCl2, and 2 N-acetyl cysteine (NAC). Individual slices were transferred to a recording chamber and visualized after allowing ∼60 min for recovery at 30–32°C in solution containing the following (in mm): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 1 CaCl2, 2 MgCl2, and 2 NAC. The ACSF used during recordings was identical to the recovery solution with the exception that it contained 2 mm CaCl2 and 1 mm MgCl2 and NAC was omitted. After the initial recovery, slices were kept at room temperature until being used for recording. dSPNs were identified visually based on expression of the tdTomato reporter expressed selectively in dSPNs by the drd1a promoter (Shuen et al., 2008). For whole-cell voltage-clamp recordings, borosilicate glass electrodes (resistance 2.5–5 MΩ) were filled with an internal solution containing the following (in mm): 95 CsF, 25 CsCl, 10 Cs-HEPES, 10 Cs-EGTA, 2 NaCl, 2 Mg-ATP, 10 QX-314, 5 TEA-Cl, and 5 4-AP, pH adjusted to 7.3 with CsOH. Cells were voltage clamped at −80 mV and corticostriatal inputs stimulated using an extracellular bipolar concentric electrode placed in cortical layers 5/6. EPSCs were isolated by the continuous presence of the GABAA receptor antagonist bicuculline (10 μm). Miniature EPSCs (mEPSCs) were recorded in the presence of bicuculline (10 μm) and 1 μm tetrodotoxin (TTX). The amplifier output and stimulus isolator was controlled using pClamp9 software. Series resistance (Rs) was monitored using a 10 mV hyperpolarizing step preceding each evoked event, which was also used to calculate input membrane resistance (Rm) and capacitance (Cm). Recordings with >25% variation in Rs or Cm were excluded from the analysis. All recordings were performed at 30–32°C.
Drugs were made as concentrated stock solutions in either water or DMSO (1:1000 or 1:10,000) and diluted to their final concentration on the day of experiments. For experiments using AM251, tetrahydrolipstatin (THL), or RHC 80267, BSA (0.05%) was used as a carrier and was added to the slices with drugs or with control DMSO solutions for at least 1 h before recording and maintained throughout the experiment. For experiments in which BAPTA or GDPβS were included in the internal pipette solution, cells were held in whole-cell configuration for at least 30 min for dialysis before beginning baseline recordings. In interleaved controls, cells were dialyzed with standard internal solution and similarly held in whole-cell configuration for 30 min before baseline acquisition.
Data analysis.
Statistical analyses were conducted with Excel (Microsoft), OriginPro 9.0 (OriginLab), and MATLAB (The MathWorks) software. Semiautomated analysis of mEPSCs was performed using the template matching algorithm in Stimfit software (Guzmán et al., 2014). Depression during time series experiments, or paired-pulse ratio (PPR) changes, were analyzed by comparing the mean EPSC amplitude and PPR 10–15 min after beginning kainate application with the baseline using a nonparametric Wilcoxon signed-rank test for each group. Comparisons between two groups were made using a Wilcoxon rank-sum test. Differences were considered significant when p < 0.05. Data are shown as mean ± SEM.
Results
Kainate receptors mobilize eCBs in dSPNs
To determine how acute activation of kainate receptors influences corticostriatal synaptic transmission, we made recordings from dSPNs using BAC transgenic mice expressing a fluorescent reporter (tdTomato) under the control of the drd1a promoter (Shuen et al., 2008). Corticostriatal EPSCs in dSPNs were evoked in a parasagittal slice preparation that preserved cortical afferents with a bipolar electrode placed in layers 5/6 of the cortex (Ding et al., 2008). Application of a low concentration of KA (100 nm) for 15 min depressed the amplitude of corticostriatal-evoked EPSCs in dSPNs. In most cases, the depression remained 10–15 min after washing out the agonist. Figure 1A illustrates a representative time course of an individual recording and Figure 1D shows the grouped data of all of the recordings in these conditions (p = 0.005, n = 12 cells from 8 animals, Wilcoxon signed-rank test; Fig. 1A,D). Depression of the EPSC was accompanied by an increase in the PPR of EPSCs measured at an interstimulus interval of 40 ms (PP40) (p = 0.012, Wilcoxon signed-rank test; Fig. 1B,C). The magnitude of the inhibition of the EPSC in response to KA application correlated significantly with the change in the PPR, suggesting that the depression of the EPSC involves a decrease in presynaptic release probability (p = 0.034, ANOVA, F = 6.051; Fig. 1E).
Figure 1.
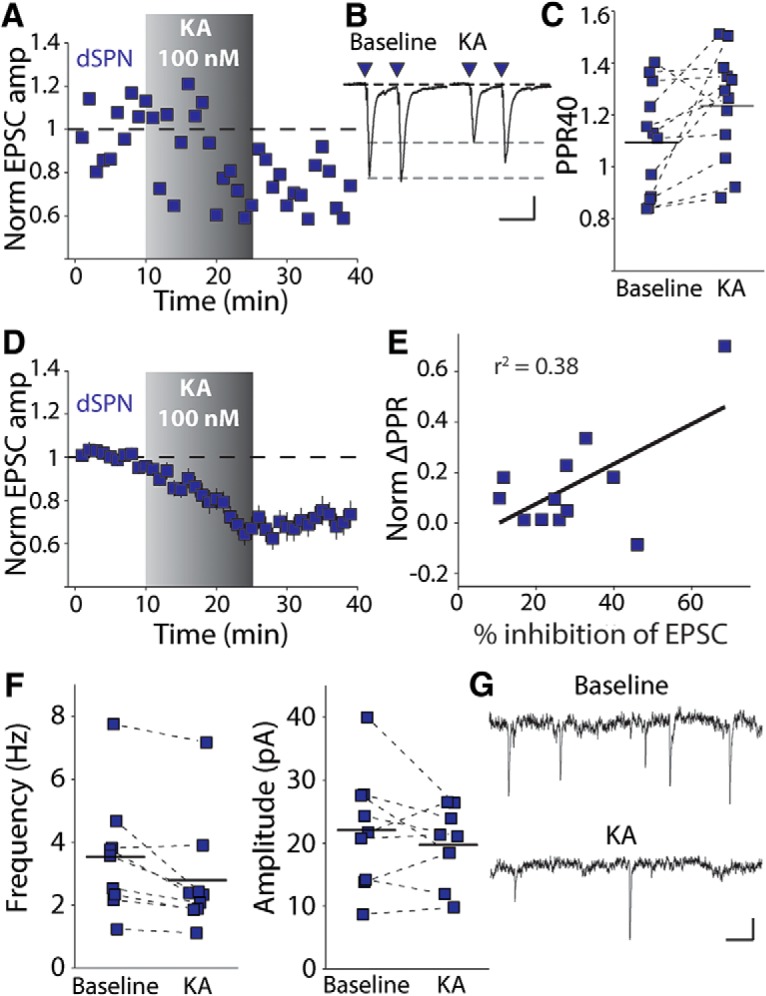
Kainate receptor activation inhibits glutamate release in dSPNs. A, Representative time course of recording from a dSPN identified with the D1tdTomato BAC transgene. A 15 min application of 100 nm KA resulted in a depression of the EPSC amplitude that persisted for the remainder of the recording. B, Representative ESPC traces from the same experiment before and after KA application. There is a decrease in amplitude and an increase in the PPR. Calibration: 50 pA, 40 ms. C, Average PPR for each recording during baseline and 10–15 min after beginning KA application. D, Grouped time course of EPSC depression by KA in dSPNs. E, EPSC inhibition is positively and significantly correlated with the change in the PPR. F, 100 nm KA consistently reduced the frequency, but not amplitude, of miniature EPSCs. G, Representative mEPSCs recorded before and after a 15 min application of 100 nm KA. Calibration: 10 pA, 100 ms.
To further confirm the effect of activation of kainate receptors on release probability at striatal synapses, we recorded mEPSCs before and after a 15 min application of KA. KA consistently decreased the frequency, but not amplitude, of mEPSCs (Hz: p = 0.012, amplitude: p = 0.477, n = 9 cells from 8 animals, Wilcoxon signed-rank test; Fig. 1F,G). Even though mEPSCs in dSPNs reflect contributions from both corticostriatal and thalamostriatal synapses (Ding et al., 2008), corticostriatal synapses are the most prominent excitatory input (Huerta-Ocampo et al., 2014). Therefore, the KA-mediated suppression of mEPSC frequency is consistent with our observations that evoked corticostriatal EPSCs are inhibited by a decrease in release probability after 100 nm KA application.
Presynaptic kainate receptors are known to inhibit glutamate release directly at a number of synapses in the cortex and hippocampus (Chittajallu et al., 1996; Contractor et al., 2000; Jouhanneau et al., 2011); however, in the striatum retrograde eCB signaling is a major mechanism for depression of glutamate release (Gerdeman et al., 2002) and pharmacological activation of kainate receptors has been shown to lead to eCB release in the hippocampus (Lourenço et al., 2011). Therefore, we tested whether the effect of KA might be mediated indirectly through an eCB signaling pathway. Application of the CB1 receptor antagonist AM251 (2 μm) 60 min before and maintained throughout the recording prevented KA-mediated depression of the corticostriatal EPSC (p = 0.240, n = 11 cells from 7 animals, Wilcoxon signed-rank test; Fig. 2A). AM251 also prevented a consistent increase in the PPR (p = 0.206, Wilcoxon signed-rank test; Fig. 2B,C). There was no significant difference between either the baseline EPSC amplitude or PP40 ratio in the DMSO- and AM251-treated groups [EPSC amplitude: 170.9 ± 21.37 (DMSO), 142.3 ± 14.5 (AM251), p = 0.465, Wilcoxon signed-rank test; PP40 ratio: 1.095 ± 0.061 (DMSO), 1.076 ± 0.070 (AM251), p = 0.700, Wilcoxon signed-rank test]. In addition, cells from slices incubated with AM251 did not show a consistent reduction in mEPSC frequency in response to kainate application (p = 0.810, n = 7 cells from 6 animals, Wilcoxon signed-rank test; Fig. 2E,F).
Figure 2.
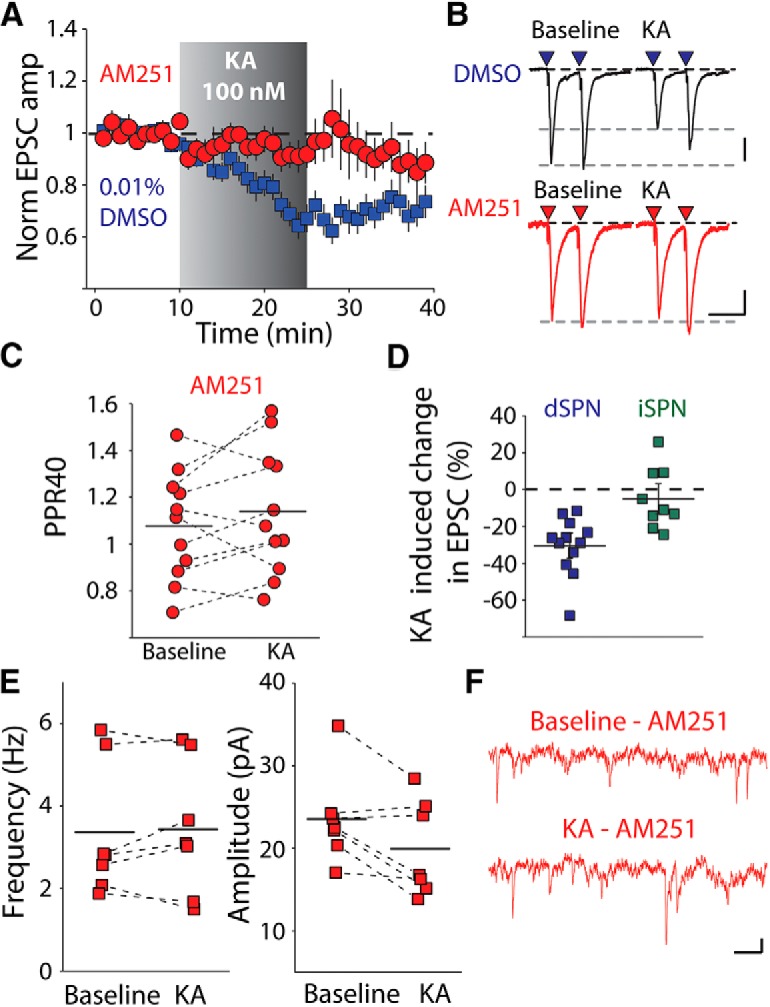
Kainate receptor activation mobilizes eCBs in dSPNs. A, Time course of KA effect on corticostriatal synapses in dSPN in the presence of the CB1 receptor antagonist AM251 (2 μm). B, Representative traces from one experiment of corticostriatal EPSCs recorded in a dSPN in the presence of AM251 before and after KA application. Calibration: 50 pA, 40 ms. C, PPR measurements of dSPN corticostriatal EPSCs at interstimulus intervals of 40 ms before and after KA in the presence of AM251. D, Average KA depression in dSPNs and iSPNs. KA-mediated effect is selective for dSPNs. E, AM251 prevents the KA-mediated depression of mEPSC frequency in dSPNs. F, Representative mEPSC traces in AM251 before and after 100 nm KA application. Calibration: 10 pA,100 ms.
In contrast to our results from dSPNs, in recordings from iSPNs, application of kainate did not significantly depress corticostriatal EPSCs, demonstrating a pathway- and cell-type-specific mechanism (p = 0.359, n = 9 cells, 4 animals, Wilcoxon signed-rank test; Fig. 2D) Together, this demonstrates that activation of kainate receptors by a low concentration of agonist leads to an eCB-dependent presynaptic depression of corticostriatal EPSCs in dSPNs but not iSPNs.
Kainate receptors mobilize 2-AG via a Gα pathway
We next sought to address which eCB species might underlie kainate-mediated EPSC depression in dSPNs. 2-AG is produced exclusively by the enzyme diacylglycerol lipase (DAG-L), which hydrolyzes diacylglycerol into 2-AG (Gao et al., 2010). Incubating slices with the DAG-L inhibitor RHC 80267 (50 μm) starting at least 60 min before and during recording prevented the KA-mediated depression of corticostriatal EPSCs (p = 0.562, n = 7 cells from 3 animals, Wilcoxon signed-rank test; Fig. 3A,B). An alternate DAG-L inhibitor, THL (10 μm), had the same effect when it was coapplied to slices, fully blocking the KA-mediated depression of the EPSC (p = 0.831, n = 11 cells from 10 animals, Wilcoxon signed-rank test; Fig. 3A,B). Therefore, kainate receptor activation leads to the mobilization of 2-AG and CB1 receptor activation, causing depression of EPSCs in striatal dSPNs.
Figure 3.
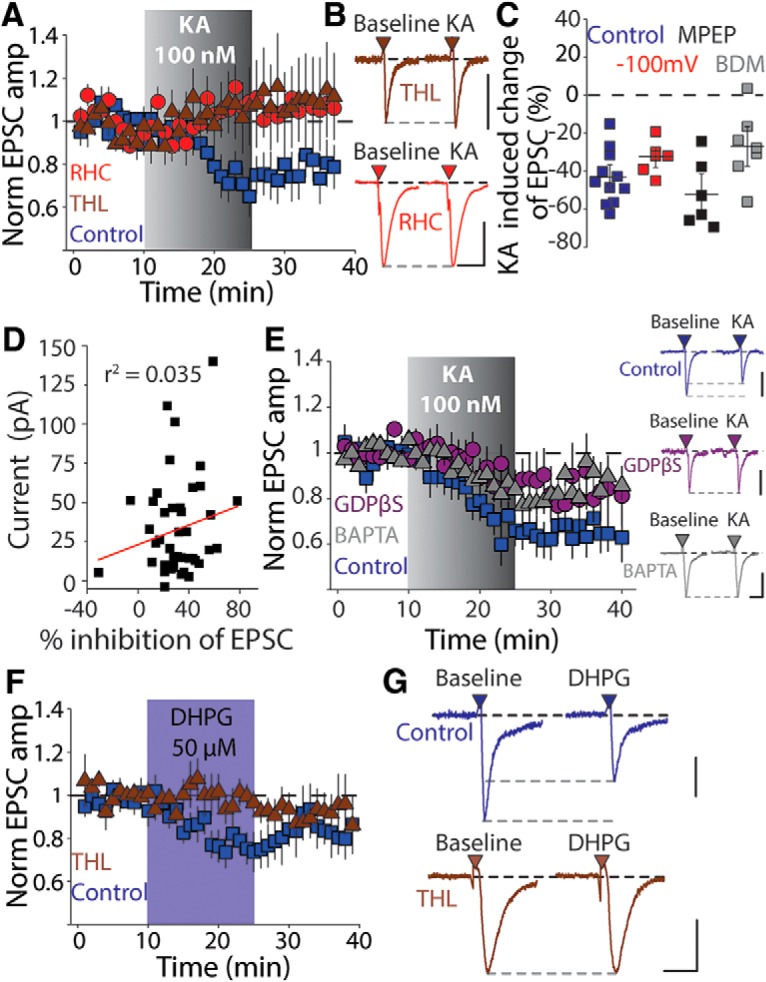
Glutamate receptor activation in dSPNs mobilizes 2-AG. A, Time course of KA-mediated depression in the presence of the DAG-lipase inhibitors RHC (50 μm) or THL (100 μm). B, Representative EPSCs from individual recordings before and after KA application recorded in the presence of THL (top) and RHC (bottom). Calibration: 100 pA, 25 ms. C, Summary plot of KA-mediated depression in experiments in which cells were held at hyperpolarized potentials during KA or the recordings were performed in the presence of the group I mGluR antagonist MPEP (10 μm) or the cell-permeable PKC inhibitor bisindolylmaleimide (BDM, 1 μm). D, Plot of KA-mediated inhibition of corticostriatal EPSCs against the agonist-induced depolarization of dSPNs by 100 nm KA. E, Time course of KA-mediated effects on corticostriatal EPSCs and representative EPSCs in experiments in which the postsynaptic dSPN was dialyzed for 30 min with either GDPβS or BAPTA (17 mm). Calibration: 200 pA, 20 ms (control), 100 pA, 20 ms (GDPβS), 150 pA, 20 ms (BAPTA). F, Time course for experiments in which the group 1 mGluR agonist DHPG (50 μm) was applied in the presence or absence of the DAG-L inhibitor THL (10 μm). Group 1 mGluR activation produced an inhibition of the corticostriatal EPSC amplitude in dSPNs similar to KA. G, Representative EPSCs recorded before and after DHPG application in the absence (top) or presence (bottom) of THL. Calibration: 50 pA (top) and 100 pA (bottom), 20 ms.
DAG-L activation and eCB mobilization are dependent upon signaling pathways that are activated by G-protein-coupled receptor activation, in particular the Gq linked group 1 metabotropic glutamate receptor 5 (mGluR5) in SPNs. Therefore, we tested whether the effect of application of KA might be indirectly mediated through mGluR5. The depression of the EPSC measured 10–15 min after the start of KA application was still intact in the presence of the mGluR5 antagonist MPEP (10 μm) (p = 0.031, n = 6, Wilcoxon signed-rank test; Fig. 3C) In addition, hyperpolarizing the neuron to −100 mV to reduce potential effects from depolarization of the postsynaptic neuron also failed to reduce the KA-mediated depression of the EPSC (p = 0.031, n = 6, Wilcoxon signed-rank test; Fig. 3C). Kainate receptors are known to exert their effects both though ionotropic actions, requiring current flux through an integral channel pore, and through activation of noncanonical signaling pathways in a manner not strictly correlated with current flux through the channel (Rozas et al., 2003). The very low concentration of KA (100 nm) that we used in these experiments produced only a very small inward current (32.7 ± 4.59 pA, n = 45) and there was no correlation between the KA-induced current and the amount of depression of the corticostriatal EPSC (p = 0.221, F = 1.54, ANOVA; Fig. 3D).
To further test whether kainate-mediated EPSC depression involved activation of G-protein signaling, specifically Gα subunits, we included the nonhydrolyzable GDP analog GDPβS (2 mm) in the patch pipette and dialyzed cells for at least 30 min before recording corticostriatal EPSCs. In recordings in which we included GDPβS, KA-mediated depression, although still observed in some recordings, was significantly reduced (p = 0.102, n = 11 cells, 9 animals, Wilcoxon signed-rank test; Fig. 3E), whereas in interleaved recordings in which the cells were dialyzed for the same time with a normal internal solution, the KA-mediated depression was intact (p = 0.002, Wilcoxon signed-rank test, n = 10 cells, 7 animals, Wilcoxon signed-rank test; Fig. 3E). Several prior reports have functionally linked kainate receptor effects to G-protein signaling, including direct effects on transmitter release (Rodríguez-Moreno and Lerma, 1998; Frerking et al., 2001) and postsynaptic effects of kainate receptor activation on ion channels (Melyan et al., 2002; Fernandes et al., 2009). In addition, coimmunoprecipitation and proteomic analysis have detected interactions between kainate receptor subunits GluK1 and GluK5 and Gαo and Gαq G-proteins (Ruiz et al., 2005; Rutkowska-Wlodarczyk et al., 2015). Activation of Gα subunits are known to stimulate eCB synthesis (Olmo et al., 2016), therefore, the reduction in EPSC inhibition seen here with postsynaptic loading of GDPβS is consistent with kainate receptor stimulation of a G-protein pathway to mobilize 2-AG.
The DAG-L enzyme can be activated in a Ca2+-dependent manner to mobilize 2-AG (Kondo et al., 1998; Shonesy et al., 2015). In recordings in which we included the kinetically fast calcium chelator BAPTA (17 mm) in the patch pipette, KA-mediated depression was significantly reduced (p = 0.006, n = 8 cells, 4 animals, Wilcoxon rank-sum test; Fig. 3E) compared with dialysis with control internal solutions containing EGTA. We also tested whether the KA-mediated depression was dependent on the protein kinase C (PKC) pathway, which has been implicated in kainate receptor-dependent inhibition of the slow afterhyperpolarization potential (Fisahn et al., 2005). Incubation of slices with the PKC inhibitor bisindolymaleimide (0.1 μm) failed to prevent the KA-mediated EPSC depression (p = 0.031, n = 7, Wilcoxon signed-rank test; Fig. 3C). Together, our results demonstrate that application of a low concentration of KA mobilizes the eCB 2-AG from dSPNs via a G-protein and Ca2+-dependent activation of the enzyme DAG-L.
Because we had found that kainate receptor activation mobilizes 2-AG, we wanted to determine whether group 1 mGluR activation linked to the same signaling pathway. In the striatum, mGluR activation is required for high-frequency eCB-dependent long-term depression (eCB-LTD) in iSPNs (Kreitzer and Malenka, 2005) and, in dSPNs in the dorsolateral striatum, a spike-timing-dependent mGluR-mediated eCB-LTD has also been described at corticostriatal synapses (Shen et al., 2008). However, direct activation of group 1 mGluRs by the agonist dihydroxyphenylglycine (DHPG) was demonstrated to cause only a small transient depression of EPSCs in dSPNs (Kreitzer and Malenka, 2007). We found that the mGluR agonist DHPG (50 μm) depressed the corticostriatal EPSC amplitude in dSPNs (p = 0.008, n = 7 cells, 5 animals, Wilcoxon signed-rank test; Fig. 3F,G). There was partial washout of the DHPG-mediated depression (the amplitudes were not different from baseline at 10–15 min after washout, p = 0.156, Wilcoxon signed-rank test) unlike the effect of KA, which persisted after washout of the drug. The effect of DHPG was blocked by preapplication of the DAG-L inhibitor THL (p = 0.844, n = 7 cells, 4 animals, Wilcoxon signed-rank test; Fig. 3F,G), demonstrating that group 1 mGluR activation can mobilize 2-AG in dSPNs, as does the activation of kainate receptors.
Postsynaptic GluK2 containing kainate receptors mobilize 2-AG in dSPNs
The GluK2 subunit is required for functional kainate receptor expression in the mature striatum (Chergui et al., 2000), although the exact kainate receptor stoichiometry in dSPNs and iSPNs is unknown. Kainate receptors are heterotetramers formed from combinations of the GluK1–5 subunits and likely exist in complexes requiring the low-affinity GluK1–3 subunits combined with at least one of the high-affinity GluK4 and 5 subunits (Fernandes et al., 2009). To determine whether postsynaptic GluK2-containing receptors in dSPN mediate the mobilization of eCBs, we generated conditional knock-out mice that lack expression of the gene Grik2, encoding the kainate receptor subunit GluK2 (Grik2f/f mice), selectively in dSPNs (D1-Cre;Grik2f/f) (Gong et al., 2007). Further intercrossing mice expressing the Ai9 allele (Madisen et al., 2010) allowed for identification of dSPNs (D1-Cre;Grik2f/f;Ai9). The current mediated by 10 μm KA was eliminated in dSPNs in these mice but present in neighboring iSPNs from the same slices (Fig. 4A,B). In addition, we found that D1-Cre;Grik2f/f;Ai9 mice lacked significant KA-mediated EPSC depression in response to 100 nm KA and also lacked a consistent increase in the PPR, confirming that postsynaptic GluK2-containing kainate receptors are required for kainate-mediated presynaptic depression of corticostriatal synapses in dSPNs (EPSC amplitude: p = 0.110 n = 12 cells, 9 animals; PPR: p = 0.424 Wilcoxon signed-rank test; Fig. 4C,D). In these experiments, there was a small, but not statistically significant, depression of the EPSC amplitude seen in some recordings from the D1-Cre;Grik2f/f mice. However, unlike in control experiments, this was not long lasting, leading us to conclude that GluK2-containing kainate receptors on dSPNs are largely responsible for the depression of corticostriatal EPSCs. We also observed a higher baseline PPR in the D1-Cre;Grik2f/f mice, which could indicate adaptive changes at the corticostriatal synapse to compensate for the loss of GluK2-containing receptors in dSPNs [p = 0.010, n = 12 cells, 9 animals (D1-Cre;Grik2f/f), n = 9 cells, 5 animals (D1-Cre;Grik2 w/w mice), Wilcoxon rank-sum test].
Figure 4.
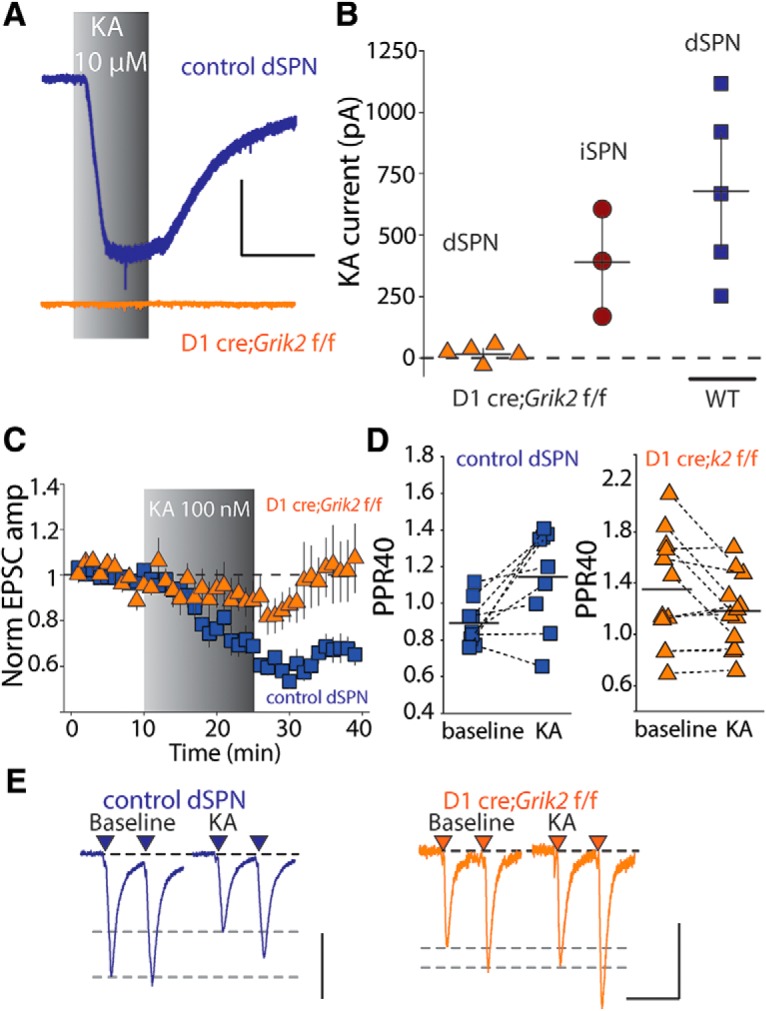
GluK2-containing receptors are required for kainate-mediated eCB depression. A, Example whole-cell current responses to saturating KA (10 μm) application of dSPNs from control mice (top) and from mice in which GluK2 is specifically ablated in dSPNs (D1cre;Grik2 f/f) (bottom). Calibration: 200 pA, 300 s. B, Summary of whole-cell current amplitudes in dSPNs and iSPNs from D1cre; Grik2 f/f mice and dSPNs from control mice. C, KA-mediated effects on corticostriatal EPSCs in dSPNs in control and conditional GluK2 KO mice. No significant kainate-mediated depression is seen in slices from dSPNs in conditional GluK2 KO mice (D1-Cre;Grik2 w/w mice; Ai9: p = 0.008, n = 9 cells from 5 animals, Wilcoxon signed-rank test). D, KA increases the PPR in control dSPNs (p = 0.027, Wilcoxon signed-rank test), but not dSPNs from D1-Cre;Grik2f/f mice. E, Representative traces from individual experiments showing EPSCs before and after KA application from dSPNs in control mice and conditional GluK2 KO mice. Calibration: 150 pA (left) and 100 pA (right), 50 ms.
Synaptic activation of kainate receptors inhibits corticostriatal synapses in dSPNs
To determine whether dSPN kainate receptors could mobilize eCBs in response to synaptically released endogenous glutamate, we stimulated cortical afferents with trains at 25 Hz for 1 s (Fig. 5A). When these trains were repeated with a 20 s intertrain interval, the initial EPSC of the final train (E5) was significantly depressed compared with the average EPSC during the baseline period (p = 0.017, n = 18 cells, 10 animals, Wilcoxon signed-rank test; Fig. 5B,C). Although a reduction in EPSC amplitude due to vesicle depletion occurs within the 1 s trains, the depression that accrued over the course of multiple trains from the first to the fifth train was due to eCB mobilization because it was completely eliminated by the CB1R antagonist AM251. In fact, recordings in AM251 showed a slight potentiation of E5 (p = 0.016, n = 8 cells, 5 animals, Wilcoxon signed-rank test; Fig. 5B,C). The E5:E1 ratio was <1 in the control group (dSPN), but not in the AM251 group, demonstrating that the depression of the EPSC was eliminated by CB1R antagonism (dSPN: p = 0.001, AM251: p = 0.844, Wilcoxon signed-rank test; Fig. 5D). This train-mediated effect was a short-term depression and, after cessation of the train, the EPSC amplitude returned to the baseline value within 1 min (p = 0.306, Wilcoxon signed-rank test). To further establish whether this short-term synaptic depression occurred through activation of postsynaptic kainate receptors or mGluRs, we first recorded 25 Hz trains in D1-Cre;Grik2f/f;Ai9 conditional GluK2 knock-out mice. In these recordings, the trains failed to cause depression of E5 (p = 0.946, n = 13 cells, 4 animals, Wilcoxon signed-rank test; Fig. 5E). This is consistent with kainate receptors being preferentially activated by these synaptic trains; however, it is possible that mGluRs localized at these synapses could also play a role in eCB mobilization. We therefore performed experiments in the presence of the group 1 antagonist MPEP. We observed a depression of the E5 EPSC in recordings from WT mice (E5: p = 0.001, n = 14 cells, 7 animals, Wilcoxon signed-rank test; E5:E1 ratio: p = 0.004, Wilcoxon signed-rank test; Fig. 6A,B), similar to recordings without MPEP. We also found that, in recordings from D1-Cre;Grik2f/f mice in which we included MPEP, there was no depression of the EPSC E5, as had been observed previously (E5: p = 0.890, n = 16 cells, 6 animals, Wilcoxon signed-rank test; E5:E1 ratio: p = 0.109, Wilcoxon signed-rank test; Fig. 6A,B). Therefore, in dSPNs, repetitive 25 Hz trains of corticostriatal synaptic stimulation engage kainate-receptor-mediated eCB mobilization, causing short-term synaptic depression, whereas group 1 mGluRs, which also have the potential to link to the same signaling pathways, do not participate in synaptic depression during repetitive 25 Hz trains.
Figure 5.
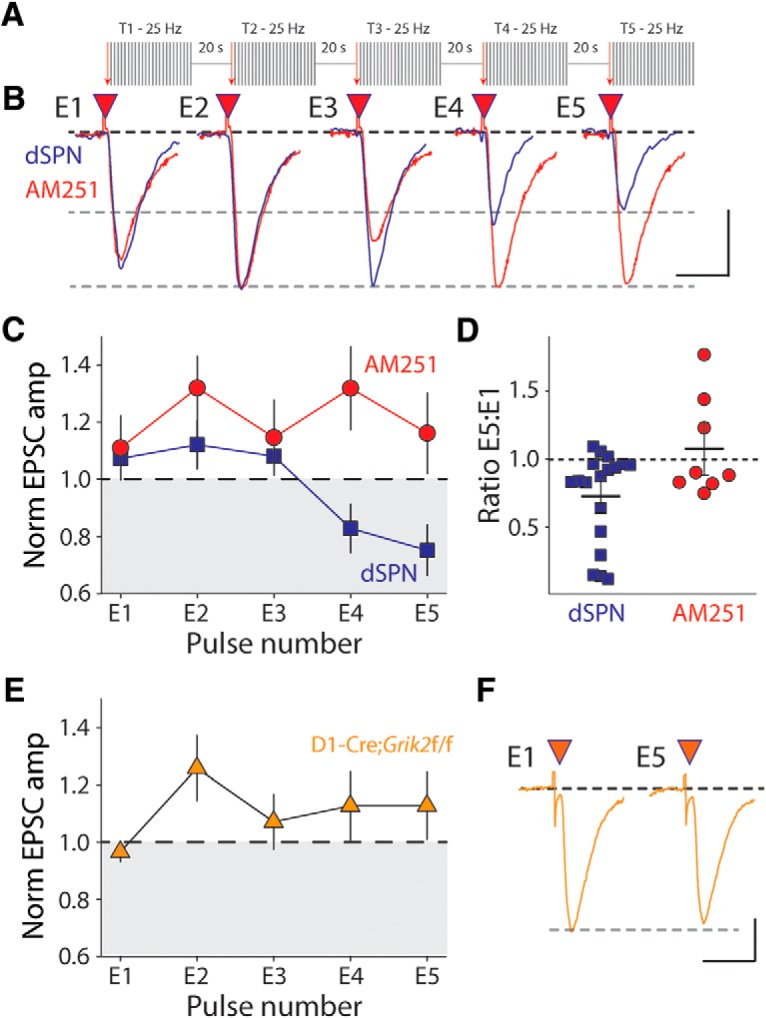
Synaptic activation of kainate receptors causes an eCB-mediated short-term depression of corticostriatal EPSCs during presynaptic trains of activity. A, Schematic of train stimulation protocol. After 5 min of baseline recording, five trains (T1–T5) of synaptic stimulation are presented. Each train consists of 25 stimuli at 25 Hz and is separated by 20 ms. The amplitude of the first EPSC in each of the trains (E1–E5) is a measure of the accrued depression. B, Representative EPSC traces E1–E5 for individual experiments recorded from dSPN in control solution or in the presence of AM251 (2 μm). Calibration: 50 pA (control) and 100 pA (AM251), 25 ms. C, Grouped data of E1–E5 amplitudes during train stimulation for recordings from dSPNs in control and slices incubated with AM251. D, Ratio of the first EPSC from the fifth train (E5) to the first EPSC from the first train (E1). E, E1–E5 amplitudes during trains recorded from dSPNs in D1cre;Grik2 f/f mice. F, Representative EPSCs from a single experiment of the first and fifth train in D1cre;Grik2f/f mice. Calibration: 50 pA, 25 ms.
Figure 6.
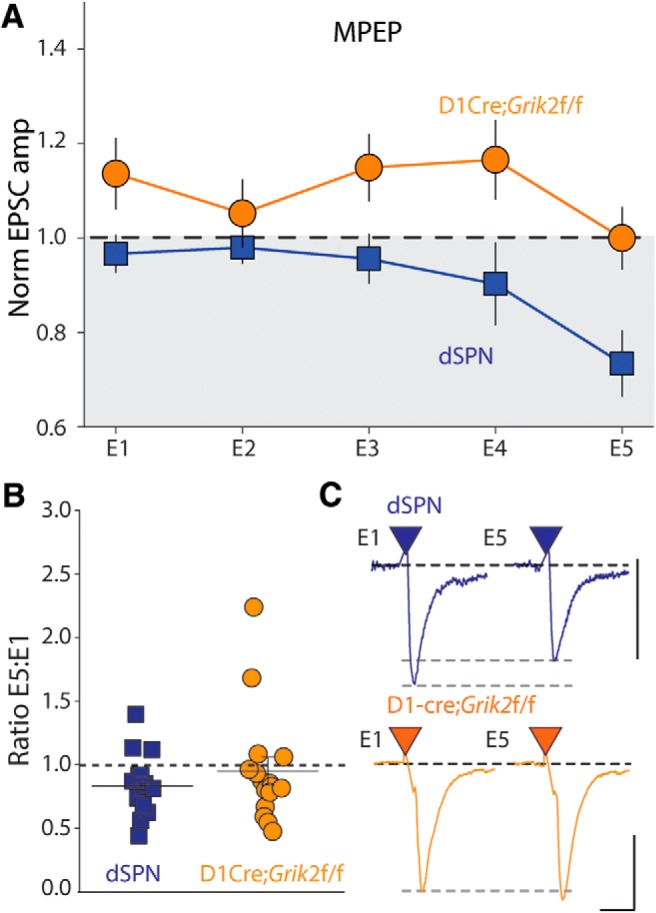
Synaptic 25 Hz trains do not engage group I mGluRs in dSPNs. A, Grouped data of normalized amplitudes of EPSCs E1–E5 during 25 Hz trains in the presence of MPEP in dSPNs from WT and D1cre;Grik2 f/f mice. B, E5/E1 ratio in dSPNs from control mice and D1cre;Grik2 f/f mice. C, Representative E1 and E5 traces from individual experiments in control and conditional GluK2 KO mice in the presence of MPEP. Calibration: 200 pA (top), 400 pA (bottom), 10 ms.
Discussion
Kainate receptors mobilize eCBs at striatal dSPN synapses
Kainate receptor subunits are expressed throughout the brain and have a number of diverse roles in regulating synapses and neuronal excitability (Contractor et al., 2011). Several mutations and allelic variations in the GRIK genes have been associated with neuropsychiatric and neurological disorders, including intellectual disability (Motazacker et al., 2007; Guzmán et al., 2017), bipolar disorder (Pickard et al., 2008), and OCD (Delorme et al., 2004). Alterations in striatal function have been well studied in animal models of OCD, strongly implicating alterations in this brain region in OCD pathology (Burguière et al., 2015; Monteiro and Feng, 2016). We recently demonstrated that mice lacking all five subunits of the kainate receptor have pronounced striatal-dependent phenotypes, including perseverative behaviors, as well as physiological and morphological disruptions of SPNs (Xu et al., 2017). However, it remained unclear whether kainate receptors expressed in mature SPNs regulated the striatal circuit and, if so, if specific receptor subunits were required for ongoing kainate receptor activity.
In the current work, we tested the consequence of acute activation of kainate receptors on corticostriatal synaptic transmission. Consistent with what has been seen at synapses in the hippocampus, activation of kainate receptors with a low concentration of agonist depressed the EPSC amplitude (Contractor et al., 2000). The reduction in EPSC amplitude involved a decrease in presynaptic glutamate release indicated by a concomitant increase in the PPR. In addition, 100 nm KA led to a consistent reduction in mEPSC frequency but not amplitude in dSPNs. However, unlike at other synapses, where presynaptic kainate receptors inhibit transmitter release directly (Kullmann, 2001), we found that dSPN kainate receptors mediated this depression through release of eCBs. The KA-mediated depression was prevented by antagonizing the presynaptic CB1 receptor and also by inhibitors of DAG-L, demonstrating that 2-AG synthesis was necessary. This occurred selectively in dSPNs and not iSPNs, demonstrating functional heterogeneity and potentially reflecting molecular diversity between the two SPN types.
Kainate receptor eCB release involves postsynaptic receptors and activation of a G-protein signaling pathway
Prior work has demonstrated that activation of GluK2 containing kainate receptors can mobilize anandamide (AEA) and modulate GABA release at synapses onto CA1 neurons in the hippocampus (Lourenço et al., 2011). However, those studies used relatively high concentrations of KA that depolarized the cell and there was a clear correlation between the amount of KA-induced inward current and the depression of the EPSC, suggesting that depolarization was necessary for the eCB release. Previous work in SPNs has shown that eCBs are released in response to depolarization and Ca2+ influx through voltage gated Ca2+ channels (Kreitzer and Regehr, 2001; Adermark and Lovinger, 2007). Therefore, the extent to which substantial depolarization is required for KAR-eCB mobilization was unclear (Lourenço et al., 2011).
In this study, we found that a low concentration of KA that does not cause large changes in the holding current was sufficient to mobilize 2-AG. The magnitude of EPSC depression mediated by KA was not significantly correlated with alterations in the membrane current. In addition, the KA-mediated depression was reduced after dialysis of the postsynaptic cell with BAPTA to chelate calcium and by including GDPβS in the patch pipette to inhibit postsynaptic G-protein signaling. These data demonstrate that kainate-receptor-mediated eCB release in dSPNs depends on kainate receptor activation of metabotropic signaling pathways.
This noncanonical signaling by a nominally ionotropic ligand gated receptor adds to a growing list of effects linking kainate receptors and other ligand gated ion channels to activation of G-protein signaling pathways (Valbuena and Lerma, 2016). For kainate receptors, these diverse effects range from G-protein-mediated mechanism of direct regulation of transmitter release in the hippocampus (Lauri et al., 2006), modulation of ion channels that control excitability in neurons of the dorsal root ganglion (DRG) (Rozas et al., 2003) and in hippocampal pyramidal neurons (Melyan et al., 2002), and regulation of filopodial motility in the hippocampus (Tashiro et al., 2003), as well as neurite outgrowth in DRGs (Marques et al., 2013). The structural basis of kainate receptor activation of G-protein pathways remains unclear. Biochemical studies have demonstrated association through coimmunoprecipitation of Gαq proteins with GluK2/K5 receptor complexes (Ruiz et al., 2005) and through proteomic analysis of Gαo proteins and GluK1 (Rutkowska-Wlodarczyk et al., 2015). There is also evidence that the kainate receptor auxiliary subunit NETO1 is required for coupling of kainate receptors to metabotropic signaling, although, again, the structural basis for this interaction is still lacking (Wyeth et al., 2014).
We also found that mice lacking GluK2 receptors in dSPNs (D1-Cre:Grik2f/f mice) do not show a significant long-lasting reduction in EPSC amplitude in response to 100 nm KA. In recordings from D1-Cre:Grik2f/f mice, we did see a small, reversible depression of the EPSC amplitude with KA application. This could reflect activation of kainate receptors on other cell populations that could also contribute to corticostriatal EPSC depression; however, in this case, there was no change in the PPR, arguing against a presynaptic mechanism.
Metabotropic glutamate receptors mobilize 2-AG in dSPNs
eCBs are important for regulating corticostriatal synaptic strength (Gerdeman et al., 2002). Neuromodulatory receptors such as dopamine receptors, muscarinic acetylcholine receptors, and adenosine A2A receptors interact with the eCB system in complex ways depending on the SPN cell type (Calabresi et al., 1997; Kreitzer and Malenka, 2005; Orrú et al., 2011; Shen et al., 2015). However, mobilization of eCBs has been thought to be primarily mediated by activation of group 1 mGluRs (Lüscher and Huber, 2010). dSPNs and iSPNs are functionally and molecularly heterogenous. In iSPNs, high-frequency stimuli and low-frequency stimuli engage eCB-LTD with different mechanisms, both requiring mGluRs, but one mobilizing 2-AG and the other AEA (Lerner and Kreitzer, 2012). In contrast, much less is known about the pathways that lead to eCB mobilization in dSPNs or which eCB species can be mobilized in these neurons. We found that kainate-receptor-mediated depression of corticostriatal EPSCs was only observed in dSPNs, prompting us to focus on this cell type and determine how glutamate receptors, both mGluRs and kainate receptors, couple to the same or different eCB signaling pathways. As with kainate, application of the mGluR antagonist DHPG led to a reduction of EPSC amplitude at corticostriatal synapses that was blocked by a DAG-L inhibitor. Therefore, in dSPNs, 2-AG is mobilized by activation of either kainate or metabotropic glutamate receptors.
Kainate receptors regulate short-term plasticity at corticostriatal dSPN synapses
Previous studies have demonstrated that high-frequency (100 Hz) stimulation and mGluR-dependent eCB release is crucial for the induction of LTD at striatal synapses, which may help to ensure balanced basal ganglia output in response to different patterns of cortical afferent activity (Gremel et al., 2016). We found that synaptic GluK2-containing kainate receptors in dSPNs are engaged by trains of 25 Hz activity and regulate synaptic transmission via releasing eCBs. In vivo recordings in anesthetized rats have demonstrated that corticostriatal projection neurons fire during periods of membrane depolarization known as “UP” states that last from a few hundred milliseconds to approximately 1 s (Stern et al., 1997). Within “UP” states, periodigrams of the instantaneous firing frequency indicate a mean value in the range of 25 Hz. Therefore, these trains, which activate kainate receptor short-term plasticity, fall within the range of physiological afferent stimuli dSPNs receive in vivo. Although these specific experiments used a 1 s 25 Hz train repeated at 0.05 Hz, the complete range of stimulation frequencies and patterns that activate kainate receptors at this synapse are not known. In addition, the intervals at which UP states occur in vivo in striatal projecting neurons are not fully characterized and likely depend on the behavioral state of the animal (Wilson, 1993). In our experiments, we selected a 20 s interstimulus interval to allow for vesicle recovery after train stimulation (Stevens and Tsujimoto, 1995). Despite the remaining questions, short-term eCB-dependent inhibition of glutamate release by kainate receptor activation represents a novel mechanism for regulating excitatory dSPN synapses in response to cortical input. This regulation of glutamate release could in turn affect the probability of action potential generation in SPNs in response to certain patterns of cortical input, ultimately influencing basal ganglia output.
Modification of behavioral sequences through a type of reinforcement learning is thought to be key to understanding the functional role of the cortical–basal ganglia loop (Graybiel and Grafton, 2015). How cortical information is transmitted into the basal ganglia circuit has implications for how this neural circuit promotes effective and discourages ineffective behavioral sequences (Desrochers et al., 2015; Nakamura et al., 2017). In particular, a selective knock-out of the CB1 receptor from orbitofrontal cortex–dorsal striatum projections prevents the normal emergence of habit-like behavior in an operant task (Gremel et al., 2016). Our results demonstrate that synaptic GluK2-containing kainate receptors in dSPNs couple to downstream eCB signaling pathways to regulate the strength of corticostriatal synapses. These findings add to growing evidence, both from genetic studies associating variants in the GRIK2 gene to disorders involving striatal pathology and behavioral studies in knock-out mice, that kainate receptors in the striatum play an important role in regulating basal ganglia circuits.
Footnotes
This work was funded by the National Institute of Mental Health–National Institutes of Health (Grant R01MH099114 to A.C., Fellowship 1F31MH099807 to J.J.M., and Grant K01MH094464 to J.X.). We thank Prof. Geoffrey Swanson for comments on the manuscript.
The authors declare no competing financial interests.
References
- Adermark L, Lovinger DM (2007) Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci 27:6781–6787. 10.1523/JNEUROSCI.0280-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL (1986) Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 9:357–381. 10.1146/annurev.ne.09.030186.002041 [DOI] [PubMed] [Google Scholar]
- Burguière E, Monteiro P, Mallet L, Feng G, Graybiel AM (2015) Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr Opin Neurobiol 30:59–65. 10.1016/j.conb.2014.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E (1997) Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci 17:4536–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA (1997) Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388:182–186. 10.1038/40645 [DOI] [PubMed] [Google Scholar]
- Chergui K, Bouron A, Normand E, Mulle C (2000) Functional GluR6 kainate receptors in the striatum: indirect downregulation of synaptic transmission. J Neurosci 20:2175–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM (1996) Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 379:78–81. 10.1038/379078a0 [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson GT, Sailer A, O'Gorman S, Heinemann SF (2000) Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J Neurosci 20:8269–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF (2001) Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 29:209–216. 10.1016/S0896-6273(01)00191-X [DOI] [PubMed] [Google Scholar]
- Contractor A, Mulle C, Swanson GT (2011) Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci 34:154–163. 10.1016/j.tins.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme R, Krebs MO, Chabane N, Roy I, Millet B, Mouren-Simeoni MC, Maier W, Bourgeron T, Leboyer M (2004) Frequency and transmission of glutamate receptors GRIK2 and GRIK3 polymorphisms in patients with obsessive compulsive disorder. Neuroreport 15:699–702. 10.1097/00001756-200403220-00025 [DOI] [PubMed] [Google Scholar]
- Desrochers TM, Amemori K, Graybiel AM (2015) Habit learning by naive macaques is marked by response sharpening of striatal neurons representing the cost and outcome of acquired action sequences. Neuron 87:853–868. 10.1016/j.neuron.2015.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Peterson JD, Surmeier DJ (2008) Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci 28:6483–6492. 10.1523/JNEUROSCI.0435-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R. (1991) New wave of non-NMDA excitatory amino acid receptors. Trends Pharmacol Sci 12:360–362. 10.1016/0165-6147(91)90602-O [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Catches JS, Petralia RS, Copits BA, Xu J, Russell TA, Swanson GT, Contractor A (2009) High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 63:818–829. 10.1016/j.neuron.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Heinemann SF, McBain CJ (2005) The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J Physiol 562:199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA (2001) Kainate receptors depress excitatory synaptic transmission at CA3->CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci 21:2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017–2024. 10.1523/JNEUROSCI.5693-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. 10.1038/nn832 [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR (2007) Targeting cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27:9817–9823. 10.1523/JNEUROSCI.2707-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Grafton ST (2015) The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol 7:a021691. 10.1101/cshperspect.a021691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Chancey JH, Atwood BK, Luo G, Neve R, Ramakrishnan C, Deisseroth K, Lovinger DM, Costa RM (2016) Endocannabinoid modulation of orbitostriatal circuits gates habit formation. Neuron 90:1312–1324. 10.1016/j.neuron.2016.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán SJ, Schlögl A, Schmidt-Hieber C (2014) Stimfit: quantifying electrophysiological data with python. Front Neuroinform 8:16. 10.3389/fninf.2014.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán YF, Ramsey K, Stolz JR, Craig DW, Huentelman MJ, Narayanan V, Swanson GT (2017) A gain-of-function mutation in the GRIK2 gene causes neurodevelopmental deficits. Neurol Genet 3:e129. 10.1212/NXG.0000000000000129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Ocampo I, Mena-Segovia J, Bolam JP (2014) Convergence of cortical and thalamic input to direct and indirect pathway medium spiny neurons in the striatum. Brain Struct Funct 219:1787–1800. 10.1007/s00429-013-0601-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouhanneau JS, Ball SM, Molnár E, Isaac JT (2011) Mechanisms of bi-directional modulation of thalamocortical transmission in barrel cortex by presynaptic kainate receptors. Neuropharmacology 60:832–841. 10.1016/j.neuropharm.2010.12.023 [DOI] [PubMed] [Google Scholar]
- Kondo S, Kondo H, Nakane S, Kodaka T, Tokumura A, Waku K, Sugiura T (1998) 2-arachidonoylglycerol, an endogenous cannabinoid receptor agonist: identification as one of the major species of monoacylglycerols in various rat tissues, and evidence for its generation through Ca2+-dependent and -independent mechanisms. FEBS Lett 429:152–156. 10.1016/S0014-5793(98)00581-X [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2005) Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci 25:10537–10545. 10.1523/JNEUROSCI.2959-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2007) Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 445:643–647. 10.1038/nature05506 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2008) Striatal plasticity and basal ganglia circuit function. Neuron 60:543–554. 10.1016/j.neuron.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21:RC174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. (2001) Presynaptic kainate receptors in the hippocampus: slowly emerging from obscurity. Neuron 32:561–564. 10.1016/S0896-6273(01)00507-4 [DOI] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T (2006) Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate-receptor-mediated inhibition of glutamate release. Neuron 50:415–429. 10.1016/j.neuron.2006.03.020 [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC (2012) RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73:347–359. 10.1016/j.neuron.2011.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço J, Matias I, Marsicano G, Mulle C (2011) Pharmacological activation of kainate receptors drives endocannabinoid mobilization. J Neurosci 31:3243–3248. 10.1523/JNEUROSCI.3512-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. (2007) Endocannabinoid liberation from neurons in transsynaptic signaling. J Mol Neurosci 33:87–93. 10.1007/s12031-007-0043-2 [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65:445–459. 10.1016/j.neuron.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JM, Rodrigues RJ, Valbuena S, Rozas JL, Selak S, Marin P, Aller MI, Lerma J (2013) CRMP2 tethers kainate receptor activity to cytoskeleton dynamics during neuronal maturation. J Neurosci 33:18298–18310. 10.1523/JNEUROSCI.3136-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, Geller DA, Murphy DL, Knowles JA, Grados MA, Riddle MA, Rasmussen SA, McLaughlin NC, Nurmi EL, Askland KD, Qin HD, Cullen BA, Piacentini J, Pauls DL, Bienvenu OJ, Stewart SE, et al. (2015) Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry 20:337–344. 10.1038/mp.2014.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B (2002) Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34:107–114. 10.1016/S0896-6273(02)00624-4 [DOI] [PubMed] [Google Scholar]
- Monteiro P, Feng G (2016) Learning from animal models of obsessive-compulsive disorder. Biol Psychiatry 79:7–16. 10.1016/j.biopsych.2015.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motazacker MM, Rost BR, Hucho T, Garshasbi M, Kahrizi K, Ullmann R, Abedini SS, Nieh SE, Amini SH, Goswami C, Tzschach A, Jensen LR, Schmitz D, Ropers HH, Najmabadi H, Kuss AW (2007) A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Human Genet 81:792–798. 10.1086/521275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392:601–605. 10.1038/33408 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nagata M, Yagi T, Graybiel AM, Yamamori T, Kitsukawa T (2017) Learning new sequential stepping patterns requires striatal plasticity during the earliest phase of acquisition. Eur J Neurosci 45:901–911. 10.1111/ejn.13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmo IG, Ferreira-Vieira TH, Ribeiro FM (2016) Dissecting the signaling pathways involved in the crosstalk between metabotropic glutamate 5 and cannabinoid type 1 receptors. Mol Pharmacol 90:609–619. 10.1124/mol.116.104372 [DOI] [PubMed] [Google Scholar]
- Orrú M, Quiroz C, Guitart X, Ferré S (2011) Pharmacological evidence for different populations of postsynaptic adenosine A2A receptors in the rat striatum. Neuropharmacology 61:967–974. 10.1016/j.neuropharm.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls DL, Abramovitch A, Rauch SL, Geller DA (2014) Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci 15:410–424. 10.1038/nrn3746 [DOI] [PubMed] [Google Scholar]
- Pickard BS, Knight HM, Hamilton RS, Soares DC, Walker R, Boyd JK, Machell J, Maclean A, McGhee KA, Condie A, Porteous DJ, St Clair D, Davis I, Blackwood DH, Muir WJ (2008) A common variant in the 3′UTR of the GRIK4 glutamate receptor gene affects transcript abundance and protects against bipolar disorder. Proc Natl Acad Sci U S A 105:14940–14945. 10.1073/pnas.0800643105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Lerma J (1998) Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 20:1211–1218. 10.1016/S0896-6273(00)80501-2 [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J (2003) Noncanonical signaling by ionotropic kainate receptors. Neuron 39:543–553. 10.1016/S0896-6273(03)00436-7 [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C (2005) Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci 25:11710–11718. 10.1523/JNEUROSCI.4041-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowska-Wlodarczyk I, Aller MI, Valbuena S, Bologna JC, Prézeau L, Lerma J (2015) A proteomic analysis reveals the interaction of GluK1 ionotropic kainate receptor subunits with go proteins. J Neurosci 35:5171–5179. 10.1523/JNEUROSCI.5059-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio AS, Fagerness J, Crane J, Leboyer M, Delorme R, Pauls DL, Stewart SE (2011) Association between polymorphisms in GRIK2 gene and obsessive-compulsive disorder: a family-based study. CNS Neurosci Ther 17:141–147. 10.1111/j.1755-5949.2009.00130.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA (2001) Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291:1972–1976. 10.1126/science.1057105 [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Maeng S, Malkesman O, Pearson B, Schloesser RJ, Tragon T, Rogawski M, Gasior M, Luckenbaugh D, Chen G, Manji HK (2008) Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry 13:858–872. 10.1038/mp.2008.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ (2008) Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321:848–851. 10.1126/science.1160575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Plotkin JL, Francardo V, Ko WK, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, Conn PJ, Greengard P, Bezard E, Cenci MA, Surmeier DJ (2015) M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron 88:762–773. 10.1016/j.neuron.2015.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Winder DG, Patel S, Colbran RJ (2015) The initiation of synaptic 2-AG mobilization requires both an increased supply of diacylglycerol precursor and increased postsynaptic calcium. Neuropharmacology 91:57–62. 10.1016/j.neuropharm.2014.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, Calakos N (2008) Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci 28:2681–2685. 10.1523/JNEUROSCI.5492-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Kincaid AE, Wilson CJ (1997) Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol 77:1697–1715. 10.1152/jn.1997.77.4.1697 [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T (1995) Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci U S A 92:846–849. 10.1073/pnas.92.3.846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA, Arnold PD, Evans PD, Gamazon ER, Davis LK, Osiecki L, McGrath L, Haddad S, Crane J, Hezel D, Illman C, Mayerfeld C, Konkashbaev A, Liu C, Pluzhnikov A, et al. (2013) Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry 18:788–798. 10.1038/mp.2012.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Graves SM, Shen W (2014) Dopaminergic modulation of striatal networks in health and Parkinson's disease. Curr Opin Neurobiol 29:109–117. 10.1016/j.conb.2014.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Dunaevsky A, Blazeski R, Mason CA, Yuste R (2003) Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron 38:773–784. 10.1016/S0896-6273(03)00299-X [DOI] [PubMed] [Google Scholar]
- Valbuena S, Lerma J (2016) Non-canonical signaling, the hidden life of ligand-gated ion channels. Neuron 92:316–329. 10.1016/j.neuron.2016.10.016 [DOI] [PubMed] [Google Scholar]
- Wilson CJ. (1993) The generation of natural firing patterns in neostriatal neurons. Prog Brain Res 99:277–297. 10.1016/S0079-6123(08)61352-7 [DOI] [PubMed] [Google Scholar]
- Wyeth MS, Pelkey KA, Petralia RS, Salter MW, McInnes RR, McBain CJ (2014) Neto auxiliary protein interactions regulate kainate and NMDA receptor subunit localization at mossy fiber-CA3 pyramidal cell synapses. J Neurosci 34:622–628. 10.1523/JNEUROSCI.3098-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, Heinemann SF (2009) mGluR5 has a critical role in inhibitory learning. J Neurosci 29:3676–3684. 10.1523/JNEUROSCI.5716-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Marshall JJ, Fernandes HB, Nomura T, Copits BA, Procissi D, Mori S, Wang L, Zhu Y, Swanson GT, Contractor A (2017) Complete disruption of the kainate receptor gene family results in corticostriatal dysfunction in mice. Cell Rep 18:1848–1857. 10.1016/j.celrep.2017.01.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ (2006) The role of the basal ganglia in habit formation. Nat Rev Neurosci 7:464–476. 10.1038/nrn1919 [DOI] [PubMed] [Google Scholar]