

Abstract
Gain control of the auditory system operates at multiple levels. Cholinergic medial olivocochlear (MOC) fibers originate in the brainstem and make synaptic contacts at the base of the outer hair cells (OHCs), the final targets of several feedback loops from the periphery and higher-processing centers. Efferent activation inhibits OHC active amplification within the mammalian cochlea, through the activation of a calcium-permeable α9α10 ionotropic cholinergic nicotinic receptor (nAChR), functionally coupled to calcium activated SK2 potassium channels. Correct operation of this feedback requires careful matching of acoustic input with the strength of cochlear inhibition (Galambos, 1956; Wiederhold and Kiang, 1970; Gifford and Guinan, 1987), which is driven by the rate of MOC activity and short-term facilitation at the MOC–OHC synapse (Ballestero et al., 2011; Katz and Elgoyhen, 2014). The present work shows (in mice of either sex) that a mutation in the α9α10 nAChR with increased duration of channel gating (Taranda et al., 2009) greatly elongates hair cell-evoked IPSCs and Ca2+ signals. Interestingly, MOC–OHC synapses of L9′T mice presented reduced quantum content and increased presynaptic facilitation. These phenotypic changes lead to enhanced and sustained synaptic responses and OHC hyperpolarization upon high-frequency stimulation of MOC terminals. At the cochlear physiology level these changes were matched by a longer time course of efferent MOC suppression. This indicates that the properties of the MOC–OHC synapse directly determine the efficacy of the MOC feedback to the cochlea being a main player in the “gain control” of the auditory periphery.
SIGNIFICANCE STATEMENT Plasticity can involve reciprocal signaling across chemical synapses. An opportunity to study this phenomenon occurs in the mammalian cochlea whose sensitivity is regulated by efferent olivocochlear neurons. These release acetylcholine to inhibit sensory hair cells. A point mutation in the hair cell's acetylcholine receptor that leads to increased gating of the receptor greatly elongates IPSCs. Interestingly, efferent terminals from mutant mice present a reduced resting release probability. However, upon high-frequency stimulation transmitter release facilitates strongly to produce stronger and far longer-lasting inhibition of cochlear function. Thus, central neuronal feedback on cochlear hair cells provides an opportunity to define plasticity mechanisms in cholinergic synapses other than the highly studied neuromuscular junction.
Keywords: α9α10 nAChR, cochlea, efferent inhibition, hair cells, synaptic plasticity
Introduction
Sensory systems have evolved to meet environmental demands, combining high sensitivity with selectivity over a wide dynamic range. For example, the mammalian auditory system has to discriminate sounds whose intensity varies several orders of magnitude (Robinson and McAlpine, 2009; Hudspeth, 2014) and to distinguish frequencies from 20 Hz to 20 kHz (in humans) with high resolution (Hudspeth, 2014). These demands must be met in a way that enables communication in the presence of background noise (Kawase et al., 1993; Guinan, 1996). The mammalian auditory system accomplishes these tasks in part by an active process within the inner ear, an amplifier that increases the gain of acoustic inputs (Brownell et al., 1985; Dallos, 1992, 2008) and by centrifugal efferent gain control of that process (Guinan, 1996; Robinson and McAlpine, 2009). Outer hair cells (OHCs) are the source of the active mechanism. They undergo voltage-dependent changes in length and stiffness, a process known as electromotility (Brownell et al., 1985). This amplifies movement of the basilar membrane, increasing the gain and tuning for acoustic inputs (Ashmore, 1987; Dallos, 1992, 2008).
Centrifugal auditory gain control operates at multiple levels. Descending direct or indirect pathways from all regions of the auditory cortex to subcortical brain structures all the way to the auditory periphery most likely come into play. Medial olivocochlear fibers that originate in the ventromedial portion of the superior olivary complex are the final targets of several feedback loops from both the periphery (Guinan, 1996) and higher-processing centers (Suga et al., 2000). They make direct synaptic contacts at the base of the OHCs and inhibit electromotility. This is mediated by acetylcholine (ACh), through the activation of calcium-permeable α9α10 ionotropic cholinergic nicotinic receptors (nAChRs) present at the basal pole of OHCs (Elgoyhen et al., 1994, 2001), functionally coupled to calcium-activated SK2 potassium channels (Housley and Ashmore, 1991; Fuchs and Murrow, 1992; Blanchet et al., 1996; Dulon et al., 1998; Oliver et al., 2000). The strength of cochlear inhibition is proportional to the rate of MOC activity (Galambos, 1956; Wiederhold and Kiang, 1970; Gifford and Guinan, 1987). MOC efferents respond to sound with sensitivity and tuning like that of type I cochlear afferents (Robertson and Gummer, 1985; Liberman and Brown, 1986), so providing a feedback loop for cochlear gain control. Recent work showed for the first time that short-term facilitation at the MOC–OHC synapse constitutes a mechanism by which OHCs encode MOC firing frequency to adjust the sensitivity of the auditory periphery (Ballestero et al., 2011). Therefore, changes in the synaptic properties of the efferent synapse should be matched by changes in the efferent physiological response.
Genetically modified mice bearing a threonine for a leucine substitution at position 9′ of the second transmembrane domain of the α9 nAChR subunit (Chrna9L9′T/ L9′T, L9′T knock-in) have stronger MOC suppression (Taranda et al., 2009). The mechanisms underlying these prolonged in vivo effects were left unknown. The present study shows that the postsynaptic effect of each ACh quantum was enhanced in the L9′T mice due to a slower decay time of evoked postsynaptic currents and an increase in the total charge accumulated in the hair cell upon each release event. In addition, evoked synaptic responses occurred with lower probability and presented more facilitation, building up and decaying over a longer time course. Together, these presynaptic and postsynaptic effects lead to enhanced and prolonged synaptic responses and OHC hyperpolarization upon high-frequency stimulation of MOC terminals in L9′T mice. Noticeably, these synaptic changes correlated with the in vivo longer time course of efferent MOC suppression. This indicates that the dynamics of the MOC–OHC synapse determines the efficacy of the response to high-frequency MOC stimulation and is a key player in the “gain control” of the auditory periphery.
Materials and Methods
All experimental protocols were performed in accordance with American Veterinary Medical Association's AVMA Guidelines on Euthanasia (2013) and approved by the IACUC at INGEBI. L9′T knock-in mice were backcrossed with congenic FVB.129P2-Pde6bþ Tyrc-ch/AntJ stock for 17 generations (i.e., N-17).
Isolation of the organ of Corti.
Procedures for preparing and recording from the mouse organ of Corti were essentially identical to those published previously (Glowatzki and Fuchs, 2000; Ballestero et al., 2011; Wedemeyer et al., 2013). Briefly, apical turns of the organ of Corti were excised from homozygous L9′T knock-in mice (Taranda et al., 2009) and their wild-type (wt) littermates, placed in the chamber for electrophysiological recordings mounted on the stage of a Zeiss Axioskop or Leica LFS microscope and viewed with differential interference contrast (DIC) using a 40× water-immersion objective and a camera with contrast enhancement (Dage-MTI, or Hamamatsu C275410, Hamamatsu Photonics). Mice of either sex were used between postnatal day (P)9 (day of birth was considered P0) and P11 for inner hair cell (IHC), and P11–P13 for OHC recordings. The preparations were used within 3 h.
Electrophysiological recordings.
IHCs were identified visually by their characteristic shape and by the size of their capacitance (7–12 pF), whereas OHCs were identified by their shape and their three-row arrangement. The cochlear preparation was continuously superfused by means of a peristaltic pump (Gilson Minipulse 3, with 8 channels, Bioesanco) containing an extracellular saline solution of an ionic composition similar to that of the perilymph (in mm): 155 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, and 10 HEPES buffer; pH 7.4, 295–300 mOsm. The pipette solution contained the following (in mm): 140 KCl, 3.5 MgCl2, 0.1 CaCl2, 5 mm EGTA, 5 HEPES buffer, 2.5 Na2ATP; pH 7.2, 280–283 mOsm. When performing experiments in the absence of SK2 channel activation, EGTA in the pipette solution was replaced by 5 mm of the fast calcium chelator 1,2-bis(2-aminophenoxy) ethane-N,N,N′,N′tetra-acetic acid (BAPTA) to preclude the activation of SK2 currents by Ca2+. Some cells were removed to access the IHCs, but mostly the pipette moved through the tissue using positive fluid flow to clear the tip. Currents in IHCs and OHCs were recorded in the whole-cell patch-clamp mode at holding potentials of −90 mV (IHCs) or −40 mV (OHCs) using an Axopatch 200B amplifier (Molecular Devices), low-pass filtered at 2–10 kHz and digitized at 5–20 kHz with a Digidata 1322A board (Molecular Devices). Recordings were made at room temperature (22–25°C). Borosilicate glass pipettes (World Precision Instruments) had resistances of 6–8 MΩ. Indicated holding potentials were not corrected for liquid junction potentials (−4 mV).
Electrical stimulation of MOC efferent axons.
Neurotransmitter release was evoked by electrical stimulation of the MOC efferent axons as previously described (Goutman et al., 2005; Ballestero et al., 2011). Briefly, the electrical stimulus for IHCs was delivered via a 20–80 μm diameter theta glass pipette placed at 20–60 μm modiolar to the base of the IHC under study, while for OHCs the electrical stimulus was applied with a borosilicate glass pipette of 0.5–1 MΩ at the base of the IHC that was aligned with the OHC under study. For optimal stimulation, some supporting cells were removed by applying negative pressure. The position of the pipette was adjusted until postsynaptic currents in the IHCs or OHCs were consistently activated. An electrically isolated constant voltage source (Grass model S48 stimulator coupled to a SIU5 isolation unit) was triggered via the data acquisition computer to generate 100–300 μA pulses of 0.1–1 ms duration.
Estimation of the quantum content of transmitter release.
To obtain the quantum content of transmitter release (m), protocols of 100 stimuli were applied at a frequency of 1 Hz. Under the assumption that evoked synaptic events follow a Poisson distribution, m was calculated by the “method of failures” [m = ln (N/N0), where N is the total number of stimuli and N0 is the number of failures; Del Castillo and Katz, 1954].
Stimulation protocols.
Train stimulation protocols composed of 10 pulses at frequencies ranging from 10 to 80 Hz, were repeated 30 times at 2 s-intervals. For every pulse, the current amplitude was computed as the difference between the peak of the response and the baseline, which was considered as the current value before the pulse. The probability of release (P) for each pulse was computed as the ratio between the number of evoked IPSCs (eIPSCs) and the number of sweeps. The average amplitude (A) was obtained by averaging the successful eIPSCs amplitudes after each pulse. The average response (S) for each stimulus was computed as the average of all recorded amplitudes, including failures of response. To determine the extent of facilitation or depression during a train, values for the parameters S, P, and A were normalized to their values in the first pulse. For paired-pulse experiments, two consecutive shocks with interstimulus intervals (ISIs) of 25 and 50 ms were applied 50–100 times, and values for the parameters S, P, and A were normalized to their values in the first pulse. To calculate the facilitation index (FI), S1 values were obtained by measuring the maximum current amplitude reached after the end of the first stimulation artifact. S2 was obtained by measuring the current amplitude after the second stimulation artifact, with the same time interval used to measure S1 (even if this point is not the maximum peak of the response after the second pulse). For long-lasting trains, stimulation pulses at 80 Hz were applied once during 10 s. For this protocol, accumulated current amplitudes were evaluated at every second during the train. There were no significant changes either in the profile of the responses or in the average amplitude of the first pulse in each train upon application of successive trains. In experiments in current-clamp mode, current injections of 30–50 pA for 10 s were applied to induce repetitive firing of calcium action potentials in IHCs. Synaptic current analysis was made off-line, with Clampfit 9.2 (Molecular Devices) and custom routines implemented in Igor Pro 6.37 (Wavemetrics, Igor Pro, RRID:SCR_000325) with a threshold of 4–5 times the RMS noise (7–12 pA). Rise (20–80%, τrise) and decay (10–90%, τdecay) time constants were analyzed only in eIPSCs that were not compromised in shape by the stimulus artifact or following events. The decay of eIPSCs was fitted with a monoexponential function using Clampfit 9.2.
Ca2+ imaging.
Ca2+ imaging experiments were performed as previously reported (Moglie et al., 2018). The intracellular solution was made from a 2× stock to reach a final concentration of the following (in mm): 140 KCl, 5 HEPES buffer, 2.5 Na2ATP, 5 phosphocreatine-Na2, 3.5 MgCl2, 0.5 EGTA, pH 7.2. The Ca2+ indicator Fluo-4 was added to reach a final concentration of 0.4 mm. The cochlear preparation was placed in a chamber on the stage of an upright microscope (Olympus BX51WI) and illuminated with a blue LED system (Tolket). Cells were loaded with the intracellular solution containing the Ca2+ indicator via the patch-pipette. Image acquisition started 5 min after whole-cell break-in to ensure the proper dialysis of the cell content. Images were acquired using an Andor iXon 885 camera controlled through a Till Photonics interface system. The focal plane was located close to the basal pole of the IHC and the image size was set to 100 × 100 pixels, which allowed an acquisition rate of 140 frames/s. The signal-to-noise ratio was improved with a chip binning of 4 × 4, giving a resolution of 0.533 μm per pixel using a 60× water-immersion objective. Simultaneously, electrophysiological recordings were performed using a MultiClamp 700B amplifier (Molecular Devices) and digitized at 50 kHz via a National Instruments board. Data were acquired using WinWCP (RRID:SCR_014713). Images were analyzed with custom-written routines in IgorPro 6.37 (Wavemetrics; RRID:SCR_000325). Briefly, a region-of-interest (ROI), comprising the highest fluorescence region of the IHC, was defined and the average fluorescence calculated for each time frame. Photobleaching was corrected by fitting a line between prestimulus baseline and final fluorescence. Finally, the fluorescence signal, computed as ΔF/F0, was normalized to its maximum value.
Distortion product otoacoustic emissions.
Six-week-old mice of either sex were anesthetized with urethane (1.20 g/kg, i.p.) and xylazine (20 mg/kg, i.p.). Distortion product otoacoustic emissions (DPOAEs) at 2f1-f2 were recorded with a custom acoustic assembly consisting of two electrostatic drivers (TDT EC-1, Tucker-Davis Technologies) to generate primary tones (f1 and f2 with f2/f1 = 1.2 and f2 level 10 dB < f1 level) and a Knowles miniature microphone (EK3103) to record ear-canal sound pressure. Stimuli were generated digitally, whereas resultant ear-canal sound pressure was amplified and digitally sampled at 4 μs (16 bit DAQ boards, NI 6052E, National Instruments). Fast Fourier Transforms were computed and averaged over five consecutive waveform traces, and 2f1-f2 DPOAE amplitude and surrounding noise floor were extracted.
Medial olivocochlear assay.
A posterior craniotomy and partial cerebellar aspiration were performed to expose the floor of the IVth ventricle. To stimulate the olivocochlear (OC) bundle, shocks (monophasic pulses, 150 μs duration, 200/s) were applied through fine silver wires (0.4 mm spacing) placed along the midline, spanning the OC decussation. Shock threshold for facial twitches was determined, muscle paralysis induced with α-d-tubocurarine (1.25 mg/kg, i.p.), and the animal connected to a respirator via a tracheal cannula. Shock levels were raised to 6 dB above twitch threshold. During the OC suppression assay, f2 level was set to produce a DPOAE 10–15 dB > noise floor. To measure OC effects, repeated measures of baseline DPOAE amplitude were first obtained (n = 56), followed by a series of 70 contiguous periods in which DPOAE amplitudes were measured with simultaneous shocks to the OC bundle and additional periods during which DPOAE measures continued after the termination of the shock train.
Drugs and reagents.
All drugs and reagents were purchased from Sigma-Aldrich with the exception of Fluo-4 which was purchased from Invitrogen.
Statistics.
All statistical tests were performed with GraphPad Prism 6 (RRID:SCR_002798). Before performing any analysis, data were tested for normal distribution using the Shapiro–Wilk normality test, and parametric or nonparametric tests were applied accordingly. When the normality test could not be applied, data were assumed to be not normally distributed. For statistical analyses with two datasets, two-tailed paired and unpaired t tests or two-sided Wilcoxon rank sum and Kolmogorov–Smirnov tests were used. For comparisons of more than two datasets, Kruskal–Wallis one-way ANOVA followed by Dunn's multiple-comparison test, and two-way ANOVA repeated-measures followed by Sidak's multiple-comparison test were used. Values of p < 0.05 were considered significant. All data were expressed as mean ± SEM, unless otherwise stated. In all cases n indicates the number of cells tested, with exception of Table 2, where n indicates the number of animals tested.
Table 2.
MOC-mediated suppression (sup) of cochlear DPOAEs
| 16 kHz |
22 kHz |
|||
|---|---|---|---|---|
| wt (n = 5) | L9′T (n = 4) | wt (n = 3) | L9′T (n = 7) | |
| Max sup: effect vs baseline, dB | −10.0 ± 1.4 | −18.4 ± 1.2** | −9.6 ± 2.1 | −20.4 ± 1.7* |
| Reached within, s | 5.3 ± 1.0 | 30.8 ± 4.1* | 1.5 ± 0.4 | 44.1 ± 3.1*** |
| Mean sup: effect under shocks, dB | −4.2 ± 1.5 | −11.6 ± 1.3* | −1.4 ± 1.2 | −10.1 ± 2.3* |
| Return to baseline (after shocks off) | ≤ 1 s | > 5 min | ≤ 1 s | > 5 min |
*p < 0.05,
**p < 0.01,
***p < 0.001; two-tailed Kolmogorov–Smirnov test.
Results
Properties of synaptic transmission at MOC–hair cell synapses in L9′T mice
In mammals, both the MOC–OHC and the transient MOC–IHC synapses are mediated by ACh acting on the α9α10 nAChR functionally coupled to the opening of Ca2+-activated SK2 K+ channels that hyperpolarize hair cells (Glowatzki and Fuchs, 2000; Oliver et al., 2000; Elgoyhen et al., 2001; Katz et al., 2004; Wersinger et al., 2010; Fig. 1A). Figure 1B illustrates the responses obtained in IHCs and OHCs from both genotypes (L9′T and wt) upon 1 Hz stimulation of the MOC efferent fibers. Representative eIPSCs show that postsynaptic responses in both cell types from L9′T mice present slower kinetics (τrise L9′T/wt IHCs ∼3-fold increase; OHCs ∼6-fold increase; τdecay L9′T/wt IHCs ∼4.5-fold increase; OHCs ∼3-fold increase; Table 1) and smaller amplitude (L9′T/wt IHCs 0.52; OHCs 0.52). The slower decay time leads to a significantly prolonged duration of the eIPSC with the consequent increase in the total charge (Q) accumulated in the hair cell upon each release event (IHCs wt: 1.1 ± 0.2 pC, n = 6, L9′T: 2.7 ± 0.2 pC, n = 5, p = 0.016; OHCs wt: 2.3 ± 0.32 pC, n = 12, L9′T: 3.23 ± 0.39 pC, n = 15, p = 0.046; Kolmogorov–Smirnov test). A detailed quantification of these parameters is reported in Table 1. The slower time course and smaller amplitude of eIPSCs are in agreement with what was reported for spontaneous and K+-evoked IPSCs in L9′T IHCs and OHCs (Taranda et al., 2009).
Figure 1.

Characteristics of evoked synaptic currents in OHCs and IHCs from L9′T mice. A, Schematic representation of the cholinergic MOC–hair cell synapse. MOC efferent neurons located in the superior olivary complex of the brainstem project to the cochlea, where they contact the OHCs in hearing mice. Since birth to hearing onset, MOC fibers also transiently innervate the IHCs. Upon nerve stimulation, ACh released from MOC terminals activates the α9α10 nicotinic nAChR. This leads to Ca2+ influx and the subsequent activation of Ca2+-dependent Ca2+-activated SK2 channels. B, Representative traces of eIPSCs in OHCs (left) and IHCs (right) from wt (black) and L9′T (red) mice in response to MOC stimulation at 1 Hz. Vhold was −40 mV for OHCs and −90 mV for IHCs.
Table 1.
Evoked synaptic currents in IHCs and OHCs
| IHCs |
OHCs |
|||
|---|---|---|---|---|
| wt (n = 6) | L9′T (n = 5) | wt (n = 12) | L9′T (n = 15) | |
| Quantum content | 1.29 ± 0.21 | 0.83 ± 0.12* | 0.23 ± 0.04 | 0.14 ± 0.02* |
| Amplitude, pA | 28.4 ± 4.6 | 14.9 ± 2.2* | 42.3 ± 4.3 | 21.9 ± 2.8*** |
| τrise, ms (20–80%) | 5.3 ± 0.6 | 16.3 ± 2.2** | 8.5 ± 0.4 | 51.5 ± 6.7*** |
| τdecay, ms (10–90%) | 63.6 ± 9.7 | 290.4 ± 62.3* | 55.2 ± 7.4 | 166.7 ± 21.5** |
| H-W, ms | 38.3 ± 4.7 | 175.2 ± 24.9* | 48.5 ± 2.7 | 150.3 ± 18.8*** |
| Q, pC | 1.1 ± 0.2 | 2.7 ± 0.2* | 2.3 ± 0.32 | 3.23 ± 0.39* |
For IHCs, four and five different mice were used for each genotype, whereas for OHCs, 9 and 11 mice were used for each genotype.
*p < 0.05,
**p < 0.01,
***p < 0.001; two-tailed unpaired t test or Kolmogorov–Smirnov test.
In addition, the quantum content of evoked release was found to be significantly lower both at the L9′T MOC–IHC and the MOC–OHC synapses (IHCs wt: 1.29 ± 0.21, n = 6, L9′T: 0.83 ± 0.12, n = 5, p = 0.041; OHCs wt: 0.23 ± 0.04, n = 12, L9′T: 0.14 ± 0.02, n = 15, p = 0.035; Kolmogorov–Smirnov test; Table 1). This reduction in quantum content shows that a point mutation in the postsynaptic receptor not only modifies the kinetics of postsynaptic responses but also alters the presynaptic terminal with the consequent reduction in the number of quanta released upon nerve stimulation.
Postsynaptic Ca2+ signals in response to MOC efferent fibers stimulation are prolonged in L9′T mice
To study the Ca2+ dynamics in response to activation of the α9α10 nAChR in both wt and L9′T mice, IHC electrical activity was recorded while performing Ca2+ imaging experiments (Fig. 2). The representative sequence of wide-field microscopy images shown in Figure 2A indicates that, after efferent electrical stimulation (10 pulses at 80 Hz), Ca2+ signals in IHCs from L9′T mice returned to baseline long after the signal disappeared from wt IHCs. Figure 2B illustrates representative synaptic responses (top) and the concomitant changes in fluorescence (bottom) upon MOC stimulation by 10-shock trains at 20 (left) and 80 Hz (right) obtained in a wt (black) and L9′T (red) mice IHC. Quantification of the half-width (H-W) and decay kinetics (20–80% decay time of the peak response) of the Ca2+ transients obtained during the stimulation protocols are plotted in Figure 2, C and D, respectively (H-W wt: 0.45 ± 0.12 s and L9′T: 1.08 ± 0.03 s for 20 Hz, p = 0.0082; wt: 0.36 ± 0.06 s and L9′T: 1.26 ± 0.16 s for 80 Hz, p = 0.0012; τdecay 20–80% wt: 1.07 ± 0.20 s and L9′T: 2.56 ± 0.34 s for 20 Hz, p = 0.026; wt: 0.97 ± 0.18 s and L9′T: 2.76 ± 0.40 s for 80 Hz, p = 0.0082; Kolmogorov–Smirnov test for all datasets, n = 7 for wt and n = 6 for L9′T). These results show that in agreement with the slower decay times of synaptic responses obtained in IHCs from L9′T mice (Fig. 1B; Table 1), Ca2+ signals obtained in IHCs from L9′T mice upon MOC stimulation are wider and more prolonged than those obtained in cells from wt mice.
Figure 2.

Ca2+ signals in IHCs after MOC fiber activation exhibit slower kinetics in L9′T mice compared with wt mice. A, Sequence of wide-field microscopy images of IHCs from wt and L9′T mice filled with Fluo-4 and illuminated with 488 nm LED light during efferent electrical stimulation. “Stim” indicates the time when a 10-shock train at 80 Hz was applied. Fluorescence signal was normalized to its maximum value. B, Representative synaptic responses (top) and changes in fluorescence expressed as ΔF/F0 (bottom), obtained in a wt (black) and L9′T (red) mice IHC during efferent fiber stimulation by 10-shock trains at 20 and 80 Hz. Fluorescence was measured in the region of the cell where the signal reached its maximum value. C, H-W of the Ca2+ transients obtained during the stimulation protocols like those illustrated in B. D, Decay kinetics of Ca2+ transients computed as the decay time between 20 and 80% of the peak response. *p < 0.05, **p < 0.01, Kolmogorov–Smirnov test; n = 7 for wt and n = 6 for L9′T.
Short-term synaptic plasticity of MOC–OHC synapses
Synapses with low-quantum content tend to facilitate upon high-frequency stimulation (Zucker and Regehr, 2002; Fioravante and Regehr, 2011; Jackman and Regehr, 2017). Previous work showed that the probability of release is low upon 1 Hz stimulation at the MOC–OHC synapse, but that presynaptic facilitation raises the reliability of release at higher stimulation frequencies (Ballestero et al., 2011).
Because the quantum content of MOC–OHC synapses from L9′T mice was even lower than that of their wt littermates (Table 1), facilitation might be more prominent in this genotype. To evaluate synaptic facilitation, paired-pulse protocols were applied at interstimuli intervals (ISIs) of 50 and 25 ms, corresponding to frequencies of 20 and 40 Hz, respectively (Fig. 3A,B). The lowest frequency (20 Hz), was chosen because MOC–OHCs from wild-type mice do not facilitate (Ballestero et al., 2011). The 40 Hz frequency was selected to compare the degree of facilitation between genotypes. Paired-pulse experiments at higher frequencies cannot be confidently analyzed due to the slow kinetics of the L9′T mice. The facilitation index (FIS) was defined as the ratio between S2/S1, where S1 and S2 are the average responses to the first and second pulse, respectively. FIS values >1 indicate facilitation, whereas FIS values <1, indicate depression (Del Castillo and Katz, 1954; Katz and Miledi, 1968; Goutman et al., 2005; Ballestero et al., 2011). At an ISI of 25 ms (Fig. 3A,B, right), FIS were >1 for MOC–OHC synapses from both wt and L9′T mice (S2/S1 wt: 1.37 ± 0.16, n = 6, p = 0.031 and L9′T: 3.47 ± 0.44, n = 8, p = 0.008; Wilcoxon rank sum test). Facilitation at this ISI (25 ms) was significantly stronger in the L9′T animals (Fig. 3B). At ISIs of 50 ms, only MOC–OHC synapses from L9′T animals presented facilitation upon paired-pulse protocols (S2/S1 wt: 1.00 ± 0.08, n = 4, p = 0.625 and L9′T: 2.87 ± 0.58, n = 7, p = 0.016; Wilcoxon rank sum test).
Figure 3.

Paired-pulse facilitation is enhanced at MOC–OHC synapses from L9′T mice. A, Representative traces of 10 individual responses from L9′T (red tones) and wt (black tones) MOC–OHC synapses at ISIs of 50 ms (left) and 25 ms (right; corresponding to stimulation frequencies of 20 and 40 Hz, respectively). B, Bar graphs showing the ratio between the second and first pulse for the total average response (S2/S1), the release probability (P2/P1) and the amplitude of the successfully evoked postsynaptic responses (A2/A1) for ISIs of 50 ms (left; wt n = 7 cells from 6 different mice and L9′T n = 4 cells from 4 different mice) and 25 ms (right; wt n = 6 cells from 4 different mice and L9′T n = 8 cells from 8 different mice). S2/S1 is significantly >1 at MOC–OHC synapses from L9′T mice for both ISIs. At wt MOC–OHC synapses, S2/S1 is > 1 only at ISIs of 25 ms. Note that the increase in S2/S1 is matched by an increase in P2/P1, whereas A2/A1 is = 1 for both genotypes at the two ISIs tested. *p < 0.05, **p < 0.01, Kolmogorov–Smirnov test when comparing L9′T mice FI values to those of their wt littermates.
As described for most synapses (Fioravante and Regehr, 2011; Jackman and Regehr, 2017), facilitation at MOC–OHC synapses from wt mice upon paired-pulse stimulation is of presynaptic origin and solely due to an increase in the number of quanta released upon nerve stimulation (Ballestero et al., 2011). To evaluate the presynaptic origin of the observed facilitation, a facilitation index was calculated for both the amplitude of the successfully evoked events (FIA = A2/A1) and the probability of release (FIP = P2/P1). No significant effect was found for FIA (p > 0.05) for both wt and L9′T mice at either 50 or 25 ISIs (Fig. 3B). However, changes in S2/S1 were matched by changes in P2/P1 both in the L9′T mice and their wt littermates. For ISIs of 50 ms: P2/P1, wt: 0.93 ± 0.21, n = 4, p = 0.75 and L9′T: 2.60 ± 0.47, n = 7, p = 0.016; A2/A1, wt: 1.21 ± 0.41, n = 4, p = 0.88 and L9′T: 1.09 ± 0.10, n = 7, p = 0.52 (Wilcoxon rank sum test; Fig. 3B, left). For ISIs of 25 ms: P2/P1, wt: 1.59 ± 0.13, n = 6, p = 0.003 and L9′T: 3.25 ± 0.56, n = 8, p = 0.016; A2/A1, wt: 0.94 ± 0.07, n = 6, p = 0.07 and L9′T: 1.08 ± 0.08, n = 8, p = 0.89 (two-tailed paired t test and Wilcoxon rank sum test, respectively; Fig. 3B, right). These results suggest that the strong facilitation observed at MOC–OHC synapses from L9′T mice is due to changes in the probability of release.
In the previous experiments transmitter release was measured at −40 mV, where the response is dominated by the secondary activation of the SK2 channel following Ca2+ entry through the α9α10 nAChR (Oliver et al., 2000; Ballestero et al., 2011) and possibly by Ca2+ released from nearby subsynaptic cisterns (Lioudyno et al., 2004; Fuchs et al., 2014). Because in L9′T animals Ca2+ dynamics after α9α10 nAChR activation is changed (Fig. 2), to further evaluate the presynaptic origin of facilitation, in the following experiments nAChR currents were measured in isolation from SK2 channels. Thus, the contribution of SK2 channels was minimized by using by BAPTA (5 mm) in the recording pipette and voltage-clamping the OHCs at −82 mV (Erev for K+ in these recording conditions; see Materials and Methods).
Figure 4A shows representative recordings of isolated nAChR responses in the MOC–OHC synapse from L9′T mice obtained upon paired-pulse protocols at ISIs of 50 and 25 ms, corresponding to 20 and 40 Hz, respectively. Even though isolated nAChR synaptic currents are very small and the recordings are somewhat noisy, they can be clearly distinguished from noise and analyzed. The bar graphs in Figure 4B show that MOC–OHC synapses facilitated (FIS > 1) at both 20 and 40 Hz (S2/S1 = 2.0 ± 0.25 for ISIs of 50 ms, n = 6, p = 0.031 and 2.38 ± 0.46 for ISIs of 25 ms, n = 8, p = 0.001; Wilcoxon rank sum test and two-tailed paired t test, respectively). In addition, this facilitation was due to an increment in the probability of release without any changes in the amplitude of successfully evoked postsynaptic currents (P2/P1 = 2.15 ± 0.28, p = 0.031; A2/A1 = 1.04 ± 0.04, p = 0.84 for ISIs of 50 ms, and P2/P1 = 2.38 ± 0.36, p = 0.0003; A2/A1 = 0.95 ± 0.03, p = 0.31 for ISIs of 25 ms; Wilcoxon rank sum test for P2/P1 datasets and two-tailed paired t test for A2/A1 datasets).
Figure 4.

Facilitation by paired-pulse stimuli at L9′T MOC–OHC synapses is observed even in the absence of SK2 activity. A, Representative individual traces of synaptic responses obtained at MOC–OHC synapses from L9′T mice at ISIs of 50 and 25 ms (corresponding to frequencies of 20 and 40 Hz) in the presence of 5 mm BAPTA in the recording pipette and voltage-clamping the OHCs at a Vhold of −82 mV. B, Bar graphs showing the ratio between the second and first pulse for the total average response (S2/S1), the release probability (P2/P1) and the amplitude of the successfully evoked postsynaptic responses (A2/A1) for ISIs of 50 ms (left; n = 6 cells from 4 different mice) and 25 ms (right; n = 8 cells from 5 different mice) at MOC–OHC nAChR + SK (EGTA in the recording pipette, Vhold = −40 mV; solid bars) and isolated nAChR (BAPTA in the recording pipette, Vhold = −82 mV; striped bars) synapses from L9′T mice.
When comparing the results of paired-pulse experiments obtained when synaptic currents were dominated by the secondary activation of the SK2 channels (Fig. 3B), with those obtained with the nAChR in isolation (Fig. 4B), no significant differences in the degree of facilitation at either frequency were found (ISIs of 50 ms: EGTA S2/S1 = 2.73 ± 0.62, BAPTA S2/S1 = 2.0 ± 0.25, p = 0.43; ISIs of 25 ms: EGTA S2/S1 = 3.47 ± 0.44, BAPTA S2/S1 = 2.38 ± 0.46, p = 0.08; Kolmogorov–Smirnov test).
These results confirm that the increased facilitation at MOC–OHC synapses from L9′T mice can be solely accounted for by changes at the presynapse and supports the notion that the presence of the L9′T mutation in the α9 nAChR subunit somehow alters the presynaptic machinery and/or the mechanisms involved in ACh release.
Synaptic currents evoked at MOC–OHC synapses from L9′T mice at physiological MOC firing frequencies
In vivo MOC fibers fire regularly at frequencies that range from 1 to 120 Hz (Robertson and Gummer, 1985; Liberman and Brown, 1986; Brown, 1989) and cochlear inhibition increases with the frequency of MOC fiber activation (Galambos, 1956; Wiederhold and Kiang, 1970; Gifford and Guinan, 1987). In addition, the combined effect of presynaptic facilitation and postsynaptic summation of IPSCs results in a frequency-dependent increment in the amplitude of IPSCs in OHCs, thus scaling inhibition to the frequency of MOC activity (Ballestero et al., 2011).
The postsynaptic effect of repetitive efferent activity in L9′T mice was evoked by trains of 10 shocks at frequencies ranging from 10 to 80 Hz (corresponding to ISIs of 100–12.5 ms). Representative responses of OHCs to these trains are illustrated in Figure 5A. Averaged representative traces at 10, 20, 40, and 80 Hz, show that synaptic responses at MOC–OHC synapses from L9′T mice remained strongly potentiated after the end of the 10 stimulation shocks. As illustrated in the bar graph of Figure 5B, the total charge accumulated in the OHC during this protocol, measured at the end of the recording period, was significantly larger in the L9′T mice than in wt at all frequencies tested (10 Hz, p = 0.024; 20 Hz, p = 0.019; 40 Hz, p = 0.024; 80 Hz, p = 0.048; Kolmogorov–Smirnov test). Another salient feature of L9′T MOC–OHC synapses was that the accumulated charge reached 50% of its maximum value with a significant delay compared with those of wt mice at all tested frequencies (Fig. 5C): 10 Hz, wt: 0.69 ± 0.09 s, n = 3; L9′T: 0.85 ± 0.02, n = 7, Δt = 160 ms, p = 0.017; 20 Hz, wt: 0.40 ± 0.02 s, n = 6, L9′T: 0.58 ± 0.01 s, n = 10, Δt = 180 ms, p = 0.0002; 40 Hz, wt: 0.26 ± 0.03 s, n = 4, L9′T: 0.48 ± 0.07 s, n = 7, Δt = 220 ms, p = 0.006; 80 Hz, wt: 0.17 ± 0.01 s, n = 5, L9′T: 0.40 ± 0.02 s, n = 11, Δt = 230 ms, p = 0.0005 (Kolmogorov–Smirnov test for all datasets). Taking into account that synaptic currents in L9′T mice are significantly prolonged (Fig. 1B; Table 1) and that both presynaptic facilitation and postsynaptic summation of IPSCs result in a frequency-dependent increment in the amplitude of IPSCs in OHCs (Ballestero et al., 2011), the enhanced sustained potentiation observed in the L9′T mice, even at the lowest frequencies tested (10 and 20 Hz), is likely due to a higher contribution of IPSCs summation at the postsynapse in addition to facilitation due to a higher probability of release during the stimulation trains.
Figure 5.
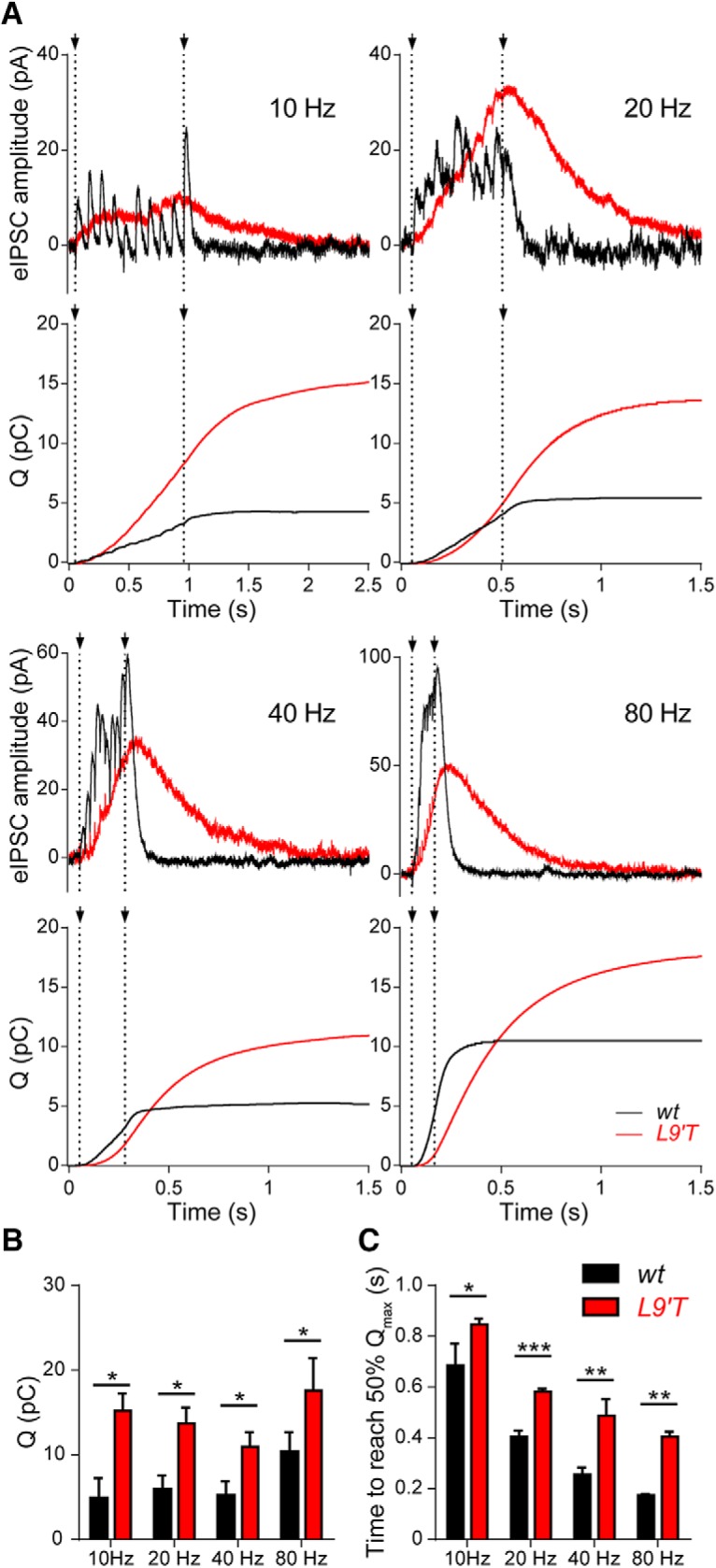
Synaptic responses and total charge in OHCs from wt and L9′T mice upon stimulating the MOC fibers at different frequencies. A, Representative averaged traces of 30 individual synaptic responses in OHCs from both wt (black) and L9′T (red) mice upon a 10-pulse stimulation to the MOC fibers at frequencies of 10, 20, 40, and 80 Hz. Dotted lines indicate the beginning and the end of the stimulation period. Below every pair of traces, the cumulative charge (in pC) during the complete protocol is plotted as a function of time. Note that the total charge is larger and the time required to reach the maximum charge is longer in L9′T synapses compared with wt ones. B, The bar diagram shows the total charge (Q) computed at the end of the protocol for both genotypes (10 Hz, wt: 4.85 ± 2.42 pC, n = 3 cells from 3 different mice, L9′T: 16.84 ± 1.4,1 pC, n = 7 cells from 7 different mice; 20 Hz, wt: 5.91 ± 1.66 pC, n = 6 cells from 4 different mice, L9′T: 13.69 ± 1.87 pC, n = 10 cells from 7 different mice; 40 Hz, wt: 3.71 ± 1.1 pC, n = 4 cells from 4 different mice, L9′T: 11.77 ± 1.77 pC, n = 7 cells from 6 different mice; 80 Hz, wt: 10.34 ± 2.33, n = 5 cells from 5 different mice, L9′T: 17.59 ± 3.86 n = 11 cells from 6 different mice; p < 0.05 for all frequencies tested, Kolmogorov–Smirnov test). C, The bar diagram shows the time at which the accumulated charge reaches 50% of its maximum value in both genotypes at frequencies of 10, 20, 40, and 80 Hz (*p < 0.05, **p < 0.01, ***p < 0.001, Kolmogorov–Smirnov test for all datasets).
Facilitation during repetitive stimulation was assessed by measuring the probability of release (P) and the average amplitude of the successfully evoked responses (A) for each successive stimulus (Fig. 6A). The total averaged response (S) is the product of P × A along the 10-shock stimulation train. These parameters were only analyzed for the 10 and 20 Hz stimulation trains. Trains at higher frequencies were difficult to analyze in the L9′T animals due to the very slow kinetics of the evoked IPSCs (Fig. 1; Table 1). The initial probability of release was computed for each genotype and, in agreement with the quantum content values (Table 1), it was significantly higher in MOC–OHC synapses from wt animals than in those from the L9′T ones (0.16 ± 0.02, n = 43 for wt mice and 0.10 ± 0.01, n = 43 for L9′T mice, p = 0.02; two-tailed unpaired t test).
Figure 6.

MOC–OHC synapses from L9′T mice strongly facilitate upon stimulating the MOC fibers with 20 Hz trains. A, The average response (S), the probability of release (P), and the eIPSCs average amplitude (A) were computed for every pulse during 10 shock trains at frequencies of 10 (left) and 20 Hz (right). The normalized mean values for these parameters at the two stimulation frequencies are plotted versus the pulse number. Interestingly, the 20 Hz stimulation train caused a significant increase in the total average response (S) only in the L9′T mice. Note there is a significant increase in the probability of release (P) during the train, whereas the eIPSCs amplitude remains stable. B, Bar graphs showing the ratio between the tenth and the first pulse for the total average response (S10/S1), the release probability (P10/P1) and the amplitude of the successfully evoked postsynaptic responses (A10/A1). 10 Hz: wt, n = 5 cells from five different mice, L9′T, n = 7 cells from seven different mice; 20 Hz: wt, n = 4 cells from four different mice, L9′T n = 6 cells from five different mice; **p < 0.01, Kolmogorov–Smirnov test.
As illustrated in Figure 6A (left) and quantified in the bar graph of Figure 6B (left), there were no significant changes at a stimulation frequency of 10 Hz both for wt (n = 5) and L9′T (n = 7) mice when comparing the 10th pulse with the first one. Thus, the ratios S10/S1 (wt: 1.39 ± 0.45, L9′T: 1.00 ± 0.27, p = 0.434), P10/P1 (wt: 1.14 ± 0.38, L9′T: 1.08 ± 0.27, p = 0.939), and A10/A1 (wt: 1.3 ± 0.31, L9′T: 0.91 ± 0.1, p = 0.891) did not differ between genotypes (Kolmogorov–Smirnov test for all datasets). When applying the 20 Hz stimulation trains, evoked responses in the OHCs from wt mice did not differ in either the total averaged response, probability of release or amplitude of successful responses along the train (Fig. 6A,B, right). However, at MOC–OHC synapses from L9′T mice, both S and P were significantly enhanced along the train, whereas the mean eIPSC amplitude (A) remained unchanged. Thus, the ratios S10/S1 (wt: 0.92 ± 0.21, L9′T = 5.54 ± 1.13) and P10/P1 (wt: 0.95 ± 0.13, L9′T: 5.77 ± 1.56) were significantly different between wt and L9′T mice (n = 4 and n = 6, respectively; p = 0.009 for both parameters; Kolmogorov–Smirnov test), whereas A10/A1 remained unchanged (wt: 1.01 ± 0.23, L9′T: 1.07 ± 0.06, p = 0.92; Kolmogorov–Smirnov test). The lack of facilitation observed at MOC–OHC synapses from wt mice upon stimulating the MOC fibers with 10 and 20 Hz is in agreement with the results obtained with the paired-pulse experiments (Fig. 3).
High-frequency stimulation of MOC fibers evokes slow onset and prolonged synaptic responses in MOC–hair cell synapses from L9′T mice
Because in vivo MOC suppression experiments are usually performed at high- and prolonged-stimulation frequencies (Taranda et al., 2009), synaptic MOC–OHC responses upon electrical stimulation of the MOC fibers at 80 Hz for 10 s was assessed (Fig. 7). Longer-lasting stimulations and higher frequencies could not be analyzed because recordings became very unstable. As illustrated in Figure 7, A and B, synaptic responses at MOC–OHC synapses from both genotypes were significantly potentiated upon high-frequency 80 Hz 10 s stimulation. Significant differences between genotypes were found in the amplitude of synaptic responses at 2, 3, 4, and 5 s after the start of efferent stimulation (two-way ANOVA repeated measures; Fig. 7B). In OHCs from L9′T animals this enhancement took a longer time to reach its peak. Thus, the latency to obtain a postsynaptic response, measured as the time elapsed between the start of the stimulation protocol and the first detectable current above baseline, was significantly longer (wt: 0.031 ± 0.002 s, n = 9 and L9′T: 0.069 ± 0.007 s, n = 17; p = 0.005; Fig. 7C,D). Moreover, whereas in wt animals the response amplitude returned to baseline during the stimulation period, in L9′T mutants it persisted throughout the stimulation protocol.
Figure 7.

OHCs from L9′T mice exhibit slow and longer-lasting synaptic responses upon prolonged MOC stimulation at high frequency. A, Representative traces of responses in OHCs at 80 Hz 10 s stimulation. B, Averaged amplitude of synaptic responses normalized to the maximal response of each train in each cell was plotted as a function of time; *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA repeated measures. C, The dotted-line square drawn in A is presented at an expanded time scale to show the time it took to obtain an evoked response after the first stimulus (latency) in wt and L9′T MOC–OHC synapses. Arrows point to the first detectable current after the first shock (wt: black; L9′T: red). D, Latency of synaptic responses in wt and L9′T mice. The box plot represents the median value (line) and the interquartile range (box), with maximum and minimum points indicated with error bars. The “+” symbol indicates the mean values of the datasets. **p < 0.01, Kolmogorov–Smirnov test. wt, n = 9 cells from nine different mice; L9′T n = 17 cells from 12 different mice. E, Representative traces of OHCs in the current-clamp configuration upon 80 Hz stimulation of the MOC efferent fibers. Note the slow onset and sustained hyperpolarization throughout the stimulation period in L9′T mice.
To evaluate the changes in amplitude and time course of OHC hyperpolarization upon MOC high-frequency stimulation, experiments were performed in the current-clamp configuration (Fig. 7E). Consistent with the kinetics of responses observed in L9′T in the voltage-clamp configuration (Fig. 7A,C), L9′T animals showed a slower onset (wt: 0.0737 ± 0.006 s, n = 6; L9′T: 0.125 ± 0.0257 s, n = 5; p = 0.0476; Kolmogorov–Smirnov test) and a persistent degree of hyperpolarization (right) when compared with wt littermates (left), where the membrane resting potential even flickered back to baseline during the stimulation protocol.
MOC suppression of cochlear DPOAEs in L9′T mutants
Electrical stimulation of the MOC fibers leads to a reduction of sound-evoked motion in the cochlea and to a decrease in auditory nerve responses, indicating that the MOC system reduces the gain of the cochlea through a direct inhibition of OHC function (Guinan, 1996, 2006). Consistent with this notion, when OC fibers are electrically stimulated at the floor of the IVth ventricle, there is a significant reduction in the amplitude of DPOAEs (Maison et al., 2007a). Previous work (Fig. 8, modified from Taranda et al., 2009) assessed MOC function in vivo by measuring DPOAEs suppression with a 70 s train (200 Hz) of shocks to the OC bundle and found that in L9′T mice it was prolonged at a 22 kHz f2 primary frequency. In the present work, electrical shocks (70 s, 200 Hz) were used on DPOAEs to further analyze the effect at a 16 kHz primary frequency and to provide a detailed quantification of the kinetics of effects at two different f2 primary frequencies (16 and 22 kHz) in wt and L9′T mice. This allowed the comparison of these in vivo effects with the cochlear synaptic changes observed ex vivo. As shown in Table 2 the maximal suppressive effect with respect to the baseline was doubled both at 16 (wt vs L9′T, p = 0.0056) and 22 kHz (wt vs L9′T, p = 0.0302), was reached much more slowly (16 kHz, wt: 5.3 ± 1.0 s, L9′T: 30.8 ± 4.1 s; p = 0.0161; 22 kHz, wt: 1.5 ± 0.4 s, L9′T: 44.1 ± 3.1 s, p = 0.000007) and persisted for a sixfold longer time after the shocks for both 16 and 22 kHz in L9′T mice compared with their wt littermates (16 kHz: wt vs L9′T, p = 0.00466; 22 kHz: wt vs L9′T, p = 0.0018).
Figure 8.
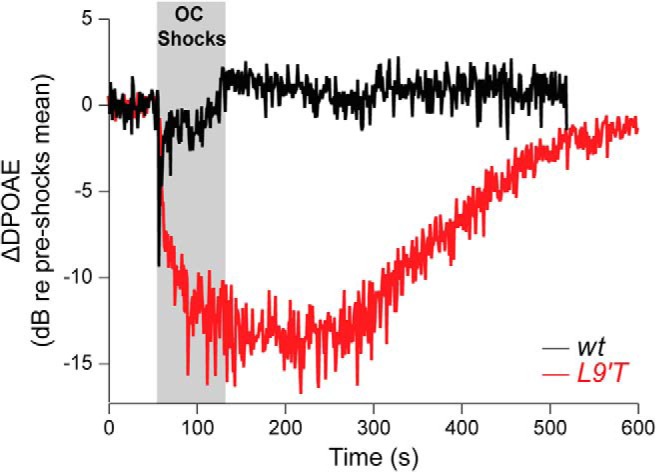
MOC-mediated DPOAE suppression is slowed, enhanced and prolonged in L9′T mice. DPOAE amplitudes were measured before, during, and after a 70 s shock train to the olivocochlear bundle. DPOAE amplitudes were normalized to the average pre-shock value and then averaged across ears within a genotype. Data acquisition times were ∼1 s/point with an f2 primary frequency at 22.6 kHz and primary levels adjusted to produce a DPOAE ∼15 dB above the noise floor. (adapted from Taranda et al., 2009, their Fig. 8).
The decrease in onset and offset together with the increase in maximal DPOAE suppressive effect of the MOC system in L9′T mice is consistent with the slow onset and prolonged synaptic MOC–OHC responses, reflected in longer latency and sustained OHC hyperpolarization at high-frequency stimulation of the MOC fibers.
Functional consequences of enhanced and sustained MOC synaptic responses in IHCs
In the mature mammalian cochlea IHCs are mainly innervated by afferent fibers. However, during postnatal development, before the onset of hearing, a transient efferent innervation is found on IHCs even before OC fibers contact their final targets, the OHCs (Simmons, 2002). This innervation, like that on mature OHCs, is cholinergic, inhibitory and is mediated by α9α10 nAChRs functionally coupled to Ca2+-activated SK2 K+ channels (Glowatzki and Fuchs, 2000; Elgoyhen et al., 2001; Katz et al., 2004; Roux et al., 2011). It has been postulated that activation of MOC fibers during this critical developmental period regulates the pattern and frequency of sensory independent, spontaneous Ca2+ action potentials (Johnson et al., 2011; Sendin et al., 2014), which drive activity in the immature auditory system, most likely as a means to direct early stages of central synapse formation (Kandler, 2004).
As illustrated in Figure 1B, IHCs from L9′T mice presented significantly slower kinetics than those of their wt littermates (Table 1, τrise and τdecay values) thus leading to prolonged IPSCs upon MOC stimulation. These might lead to altered patterns of Ca2+ action potentials in response to MOC activity in L9′T mice.
Repetitive firing of Ca2+ action potentials in IHCs from P9–P11 mice were induced by depolarizing current injections of 50 pA to the IHCs held in the current-clamp configuration. These excitatory stimuli to elicit action potentials in the IHCs were combined with 5 and 10 Hz shocks to the MOC fibers innervating the IHCs as previously described in rat IHCs (Goutman et al., 2005). Figure 9A shows representative traces in one IHC from either a wt (top, black traces) or a L9′T mouse (bottom, red traces). Firing frequencies in the IHCs were computed before (from 1.3 to 3.2 s), during (3.4–5.4 s) and after (5.5–8.5 s) stimulation of the MOC fibers. As the control firing frequency was very variable from cell to cell, both the effect of MOC activation on this parameter and the recovery to control frequency after shutting off MOC stimulation were computed in the same cell and then averaged. The bar diagram in Figure 9B (top) shows that upon stimulation of the MOC efferent fibers at 5 Hz, action potential frequency was not significantly diminished with respect to the control frequency in the IHCs from wt mice (control 5.9 ± 1.2 spikes/s, stimulation 2.4 ± 0.3 spikes/s, recovery 5.6 ± 1.46 spikes/s, n = 6, P = 0.078; Kruskal–Wallis one-way ANOVA). At a stimulation frequency of 10 Hz, in wt action potentials were significantly diminished and the control frequency was immediately recovered at the end of the stimulation period (control 6.14 ± 1.86 spikes/s, stimulation 0.2 ± 0.2 spikes/s, recovery 5.6 ± 1.2 spikes/s, n = 5, p = 0.003; Kruskal–Wallis one-way ANOVA; Fig. 9B, top). In the L9′T mice, however, activation of the MOC fibers at 5 Hz almost completely abolished action potentials in the IHCs (control 8.2 ± 2.1 spikes/s, stimulation 0.3 ± 0.2 spikes/s, recovery 3.7 ± 1.6 spikes/s, n = 6, p = 0.005; Kruskal–Wallis one-way ANOVA), and recovery to control values was only partial (Fig. 9B, bottom, bar diagrams). At a stimulation frequency of 10 Hz, action potentials were completely abolished and this silencing was sustained even after the stimulation shocks to the MOC fibers ceased. Recovery to the control frequency was almost absent in four of five IHCs tested (control 15.2 ± 6.4 spikes/s, stimulation 0.8 ± 0.8 spikes/s, recovery 1.3 ± 0.7 spikes/s, n = 6, p = 0.0009; Kruskal–Wallis one-way ANOVA), reaching ∼45% of control only in one cell (Fig. 9A, bottom right, B, bottom, bar diagram).
Figure 9.

Stimulation of the MOC efferent fibers causes a strong and persistent inhibition of Ca2+ action potentials in L9′T mice. A, Representative traces of action potential firing in one cell from a wt (top, black) and in one cell from a L9′T (bottom, red) mouse. Efferent stimulation at 5 and 10 Hz (dotted lines indicate the onset and the end of the stimulation period) caused hyperpolarization of the IHCs and diminished or suppressed firing. B, Bar graphs showing spike frequency in the IHCs at 5 and 10 Hz in both genotypes. Note that in L9′T mice, firing suppression is seen at 5 Hz and recovery after the end of the stimulation period is not complete. At 10 Hz action potentials are suppressed in both genotypes, however, in the L9′T mice, almost no recovery is observed after the stimulation period. ctrl, Control; stim, stimulation; rec, recovery. wt, n = 5 cells from three different animals; L9′T, n = 5 cells from four different mice. *p < 0.05, **p < 0.01; Kruskal–Wallis one-way ANOVA. C, Representative averaged traces of 20 individual synaptic responses in IHCs from both wt (black, n = 4 from 4 different mice) and L9′T mice (red, n = 3 from 3 different mice) upon a 10-pulse 10 Hz stimulation of the MOC fibers. Dotted lines indicate the beginning and the end of the stimulation period. D, Bar diagram depicting the total charge (Q) computed at the end of the protocol for both genotypes, at 10 Hz. *p < 0.05, Kolmogorov–Smirnov test.
This enhanced inhibition of action potentials and lack of recovery after the end of MOC fiber stimulation at 10 Hz in L9′T mice could be accounted for by a potentiated and sustained synaptic transmission at the MOC–IHC synapse, as shown for the MOC–OHC synapse (Fig. 5A). Responses of voltage-clamped IHCs to 10-pulse trains of stimulation at 10 Hz to the MOC fibers remained strongly potentiated after the end of the shocks (Fig. 9C). As shown in Figure 9D the total charge accumulated in the IHC during the recording period was significantly larger in L9′T mice (77.37 ± 7.06 pC, n = 3) when compared with wt (15.81 ± 3.16 pC, n = 5, p = 0.0357; Kolmogorov–Smirnov test). These results can account for the stronger inhibition of action potentials observed in the L9′T mice at lower frequencies (Fig. 9A,B). In addition, the lack of recovery after the end of the stimulation period in these mice (Fig. 9A,B, bottom), is consistent with the persistence of potentiated evoked responses in IHCs even after the end of the stimulation train (Fig. 9C).
Discussion
The present work shows that in mice with a mutation in the α9 nAChR subunit (Taranda et al., 2009) that confers much longer-lasting channel openings (Plazas et al., 2005), sustained high-frequency stimulation of MOC terminals to OHCs in an ex vivo cochlear explant preparation leads to slow onset, enhanced and prolonged evoked synaptic responses. At the in vivo physiological level this is translated into a slow onset, enhancement, and persistence of MOC efferent-mediated DPOAE suppression. This demonstrates that MOC–OHC synapse kinetics is sufficient to directly guide the dynamics of MOC feedback to the cochlea as previously proposed (Ballestero et al., 2011). Moreover, it suggests that pharmacological interventions which mimic the modifications produced by the α9 nAChR subunit mutation might enhance MOC activity and therefore serve as a tool to enhance MOC-mediated gain inhibition at the auditory periphery and prevent acoustic trauma (Maison et al., 2007b; Taranda et al., 2009).
The slower kinetics of evoked synaptic currents of L9′T mice, with the consequent increase in the total charge accumulated, most likely results from the nature of the point mutation introduced in the channel pore of the α9 nAChR subunit (Plazas et al., 2005). This produces a change in the α9α10 nAChR gating properties with an increase in the apparent mean open time and stabilization of the open state of the mutant receptor at the single channel level and a slow onset and decreased desensitization rate of ACh-evoked currents (Plazas et al., 2005). Although the 70 s, 200 Hz shocks applied for the in vivo MOC stimulation could not be reproduced in the ex vivo cochlear explant, since recordings in the OHCs become very unstable, the prolonged and high-frequency (10 s, 80 Hz) electrical stimulation of efferent terminals to OHCs, exhibited similar changes in response profiles. The observation that the magnitude of changes was more pronounced in vivo than ex vivo, may derive from these differences in stimulation parameters, together with the fact that synaptic responses were recorded from one OHC whereas DPOAEs arise as electrical distortions in electromechanical transduction within the inner ear, that are reverse-transduced into mechanical signals and amplified by (multiple) OHCs (Lukashkin et al., 2002). Moreover, and again due to experimental limitations of electrophysiological recordings from OHCs, these were performed at room temperature in the apical turn of the cochlea around hearing onset, whereas DPOAEs were elicited at physiological temperature, in 6-week-old mice and in the midbasal turn of the cochlea (10–20 kHz), a region with the highest density of MOC innervation (Maison et al., 2003). Moreover, in addition to the immediate effect of enhanced synaptic inhibition of OHC membrane conductance seen in L9′T mutants, the prolonged Ca2+ signals also contribute to increased DPOAE suppression. Thus, it has been reported that prolonged application of ACh causes a sustained increase in OHC length and a decrease in axial stiffness attributed to cytoplasmic Ca2+ acting on the cytoskeleton (Dallos et al., 1997). In this regard, efferent suppression of compound action potentials and enhancement of cochlear microphonic potentials occurs over fast (50–100 ms) and slow (25–50 s) time courses both of which are attributed to signaling by cytoplasmic calcium triggered by the same nAChR (Sridhar et al., 1995, 1997). Finally, as observed in α9 knock-out mice (Clause et al., 2014), some compensatory changes might occur in the central auditory pathway in L9′T mice caused by prolonged silencing of pre-hearing IHCs Ca2+ action potentials.
Morphological changes in efferent innervation to OHCs have been previously reported in L9′T and α9α10 nAChR mutant mice (Vetter et al., 1999, 2007; Murthy et al., 2009a,b; Fuchs et al., 2014). This resembles what has been described for other neuronal nAChR-mediated synapses like the superior cervical ganglia, where the genetic ablation of the α3 nAChR subunit, leads to larger presynaptic terminals and clusters (Krishnaswamy and Cooper, 2009). The present work shows that, in addition to changes in presynaptic morphology, increased nAChR gating-derived postsynaptic changes were also accompanied by physiological modifications at the presynaptic level. The mechanisms leading to these changes remain unknown. However, the increased synaptic terminal volume might in part account for the reduced quantum content of evoked release in L9′T mice as the fixed amount of current reaching the MOC synaptic terminals after an action potential would be less effective in depolarizing the enlarged synaptic membranes. Therefore, Ca2+ influx through presynaptic voltage-gated Ca2+ channels would diminish, thus causing a reduction in the release probability. In addition, similar to that proposed by Krishnaswamy and Cooper (2009), one could also speculate that a retrograde signal might originate downstream of the strong postsynaptic activity that leads to increased Ca2+ influx, and thus modify the presynaptic ACh release machinery.
Synapses with low quantum content tend to facilitate upon high-frequency stimulation (Fioravante and Regehr, 2011; Jackman and Regehr, 2017), as is the case of the efferent synapse to turtle auditory hair cells (Art et al., 1984), rat neonatal IHCs (Goutman et al., 2005), and mouse OHCs (Ballestero et al., 2011). This was confirmed by the fact that when compared with wild-type, in L9′T mutant mice the lower quantum content was accompanied by a stronger synaptic facilitation. Although the size of the readily releasable pool of vesicles is unknown, the observation that efferent endings of both animals have a large number of synaptic vesicles (Fuchs et al., 2014), suggests that vesicle availability is not the limiting factor. Moreover, the fact that release can be sustained at a very high stimulation frequency further supports the latter conclusion. In addition to an increase in presynaptic facilitation, the strengthening of transmission in L9′T mice is most likely also accounted for by summation of postsynaptic responses, as described for wt mice (Ballestero et al., 2011), due to the longer decay times of the IPSCs which lead to prolonged Ca2+ transients and the consequent sustained SK2 activity. Ca2+ transients could, in addition, be altered by the enlarged cisternal volumes in L9′T mice OHCs (Fuchs et al., 2014). Moreover, since Ca2+ transients are wider and decay with a slower time course, the Ca2+ load might be probably higher and therefore, SK2 channels could continue to be activated even after the closure of the nAChR.
The observation that in the L9′T mice facilitation is preserved and the probability of release increases in a frequency-dependent manner indicates that, as observed in wt mice (Ballestero et al., 2011), in mutant mice the MOC–OHC synapse still behaves as a high pass filter (Atluri and Regehr, 1996; Jackman and Regehr, 2017), since it fails to transmit during low-frequency action potentials but facilitates effectively during sustained firing. Thus, as reported for several species, L9′T mice OHCs will ignore spontaneous or low-frequency MOC activity and scale inhibition of OHCs (Galambos, 1956; Wiederhold and Kiang, 1970; Art and Fettiplace, 1984; Brown and Nuttal, 1984; Gifford and Guinan, 1987) as a function of sound intensity (Robertson and Gummer, 1985; Liberman and Brown, 1986). The observation that compared with wild-type, similar stimulation frequency produces enhanced MOC inhibition in L9′T mice, is in accordance with the reported protection from permanent hearing threshold shifts when exposed to loud sounds (Taranda et al., 2009). Although very few drugs activate the α9α10 nAChR (Verbitsky et al., 2000), compounds that mimic the effect of the L9′T mutation might result beneficial in the search for compounds to prevent noise-induced damage (Holley, 2005; Lynch and Kil, 2005; Elgoyhen et al., 2009). Positive allosteric modulators of other nAChRs which activate the receptor in the absence of the agonist and decrease the desensitization rate (Wang and Lindstrom, 2017) might result useful as lead molecules for drug design.
The application of ACh (Glowatzki and Fuchs, 2000) or the electrical stimulation of efferent terminals (Goutman et al., 2005) results in hyperpolarization of IHCs and reduction in the frequency of sensory-independent spontaneous Ca2+ action potentials. These spontaneous action potentials are present in developing IHCs (Kros et al., 1998; Marcotti et al., 2003) and proposed to be required for the maturation of the developing auditory pathway and the correct establishment of tonotopic maps (Brandt et al., 2003; Leao et al., 2006; Erazo-Fischer et al., 2007). Although discrepancy still exists regarding the nature of cholinergic modulation of action potential firing (Tritsch et al., 2010; Johnson et al., 2011; Sendin et al., 2014), recent findings show that the ablation of the cholinergic efferent system leads to altered establishment of central auditory synaptic connections (Clause et al., 2014) and hearing deficits (Clause et al., 2017). In line with the prolonged synaptic responses upon high-frequency MOC stimulation in IHCs of L9′T mutant mice, a much enhanced and prolonged inhibition of action potentials was observed at lower stimulation frequencies compared with wt. Thus, the L9′T mouse arises as a valuable tool to further analyze the participation of the spontaneous action potentials and efferent innervation in auditory pathway development.
Footnotes
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica, Argentina (A.B.E.), and NIH Grant R01 DC001508 (P.A.F., A.B.E.). We thank Dr. Juan D. Goutman for assistance with data analysis through routines implemented in Igor Pro and for critical comments on the paper.
The authors declare no competing financial interests.
References
- Art JJ, Fettiplace R (1984) Efferent desensitization of auditory nerve fibre responses in the cochlea of the turtle Pseudemys scripta elegans. J Physiol 356:507–523. 10.1113/jphysiol.1984.sp015480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Art JJ, Fettiplace R, Fuchs PA (1984) Synaptic hyperpolarization and inhibition of turtle cochlear hair cells. J Physiol 365:525–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore JF. (1987) A fast motile response in guinea pig outer hair cells: the cellular basis of the cochlear amplifier. J Physiol 388:323–347. 10.1113/jphysiol.1987.sp016617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG (1996) Determinants of the time course of facilitation at the granule cell to purkinje cell synapse. J Neurosci 16:5661–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestero J, Zorrilla de San Martín J, Goutman J, Elgoyhen AB, Fuchs P, Katz E (2011) Short-term synaptic plasticity regulates the level of olivocochlear inhibition to auditory hair cells. J Neurosci 31:14763–14774. 10.1523/JNEUROSCI.6788-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet C, Eróstegui C, Sugasawa M, Dulon D (1996) Acetylcholine-induced potassium current of guinea pig outer hair cells: its dependence on a calcium influx through nicotinic-like receptors. J Neurosci 16:2574–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T (2003) CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci 23:10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC. (1989) Morphology and response properties of single olivocochlear fibers in the guinea pig. Hear Res 40:93–109. 10.1016/0378-5955(89)90103-2 [DOI] [PubMed] [Google Scholar]
- Brown MC, Nuttall AL (1984) Efferent control of cochlear inner hair cell responses in guinea-pig. J Physiol 354:625–646. 10.1113/jphysiol.1984.sp015396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell WE, Bader CR, Bertrand D, de Ribaupierre Y (1985) Evoked mechanical responses of isolated cochlear hair cells. Science 227:194–196. 10.1126/science.3966153 [DOI] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rűbsamen R, Kandler K (2014) The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82:822–835. 10.1016/j.neuron.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Lauer AM, Kandler K (2017) Mice lacking the alpha9 subunit of the nicotinic acetylcholine receptor exhibit deficits in frequency difference limens and sound localization. Front Cell Neurosci 11:167. 10.3389/fncel.2017.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P. (1992) The active cochlea. J Neurosci 12:4575–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P. (2008) Cochlear amplification, outer hair cells and prestin. Curr Opin Neurobiol 18:370–376. 10.1016/j.conb.2008.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P, He DZ, Lin X, Sziklai I, Mehta S, Evans BN (1997) Acetylcholine, outer hair cell electromotility, and the cochlear amplifier. J Neurosci 17:2212–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1954) Statistical factors involved in neuromuscular facilitation and depression. J Physiol 124:574–585. 10.1113/jphysiol.1954.sp005130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon D, Luo L, Zhang C, Ryan AF (1998) Expression of small-conductance calcium-activated potassium channels (SK) in outer hair cells of the rat cochlea. Eur J Neurosci 10:907–915. 10.1046/j.1460-9568.1998.00098.x [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994) α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79:705–715. 10.1016/0092-8674(94)90555-X [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001) α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98:3501–3506. 10.1073/pnas.051622798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Katz E, Fuchs PA (2009) The nicotinic receptor of cochlear hair cells: a possible pharmacotherapeutic target? Biochem Pharmacol 78:712–719. 10.1016/j.bcp.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo-Fischer E, Striessnig J, Taschenberger H (2007) The role of physiological afferent nerve activity during in vivo maturation of the calyx of held synapse. J Neurosci 27:1725–1737. 10.1523/JNEUROSCI.4116-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21:269–274. 10.1016/j.conb.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs PA, Murrow BW (1992) A novel cholinergic receptor mediates inhibition of chick cochlear hair cells. Proc Biol Sci 248:35–40. 10.1098/rspb.1992.0039 [DOI] [PubMed] [Google Scholar]
- Fuchs PA, Lehar M, Hiel H (2014) Ultrastructure of cisternal synapses on outer hair cells of the mouse cochlea. J Comp Neurol 522:717–729. 10.1002/cne.23478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galambos R. (1956) Suppression of auditory nerve activity by stimulation of efferent fibers to the cochlea. J Neurophysiol 19:424–437. 10.1152/jn.1956.19.5.424 [DOI] [PubMed] [Google Scholar]
- Gifford ML, Guinan JJ Jr (1987) Effects of electrical stimulation of medial olivocochlear neurons on ipsilateral and contralateral cochlear responses. Hear Res 29:179–194. 10.1016/0378-5955(87)90166-3 [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA (2000) Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288:2366–2368. 10.1126/science.288.5475.2366 [DOI] [PubMed] [Google Scholar]
- Goutman JD, Fuchs PA, Glowatzki E (2005) Facilitating efferent inhibition of inner hair cells in the cochlea of the neonatal rat. J Physiol 566:49–59. 10.1113/jphysiol.2005.087460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinan JJ. (1996) Physiology of olivocochlear efferents. In: The cochlea (Dallos P, Popper AN, Fay RR, eds.), pp. 435–502. New York, NY: Springer. [Google Scholar]
- Guinan JJ., Jr (2006) Olivocochlear efferents: anatomy, physiology, function, and the measurement of efferent effects in humans. Ear Hear 27:589–607. 10.1097/01.aud.0000240507.83072.e7 [DOI] [PubMed] [Google Scholar]
- Holley MC. (2005) Keynote review: the auditory system, hearing loss and potential targets for drug development. Drug Discov Today 10:1269–1282. 10.1016/S1359-6446(05)03595-6 [DOI] [PubMed] [Google Scholar]
- Housley GD, Ashmore JF (1991) Direct measurement of the action of acetylcholine on isolated outer hair cells of the guinea pig cochlea. Proc Biol Sci 244:161–167. 10.1098/rspb.1991.0065 [DOI] [PubMed] [Google Scholar]
- Hudspeth AJ. (2014) Integrating the active process of hair cells with cochlear function. Nat Rev Neurosci 15:600–614. 10.1038/nrn3786 [DOI] [PubMed] [Google Scholar]
- Jackman SL, Regehr WG (2017) The mechanisms and functions of synaptic facilitation. Neuron 94:447–464. 10.1016/j.neuron.2017.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ, Marcotti W (2011) Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci 14:711–717. 10.1038/nn.2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K. (2004) Activity-dependent organization of inhibitory circuits: lessons from the auditory system. Curr Opin Neurobiol 14:96–104. 10.1016/j.conb.2004.01.017 [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R (1968) The role of calcium in neuromuscular facilitation. J Physiol 195:481–492. 10.1113/jphysiol.1968.sp008469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB (2014) Short-term plasticity and modulation of synaptic transmission at mammalian inhibitory cholinergic olivocochlear synapses. Front Syst Neurosci 8:224. 10.3389/fnsys.2014.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gómez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E (2004) Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci 24:7814–7820. 10.1523/JNEUROSCI.2102-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase T, Delgutte B, Liberman MC (1993) Antimasking effects of the olivocochlear reflex: II. enhancement of auditory-nerve response to masked tones. J Neurophysiol 70:2533–2549. 10.1152/jn.1993.70.6.2533 [DOI] [PubMed] [Google Scholar]
- Krishnaswamy A, Cooper E (2009) An activity-dependent retrograde signal induces the expression of the high-affinity choline transporter in cholinergic neurons. Neuron 61:272–286. 10.1016/j.neuron.2008.11.025 [DOI] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rüsch A (1998) Expression of a potassium current in inner hair cells during development of hearing in mice. Nature 394:281–284. 10.1038/28401 [DOI] [PubMed] [Google Scholar]
- Leao RN, Sun H, Svahn K, Berntson A, Youssoufian M, Paolini AG, Fyffe RE, Walmsley B (2006) Topographic organization in the auditory brainstem of juvenile mice is disrupted in congenital deafness. J Physiol 571:563–578. 10.1113/jphysiol.2005.098780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Brown MC (1986) Physiology and anatomy of single olivocochlear neurons in the cat. Hear Res 24:17–36. 10.1016/0378-5955(86)90003-1 [DOI] [PubMed] [Google Scholar]
- Lioudyno M, Hiel H, Kong JH, Katz E, Waldman E, Parameshwaran-Iyer S, Glowatzki E, Fuchs PA (2004) A “synaptoplasmic cistern” mediates rapid inhibition of cochlear hair cells. J Neurosci 24:11160–11164. 10.1523/JNEUROSCI.3674-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashkin AN, Lukashkina VA, Russell IJ (2002) One source for distortion product otoacoustic emissions generated by low- and high-level primaries. J Acoust Soc Am 111:2740–2748. 10.1121/1.1479151 [DOI] [PubMed] [Google Scholar]
- Lynch ED, Kil J (2005) Compounds for the prevention and treatment of noise-induced hearing loss. Drug Discov Today 10:1291–1298. 10.1016/S1359-6446(05)03561-0 [DOI] [PubMed] [Google Scholar]
- Maison SF, Adams JC, Liberman MC (2003) Olivocochlear innervation in the mouse: immunocytochemical maps, crossed versus uncrossed contributions, and transmitter colocalization. J Comp Neurol 455:406–416. 10.1002/cne.10490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Parker LL, Young L, Adelman JP, Zuo J, Liberman MC (2007a) Overexpression of SK2 channels enhances efferent suppression of cochlear responses without enhancing noise resistance. J Neurophysiol 97:2930–2936. 10.1152/jn.01183.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Vetter DE, Liberman MC (2007b) A novel effect of cochlear efferents: in vivo response enhancement does not require alpha9 cholinergic receptors. J Neurophysiol 97:3269–3278. 10.1152/jn.00067.2007 [DOI] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Rusch A, Kros CJ (2003) Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol 552:743–761. 10.1113/jphysiol.2003.043612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moglie MJ, Fuchs PA, Elgoyhen AB, Goutman JD (2018) Compartmentalization of antagonistic Ca2+ signals in developing cochlear hair cells. Proc Natl Acad Sci U S A 115:E2095–E2104. 10.1073/pnas.1719077115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Taranda J, Elgoyhen AB, Vetter DE (2009a) Activity of nAChRs containing α9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev Neurobiol 69:931–949. 10.1002/dneu.20753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Maison SF, Taranda J, Haque N, Bond CT, Elgoyhen AB, Adelman JP, Liberman MC, Vetter DE (2009b) SK2 channels are required for function and long-term survival of efferent synapses on mammalian outer hair cells. Mol Cell Neurosci 40:39–49. 10.1016/j.mcn.2008.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Klöcker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B (2000) Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26:595–601. 10.1016/S0896-6273(00)81197-6 [DOI] [PubMed] [Google Scholar]
- Plazas PV, De Rosa MJ, Gomez-Casati ME, Verbitsky M, Weisstaub N, Katz E, Bouzat C, Elgoyhen AB (2005) Key roles of hydrophobic rings of TM2 in gating of the alpha9alpha10 nicotinic cholinergic receptor. Br J Pharmacol 145:963–974. 10.1038/sj.bjp.0706224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D, Gummer M (1985) Physiological and morphological characterization of efferent neurones in the guinea pig cochlea. Hear Res 20:63–77. 10.1016/0378-5955(85)90059-0 [DOI] [PubMed] [Google Scholar]
- Robinson BL, McAlpine D (2009) Gain control mechanisms in the auditory pathway. Curr Opin Neurobiol 19:402–407. 10.1016/j.conb.2009.07.006 [DOI] [PubMed] [Google Scholar]
- Roux I, Wersinger E, McIntosh JM, Fuchs PA, Glowatzki E (2011) Onset of cholinergic efferent synaptic function in sensory hair cells of the rat cochlea. J Neurosci 31:15092–15101. 10.1523/JNEUROSCI.2743-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendin G, Bourien J, Rassendren F, Puel JL, Nouvian R (2014) Spatiotemporal pattern of action potential firing in developing inner hair cells of the mouse cochlea. Proc Natl Acad Sci U S A 111:1999–2004. 10.1073/pnas.1319615111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DD. (2002) Development of the inner ear efferent system across vertebrate species. J Neurobiol 53:228–250. 10.1002/neu.10130 [DOI] [PubMed] [Google Scholar]
- Sridhar TS, Liberman MC, Brown MC, Sewell WF (1995) A novel cholinergic “slow effect” of efferent stimulation on cochlear potentials in the guinea pig. J Neurosci 15:3667–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar TS, Brown MC, Sewell WF (1997) Unique postsynaptic signaling at the hair cell efferent synapse permits calcium to evoke changes on two time scales. J Neurosci 17:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga N, Gao E, Zhang Y, Ma X, Olsen JF (2000) The corticofugal system for hearing: recent progress. Proc Natl Acad Sci U S A 97:11807–11814. 10.1073/pnas.97.22.11807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs PA, Elgoyhen AB (2009) A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol 7:e18. 10.1371/journal.pbio.1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Rodríguez-Contreras A, Crins TT, Wang HC, Borst JG, Bergles DE (2010) Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat Neurosci 13:1050–1052. 10.1038/nn.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB (2000) Mixed nicotinic–muscarinic properties of the α9 nicotinic cholinergic receptor. Neuropharmacology 39:2515–2524. 10.1016/S0028-3908(00)00124-6 [DOI] [PubMed] [Google Scholar]
- Vetter DE, Lieberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kollman J, Heinemann SF, Elgoyhen AB (1999) Role of α9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23:93–103. 10.1016/S0896-6273(00)80756-4 [DOI] [PubMed] [Google Scholar]
- Vetter DE, Katz E, Maison SF, Taranda J, Turcan S, Ballestero J, Liberman MC, Elgoyhen AB, Boulter J (2007) The α10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc Natl Acad Sci U S A 104:20594–20599. 10.1073/pnas.0708545105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lindstrom J (2017) Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br J Pharmacol. Advance online publication. Retrieved Feb 15, 2017. doi: 10.1111/bph.13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer C, Zorrilla de San Martín J, Ballestero J, Gómez-Casati ME, Torbidoni AV, Fuchs PA, Bettler B, Elgoyhen AB, Katz E (2013) Activation of presynaptic GABAB(1a,2) receptors inhibits synaptic transmission at mammalian inhibitory cholinergic olivocochlear–hair cell synapses. J Neurosci 33:15477–15487. 10.1523/JNEUROSCI.2554-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wersinger E, McLean WJ, Fuchs PA, Pyott SJ (2010) BK channels mediate cholinergic inhibition of high-frequency cochlear hair cells. PLoS One 5:e13836. 10.1371/journal.pone.0013836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold ML, Kiang NY (1970) Effects of electrical stimulation of the crossed olivocochlear bundle on cat single auditory nerve fibres. J Acoust Soc Am 48:950–965. 10.1121/1.1912234 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]