

Abstract
Background
Antenatal infection (i.e., chorioamnionitis) is an important risk factor for adverse neurodevelopmental outcomes after preterm birth. Destructive and developmental disturbances of the white matter are hallmarks of preterm brain injury. Understanding the temporal effects of antenatal infection in relation to the onset of neurological injury is crucial for the development of neurotherapeutics for preterm infants. However, these dynamics remain unstudied.
Methods
Time-mated ewes were intra-amniotically injected with lipopolysaccharide at 5, 12, or 24 h or 2, 4, 8, or 15 days before preterm delivery at 125 days gestational age (term ~ 150 days). Post mortem analyses for peripheral immune activation, neuroinflammation, and white matter/neuronal injury were performed. Moreover, considering the neuroprotective potential of erythropoietin (EPO) for perinatal brain injury, we evaluated (phosphorylated) EPO receptor (pEPOR) expression in the fetal brain following LPS exposure.
Results
Intra-amniotic exposure to this single bolus of LPS resulted in a biphasic systemic IL-6 and IL-8 response. In the developing brain, intra-amniotic LPS exposure induces a persistent microgliosis (IBA-1 immunoreactivity) but a shorter-lived increase in the pro-inflammatory marker COX-2. Cell death (caspase-3 immunoreactivity) was only observed when LPS exposure was greater than 8 days in the white matter, and there was a reduction in the number of (pre) oligodendrocytes (Olig2- and PDGFRα-positive cells) within the white matter at 15 days post LPS exposure only. pEPOR expression displayed a striking biphasic regulation following LPS exposure which may help explain contradicting results among clinical trials that tested EPO for the prevention of preterm brain injury.
Conclusion
We provide increased understanding of the spatiotemporal pathophysiological changes in the preterm brain following intra-amniotic inflammation which may aid development of new interventions or implement interventions more effectively to prevent perinatal brain damage.
Keywords: Chorioamnionitis, Fetal, Preterm, Sheep, Inflammation, Brain injury, Erythropoietin, EPO receptor
Background
Antenatal infections (i.e., chorioamnionitis) are an important risk factor for preterm birth and a major contributor to neonatal morbidity and mortality [1, 2]. Intra-amniotic exposure to microorganisms and subsequent induction of inflammatory mediators in the amniotic cavity can initiate a fetal systemic immune response that is characterized by increased plasma interleukin (IL)-6 and IL-8 concentrations [3], and (persistent) changes in essential immunological organs including the fetal spleen and thymus [4, 5]. At the crosstalk between fetal peripheral blood and the brain (i.e., blood-brain barrier), this systemic inflammatory response can initiate a detrimental neuroinflammatory response which is primarily mediated by microglia and peripheral immune effector cells [6, 5]. This cerebral inflammatory response is a risk factor for preterm brain injury and concomitant adverse neurodevelopmental outcomes including cognitive, behavioral, and attentional impairments and motor dysfunctions (i.e., cerebral palsy) [7, 5].
In a pre-clinical chorioamnionitis model, we showed that short-term (2 days) intra-amniotic exposure to lipopolysaccharide (LPS) resulted in systemic inflammation, overt microgliosis, and changes in myelin basic protein (MBP) immunoreactivity (IR) in the fetal ovine brain [8]. However, this systemic and cerebral phenotype was substantially different following longer exposure time (7 days) indicating that time-dependent peripheral and cerebral changes occur following intra-amniotic inflammation. Moreover, we and others have shown that inflammation can modulate a second inflammatory stimulus through either preconditioning or sensitization of the fetal brain [9, 8, 10]. Taken together, this emphasizes that inflammation as pathogenic mediator for brain damage is not a single trigger within a short time frame but more a dynamic process over an extended period of time. Therefore, detailed studies elucidating the time-dependent effects of antenatal infection/inflammation in relation to neurological injury and development are crucial to gain insight in the pathophysiological changes in the fetal brain following antenatal stress. Importantly, such temporal insight in the induction of brain injury following antenatal stress is also essential to define the therapeutic window of opportunity for neurotherapeutics.
One of the most promising treatment options for preterm neonates at high risk for brain injury is erythropoietin (EPO), an important cytokine for brain development [11, 12]. Multiple experimental and clinical studies have demonstrated efficacy of EPO administration to prevent injury to the preterm brain, including severe periventricular leukomalacia, without adverse effects [13–21]. In contrast, other clinical trials do not report improvement in neurodevelopment following EPO treatment [22, 23]. Effects of EPO are mediated by its receptor, which is abundantly present on (pre) oligodendrocytes, astrocytes, microglia, and neurons. EPO binding triggers phosphorylation of two monomers, which in turn phosphorylates and activates the signaling kinase Jak-2 facilitating effects including its anti-inflammatory, anti-oxidative, and anti-apoptotic properties [24, 25]. In addition, EPO enhances neuro- and oligodendrogenesis, oligodendrocyte maturation, and myelin production which are indispensable events in injury repair and normal neurodevelopment [26]. We hypothesize that changes in basal levels of EPO receptor activation in response to inflammation or perinatal stress might explain at least part of the differences in clinical outcomes following EPO treatment.
Considering the clinical need for understanding the time-dependent cerebral changes following intra-amniotic inflammation, we performed a detailed analysis of the temporal dynamics of intra-amniotic LPS-induced systemic and cerebral inflammation and subsequent fetal brain injury. In addition, to optimize EPO treatment in the clinical setting, we analyzed the temporal expression of the phosphorylated EPO receptor (pEPOR) in the course of intra-amniotic inflammation.
Methods
Study approval
Animal procedures were performed with approval of the animal ethics committee of the University of Western Australia (Perth, Australia).
Experimental design
The design of this study was published previously [4]. Briefly, 52 time-mated ewes with singleton fetuses were randomly allocated in groups of 5–7 animals per group to receive an intra-amniotic injection under ultrasound guidance with an established dose [27] of 10 mg Escherichia coli-derived LPS (O55:B5; Sigma-Aldrich, St. Louis, MO) at 5, 12, or 24 h or 2, 4, 8, or 15 days before preterm delivery at 125 days of gestation (term ~ 150 days) (Fig. 1). This paradigm is based on the clinical paradigm, where we know the gestational age of the infant but not the length of exposure to inflammation. As such, all our tissues were collected at a known gestation age but inflammation was induced at various times before. The half-life time of LPS in the amniotic fluid is relatively long (1.7 days) and LPS concentrations remain detectable till 15 days after injection [28]. Moreover, intra-amniotic delivery of 0.1 mg LPS, a bolus which in this study is reached at 10 days after injection, still results in an influx of inflammatory cells in the amniotic fluid and fetal lungs [27] indicating that IA delivery of 10 mg LPS is a clinical relevant ongoing inflammatory stimulus. Fetuses of either sex were used, and previous analysis of the thymus reported no sex specific differences in this model [4]. Control animals received an equivalent volume of 0.9% saline solution (SAL; controls) at variable gestational ages comparable to LPS injections, ranging from 5 h to 15 days before preterm delivery. Within this control group, no differences were observed between different lengths of saline exposure for which we have pooled these animals in one control group (SAL). At 125 days of gestation, when ovine brain development is similar to 32–34 weeks of human gestation [29], all fetuses were surgically delivered and immediately euthanized with intravenous pentobarbitone (100 mg/kg). Fetal blood was collected and the brains were removed and immersion fixed in 4% paraformaldehyde.
Fig. 1.
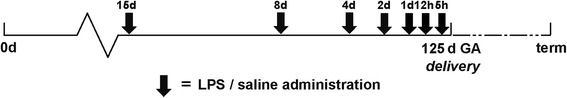
Study design. Pregnant ewes received an intra-amniotic injection with 10 mg Escherichia coli-derived lipopolysaccharide (LPS) at 5, 12, or 24 h or 2, 4, 8, or 15 days (black arrows) before preterm delivery at 122 days of gestation (term ~ 150 days). Control animals received an intra-amniotic injection with an equivalent volume of 0.9% saline solution at comparable time points to LPS injections
Analysis of blood IL-6 and IL-8 concentration
Levels of the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 were measured in fetal plasma as markers for systemic inflammation using ovine-specific sandwich enzyme-linked immunosorbent assays (ELISA) as previously described [8].
In short, a 96-wells plate was coated with a monoclonal mouse-anti IL-6 (Millipore Cat# MAB1004, working concentration 1:200) or IL-8 (Millipore Cat# MAB1044, working concentration 1:200) and incubated overnight at 4 °C. The standard curve and serum samples were diluted in PBS + 0.1% BSA in 1:1 or 1:80, respectively, for IL-6 and IL-8. Incubation with the detection antibody rabbit-anti-ovine IL-6 (Millipore Cat# AB1839, working concentration 1:500) or IL-8 (AB1040, Millipore, working concentration 1:500) was performed for 1 h, followed by incubation with a HRP-labeled antibody (Jackson ImmunoResearch Labs Cat# 111-035-045, working concentration 1:500). Next, incubation with 3,3′5,5′-tetramethylbenzidine (TMB) substrate solution was done for 10 (IL-6) or 2,5 (IL-8) minutes. The reaction was stopped by addition of H2SO4, and the optical density (OD) was measured at 450 nm in a Thermo Electron Type 1500 Multiskan Spectrum Microplate Reader. Concentrations were expressed relative to a standard curve of recombinant ovine IL-6 or IL-8 (ImmunoChemistry Technologies, Bloomington, MN, USA).
Histology and immunohistochemistry
The cerebral white matter and hippocampus are most commonly affected by intra-amniotic infections at this developmental stage [30]. Therefore, we have chosen to assess inflammatory and structural changes within these regions of interest. After fixation, a predefined region containing the posterior hippocampus/mid-thalamus of the left hemisphere was embedded in paraffin and serial coronal sections (4 μm) were cut with a Leica RM2235 microtome. Hematoxylin and eosin (H&E) staining was performed for structural and morphological analysis. Immunohistochemical staining was performed on four slides per staining per animal (every 10th consecutive slide) as previously reported [8]. Inflammatory changes were assessed by the following immunohistochemical markers: cylcooxygenase-2 (COX-2) (1:50, Cayman Chemical; aa570-598), ionized calcium-binding adapter molecule 1 (IBA-1) (1:1000, Wako Pure Chemical Industries, Osaka, Japan), and glial fibrillary acidic protein (GFAP) (1:1000, DAKO Z0334). The presence of neutrophils was assessed by myeloperoxidase (MPO) staining (1:200, DAKO A0398). Markers used to assess alterations in the white matter including oligodendrocyte differentiation were oligodendrocyte transcription factor 2 (Olig2) (1:200, Millipore, 13 AB9610), platelet-derived growth factor receptor alpha (PDGFRa) (1:100, Santa Cruz Biotechnology, sc338), 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) (1:1000, Sigma, C5922), and myelin basic protein (MBP) (1:1000, Merck Millipore, MAB386). Neuronal architecture, including cell bodies and dendrites, was determined by microtubule-associated protein-2 (MAP-2) (1:500, Sigma, M9942). Apoptotic cell death was measured as cells positive for cleaved caspase-3 (1:1000, cell signaling, #9661), and the number of mitotic cells were identified by phospho-Histone H3 (pHH3) (1:100, Santa Cruz Biotechnology, sc-101,679). The presence of the erythropoietin receptor was assessed by measuring the expression of the (phosphorylated) erythropoietin receptor (EPOR and pEPOR) (1:200, Santa Cruz, SC-365662 and SC-20236).
Deparaffinization and rehydration was performed by incubation in xylol and decreasing alcohol concentrations. Endogenous peroxidase activity was quenched via incubation with 0.3% H2O2 for 10 min. Antigen retrieval involved boiling tissues in citrate buffer (pH 6.0) for 10 min or for pEPOR proteinase K at 37 °C for 5 min. Nonspecific binding was prevented by incubation with 5% (IBA-1, GFAP, MAP2, pHH3) or 10% (MPO, CNPase) normal goat serum, 5% bovine serum albumin (COX-2, MBP, Olig2, EPOR) (Invitrogen Thermofisher Scientific), or 10% nonfat dry milk (pEPOR; Elk, Campina bv., Eindhoven, The Netherlands) for 1 h. Tissues were incubated with the primary antibody overnight at 4 °C, followed by incubation with the species specific secondary antibody at 1:200 (DAKO) for 1 h at room temperature. The antibody-specific signal was enhanced with a Vectastain ABC peroxidase Elite kit (Vector Laboratories Inc., Burlingame, CA) for 1 h and 3,3′-diaminobenzide (COX-2, IBA-1, GFAP, MPO, PDGFRa, CNPase, MBP, MAP-2, pHH3, EPOR) or nickel chloride 3,3′-diaminobenzide (Olig2, cleaved caspase-3, pEPOR) for 2–10 min. Nuclei were stained with Mayer’s hematoxylin.
Qualitative and quantitative analysis
An independent neuropathologist and two independent researchers who were blinded for the experimental conditions performed qualitative and quantitative analysis of the tissues. Analysis was performed using a light microscope (Leica DM2000) equipped with Leica QWin Pro version 3.4.0 software (Leica Microsystems, Mannheim, Germany). H&E-stained sections were scored for gliosis, hemorrhages, and structural damage-like cyst formation. Regions of interest of the white matter and hippocampus were defined as previously described [31]. In addition, gray matter alterations in the cerebral cortex were assessed within the same section. Three to five adjacent images were taken per region of interest at × 100 magnification, and analyses were performed using Leica Qwin Pro v3.4.0. software (Leica Microsystems, Wetzlar, Germany). Area fractions and integrated densities were calculated for IBA-1, GFAP, COX-2, MBP, MAP-2, and pEPOR. MPO, Olig2, PDGFRa, CNPase, pHH3, and cleaved caspase-3-positive cells were counted and expressed as total cell count per square millimeter (cells/mm2). In addition, MPO+ cells were also counted in the choroid plexus. Values per region of interest were averaged.
Statistical analysis
All values are shown as mean with 95% confidence interval (CI) or standard deviations (SD). Comparison between different experimental groups was performed with analysis of variance (ANOVA) or with a random intercept-mixed model in case of repeated measurements per animal (e.g., different sections per brain) with Bonferroni correction for multiple comparisons. We applied log transformation to obtain normal distributed data when data or variables were positively skewed before statistical testing. Statistical analysis was performed with IBM SPSS Statistics Version 22.0 (IBM Corp., Armonk, NY, USA; SPSS). Statistical significance was accepted at p < 0.05. Considering the relatively low number of animals per group, exact p values are provided and 0.05 < p < 0.1 is considered a trend.
Results
Animal characteristics
At birth, no differences in weight were found between experimental groups. Fetal blood pH and hemoglobin levels did not differ following intra-amniotic LPS exposure. No sex differences in susceptibility were observed in either readout including animal characteristics and all following readouts regarding systemic cytokine levels and immunohistochemical analysis.
Intra-amniotic LPS exposure results in a biphasic fetal systemic inflammatory response
Intra-amniotic exposure to LPS results in an acute increase of fetal systemic IL-6 concentrations at 5, 12, and 24 h after LPS exposure compared to control levels (SAL vs 5 h LPS p = 0.0157; SAL vs 12 h LPS p = 0.0011; SAL vs 24 h LPS p = 0.0035) (Fig. 2a). Subsequently, at 4 and 8 days after LPS exposure, systemic IL-8 concentrations are increased compared to controls (SAL vs. 4 days LPS p = 0.0147; SAL vs. 8 days LPS p = 0.0502) (Fig. 2b).
Fig. 2.
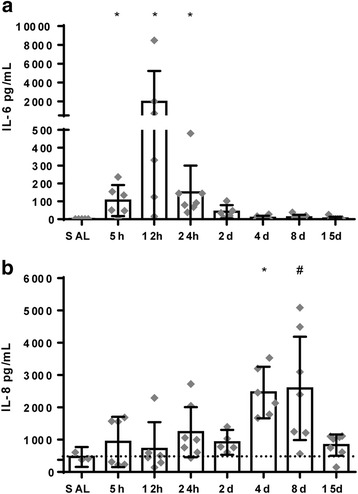
a–b Circulatory interleukin (IL)-6 and IL-8 concentrations illustrate a biphasic response following intra-amniotic LPS exposure. Undetectable values were assigned an arbitrary value of 1 pg/mL in order to perform statistical analysis. Statistical analysis was done with ANOVA, and values are expressed as mean ± 95% CI. Asterisk indicated p < 0.05 versus control group; number sign indicated 0.05 < p < 0.1 versus control
Cerebral inflammation in the fetal white matter, hippocampus, and cortex following intra-amniotic LPS exposure
A systemic fetal inflammatory response is postulated to initiate cerebral inflammation leading to subsequent injury [32–34]. Therefore, we initially measured markers of changes in neuroinflammatory processes (IBA-1, COX-2, GFAP). We found that the systemic inflammatory response following intra-amniotic LPS exposure is followed by cerebral inflammatory changes as indicated by an increase in IBA-1 IR in the white matter at 12 h, 2 days, 4 days, and 8 days following LPS exposure compared to controls (SAL vs. 12 h LPS p = 0.012; SAL vs. 2 days LPS p = 0.006; SAL vs. 4 days LPS p = 0.005; SAL vs. 8 days LPS p = 0.088) (Fig. 3a, b). In the hippocampus, inflammation is detected by an acute increase in COX-2 IR at 5, 12, and 24 h post LPS exposure (SAL vs. 5 h LPS p = 0.055; SAL vs. 12 h LPS p = 0.016; SAL vs. 24 h LPS p = 0.096) (Fig. 3c, d) and an increase in IBA-1 IR at 15 days after LPS exposure compared to controls (SAL vs. 15 days LPS p = 0.073). There were no changes of GFAP IR after LPS exposure at any of the time points. As outlined in Table 1, the number of MPO+ cells is increased in the white matter at 15 days post LPS exposure (SAL vs. 15 days LPS p = 0,027). In the choroid plexus, no significant differences of MPO+ cells were found between groups (Table 1).
Fig. 3.
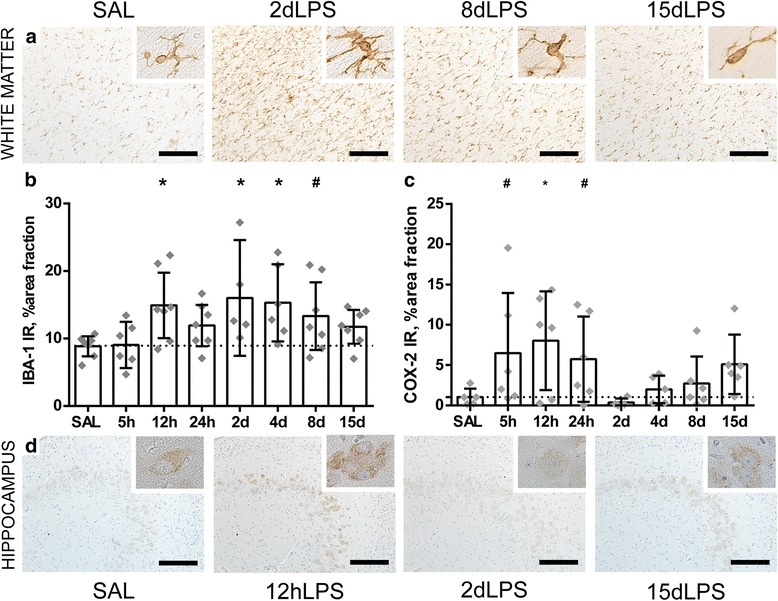
Intra-amniotic exposure to LPS induces an acute, transient cerebral inflammatory response in the preterm white matter and hippocampus. An increase of the area fraction of IBA-1 immunoreactivity (IR) was observed in the white matter at 12 h, 2 days, 4 days, and 8 days following LPS exposure compared to controls (SAL vs. 12 h LPS p = 0.012; SAL vs. 2 days LPS p = 0.006; SAL vs. 4 days LPS p = 0.005; SAL vs. 8 days LPS p = 0.088) (a, b). In the hippocampus, an increase of the area fraction of COX-2 IR was found at 5, 12, and 24 h following LPS exposure (SAL vs. 5 h LPS p = 0.055; SAL vs. 12 h LPS p = 0.016; SAL vs. 24 h LPS p = 0.096) (c, d). Representative histological figures of the IBA-1-positive microglia in animals exposed to intra-amniotic saline (SAL), 2, 8, and 15 days of LPS are shown in a. Representative histological figures of COX-2-positive neurons in the hippocampus of animals exposed to saline (SAL), 12 h, 2 days, and 15 days LPS are depicted in (d). IBA-1 IR and COX-2 IR are depicted as mean % area fraction ± 95% CI. Asterisk indicated p < 0.05 versus control (SAL); number sign indicated 0.05 < p < 0.1 versus control (SAL). Images taken at × 100 magnification (insert at × 400 magnification), scale bar = 200 μm
Table 1.
MPO-positive cells in the choroid plexus and white matter
| MPO+ cells/mm2 | SAL | 5 hours LPS | 12 hours LPS | 24 hours LPS | 2 days LPS | 4 days LPS | 8 days LPS | 15 days LPS |
|---|---|---|---|---|---|---|---|---|
| Choroid plexus | 2.44 ± 3.22 | 4.34 ± 2.86 | 3.19 ± 3.44 | 1.80 ± 1.45 | 3.88 ± 2.54 | 6.25 ± 4.25 | 4.07 ± 2.67 | 3.51 ± 4.38 |
| White matter | 0.47 ± 0.30 | 3.83 ± 3.82 | 1.14 ± 0.83 | 1.95 ± 2.46 | 1.88 ± 2.70 | 2.05 ± 1.84 | 2.67 ± 2.50 | 4.52 ± 4.22* |
Mean values ± standard deviations are represented
*p < 0.05
Cell death and proliferation in the fetal white matter, hippocampus, and cortex following intra-amniotic LPS exposure
To assess whether cerebral inflammation is followed by tissue injury, we measured the number of caspase-3-positive cells in the cerebral white matter, hippocampus, and cortex as a marker of apoptotic cell death as this is an important prognostic factor for neurological outcomes [35]. Exposure to LPS results in an increase in cleaved caspase-3-positive cells in the white matter at 8 days following LPS exposure (SAL vs 8 days LPS p = 0.004), in the hippocampus at 2, 4, 8, and 15 days following LPS exposure (SAL vs 2 days LPS p = 0.002; SAL vs 4 days LPS p = 0.030; SAL vs 8 days LPS p = 0.058; SAL vs 15 days LPS p = 0.042) and in the cortex at 8 days following LPS exposure (SAL vs 8 days LPS p = 0.041) compared to controls (Fig. 4a, b). No evidence of structural changes such as intraventricular hemorrhages and cystic lesions in all experimental groups was found. To assess the proliferation state of the brain, pHH3+ cells were counted. At 2 days following LPS exposure, a trend towards significant decrease in pHH3+ cells was found (SAL vs. 2 days LPS p = 0.100) (Fig. 4c).
Fig. 4.
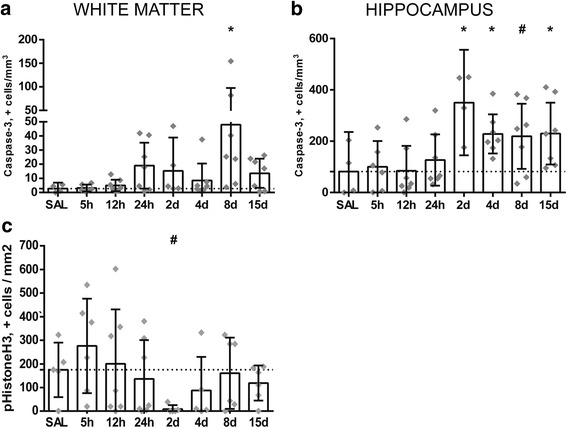
Intra-amniotic exposure to LPS results in a decrease in mitotic cells and relatively late onset of cell death in the preterm white matter and hippocampus. A significant increase of caspase-3-positive cells is observed at 8 days following LPS exposure in the white matter compared to controls (SAL vs 8 days LPS p = 0.004) (a). In the hippocampus, at 2, 4, 8, and 15 days following LPS exposure an increase in caspase-3-positive cells was found compared to controls (SAL vs 2 days LPS p = 0.002; SAL vs 4 days LPS p = 0.030; SAL vs 8 days LPS p = 0.058; SAL vs 15 days LPS p = 0.042) (b). At 2 days following LPS exposure, a decrease in pHH3+ cells was found compared to controls (SAL vs. 2 days LPS p = 0.100) (c). Caspase-3 and pHH3 are expressed as positive cells/mm2 and represented in the graphs as mean ± 95% CI. Asterisk indicated p < 0.05 versus control (SAL); number sign indicated 0.05 < p < 0.1 versus control (SAL)
Distinct time-dependent changes in numbers of oligodendrocyte lineage cells following LPS exposure
The typical histopathological substrate of brain injury in premature infants consists of injury of the developing oligodendrocyte (OL), a cell type that is abundantly present within the brain during weeks 23–32 of gestation (when preterm birth often occurs) and is prone to inflammatory insults [36]. Therefore, we have assessed oligodendrocyte development following intra-amniotic LPS exposure by studying the following oligodendrocyte lineage markers: Olig-2 as a pan-oligodendrocyte lineage marker, PDGFRα as a pre-oligodendrocyte marker (for both OL progenitor and pre-OLs), CNPase as an early oligodendrocyte differentiation marker, and MBP for mature oligodendrocytes and myelin. At 15 days after LPS exposure, a significant decrease in Olig2+ cell number was found compared to controls (SAL vs. 15 days LPS p = 0.050) (Fig. 5a, c). At this time point, the PDGFRa+ progenitor and precursor oligodendrocyte populations tended to decrease compared to controls (SAL vs. 15 days LPS p = 0.070) (Fig. 5a, d). No significant changes of CNPase+ cells and MBP IR were found following LPS exposure compared to controls at all studied time points (Fig. 5a, e, f).
Fig. 5.
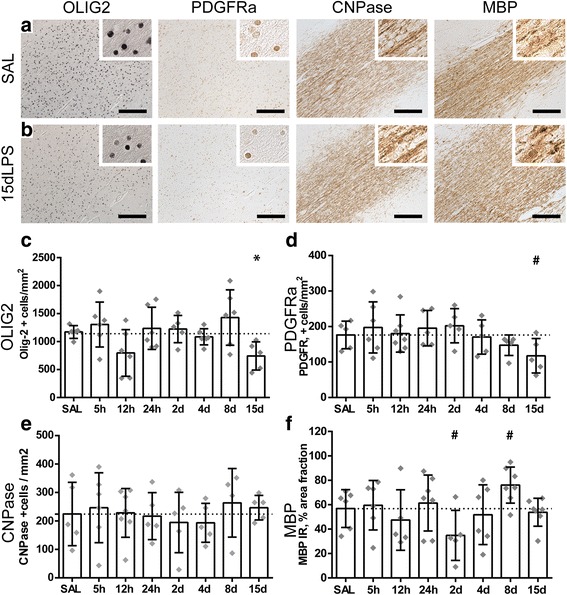
Intra-amniotic exposure to LPS induces distinct time-dependent changes in oligodendrocyte lineage cells. A significant decrease of Olig2-positive cells was observed in animals after 15 days of LPS exposure compared to controls (SAL vs. 15 days LPS p = 0.050) (c). At the same time point, a decrease in PDGFRa-positive cells was found compared to controls (SAL vs. 15 days LPS p = 0.070) (d). No significant changes of CNPase+ cells were found following LPS exposure compared to controls (e). Area fractions (%) of MBP immunoreactivity (IR) showed a decrease at 2 days and an increase at 8 days following LPS exposure compared to controls (SAL vs. 2 days LPS, p = 0.070; SAL vs. 8 days LPS p = 0.083) (f). Representative histological figures of Olig2, PDGFRa, and CNPase-positive cells and MBP IR in animals exposed to intra-amniotic saline (SAL) and 15 days of LPS are shown in a and b respectively. Images taken at × 100 magnification (insert at × 400 magnification), scale bar = 200 μm. Asterisk indicated p < 0.05 versus control (SAL); number sign indicated 0.05 < p < 0.1 versus control (SAL)
Intra-amniotic exposure to LPS resulted in altered dendritic development in gray matter regions of the fetal brain
Besides alterations in white matter development and oligodendrocyte loss, developmental disturbances of the gray matter are an increasingly important feature of perinatal brain injury [37–39]. For the assessment of dendritic maturation, we have studied MAP-2 IR as an established marker for neuronal development [40] in the hippocampus and cerebral cortex. As illustrated in Fig. 5, intra-amniotic exposure to LPS results in a significant or trend to increase in the MAP-2 IR in the hippocampus at all time points except at 2 days LPS (SAL vs 5 h LPS p = 0.007; SAL vs 12 h LPS p = 0.073; SAL vs 24 h LPS p = 0.000; SAL vs 4 days LPS p = 0.034; SAL vs 8 days LPS p = 0.000; SAL vs 15 days LPS p = 0.057) (Fig. 6a, b). At 2 days following LPS exposure, MAP-2 IR is comparable to controls (SAL vs 2 days LPS p = 0.825). In the cerebral cortex only at 24 h after LPS exposure, an increase in MAP-2 IR was found (SAL vs 24 h LPS p = 0.036) (Fig. 6a, c) and all other time points were comparable to control.
Fig. 6.
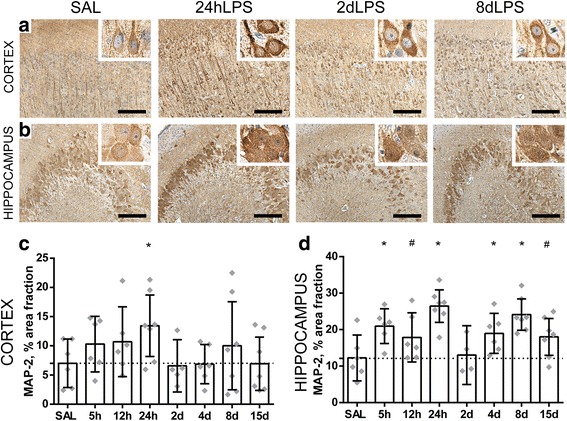
Intra-amniotic exposure to LPS results in altered dendritic development in the gray matter of the fetal brain. A significant increase of the area fraction (%) of MAP-2 immunoreactivity (IR) was found in the cerebral cortex at 24 h after LPS exposure compared to controls (SAL vs 24 h LPS p = 0.036) (a, c). In the hippocampus, an increase of area fraction (%) of MAP-2 IR was observed at 5, 12, and 24 h and at 4, 8, and 15 days after LPS exposure compared to controls (SAL vs 5 h LPS p = 0.007; SAL vs 12 h LPS p = 0.073; SAL vs 24 h LPS p = 0.000; SAL vs 4 days LPS p = 0.034; SAL vs 8 days LPS p = 0.000; SAL vs 15 days LPS p = 0.057) (b, d). Representative histological figures of MAP-2 in the cerebral cortex (a) and hippocampus (b) are depicted in control animals (SAL) and animals exposed to LPS for 24 h, 2 days, and 8 days. Images taken at × 100 magnification (insert at × 400 magnification), scale bar = 200 μm. Asterisk indicated p < 0.05 versus control (SAL); number sign indicated 0.05 < p < 0.1 versus control (SAL)
Expression of the phosphorylated erythropoietin receptor decreases 2 days following LPS exposure
There is a single EPO receptor and activation of this receptor leads to receptor phosphorylation and as such phosphorylation is a useful surrogate for EPO-induced downstream pathway activation. Analysis of the IR for total EPOR revealed no change from baseline levels at any of time points of LPS exposure. Interestingly, however, we observed that pEPOR had a distinct time-dependent switch from over-expression to under-expression. Specifically, following 5 h of LPS exposure pEPOR IR was significantly increased in the white matter (SAL vs 5 h p = 0.010) (Fig. 7a, c) and tended to increase in the cortex (SAL vs 5 h p = 0.100) (Fig. 7e) compared to controls. However, at 2 days after LPS exposure, there is a strong decrease in pEPOR IR within all brain regions compared to controls: significant in the white matter (SAL vs 2d LPS p = 0.030) and trending in the hippocampus (SAL vs 2 days LPS p = 0.088) and cortex (SAL vs 2 days LPS p = 0.010) (Fig. 7c–e). At 4 and 8 days following LPS exposure, pEPOR expression is still decreased compared to controls, trending in the white matter (SAL vs 4 days LPS p = 0.100), and significant in the hippocampus (SAL vs 4 days LPS 0.045; SAL vs 8 days LPS p = 0.014) and cortex (SAL vs 4 days LPS 0.020.; SAL vs 8 days LPS p = 0.030). When the fetus had been exposed to 15 days of LPS, there was no drop in pEPOR IR.
Fig. 7.
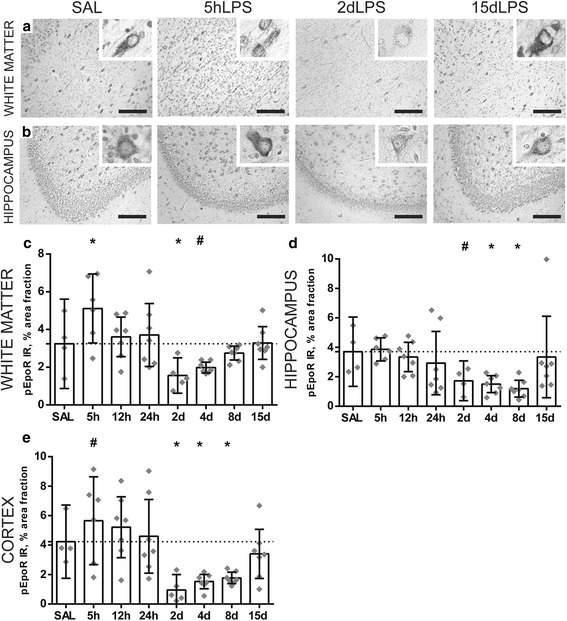
Expression of the phosphorylated erythropoietin receptor decreases 2 days following LPS exposure. An acute increase of the area fraction (%) of pEPOR immunoreactivity (IR) was observed at 5 h after LPS exposure in the white matter (SAL vs 5 h p = 0.010) and cortex (SAL vs 5 h p = 0.100) compared to controls (c, e). At 2 days after LPS exposure, there is a significant decrease in pEPOR IR within all brain regions compared to controls: white matter (SAL vs 2 days LPS p = 0.030) (c), hippocampus (SAL vs 2 days LPS p = 0.088) (d), and cortex (SAL vs 2 days LPS p = 0.010) (e). At 4 and 8 days following LPS exposure, pEPOR expression is still decreased compared to controls in the white matter (SAL vs 4 days LPS p = 0.100), hippocampus (SAL vs 4 days LPS 0.045; SAL vs 8 days LPS p = 0.014), and cortex (SAL vs 4 days LPS 0.020.; SAL vs 8 days LPS p = 0.030). When the fetus had been exposed to 15 days of LPS, there was no decrease in pEPOR IR (c–e). Representative histological figures of the pEPOR in the white matter (a) and hippocampus (b) are depicted in control animals (SAL) and animals exposed to LPS for 5 h, 2 days, and 15 days. Images taken at × 100 magnification (insert at × 400 magnification), scale bar = 200 μm. Asterisk indicated p < 0.05 versus controls (SAL), number sign indicated 0.05 < p < 0.1 versus controls (SAL)
Discussion
The main findings of this study are that intra-amniotic exposure to LPS results in (1) an acute onset and biphasic fetal systemic inflammatory response; (2) persisting microgliosis with a limited pro-inflammatory phase; (3) cell death and (pre) oligodendrocytes loss only following long exposure to inflammation (8 days +) but (4) a striking regional sensitivity to changes in neuronal architecture following short (hours) and long (days) LPS exposure, and (5) biphasic regulation of pEPOR expression. We demonstrate that fetuses, intra-amniotically exposed to LPS, develop biphasic opposing peaks in the systemic inflammatory response, illustrated by changes in IL-6 and IL-8 concentrations. This data agree with previous observations from this model on increased circulatory monocytes and neutrophils and subsequent lymphocytopenia [4] and agrees with previous clinical data showing increased levels of these cytokines, associated with adverse neurological outcomes [41, 9]. Furthermore, the systemic response to LPS is consistently associated with the production of a vast milieu of inflammatory cytokines including IL-6 [42]. These signals across the blood-brain barrier trigger a neuroinflammatory response including activation of microglia and astrocytes [6, 5]. Moreover, IL-8 is known as potent chemotactic and neutrophil-activating factor, and increased serum IL-8 levels are reported in newborns with MRI-defined cerebral abnormalities and abnormal neurodevelopmental outcomes [43, 44, 9, 45, 46]. In line, we observed an increased number of MPO+ cells within the cerebral white matter at 15 days after LPS exposure.
Our study also provides meaningful information about the cerebral inflammatory response initiated by intra-amniotic LPS exposure. We observed a rapid increase in the inflammatory mediator COX-2 and microglial marker IBA-1 in the preterm brain. COX-2 activity results in prostaglandin-E2 production which has wide ranging inflammatory actions on the brain [47], but is typically associated with a pro-inflammatory state in microglia [48] and also reported in astrocytes [49]. Weaver-Mikaere et al. demonstrated in a fetal ovine-derived mixed glial culture that COX-2 activation is the most important mechanism leading to inflammation-mediated white matter injury [50]. Blocking the COX-2 pathway has recently been shown to prevent hypomyelination and behavioral impairment in mice with neonatal white matter injury [49]. In this model, it has also been reported that there is also no change in GFAP-IR across the groups, but additional analysis revealed that despite this, astrocytes together with microglia were still an important source of oligodendrocyte-injurious COX-2 [49]. An additional injurious mechanism of increased COX-2 is that its primary product prostaglandinE2 stimulates glutamate release from astrocytes [51], which is suggested to be essential in the pathophysiological mechanism underlying neonatal encephalopathy [52]. Additionally, the dynamic temporal microglial response in our inflammatory model, indicated by increased IBA-1IR, is consistent with distinct phases of cerebral inflammation in response to other injurious factors in perinatal brain injury, including a hypoxic-ischemic or excitotoxic insult [9]. Microglial activation is proposed to be essential in cerebral injury and dysmaturation associated with exposure to maternal fetal infection/inflammation [53]. Of interest for the application of neurotherapeutics is that microglia activation based on the simple proxy of IBA-1IR was maintained for at least 8 days following LPS exposure, but increased COX2 as surrogate for a pro-inflammatory state was only elevated for 1 day. Further studies to determine a systemic surrogate of microglial activation state might re-invigorate the utility of previously discarded immunomodulatory neurotherapeutics if we understood when immunosuppression would be beneficial.
Considerable debate has raged in the field of preterm neuropathology regarding the role for cell death in encephalopathy of prematurity [54–57]. Experimental data has shown that microglia activation results in pro-inflammatory cytokine release which in turn can lead to apoptosis. However, experimental data has also demonstrated that moderate activation of microglia leads only to the maturation arrest of oligodendrocytes and not cell death [58]. Our study supports a coherent integration of clinical and experimental data as it shows that with a clinically relevant exposure paradigm, the duration of exposure is essential; oligodendrocyte death only occurs after 8 days of LPS exposure. Although outside the scope of this study, research into additional variables including pathogen type and maternal/fetal health and genetics also undoubtedly play a role understanding cohort-specific observations on neuropathology. We also wish to highlight that the significant loss of (pre) oligodendrocytes was not accompanied by myelin loss in our study. It has been previously postulated that the type of robust reduction in myelin protein levels that we would be able to measure with our analysis technique only occurs later in the course of brain injury due to normal kinetics of myelin production [59]. Furthermore, in an inflammation-induced oligodendrocyte injury model, early analysis of myelin proteins [58] and genes [60] has indeed failed to observe reductions despite later robust hypomyelination and behavioral deficits. Detailed analysis of myelin structure could be considered to investigate early markers of myelin injury in future studies.
Together with disturbed white matter development, alterations in gray matter development are implicated in long-term neurological sequelae following intra-amniotic infections and preterm birth [39, 37, 38]. During normal gray matter development, a decrease in MAP-2IR occurs which is indicative for refining of dendritic branching and spines [40]. Regulation of spine development is part of the basic homeostatic functions of microglia, which also include supporting immature cortical neuronal survival and stimulating myelination [61]. As such, increase in MAP-2 IR that we found in the acute phase following LPS exposure suggests a delay in cortical development, a phenomenon that has also been suggested from analysis of preterm infants using MRI [62]. Moreover, in humans suffering from schizophrenia, increased dendritic arborization was found in the hippocampus compared to healthy humans when stained for MAP-2 [63]. Concerning the specific mechanistic link between increased inflammation and increased MAP-2, it is noteworthy that COX-2 overexpressing neuronal-like cells showed significantly increased neurite outgrowth and branching, which was partially reverted by COX inhibitor [64].
Despite multiple studies identifying EPO as very promising neuroprotective candidate in newborn infants [65, 66, 14, 67, 13, 17, 68–70], other studies did not find therapeutic effects on neurodevelopmental outcomes [71, 72, 23]. These inconsistencies might be explained by heterogeneity between study cohorts including, as outlined in this study for the first time, differences in EPO signaling in response to inflammation. Interestingly, after an initial increase in the receptor phosphorylation levels, a distinct and long-lasting decrease of the pEPOR occurred. Activation of the EPOR phosphorylates activates the kinase Jak-2 resulting in initiation of a complex anti-apoptotic signaling cascade [73, 25]. Accordingly, we are unsurprised that a decrease in the level of pEPOR is accompanied by an increase in the number of apoptotic cells (caspase-3+) within the same region in the fetal brain. This suggests that the reduced activation of the EPO receptor in our study results in increased apoptosis in the preterm brain and in experimental paradigms this relationship between EPOR and cell death has been demonstrated [25]. In regards to the total expression of EPOR, others have also shown that this remains stable or increases [74], and this elevation occurs within hours in response to pro-inflammatory cytokines like TNFa [75], whereas the level of its parent ligand, EPO, is downregulated under pro-inflammatory conditions [76]. Thus, understanding the inflammation-induced dysregulation of EPO signaling provides a therapeutic window where treatment with EPO may be most efficacious [76]. Altogether, the considerable changes in EPOR expression within the fetal brain in the course of inflammation stresses the need for biomarkers to determine the onset of intra-amniotic infections.
One important limitation of a large animal study is the relatively low number of animals per group. Given the relatively small animal numbers per group, we reported actual p values and tended to interpret p values between 0.05 and 0.1 as biologically relevant. This assumption decreases the chance of a false-negative finding but increases the chance that one of these differences is a false-positive result.
Conclusion
Altogether, the cerebral changes as found in our translational model of inflammation-related brain injury of the preterm infant could be reasonably expected to lead to deficits in learning, memory and social skills, and/or motor disabilities that can persist into adulthood [34]. However, these outcomes need to be confirmed in longitudinal studies, incorporating behavioral analysis and MRI but nonetheless inform the development of interventions to protect and/or regenerate the preterm brain in order to allow normal development. By gaining more insight into the temporal processes underlying inflammation-induced preterm brain injury, we provide an increased understanding of the pathophysiological changes in the fetal brain with the goal of helping to design new interventions or implement interventions more effectively to prevent perinatal brain damage.
Acknowledgements
The authors would like to thank Lilian Kessels and Nico Kloosterboer for their excellent technical assistance.
Funding
The work was financially supported by Kinderonderzoeksfonds Limburg (Health Foundation Limburg) (TW).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANOVA
Analysis of variance
- CI
Confidence interval
- CNPase
2′,3′-Cyclic-nucleotide 3′-phosphodiesterase
- COX-2
Cyclo-oxygenase 2
- ELISA
Enzyme-linked immunosorbent assay
- EPO
Erythropoietin
- EPOR
Erythropoietin receptor
- GFAP
Glial fibrillary acidic protein
- H&E
Hematoxylin and eosin
- IBA-1
Ionized calcium-binding adaptor molecule-1
- IL-6
Interleukin-6
- IL-8
Interleukin-8
- IR
Immunoreactivity
- LPS
Lipopolysaccharide
- MAP2
Microtubule-associated protein 2
- MBP
Myelin basic protein
- MPO
Myeloperoxidase
- Olig2
Oligodendrocyte transcription factor 2
- PBS
Phosphate-buffered saline
- PDGFRa
Platelet-derived growth factor receptor alpha
- pEPOR
Phosphorylated erythropoietin receptor
- pHH3
Phospho-Histone H3
- SAL
Saline
- SD
Standard deviation
- TMB
3,3′,5,5′-Tetramethylbenzidine
- TNFa
Tumor necrosis factor alpha
Authors’ contributions
RG and RW analyzed the data and wrote the manuscript under the supervision of BF and TW. RG and RW performed the immunohistochemistry. RG, RW, DO, PS, SP, BK, PG, BF, and TW interpreted the data. MK, SK, BK, and AJ performed animal experiments. SK, LZ, AJ, BK, and TW significantly contributed to conception and design of the study. PS, SP, PG, BF, and TW contributed to data interpretation and editing of the manuscript. All authors read and approved the final manuscript.
Ethics approval
Animal procedures were performed with approval of the animal ethics committee of the University of Western Australia (Perth, Australia).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ruth Gussenhoven, Email: r.gussenhoven@maastrichtuniversity.nl.
Rob J. J. Westerlaken, Email: rwesterlaken@viecuri.nl
Daan R. M. G. Ophelders, Email: d.ophelders@maastrichtuniversity.nl
Alan H. Jobe, Email: alan.jobe@cchmc.org
Matthew W. Kemp, Email: matthew.kemp@uwa.edu.au
Suhas G. Kallapur, Email: skallapur@mednet.ucla.edu
Luc J. Zimmermann, Email: luc.zimmermann@mumc.nl
Per T. Sangild, Email: pts@sund.ku.dk
Stanislava Pankratova, Email: stasya@sund.ku.dk.
Pierre Gressens, Email: pierre.gressens@inserm.fr.
Boris W. Kramer, Email: b.kramer@maastrichtuniversity.nl
Bobbi Fleiss, Email: bobbi.fleiss@inserm.fr.
Tim G. A. M. Wolfs, Email: tim.wolfs@maastrichtuniversity.nl
References
- 1.Rovira N, Alarcon A, Iriondo M, Ibanez M, Poo P, Cusi V, et al. Impact of histological chorioamnionitis, funisitis and clinical chorioamnionitis on neurodevelopmental outcome of preterm infants. Early Hum Dev. 2011;87(4):253–257. doi: 10.1016/j.earlhumdev.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 2.Paton MCB, McDonald CA, Allison BJ, Fahey MC, Jenkin G, Miller SL. Perinatal brain injury as a consequence of preterm birth and intrauterine inflammation: designing targeted stem cell therapies. Front Neurosci. 2017;11:200. doi: 10.3389/fnins.2017.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goncalves LF, Chaiworapongsa T, Romero R. Intrauterine infection and prematurity. Ment Retard Dev Disabil Res Rev. 2002;8(1):3–13. doi: 10.1002/mrdd.10008. [DOI] [PubMed] [Google Scholar]
- 4.Kuypers E, Wolfs TG, Collins JJ, Jellema RK, Newnham JP, Kemp MW, et al. Intraamniotic lipopolysaccharide exposure changes cell populations and structure of the ovine fetal thymus. Reprod Sci. 2013;20(8):946–956. doi: 10.1177/1933719112472742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai JCY, Rocha-Ferreira E, Ek CJ, Wang X, Hagberg H, Mallard C. Immune responses in perinatal brain injury. Brain Behav Immun. 2017;63:210–223. doi: 10.1016/j.bbi.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 6.Malaeb S, Dammann O. Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol. 2009;24(9):1119–1126. doi: 10.1177/0883073809338066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin C, Londono I, Mallard C, Lodygensky GA. New means to assess neonatal inflammatory brain injury. J Neuroinflammation. 2015;12:180. doi: 10.1186/s12974-015-0397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gussenhoven R, Ophelders D, Kemp MW, Payne MS, Spiller OB, Beeton ML, et al. The paradoxical effects of chronic intra-amniotic Ureaplasma parvum exposure on ovine fetal brain development. Dev Neurosci. 2017; 10.1159/000479021. [DOI] [PMC free article] [PubMed]
- 9.Fleiss B, Tann CJ, Degos V, Sigaut S, Van Steenwinckel J, Schang AL, et al. Inflammation-induced sensitization of the brain in term infants. Dev Med Child Neurol. 2015;57(Suppl 3):17–28. doi: 10.1111/dmcn.12723. [DOI] [PubMed] [Google Scholar]
- 10.Hagberg H, Dammann O, Mallard C, Leviton A. Preconditioning and the developing brain. Semin Perinatol. 2004;28(6):389–395. doi: 10.1053/j.semperi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosc Off J Soc Neurosci. 2006;26(4):1269–1274. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, et al. Erythropoietin receptor signalling is required for normal brain development. Dev. 2002;129(2):505–516. doi: 10.1242/dev.129.2.505. [DOI] [PubMed] [Google Scholar]
- 13.Song J, Sun H, Xu F, Kang W, Gao L, Guo J, et al. Recombinant human erythropoietin improves neurological outcomes in very preterm infants. Ann Neurol. 2016;80(1):24–34. doi: 10.1002/ana.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leuchter RH, Gui L, Poncet A, Hagmann C, Lodygensky GA, Martin E, et al. Association between early administration of high-dose erythropoietin in preterm infants and brain MRI abnormality at term-equivalent age. JAMA. 2014;312(8):817–824. doi: 10.1001/jama.2014.9645. [DOI] [PubMed] [Google Scholar]
- 15.Fauchere JC, Koller BM, Tschopp A, Dame C, Ruegger C, Bucher HU, et al. Safety of early high-dose recombinant erythropoietin for neuroprotection in very preterm infants. J Pediatr. 2015;167(1):52–57. doi: 10.1016/j.jpeds.2015.02.052. [DOI] [PubMed] [Google Scholar]
- 16.Juul SE, McPherson RJ, Bauer LA, Ledbetter KJ, Gleason CA, Mayock DE. A phase I/II trial of high-dose erythropoietin in extremely low birth weight infants: pharmacokinetics and safety. Pediatrics. 2008;122(2):383–391. doi: 10.1542/peds.2007-2711. [DOI] [PubMed] [Google Scholar]
- 17.Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, et al. High-dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: a phase II trial. Pediatr. 2016;137(6) 10.1542/peds.2016-0191. [DOI] [PubMed]
- 18.Wassink G, Davidson JO, Dhillon SK, Fraser M, Galinsky R, Bennet L, et al. Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab. 2017;37(3):1080–1094. doi: 10.1177/0271678X16650455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao R, Tang YH, Tong JH, Yang JJ, Ji MH, Zhu SH. Systemic lipopolysaccharide administration-induced cognitive impairments are reversed by erythropoietin treatment in mice. Inflamm. 2015;38(5):1949–1958. doi: 10.1007/s10753-015-0175-4. [DOI] [PubMed] [Google Scholar]
- 20.Rees S, Hale N, De Matteo R, Cardamone L, Tolcos M, Loeliger M, et al. Erythropoietin is neuroprotective in a preterm ovine model of endotoxin-induced brain injury. J Neuropathol Exp Neurol. 2010;69(3):306–319. doi: 10.1097/NEN.0b013e3181d27138. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Zhang L, Jin Y. A meta-analysis of the protective effect of recombinant human erythropoietin (rhEPO) for neurodevelopment in preterm infants. Cell Biochem Biophys. 2015;71(2):795–802. doi: 10.1007/s12013-014-0265-1. [DOI] [PubMed] [Google Scholar]
- 22.Korzeniewski SJ, Allred E, Logan JW, Fichorova RN, Engelke S, Kuban KC, et al. Elevated endogenous erythropoietin concentrations are associated with increased risk of brain damage in extremely preterm neonates. PLoS One. 2015;10(3):e0115083. doi: 10.1371/journal.pone.0115083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natalucci G, Latal B, Koller B, Ruegger C, Sick B, Held L, et al. Effect of early prophylactic high-dose recombinant human erythropoietin in very preterm infants on Neurodevelopmental outcome at 2 years: a randomized clinical trial. JAMA. 2016;315(19):2079–2085. doi: 10.1001/jama.2016.5504. [DOI] [PubMed] [Google Scholar]
- 24.Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207(Pt 18):3233–3242. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- 25.van der Kooij MA, Groenendaal F, Kavelaars A, Heijnen CJ, van Bel F. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res Rev. 2008;59(1):22–33. doi: 10.1016/j.brainresrev.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Jantzie LL, Miller RH, Robinson S. Erythropoietin signaling promotes oligodendrocyte development following prenatal systemic hypoxic-ischemic brain injury. Pediatr Res. 2013;74(6):658–667. doi: 10.1038/pr.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer BW, Moss TJ, Willet KE, Newnham JP, Sly PD, Kallapur SG, et al. Dose and time response after intraamniotic endotoxin in preterm lambs. Am J Respir Crit Care Med. 2001;164(6):982–988. doi: 10.1164/ajrccm.164.6.2103061. [DOI] [PubMed] [Google Scholar]
- 28.Newnham JP, Kallapur SG, Kramer BW, Moss TJ, Nitsos I, Ikegami M, et al. Betamethasone effects on chorioamnionitis induced by intra-amniotic endotoxin in sheep. Am J Obstet Gynecol. 2003;189(5):1458–1466. doi: 10.1067/S0002-9378(03)00758-0. [DOI] [PubMed] [Google Scholar]
- 29.Back SA, Riddle A, Dean J, Hohimer AR. The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics J Am Soc Exp NeuroTherapeutics. 2012;9(2):359–370. doi: 10.1007/s13311-012-0108-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gavilanes AW, Strackx E, Kramer BW, Gantert M, Van den Hove D, Steinbusch H, et al. Chorioamnionitis induced by intraamniotic lipopolysaccharide resulted in an interval-dependent increase in central nervous system injury in the fetal sheep. Am J Obstet Gynecol. 2009;200(4):437. doi: 10.1016/j.ajog.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Jellema RK, Lima Passos V, Zwanenburg A, Ophelders DR, De Munter S, Vanderlocht J, et al. Cerebral inflammation and mobilization of the peripheral immune system following global hypoxia-ischemia in preterm sheep. J Neuroinflammation. 2013;10:13. doi: 10.1186/1742-2094-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bashiri A, Burstein E, Mazor M. Cerebral palsy and fetal inflammatory response syndrome: a review. J Perinat Med. 2006;34(1):5–12. doi: 10.1515/JPM.2006.001. [DOI] [PubMed] [Google Scholar]
- 33.Duggan PJ, Maalouf EF, Watts TL, Sullivan MH, Counsell SJ, Allsop J, et al. Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet. 2001;358(9294):1699–1700. doi: 10.1016/S0140-6736(01)06723-X. [DOI] [PubMed] [Google Scholar]
- 34.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71(4):444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 35.Rosell A, Cuadrado E, Alvarez-Sabin J, Hernandez-Guillamon M, Delgado P, Penalba A, et al. Caspase-3 is related to infarct growth after human ischemic stroke. Neurosci Lett. 2008;430(1):1–6. doi: 10.1016/j.neulet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int J Dev Neurosci: Off J Int Soc DevNeurosci. 2011;29(4):423–440. doi: 10.1016/j.ijdevneu.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dean JM, van de Looij Y, Sizonenko SV, Lodygensky GA, Lazeyras F, Bolouri H, et al. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Ann Neurol. 2011;70(5):846–856. doi: 10.1002/ana.22480. [DOI] [PubMed] [Google Scholar]
- 38.Mottahedin A, Ardalan M, Chumak T, Riebe I, Ek J, Mallard C. Effect of neuroinflammation on synaptic organization and function in the developing brain: implications for neurodevelopmental and neurodegenerative disorders. Front Cell Neurosci. 2017;11:190. doi: 10.3389/fncel.2017.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volpe JJ. The encephalopathy of prematurity—brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol. 2009;16(4):167–178. doi: 10.1016/j.spen.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czikk MJ, Totten S, Hammond R, Richardson BS. Microtubule-associated protein 2 and synaptophysin in the preterm and near-term ovine fetal brain and the effect of intermittent umbilical cord occlusion. Reprod Sci. 2015;22(3):367–376. doi: 10.1177/1933719114529371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu HY, Zhang Q, Wang QX, Lu JY. Contribution of histologic chorioamnionitis and fetal inflammatory response syndrome to increased risk of brain injury in infants with preterm premature rupture of membranes. Pediatr Neurol. 2016;61:94–98. doi: 10.1016/j.pediatrneurol.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol. 2015;11(4):192–208. doi: 10.1038/nrneurol.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartha AI, Foster-Barber A, Miller SP, Vigneron DB, Glidden DV, Barkovich AJ, et al. Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr Res. 2004;56(6):960–966. doi: 10.1203/01.PDR.0000144819.45689.BB. [DOI] [PubMed] [Google Scholar]
- 44.Ramaswamy V, Horton J, Vandermeer B, Buscemi N, Miller S, Yager J. Systematic review of biomarkers of brain injury in term neonatal encephalopathy. Pediatr Neurol. 2009;40(3):215–226. doi: 10.1016/j.pediatrneurol.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 45.Huang HC, Wang CL, Huang LT, Chuang H, Liu CA, Hsu TY, et al. Association of cord blood cytokines with prematurity and cerebral palsy. Early Hum Dev. 2004;77(1–2):29–36. doi: 10.1016/j.earlhumdev.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Carlo WA, McDonald SA, Tyson JE, Stoll BJ, Ehrenkranz RA, Shankaran S, et al. Cytokines and neurodevelopmental outcomes in extremely low birth weight infants. J Pediatr. 2011;159(6):919–925. doi: 10.1016/j.jpeds.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63(9):901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- 48.Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013;32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiow LR, Favrais G, Schirmer L, Schang AL, Cipriani S, Andres C, et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia. 2017;65(12):2024–2037. doi: 10.1002/glia.23212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weaver-Mikaere L, Gunn AJ, Mitchell MD, Bennet L, Fraser M. LPS and TNF alpha modulate AMPA/NMDA receptor subunit expression and induce PGE2 and glutamate release in preterm fetal ovine mixed glial cultures. J Neuroinflammation. 2013;10:153. doi: 10.1186/1742-2094-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dave KA, Platel JC, Huang F, Tian D, Stamboulian-Platel S, Bordey A. Prostaglandin E2 induces glutamate release from subventricular zone astrocytes. Neuron Glia Biol. 2010;6(3):201–207. doi: 10.1017/S1740925X10000244. [DOI] [PubMed] [Google Scholar]
- 52.Evrard P, Gressens P, Volpe JJ. New concepts to understand the neurological consequences of subcortical lesions in the premature brain. Biol Neonate. 1992;61(1):1–3. doi: 10.1159/000243525. [DOI] [PubMed] [Google Scholar]
- 53.Baburamani AA, Supramaniam VG, Hagberg H, Mallard C. Microglia toxicity in preterm brain injury. Reprod Toxicol. 2014;48:106–112. doi: 10.1016/j.reprotox.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verney C, Pogledic I, Biran V, Adle-Biassette H, Fallet-Bianco C, Gressens P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J Neuropathol Exp Neurol. 2012;71(3):251–264. doi: 10.1097/NEN.0b013e3182496429. [DOI] [PubMed] [Google Scholar]
- 55.Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH, et al. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 2008;18(2):153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62(5):441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 57.Back SA, Luo NL, Mallinson RA, O'Malley JP, Wallen LD, Frei B, et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58(1):108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- 58.Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70(4):550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- 59.Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59(3):428–433. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- 60.Krishnan ML, Van Steenwinckel J, Schang AL, Yan J, Arnadottir J, Le Charpentier T, et al. Integrative genomics of microglia implicates DLG4 (PSD95) in the white matter development of preterm infants. Nat Commun. 2017;8(1):428. doi: 10.1038/s41467-017-00422-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tay TL, Savage JC, Hui CW, Bisht K, Tremblay ME. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. 2017;595(6):1929–1945. doi: 10.1113/JP272134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ball G, Srinivasan L, Aljabar P, Counsell SJ, Durighel G, Hajnal JV, et al. Development of cortical microstructure in the preterm human brain. Proc Natl Acad Sci U S A. 2013;110(23):9541–9546. doi: 10.1073/pnas.1301652110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cotter D, Wilson S, Roberts E, Kerwin R, Everall IP. Increased dendritic MAP2 expression in the hippocampus in schizophrenia. Schizophr Res. 2000;41(2):313–323. doi: 10.1016/S0920-9964(99)00068-7. [DOI] [PubMed] [Google Scholar]
- 64.Kadoyama K, Takahashi Y, Higashida H, Tanabe T, Yoshimoto T. Cyclooxygenase-2 stimulates production of amyloid beta-peptide in neuroblastoma x glioma hybrid NG108-15 cells. Biochem Biophys Res Commun. 2001;281(2):483–490. doi: 10.1006/bbrc.2001.4357. [DOI] [PubMed] [Google Scholar]
- 65.O'Gorman RL, Bucher HU, Held U, Koller BM, Huppi PS, Hagmann CF, et al. Tract-based spatial statistics to assess the neuroprotective effect of early erythropoietin on white matter development in preterm infants. Brain J Neurol. 2015;138(Pt 2):388–397. doi: 10.1093/brain/awu363. [DOI] [PubMed] [Google Scholar]
- 66.Ohls RK, Cannon DC, Phillips J, Caprihan A, Patel S, Winter S, et al. Preschool assessment of preterm infants treated with darbepoetin and erythropoietin. Pediatrics. 2016;137(3):e20153859. doi: 10.1542/peds.2015-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neubauer AP, Voss W, Wachtendorf M, Jungmann T. Erythropoietin improves neurodevelopmental outcome of extremely preterm infants. Ann Neurol. 2010;67(5):657–666. doi: 10.1002/ana.21977. [DOI] [PubMed] [Google Scholar]
- 68.Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics. 2009;124(2):e218–e226. doi: 10.1542/peds.2008-3553. [DOI] [PubMed] [Google Scholar]
- 69.Brown MS, Eichorst D, Lala-Black B, Gonzalez R. Higher cumulative doses of erythropoietin and developmental outcomes in preterm infants. Pediatrics. 2009;124(4):e681–e687. doi: 10.1542/peds.2008-2701. [DOI] [PubMed] [Google Scholar]
- 70.McAdams RM, McPherson RJ, Mayock DE, Juul SE. Outcomes of extremely low birth weight infants given early high-dose erythropoietin. J Perinatol: Off J California Perinatal Association. 2013;33(3):226–230. doi: 10.1038/jp.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bierer R, Peceny MC, Hartenberger CH, Ohls RK. Erythropoietin concentrations and neurodevelopmental outcome in preterm infants. Pediatrics. 2006;118(3):e635–e640. doi: 10.1542/peds.2005-3186. [DOI] [PubMed] [Google Scholar]
- 72.El Shimi MS, Awad HA, Hassanein SM, Gad GI, Imam SS, Shaaban HA, et al. Single dose recombinant erythropoietin versus moderate hypothermia for neonatal hypoxic ischemic encephalopathy in low resource settings. J Matern-Fetal Neonatal Med. 2014;27(13):1295–1300. doi: 10.3109/14767058.2013.855894. [DOI] [PubMed] [Google Scholar]
- 73.Nguyen AQ, Cherry BH, Scott GF, Ryou MG, Mallet RT. Erythropoietin: powerful protection of ischemic and post-ischemic brain. Exp Biol Med. 2014;239(11):1461–1475. doi: 10.1177/1535370214523703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castillo-Melendez M, Yan E, Walker DW. Expression of erythropoietin and its receptor in the brain of late-gestation fetal sheep, and responses to asphyxia caused by umbilical cord occlusion. Dev Neurosci. 2005;27(2–4):220–227. doi: 10.1159/000085995. [DOI] [PubMed] [Google Scholar]
- 75.Taoufik E, Petit E, Divoux D, Tseveleki V, Mengozzi M, Roberts ML, et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc Natl Acad Sci U S A. 2008;105(16):6185–6190. doi: 10.1073/pnas.0801447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brines M, Cerami A. The receptor that tames the innate immune response. Mol Med. 2012;18:486–496. doi: 10.2119/molmed.2011.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.