

Abstract
Molecular recognition, binding and catalysis are often mediated by non-covalent interactions involving aromatic functional groups. Although the relative complexity of these so-called π interactions has made them challenging to study, theory and modelling have now reached the stage at which we can explain their physical origins and obtain reliable insight into their effects on molecular binding and chemical transformations. This offers opportunities for the rational manipulation of these complex non-covalent interactions and their direct incorporation into the design of small-molecule catalysts and enzymes.
Products of billions of years of evolutionary pressure, enzyme active sites are flawlessly engineered spaces, with amino acid residues precisely positioned to be complementary to the transition states of the reactions they catalyse with regard to size, shape, charge and any number of additional features1. Although the diversity of reactions observed in nature is vast, the underlying principles of molecular recognition of substrate and transition state are similar, and typically emanate from the concerted action of multiple non-covalent substrate–catalyst interactions (such as Van der Waals forces, the hydrophobic effect, electrostatic interactions) that are individually weak (about 0.5–3 kcal mol−1) but collectively important2–4. These interactions lower the free energy (Δ G) as the catalyst–substrate complex (C–S) forms and approaches the transition state (TS in Fig. 1a), which partially compensates for the energetic uptake required to form the activated complex (C–S‡) and thereby leads to catalysis (Fig. 1a). When designing synthetic catalysts that operate by way of such Δ G lowering, the array of attractive non-covalent interactions (NCIs) that exist afford seemingly endless approaches to realize it through ground-state destabilization and/ or transition-state stabilization.
Figure 1. Overview of catalysis and non-covalent π interactions.
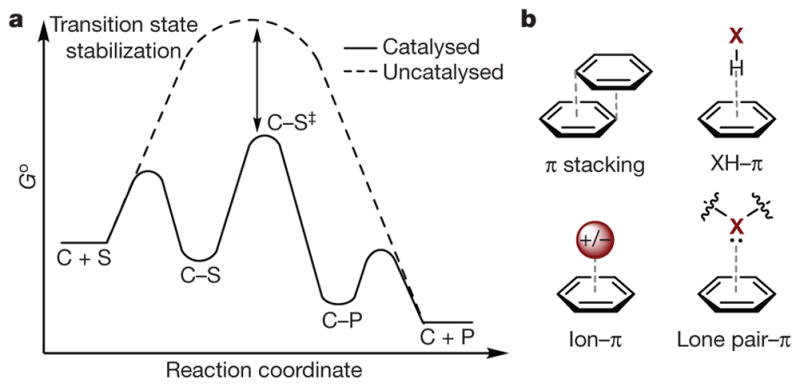
a, Qualitative depiction of catalysis via transition-state (TS) stabilization, where C is catalyst, S is substrate, and P is product. Go, Gibbs free energy in standard state; reaction coordinate indicates the progression of the reaction; C–S‡, activated complex. b, Featured interactions (grey dashed lines) of π systems in this Review. X = B, C, N, O.
Studying molecular binding in the ground state has delivered a relatively sophisticated view of the physical principles underlying NCIs, to the extent that they are often rationally built into supramolecular architectures5. As transition-state theory fundamentally relies on the treatment of the ground and transition states as being in equilibrium, the factors that govern binding in the former (change in free energy Δ G, equilibrium constant Keq) are perfectly analogous to those in the latter (free energy of activation Δ G‡, rate constant k)6. It thus seems entirely reasonable to inform the rational design of synthetic catalysts that need to bind the transition state by considering the factors that influence various ground-state NCIs (distance, orientation, substituent effects, and so on) and the strength of binding7, as illustrated by the development of small-molecule catalysts that successfully control stereo- or regioselectivity8 in numerous transformations via hydrogen-bonding9 or ion-pairing10.
Here we survey within the context of catalysis a class of NCIs that often influence molecular structure, recognition and binding as well as reaction outcomes: aromatic or π interactions11. Drawing from knowledge in fields as diverse as crystal engineering, supramolecular chemistry, structural biology, organic synthesis and computational chemistry, we give illustrative examples of π–π, XH–π (X = B, C, N, O), cation–π, anion–π, and lone pair–π interactions (Fig. 1b) and examine how they can mediate the acceleration of chemical transformations. As our interest is in mechanistic understanding, we focus on examples with rigorously quantified NCIs that can reveal the underlying physical organic principles. In the final section, we consider the prospect of truly de novo catalyst design affecting the field of synthetic organic chemistry via the paradigm of transition-state recognition using π interactions as a design element.
π–π interactions
Among the most studied interactions of π systems, the non-covalent attraction between neutral, closed shell aromatic rings is often characterized using the terms π–π interaction or π stacking12–15. Four geometries are characteristic of π-stacking interactions: parallel stacked (PS), parallel displaced (PD) and edge-to-face or ‘T’-shaped (Fig. 2a). For the benzene dimer, the ‘T’-shaped and PD geometries have been calculated to be approximately equal in stability (about–2.5 kcal mol−1) with the PS geometry much less so ( approximately–1.6 kcal mol−1). Qualitatively, this geometric preference can be understood in terms of molecular quadrupole moments. The six radially oriented C–H bonds of benzene confer a quadrupole moment to the molecule (described by the z2 component of the quadrupole moment tensor, Qzz) such that regions of negative electrostatic potential are found above and below the ring plane, and regions of positive potential around the ring periphery. From this perspective, the ‘T’-shaped and PD geometries reflect an interaction between regions of positive and negative potential, whereas the PS geometry is electrostatically repulsive owing to overlapping regions of negative potential. However, based on a number of high level ab initio studies, it is now generally accepted that dispersion15 plays a major role in the attractive nature of π–π interactions but is largely cancelled by exchange repulsion16,17. As such, the electrostatic contribution is important when comparing the interaction strengths across a series of substituted homologues.
Figure 2. Overview of π-stacking geometries and evidence for the direct interaction model.

a, π–π interaction geometries (see text for details). b, Hammett correlations (interaction energy versus σmeta, the meta-Hammett substituent parameter) supporting the direct interaction model. Broken red line in insets indicates interaction. Adapted from ref. 26, American Chemical Society. c, Correlations demonstrating geometric consequences of the direct interaction model. Insets show Ar1 (top) and Ar2 (bottom); broken red lines in insets indicate interactions. On axes, Eint(Ar1 × Ar2) denotes interaction energy between Ar1 and Ar2, and so on. Adapted from ref. 28, American Chemical Society. In b and c, regression lines and their equations are shown: R, correlation coefficient.
There have been numerous studies in the past two decades devoted to understanding the physical origins of substituent effects in tuning the strength of π–π interactions. Until recently, the prevailing viewpoint, initially put forth by Hunter and Sanders18, was based on a π-polarization argument, wherein electron-withdrawing substituents were proposed to remove electron density from an arene’s π-system via π-resonance effects (and vice versa for electron-donating substituents) 19–22. As such, this model predicts that the interaction between two rings in a PS geometry will be strengthened upon introduction of an electron-withdrawing substituent into one of the partners owing to a decrease in repulsion between the two π systems, whereas introduction of an electron-donating substituent should weaken the interaction by the opposite mechanism. An early experimental study corroborating the Hunter–Sanders model was reported by Cozzi and Siegel and co-workers, who prepared a series of 1,8-diaryl naphthalenes, in which the aryl groups were forced in a faceto-face stacking geometry in the ground state by steric constraints21. Assuming that the π–π interaction would be completely attenuated at the transition state corresponding to rotation about the aryl–naphthyl bond, measurement of the rotation barrier would be reflective of the ground-state stabilization of the stacked geometry. In practice, an excellent correlation was found between Δ G‡ for ring rotation and the para Hammett substituent parameter σpara, consistent with the notion that π-resonance effects were the dominant factor underlying the observed substituent effect.
Recently, however, the π-polarization model has been a source of controversy. Ab initio studies have suggested that the introduction of any substituent (that is, electron-donating or electron-withdrawing) should increase the strength of π–π interactions compared with the benzene dimer23. In fact, the notion that the π systems of the interacting partners are even involved in “π–π interactions” has been increasingly called into question14,24,25. For example, computations by Grimme have suggested that the interaction energy of cyclohexane ‘stacking’ (−3.09 kcal mol−1) is in fact stronger than that of benzene (PD orientation, −2.62 kcal mol−1)24, and Bloom and Wheeler have shown that the interaction between benzene and cyclohexane (−2.91 kcal mol−1) is stronger than that of the benzene dimer (PS orientation, −1.63 kcal mol−1)25. It has thus been proposed that the π–π terminology be reserved for larger aromatic systems in which special π-electron correlation effects do appear to exist. Nonetheless, as it is not our goal to coin new terminology but rather to describe the current state of the art, we have chosen to use the terms ‘π–π ’ or ‘π stacking’ fully aware of their shortcomings.
A more recent model that is consistent with the majority of available data, popularized by Wheeler and Houk, posits that the direct interactions between the local C–X/H dipoles of substituted aromatic rings determine the strength of a ‘π–π ’ interaction16,23,26–28. Viewed from this perspective, aromatic rings primarily serve as platforms upon which to place substituents in a spatial arrangement that will either be attractive or repulsive depending on the relative geometries between the interacting dipoles. The calculated interaction energy between benzene and a monosubstituted benzene derivative was found to correlate strongly with the meta Hammett substituent parameter σmeta and was attractive for all substituents evaluated (Fig. 2b, left), including those considered to be inductively electron-donating (negative σmeta)28. Retaining the same intermolecular separation but replacing the substituted benzene ring with a hydrogen atom (Fig. 2b, right) afforded an essentially identical correlation, suggesting that the aromatic ring itself is not truly involved in the interaction. Another prediction of the Wheeler–Houk direct interaction model is that substituent effects should be additive—that is, the interaction between two substituted benzene derivatives should be equal to the sum of the individual interaction energies of each substituted ring with benzene. This hypothesis is supported by the excellent 1:1 correlation between these interaction energies shown in Fig. 2c (left). However, this relationship is expected to fail if the substituents on the two substituted rings interact directly as other intermolecular interactions—such as steric repulsion—become relevant (Fig. 2c, right). Additional computational studies by Parrish and Sherrill have also demonstrated the validity of this hypothesis29.
A recent study by Shimizu and co-workers provided direct experimental support for the Wheeler–Houk additivity hypothesis (Fig. 3a)30. Owing to restricted rotation about the imide N–aryl bond, 3.1 (a ‘ molecular torsion balance’) was found to exhibit discrete unfolded and folded conformations in solution. Furthermore, it was demonstrated that the phenyl ether substituent and phenanthrene shelf were forced into a PD stacking interaction in the folded conformation. Thus, measuring the unfolded to folded ratio as a function of phenyl ether substitution allowed the authors to quantify the strength of the π–π interaction in the ground state. By comparison with a control molecular torsion balance in which the π–π interaction was precluded, the authors defined a substituent effect for several electronically diverse substituents (Fig. 3a). Notably, the magnitude of the substituent effects for monosubstituted aryl ethers were dependent on the positions of the substituents on the ring (compare 3-Cl and 4-Cl), as would be expected on the basis of the direct interaction model. Furthermore, using the calculated substituent effects for the monosubstituted derivatives, the authors were able to predict those for di- and tri-substituted analogues with reasonable accuracy (3,5-Cl2). As the Hunter–Sanders π-polarization model does not predict that substituent effects will be additive, these results, along with the observation of a positional dependence of the magnitude of the substituent effect, were taken as evidence to support the Wheeler–Houk direct interaction model.
Figure 3. Experiments quantifying effects on π-stacking interactions.

a, Substituent effects on π-stacking interactions detected using molecular torsion balance 3.1 (shown unfolded and folded); Δ Go = –RTlnKeq. b, Transition-state interaction energies (Δ GTS) of rings 2 and 3 during conversion of pyridine of 3.2 (left) to methyl pyridinium (right) using methyl iodide (MeI).
Considering the manner in which π–π interactions can be employed for ground-state stabilization, it stands to reason that they might analogously be used to effect transition-state binding for catalysis. An excellent example of this phenomenon was reported by Hunter and co-workers for the alkylation of pyridine in a supramolecular zipper complex (3.2, Fig. 3b)19. It was hypothesized that a π-interaction between the pyridine ring (ring 3) and ring 2 would be more stabilizing in the polarizable SN2 transition state than in the ground-state reactant or product complexes. Furthermore, it was anticipated that tuning the electronics of ring 2 might allow for the modulation of the putative transition-state π interaction, thereby controlling the reaction rate. Measurement of the ground-state binding constants (calculated from Δ GGS) for a series of zipper complexes (X = NO2, H, NMe2) and the rates of both the uncatalysed and catalysed alkylation reactions (calculated from Δ G‡free and Δ G‡bound) enabled the authors19 to estimate the binding constant between rings 2 and 3 at the alkylation transition state (Δ GTS, Fig. 3b). Using this information, a double mutant cycle analysis20,31 allowed for the determination of the isolated contribution of a π interaction between rings 2 and 3 to transition-state stabilization, as well as stabilization of both the reactant and product complexes, in the absence of any additional factors contributing to complex stability (such as dispersion, electrostatic repulsion, hydrogen-bonding). When X = NO2, rings 2 and 3 were engaged in stronger π–π interactions in the reactant and product complexes than in the transition state, implicating negative catalysis. In contrast, when X = H or NMe2, the complexes exhibited stronger π–π interactions between rings 2 and 3 in the transition state relative to the ground state, definitively demonstrating the contribution of these interactions to the acceleration of pyridine methylation. The nature of the substituent effect is difficult to discern given the aforementioned geometric complexities of the direct interaction model. However, the clear electronic trend lends credence to the notion that π–π interactions can be substantially modulated through subtle electronic perturbations of the structures of the interacting partners to affect chemical reactivity.
XH–π interactions
The interaction between the C–H bond of an arene (that is, X–H) and the face of another, resulting in a ‘T’-shaped geometry (Fig. 1a), has physical origins similar to those described for the attraction between two aryl rings and is often incorporated into the π-stacking category or even characterized as a type of hydrogen-bonding32. We elected to separate CH–π interactions to highlight distinct features from the former class and explore additional XH–π interactions, where X may be C(sp hybridized), C(sp3), B, N, O or a halogen. For example, the computed interaction energies of the C–H bonds in ethane, ethylene and acetylene complexed with the face of benzene showed a stronger interaction as the carbon exhibited more s character33. Thus, the identity of the interacting X component is as important as the nature of the π system. To investigate the origins of these NCIs, Wheeler and co-workers examined both X atom identity (B, C(sp3), N, O and F) and arene substituent effects in a systematic computational study, which revealed that the sensitivity to aryl substitution and the contributions to the interaction energy from dispersion and electrostatic forces varied depending on the identity of the arene substituent (S, Fig. 4a)34. Overall, electron-donating substituents on the aromatic ring enhanced the interactions for each XH–π combination examined, as established through linear correlations of the computed interaction energies with Hammett σmeta values. More electronegative atoms (F and O) demonstrated greater sensitivity to the electronic nature of the arene, and the interaction energies were thus dominated by an electrostatic term. Conversely, the CH–π and BH–π interactions were much less sensitive to the substituent effects, revealing that dispersion dominates in these attractions. Although these computational studies provide detailed insight into the more elusive XH–π interactions, most experimental work has focused on aryl and alkyl CH donors, especially in the context of biological and supramolecular systems.
Figure 4. Studies of XH–π interactions.

a, Effect of X and arene identity on XH–π interactions. The structure that was computed to determine Eint in each case is shown boxed at right; S is a variable substituent. Adapted from ref. 34, American Chemical Society. b, Double mutant cycles in β-hairpin oligopeptide to quantify CH–π interactions. Left, the oligopeptide under evaluation, where X and Y are variable substituents. Previous studies support the CH–π interactions as shown in the box on the right. c, Chiral host (left) binds amino acid guests (right) selectively through a CH–π interaction, as measured by KD/KL. d, Rationalization of enantioselectivity in asymmetric transfer hydrogenation of aryl ketones. Top, reaction studied; bottom, calculated diastereomeric transition states (Si and Re isomers shown left and right, respectively). e.e., enantiomeric excess.
Alkyl CH–π interactions have been extensively acknowledged as an important element in molecular recognition between carbohydrates and proteins35. To quantify the stabilization gained from such interactions in the ground state, Waters and co-workers applied double mutant cycle analysis to a β-hairpin oligopeptide, which was designed to allow positions X and Y to interact directly when the β-sheet was folded (Fig. 4b)36. The points of interaction between Ac4Glc (Glc = glucose) and the aromatic residues were identified through upfield 1H NMR shifts of H1, H3, H5, and H6,6′ as well as nuclear Overhauser effect (NOE) experiments (Fig. 4b, right). Double mutant cycles for four aromatic residues revealed weaker interaction energies as the surface area of the π system decreased. In a corresponding trend, a reduced extent of β-sheet folding resulted, indicating lower stability of the folded oligopeptide with decreasing surface area. When a non-aromatic substituent was included at the X position (cyclohexane), the upfield shifts and NOE signals characteristic of the aromatic groups were not identified, suggesting the two aliphatic groups did not interact. Correspondingly, a low degree of β-sheet folding was measured, and double mutant cycle analysis suggested a trivial interaction energy. Overall, these data suggest that the stabilization of the β-hairpin results from CH–π interactions between the carbohydrate and the aromatic residues. Thus, this model system reveals that an attractive interaction between alkyl C–H bonds and π networks can have a substantial role in the stabilization required for the tertiary structure of a polypeptide.
The potential for CH–π interactions to enable the enantioselective recognition of chiral molecules was also explored by Martín and co-workers, who demonstrated the ability of chiral receptor 4a to distinguish between the d and l forms of several aromatic amino acids (Fig. 4c)37. Using temperature-dependent and 2D-NMR experiments, points of interaction between the host and guest molecules were assigned, revealing that protons R in the ethylene glycol spacer of host 4a were probably interacting directly with the π systems of the aromatic amino acids. Of note, intermolecular rotating frame Overhauser effects (ROEs) were observed between host 4a (protons R) and d-tryptophan (Trp) but not with l-Trp, suggesting this interaction was important for host–guest selectivity. When ethylene protons were exchanged for fluorine atoms (host 4b), thus eliminating the putative CH–π interactions, diminished binding was observed for the favoured amino acids. In order to quantify the stabilization gained from this CH–π interaction, a double mutant cycle analysis was performed. The interaction energy between the receptor protons and l and d-Trp was determined to be −0.97 kcal mol−1, which accounted for 70% of the stabilization of the host:d-Trp complex when the selectivity for d-Trp versus l-Trp was considered (10.4:1 or Δ Δ GD/L = −1.4 kcal mol−1). This example of selective ground-state binding clearly demonstrates how a single CH–π interaction might be analogously manipulated to affect selectivity through transition-state interactions.
Despite their prevalence in the ground state, CH–π interactions have been less frequently applied explicitly for transition-state stabilization. Nonetheless, a well-known example of aromatic CH–π interactions in small-molecule catalysis is the rationalization of the origin of enantioselectivity in the Noyori-type asymmetric transfer hydrogenation of aryl ketones38,39. Through a detailed computational study, the aryl ligand was proposed to donate a C–H bond that interacts with the aryl group on the ketone, thus providing stabilization of the ‘Si’ diastereomeric transition state, which led to the observed product (Fig. 4d). Favoured by 2.1 kcal mol−1, this calculated transition state (left structure) revealed an interatomic distance of 3.0 Å between a C–H bond in the cyclopentadienyl ring and the aryl group in the substrate, well within the boundaries of a CH–π interaction. Although this specific interaction was not quantified in this example, we view this a posteriori justification of a stereochemical outcome as a foundation for future rational design of such interactions in catalytic systems.
Cation–π interactions
The cation–π interaction describes the association between a cation and the face of a molecule containing a π system40–45. First reported for the binding of K+ to benzene in the gas phase, subsequent experimental studies demonstrated association free enthalpies (Δ Ho) for the alkali metals with benzene of 38.3, 28.0 and 19.2 kcal mol−1 for Li+, Na+ and K+, respectively, placing the cation–π interaction among the strongest NCIs known40. Rationalized by the quadrupole moment of benzene (see above), electrostatic potential (ESP) maps generally serve as good qualitative predictors of the strength of the cation–π interaction as a function of ring substitution46,47, with increasingly electron-donating substituents affording increasingly negative ESPs above and below the ring plane, predicting a stronger interaction with a cation. Thus, to a first approximation, the cation–π interaction can be understood as one between an ion and an electric quadrupole (although it cannot be modelled quantitatively in this manner) and is primarily electrostatic in nature. Yet, in certain instances, particularly those involving large, polarizable π systems, the role of induction has also been suggested to be important41,48,49. Wheeler and Houk have warned against misinterpretation of the correlation of ESP maps with stronger/weaker interactions, suggesting that it is the orientations of the local dipoles of the C–H/X bonds around the ring periphery, not the donation or withdrawal of π-electron density from the ring centre by the substituent, that are responsible for the negative ESPs in the case of electron-rich aromatics46. In spite of the physical origins of these effects, ring substitution and adjustment of the spatial extent of a π system can both serve as ways to produce predictable modulation of the strength of cation–π interactions.
Nature provides several excellent platforms for studying cation–π interactions in both ground and transition states. A recurring motif in proteins for the recognition of cationic moieties is the so-called aromatic box42,47—a term that characterizes binding pockets composed of various combinations of three to four tryptophan (Trp), tyrosine (Tyr) or phenylalanine (Phe) residues precisely arranged to stabilize positive charge via cation–π interactions (Fig. 5a). A prototypical example that has been studied by the groups of Lester and Dougherty is that of the nicotinic acetylcholine receptor (nAChR), a ligand gated ion channel that is modulated by the binding of the ammonium group of acetylcholine, ACh50,51. Using classical physical organic techniques, the authors have demonstrated the half-maximum effective concentration (EC50) of nAChR resulting from ground-state ACh binding can be modulated over 2 orders of magnitude (1.2–114 μM, > 2.0 kcal mol−1) simply through the mutation of a single aromatic box constituent (α Trp 149) to substituted analogues with attenuated cation binding affinity (such as F-Trp, CN-Trp).
Figure 5. Examples of cation–π interactions.

a, Left, depiction of the aromatic box in the HP1 chromodomain binding trimethyllysine (KMe3) of histone 3 (H3: Y, tyrosine; W, tryptophan). Green, HP1 chromodomain carbon; blue, nitrogen; red, oxygen; black, H3 carbon. Right, binding of HP1 chromodomain to wild-type H3 (unmodified) and a series of mutants (tBuNle9, tert-butyl norleucine; K9Me1, methyllysine; K9Me2, dimethyllysine; K9Me3 trimethyllysine). Reprinted from ref. 52, National Academy of Sciences, USA. b, Proposed mechanism for hopene synthesis from squalene. c, Nucleophilic ring opening of episulfonium ions is stabilized by a cation–π interaction: top, reaction; middle, transition state; bottom, effect of catalyst substituents on product enantiomeric excess.
Also featured among the many classes of proteins known to possess the aromatic box motif are those that recognize methylated lysine, a post-translational modification implicated in gene regulation. In 2007, Waters and co-workers reported a study52 seeking to elucidate the physical origins of the recognition of a cationic methylated lysine (Lys or K) residue on the tail of histone 3 (H3) by the aromatic box of HP1 chromodomain (Fig. 5a). Although the interaction between H3 and HP1 was known to increase with increasing methylation (that is, KMe3 > KMe2 > KMe), it was unclear if this primarily was due to a cation–π interaction or a hydrophobic effect, as the latter may be expected considering the increase in lipophilicity with each methylation. To address this ambiguity, three H3 mutants were prepared varying in the extent of K9 methylation (H3K9Me3, H3K9Me2, H3K9Me1) and one in which K9 had been mutated to a neutral isostere, tert-butyl norleucine (t-BuNle, H3K9tBuNle). Compared with the wild-type H3 protein, diminished binding was observed with each successive methyl removal, with uncharged mutant H3K9tBuNle displaying 31-fold weaker binding relative to H3K9Me3. Given the similar sizes and shapes of these mutants, this result was taken as evidence of a cation–π interaction between the cationic H3 mutants and the aromatic residues in HP1 chromodomain.
In the same manner that the ground-state recognition of methyllysine is dictated by cation–π interactions, transition-state stabilization can also be manipulated. A field where this paradigm is frequently invoked is that of terpene biosynthesis53. In the first definitive example, Hoshino and co-workers studied54 the enzyme squalene-hopene cyclase (SHC)55, which catalyses the formation of hopene (5.4) via a complex cationic polycyclization cascade from the acyclic precursor squalene (5.1, Fig. 5b). Site-directed mutagenesis studies were conducted to study the electronic effects on Phe 356 and Phe 605, which are components of different aromatic box regions in SHC, implicated in stabilizing cationic intermediates 5.2 and 5.3, respectively. Estimation of Michaelis–Menten parameters revealed that, although the mutants displayed looser binding relative to the wild type, catalytic activity increased in the expected order based on the ability of the mutated residue to favourably interact with positive charge. Thus, Phe 605 mutants with electron-rich aromatic residues (Phe605Tyr, Phe605OMe-Tyr, Phe605Trp) displayed higher specific activities than the wild-type enzyme, and conversely, fluorinated Phe mutants (F1-Phe, F2-Phe, F3-Phe) at positions 365 and 605 demonstrated progressively diminished catalytic activity with each successive fluorination. A linear correlation was observed between the specific activity of the SHC mutants and the previously computed cation–π binding energies46 of the FnPhe residues. These results convincingly support in a quantitative manner the hypothesis that cation–π interactions at both positions within SHC are essential to hopene biosynthesis, presumably via both ground- and transition-state stabilization.
Considering the lessons learned from examples such as those presented above, it should in principle be possible to design synthetic catalysts that accelerate novel reactions via stabilizing cation–π interactions in the transition state. To this end, Jacobsen and co-workers have hypothesized that enantioselectivity, a kinetic phenomenon, can be dictated by aromatic substituents deliberately incorporated into synthetic catalyst scaffolds56–58. In an excellent example disclosing the enantioselective nucleophilic ring opening of episulfonium ions with indole58, the authors proposed that a meso-episulfonium ion was generated upon trichloroacetimidate protonation by 4-nitrobenzenesulfonic acid (4-NBSA, Fig. 5c). A bifunctional chiral thiourea catalyst (5.6) was proposed to bind the conjugate base of 4-NBSA and the episulfonium ion in a spatially resolved ion pair through a combination of hydrogen-bonding and cation–π interactions, respectively, allowing an enantioselective nucleophilic attack by indole to occur. The presence of the latter interaction was hypothesized on the basis of the observation that reaction enantioselectivity generally correlated with the size of extended π systems of the catalyst aryl substituent. To distinguish unambiguously whether enantioselection was occurring because of a transition-state-stabilizing cation–π interaction in the major pathway or a destabilizing steric interaction in the minor pathway, the authors took advantage of the observation that indole addition to the episulfonium–thiourea complex (5.5) was both rate-determining and enantio-determining. The rate constants for the pathways leading to the major and minor enantiomers could be distinguished, and both were shown to independently correlate with reaction enantioselectivity. This experiment rigorously demonstrates that the cation–π interaction can be used to achieve transition-state stabilization in a completely designed system that operates by the same principles observed in fundamental, ground-state studies (see above).
Anion–π interactions
Superficially, anion–π interactions emerge as the natural complement to their cationic counterparts, typically describing the attractive association between a negatively charged atom or molecule and the π face of an electron-deficient (hetero)arene59–62. However, this interaction is somewhat counterintuitive considering the expected electron repulsion between an anion and the π-electron cloud of an arene. Since its original proposal in a series of ab initio studies in 200263–65, the anion–π interaction has been extensively studied theoretically and experimentally. Compared with cation–π, anion–π interactions have more shallow potential energy surfaces resulting in relaxed geometric requirements60,61,66. As such, in the solid state, as opposed to residing directly above the ring centroid (6.3), anions are more frequently observed either in the same plane as the aromatic ring interacting with polarized C–H bonds (6.1), or interacting with a ring π * orbital as in a Meisenheimer complex (6.2, Fig. 6a)61,67.
Figure 6. Examples of anion–π interactions.

a, Anion–π geometries exemplified in 6.1, 6.2 and 6.3. b, Effect of polarizability on anion–π interaction. Boxed structure, interaction geometry. Values of parameters are shown under the relevant structures: Qzz, z2 component of the quadrupole moment tensor; α, molecular polarizability; E, energy. c, Neutral receptors for anion binding. Top, depiction of bimolecular association between neutral receptors 6.4–6.6 and halide anions Cl−, Br− and I−; bottom, the free energies of association (Δ Go).
The stabilizing nature of anion–π interactions is typically attributed to a combination of electrostatic and ion-induced polarization effects, which can be described by the Qzz component of the arene’s quadrupole moment and its polarizability, respectively. Notably, these features can display a compensatory effect. As illustrated in Fig. 6b, Frontera and co-workers68 demonstrated computationally that the interaction energy of a chloride anion with a series of (thio)isocyanuric acid derivatives was essentially constant despite a decrease in the Qzz because of a systematic increase in polarizability (α) with each replacement of oxygen with sulphur. Additionally, the nature of the anion has a substantial impact on the interaction: smaller anions induce greater polarization in the π system and strengthen the association; and planar, polyatomic ions, such as NO3−, benefit from a π–π contribution to the overall interaction.
For benzene derivatives, substituent effects generally follow the trends expected on the basis of electrostatics, with increasingly electron-withdrawing substituents contributing to an enhanced positive electrostatic potential above and below the π face, strengthening the interaction. Like other NCIs involving aromatic rings, this has traditionally been attributed to π-polarization effects, but has more recently been explained on the basis of the Wheeler–Houk direct interaction model26, in which an anion experiences direct, stabilizing interactions with the local dipoles of the C–X bonds located around the periphery of the arene69,70. Electron-deficient nitrogenous heterocycles have featured particularly prominently in both theoretical and experimental explorations of anion–π interactions, given their intrinsically positive Qzz and molecular ESP values63–65,71. It was recently suggested that these last features are consequences of the influx of positive nuclear charge towards the ring centre upon nitrogen incorporation, rather than π-polarization effects70. Regardless of their physical origin, both quadrupole moments and ESP maps provide reasonable qualitative guides to tuning the strength of anion–π interactions.
In a study quantifying the ground-state association between anions and electron-poor arenes in solution, Johnson and co-workers measured the association constants (Ka) between the n-heptylammonium salts of Cl−, Br− and I− and a series of neutral tripodal receptors (6.4–6.6, Fig. 6c)72. The authors anticipated that 2,4-dinitro substituted host 6.4 would interact with the halide series via C–H hydrogen-bonding. However, owing to steric constraints, this binding mode was expected to be precluded for 3,5-dinitro analogue 6.6, necessitating involvement of the arene faces in anion recognition. Strong association with all three halides was apparent by NMR titration in C6D6 (Ka = 11–53 M−1, Δ G = −1.4 to −2.4 kcal mol−1), with the 3,5-disubstituted receptor exhibiting superior binding in all cases in the expected order based on size (that is, Cl− > Br− > I−). Electron-rich control receptor 6.5 did not exhibit measurable binding. On the basis of both NMR studies and DFT calculations, it was proposed that receptor 6.6 bound the halides via a σ complex rather than a true anion–π complex, demonstrating the ambiguity often associated with the geometric nature of anion–π interactions.
The ability of anion–π interactions to stabilize both ground and transition states has been investigated intensively by Matile and co-workers73–77. Illustrative examples can be drawn from the realm of enolate chemistry74–78. A model system was developed in which a malonate moiety was situated near the surface of a naphthalenediimide (NDI) scaffold, which was expected to engage in stabilizing anion–π interactions, given the parent NDI’s notably positive (Qzz = + 19 B, where 1 buckingham (B) = 1 D Å) quadrupole moment (Fig. 7a). It was shown that tuning of the NDI’s electronic character via manipulations of the substituents around its periphery enabled the malonate’s pKa to be adjusted over six orders of magnitude (Δ Δ GGS = −7.48 ± 0.287 kcal mol−1) compared with diethyl malonate (7.1) as a control (pKa = 16.4)78. An excellent correlation was observed between the LUMO (lowest unoccupied molecular orbital) energies of the substituted NDIs and the observed pKas. These results were attributed to the ability of the NDI surfaces to engage the enolate conjugate bases in attractive anion–π interactions in a manner that could be precisely controlled.
Figure 7. Substituent effects on anion–π interactions.

a, Modulation of pKa values of malonate moiety embedded within naphthalenediimide (NDI) scaffold (7.2, bottom) through anion–π interactions compared with diethylmalonate as a control (7.1, top). b, Top, depiction of differential stabilization of planar (leading to addition product 7.4) and non-planar (leading to decarboxylation product 7.5) tautomers of malonic acid half thioester (7.3) by NDI scaffold. Bottom, effect of modulation of NDI (7.7 and 7.9) electronic properties on product distribution (addition versus decarboxylation, a/d) and comparison with controls (7.6 and 7.8) incapable participating in anion–π interactions. Differential transition-state stabilization is tuned over a range of 1.3 kcal mol−1.
The observation that NDI surfaces could stabilize enolates in the ground state was exploited for transition-state stabilization to control catalytic selectivity (Fig. 7b)76,79. The addition of malonic acid half thioester (MAHT) 7.3 to β-nitrostyrene proceeded in low yields when catalysed by triethylamine owing to competitive decarboxylation (addition/decarboxylation = 0.6). It was anticipated that, in the presence of an appropriate NDI catalyst, the planar enolate tautomer leading to the desired addition (a) would be stabilized to a greater degree than the non-planar tautomer leading to decarboxylation (d) and, correspondingly, that the transition state for each would be affected to differing degrees, affecting selectivity (a/d). This hypothesis was verified in practice, as illustrated for catalysts 7.7 and 7.9 (Fig. 7b, bottom)75. Increasing the π-acidity of the NDI surface through oxidation of the sulfur substituents was observed to progressively improve selectivity in favour of the addition product from a ratio of 1.25 up to 9.6. Furthermore, it was noted that rigidification of the ‘Leonard turn’ connecting the amine functionality to the NDI surface (7.7) led to improved selectivity (compare with 7.9), presumably by enabling closer contact between the surface and the transition state leading to the major product. Comparison with control catalysts 7.6 and 7.8 provides further support for the notion that anion–π interactions were responsible for the inversion in product selectivity, and initial rate experiments led the authors to conclude that this observation was due to simultaneous deceleration of decarboxylation and acceleration of addition. Overall, these examples embody the means by which the principles underlying ground-state association can be applied to the challenge of rational catalyst design.
Lone pair–π interactions
The lone pair–π (lp–π, also referred to as n to π* ) interaction80 describes the stabilizing association between a lone pair of electrons and the face of a π system, and is somewhat counterintuitive considering the presumed electron–electron repulsion between these components. Like the anion–π interaction, it is typically expected to be more important for electron-deficient π-systems, and can qualitatively be understood as the interaction between regions of negative (the lone pair) and positive (the Qzz component of the traceless quadrupole moment tensor above and below the face of the π system) electrostatic potential81,82. However, several studies, both theoretical83–85 and experimental, have noted the shortcomings of this picture and acknowledge the likely significance of electron correlation, or dispersion effects, with weak attractive interactions having been observed for electron-rich aromatic systems with negative quadrupole moments86. Although it is expected to be individually quite weak, the significance of the lp–π interaction has been noted in cooperation with other NCIs such as hydrogen-bonding (see below)87,88. The first lp–π interaction to be acknowledged explicitly originates in structural biology as a stabilizing structural element for Z-DNA89,90. As shown in Fig. 8a, the O4′ oxygen atom of cytidine was crystallographically observed to lie within 2.9 Å of guanine C2 for each d(CpG) step89, and a subsequent ab initio study90 estimated the stabilization gained from this interaction to be of the order of 2 kcal mol−1 when guanine was coordinated by Mg2+ at N7. The significance of the lone pair–π interaction in crystal engineering has been noted by Reedijk and co-workers, based on a thorough analysis of the crystallographic database91.
Figure 8. Examples of lone pair–π interactions.

a, Example of lone pair(lp)–π interaction in Z-DNA (broken red line in magnified inset at right). Adapted from ref. 80, PCCP Owner Societies, and refs 90 and 96, American Chemical Society. b, Quantification of lp–π interaction in solution. Box at left, depiction of the dimeric solid-state structure of amino alcohol 8.1 implicating lp–π interactions as important structural elements. Middle, association between electronically diverse dimethylamino arylethylamines (8.2 and 8.3) and methanol in solution, and (right) correlation between enthalpy of association (Δ Hobs) and the Taft parameter (σ* ). c, Quantification of lp–π interactions using molecular torsion balances (anti and syn isomers of 8.4). Δ Go = –RTlnKeq, where Keq = [syn]/[anti].
Several key experimental studies have contributed to the understanding of the strength and nature of lp–π interactions. On the basis of observance of a close contact (3.08 Å) between the alcohol oxygen and C6F6 ring centroid in the crystal structure of amino alcohol 8.1 (Fig. 8b)92, Korenaga and co-workers conducted a study on a simplified system to assess the existence of a bona fide lp–π interaction93. By studying the association between a series of N,N-dimethylamino arylethylamines and methanol by 1H NMR spectroscopy, the authors expected to observe a linear relationship between amine basicity and enthalpy of association (Δ Hobs) if hydrogen-bonding was solely responsible for the interaction. As shown in Fig. 8b (right), a good correlation was found between Δ Hobs and Taft’s σ* parameter for a range of electronically diverse arylethylamines (8.2a–8.2e, −6.09 to −5.72 kcal mol−1), with the exception of pentafluorophenyl derivative 8.3 (−6.15 kcal mol−1). This outlier was rationalized on the basis of an intramolecular lp–π interaction supplementing the otherwise weaker hydrogen bond in this complex (8.3), whose electron-deficient aryl substituent would be expected to interact favourably with a lone pair. This proposal was corroborated by a notably negative entropy of association (Δ Sobs = −19.9 cal mol−1 K−1) as might be expected for the proposed conformationally restricted complex 8.3, and by a computational analysis that showed good qualitative agreement between the predicted and measured Δ Hobs for the full data set.
Gung and Amicangelo have investigated lp–π interactions using a molecular torsion balance based on a triptycene scaffold (8.4, Fig. 8c)86,94. The relative populations of the syn and anti conformers, which were readily determined by 1H NMR spectroscopy, were expected to be dictated by the lp–π interaction strength between the C9 benzyl and the C1 methoxymethyl (MOM) substituent. As shown in Fig. 8c, the expected trend based on electrostatic reasoning (Δ Go = 1.2 kcal mol−1 for C6F5 to −0.19 for 4-NMe2) was observed. However, the fact that the interaction was attractive even for electron-neutral to electron-rich derivatives (H, Me, NMe2) could not be explained by electrostatic reasoning alone, leading the authors to invoke dispersion as the dominant force in these latter cases; they concluded that aromatic rings cannot be treated as simple quadrupolar groups at the short distances required for many NCIs86. A recent experimental study by Aliev and Motherwell demonstrated that heteroatom substitution might also serve as a means to tune the strength of lp–π interactions in a rationally designed system95.
To our knowledge, no explicit examples exist of rational catalyst design using lessons from ground-state studies of lp–π interactions such as those highlighted. However, by direct analogy with the situation for π–π, cation–π and anion–π interactions, we fully anticipate examples to be forthcoming. Although not rigorously verified as such, Toste and Sigman invoked a transition-state lp–π interaction between a chiral catalyst and an achiral additive to rationalize an inversion in the sense of enantioselection in an asymmetric fluorination of allylic alcohols (77% enantiomeric excess (e.e.) of R to 92% e.e. of S), based on a series of structure–selectivity studies96.
Conclusions and outlook
The preceding paragraphs have provided a limited subset of examples illustrating the analogy between binding in the ground and transition states within the spectrum of specific non-covalent π interactions. Given the seemingly sophisticated knowledge surrounding the physical underpinnings of each interaction discussed, one might inquire as to why genuine de novo design of catalysts for novel synthetic transformations is not commonplace. One oft-cited reason is that, although a given NCI’s spatial and geometric requirements may be well-understood, its individual contribution to binding may be small and thus in competition with many other interactions, rendering its ability to be manipulated in isolation highly context dependent97. Additionally, the design of a catalyst at the outset of a synthetic methodology project would require a detailed understanding of the rate/selectivity-determining transition state—a challenge frequently beyond current capabilities. Indeed, this prevailing viewpoint was recently expressed in a comprehensive review on NCIs in supramolecular catalysis: “non-covalent intermolecular forces are hardly predictable and so far cannot be used by a chemist who wishes to design ‘a priori’ a catalyst with assembling properties.”98
However, as is so often the case when a dead end seems imminent, perhaps a shift in perspective is required. Although it may not yet be possible to design catalysts that are perfectly complementary to any transition state, it should become increasingly feasible to identify where certain interactions are operative in a mechanism and to use the lessons from ground-state binding to manipulate them rationally. Essential to this goal will be modularity with respect to catalyst design, such that aromatic motifs may be readily installed with varying steric and electronic properties at multiple catalyst positions. To this end, organocatalysts (for example, chiral phosphoric acids, thioureas, amino acids) represent a catalyst class particularly ripe for investigation. Furthermore, in addition to the techniques described above, kinetically controlled product ratios should see increasing use as energetic probes, as these values represent exquisitely sensitive measurements of relative rates arising from subtle transition-state interactions (a 99:1 product ratio results from a 2.7 kcal mol−1 energy difference between competing transition states). Our groups have recently adopted this approach to deliberately influence enantioselectivity in the realm of chiral phosphate catalysis—this strategy is generalizable to any scenario in which catalyst activity varies as a function of molecular structure96,99. By evaluating data sets of enantioselectivity values (that is, relative rates of formation of R and S enantiomers) from different catalyst and substrate combinations, we have developed hypotheses regarding the NCIs underlying selectivity, allowing us to subsequently manipulate these interactions in explicit, predictable ways. More generally, if this sort of approach were to be adopted at the outset of a catalysis project—that is, NCIs were acknowledged to be possibly relevant—such interactions would be considered as explanations when unexpected results arose.
Given recent advances in high-throughput experimentation capabilities100, ever-more creative metrics for describing molecular structure101, and computational methods tuned to model NCIs102, the prospects for identifying causal structure–activity/selectivity relationships in catalysis seem bright. It is our hope that these modern technologies can continue to be integrated with classical physical organic methodologies to enable the ultimate goal of truly rational catalyst design to be achieved.
Acknowledgments
We thank A. Milo for discussions. M.J.H. and M.S.S. thank the NSF (CHE-1361296) for financial support; A.J.N. and F.D.T. thank the NIHGMS (R35 GM118190) for financial support.
Footnotes
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author Contributions Each author contributed to the planning and writing of this manuscript.
Reviewer Information Nature thanks S. Wheeler and J. Reek for their contribution to the peer review of this work.
References
- 1.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:939–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 2.Kirby AJ. Enzyme mechanisms, models, and mimics. Angew Chem Int Edn Engl. 1996;35:706–724. [Google Scholar]
- 3.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 4.Biedermann F, Schneider HJ. Experimental binding energies in supramolecular complexes. Chem Rev. 2016;116:5216–5300. doi: 10.1021/acs.chemrev.5b00583. [DOI] [PubMed] [Google Scholar]
- 5.Schneider HJ. Binding mechanisms in supramolecular complexes. Angew Chem Int Ed. 2009;48:3924–3977. doi: 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
- 6.Mader MM, Bartlett PA. Binding energy and catalysis: the implications for transition-state analogs and catalytic antibodies. Chem Rev. 1997;97:1281–1302. doi: 10.1021/cr960435y. [DOI] [PubMed] [Google Scholar]
- 7.Knowles RR, Jacobsen EN. Attractive noncovalent interactions in asymmetric catalysis: links between enzymes and small molecule catalysts. Proc Natl Acad Sci USA. 2010;107:20678–20685. doi: 10.1073/pnas.1006402107. Thought-provoking discussion of the rational implementation of attractive NCIs in asymmetric catalysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis HJ, Phipps RJ. Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem Sci. 2017;8:864–877. doi: 10.1039/c6sc04157d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle AG, Jacobsen EN. Small-molecule H-bond donors in asymmetric catalysis. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 10.Brak K, Jacobsen EN. Asymmetric ion-pairing catalysis. Angew Chem Int Ed. 2013;52:534–561. doi: 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krenske EH, Houk KN. Aromatic interactions as control elements in stereoselective organic reactions. Acc Chem Res. 2013;46:979–989. doi: 10.1021/ar3000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinnokrot MO, Sherrill CD. High-accuracy quantum mechanical studies of π-π interactions in benzene dimers. J Phys Chem A. 2006;110:10656–10668. doi: 10.1021/jp0610416. [DOI] [PubMed] [Google Scholar]
- 13.Lee EC, et al. Understanding of assembly phenomena by aromatic-aromatic interactions: benzene dimer and the substituted systems. J Phys Chem A. 2007;111:3446–3457. doi: 10.1021/jp068635t. [DOI] [PubMed] [Google Scholar]
- 14.Martinez CR, Iverson BL. Rethinking the term “pi-stacking”. Chem Sci. 2012;3:2191–2201. Interesting discussion of relevance of π systems to interactions between aromatic rings. [Google Scholar]
- 15.Wagner JP, Schreiner PR. London dispersion in molecular chemistry—reconsidering steric effects. Angew Chem Int Ed. 2015;54:12274–12296. doi: 10.1002/anie.201503476. Intriguing discussion of various manifestations of London dispersion forces in molecular interactions. [DOI] [PubMed] [Google Scholar]
- 16.Raju RK, Bloom JWG, An Y, Wheeler SE. Substituent effects on non-covalent interactions with aromatic rings: insights from computational chemistry. ChemPhysChem. 2011;12:3116–3130. doi: 10.1002/cphc.201100542. [DOI] [PubMed] [Google Scholar]
- 17.Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K. Origin of attraction and directionality of the π/π interaction: model chemistry calculations of benzene dimer interaction. J Am Chem Soc. 2002;124:104–112. doi: 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- 18.Hunter CA, Sanders JKM. The nature of π–π interactions. J Am Chem Soc. 1990;112:5525–5534. [Google Scholar]
- 19.Hunter CA, Low CMR, Vinter JG, Zonta C. Quantification of functional group interactions in transition states. J Am Chem Soc. 2003;125:9936–9937. doi: 10.1021/ja034767d. [DOI] [PubMed] [Google Scholar]
- 20.Cockroft SL, Hunter CA, Lawson KR, Perkins J, Urch CJ. Electrostatic control of aromatic stacking interactions. J Am Chem Soc. 2005;127:8594–8595. doi: 10.1021/ja050880n. [DOI] [PubMed] [Google Scholar]
- 21.Cozzi F, Cinquini M, Annunziata R, Dwyer T, Siegel JS. Polar/π interactions between stacked aryls in 1,8-diarylnaphthalenes. J Am Chem Soc. 1992;114:5729–5733. [Google Scholar]
- 22.Cozzi F, et al. Through-space interactions between face-to-face, center-to-edge oriented arenes: importance of polar-π effects. Org Biomol Chem. 2003;1:157–162. doi: 10.1039/b208871a. [DOI] [PubMed] [Google Scholar]
- 23.Sinnokrot MO, Sherrill CD. Substituent effects in π-π interactions: sandwich and T-shaped configurations. J Am Chem Soc. 2004;126:7690–7697. doi: 10.1021/ja049434a. [DOI] [PubMed] [Google Scholar]
- 24.Grimme S. Do special noncovalent π-π stacking interactions really exist? Angew Chem Int Ed. 2008;47:3430–3434. doi: 10.1002/anie.200705157. Computational study exploring physical reality of π–π terminology. [DOI] [PubMed] [Google Scholar]
- 25.Bloom JWG, Wheeler SE. Taking the aromaticity out of aromatic interactions. Angew Chem Int Ed. 2011;50:7847–7849. doi: 10.1002/anie.201102982. [DOI] [PubMed] [Google Scholar]
- 26.Wheeler SE, Houk KN. Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene. J Am Chem Soc. 2008;130:10854–10855. doi: 10.1021/ja802849j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler SE, Houk KN. Origin of substituent effects in edge-to-face aryl–aryl interactions. Mol Phys. 2009;107:749–760. doi: 10.1080/00268970802537614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheeler SE. Local nature of substituent effects in stacking interactions. J Am Chem Soc. 2011;133:10262–10274. doi: 10.1021/ja202932e. Early espousal of the importance of direct interaction between substituents in tuning strengths of aromatic interactions. [DOI] [PubMed] [Google Scholar]
- 29.Parrish RM, Sherrill CD. Quantum-mechanical evaluation of π-π versus substituent-π interactions in π stacking: direct evidence for the Wheeler-Houk picture. J Am Chem Soc. 2014;136:17386–17389. doi: 10.1021/ja5101245. [DOI] [PubMed] [Google Scholar]
- 30.Hwang J, et al. Additivity of substituent effects in aromatic stacking interactions. J Am Chem Soc. 2014;136:14060–14067. doi: 10.1021/ja504378p. [DOI] [PubMed] [Google Scholar]
- 31.Cockroft SL, Hunter CA. Chemical double-mutant cycles: dissecting non-covalent interactions. Chem Soc Rev. 2007;36:172–188. doi: 10.1039/b603842p. Review of an essential technique that has been used to experimentally quantify weak (< 3 kcal mol−1) interactions. [DOI] [PubMed] [Google Scholar]
- 32.Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabarti P. CH-π hydrogen bonds in biological macromolecules. Phys Chem Chem Phys. 2014;16:12648–12683. doi: 10.1039/c4cp00099d. [DOI] [PubMed] [Google Scholar]
- 33.Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K. The magnitude of the CH/π interaction between benzene and some model hydrocarbons. J Am Chem Soc. 2000;122:3746–3753. [Google Scholar]
- 34.Bloom JWG, Raju RK, Wheeler SE. Physical nature of substituent effects in XH/π interactions. J Chem Theory Comput. 2012;8:3167–3174. doi: 10.1021/ct300520n. [DOI] [PubMed] [Google Scholar]
- 35.Asensio JL, Ardá A, Cañada FJ, Jiménez-Barbero J. Carbohydrate-aromatic interactions. Acc Chem Res. 2013;46:946–954. doi: 10.1021/ar300024d. [DOI] [PubMed] [Google Scholar]
- 36.Laughrey ZR, Kiehna SE, Riemen AJ, Waters ML. Carbohydrate-π interactions: what are they worth? J Am Chem Soc. 2008;130:14625–14633. doi: 10.1021/ja803960x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrillo R, López-Rodríguez M, Martín VS, Martín T. Quantification of a CH-π interaction responsible for chiral discrimination and evaluation of its contribution to enantioselectivity. Angew Chem Int Ed. 2009;48:7803–7808. doi: 10.1002/anie.200903281. [DOI] [PubMed] [Google Scholar]
- 38.Noyori R, Hashiguchi S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 39.Yamakawa M, Yamada I, Noyori R. CH/π attraction: the origin of enantioselectivity in transfer hydrogenation of aromatic carbonyl compounds catalyzed by chiral η 6-arene-ruthenium(II) complexes. Angew Chem Int Ed. 2001;40:2818–2821. doi: 10.1002/1521-3773(20010803)40:15<2818::AID-ANIE2818>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Ma JC, Dougherty DA. The cation–π interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 41.An Y, Wheeler SE. Encyclopedia of Inorganic and Bioinorganic Chemistry. Wiley & Sons; 2011. Cation–π interactions. [Google Scholar]
- 42.Dougherty DA. The cation-π interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennedy CR, Lin S, Jacobsen EN. The cation-π interaction in small-molecule catalysis. Angew Chem Int Ed. 2016;55:12596–12624. doi: 10.1002/anie.201600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mecozzi S, West AP, Jr, Dougherty D. A Cation-π interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci USA. 1996;93:10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mecozzi S, West AP, Dougherty DA. Cation–π interactions in simple aromatics: electrostatics provide a predictive tool. J Am Chem Soc. 1996;118:2307–2308. [Google Scholar]
- 46.Wheeler SE, Houk KN. Through-space effects of substituents dominate molecular electrostatic potentials of substituted arenes. J Chem Theory Comput. 2009;5:2301–2312. doi: 10.1021/ct900344g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daze KD, Hof F. The cation-π interaction at protein-protein interaction interfaces: developing and learning from synthetic mimics of proteins that bind methylated lysines. Acc Chem Res. 2013;46:937–945. doi: 10.1021/ar300072g. [DOI] [PubMed] [Google Scholar]
- 48.Cubero E, Luque FJ, Orozco M. Is polarization important in cation-π interactions? Proc Natl Acad Sci USA. 1998;95:5976–5980. doi: 10.1073/pnas.95.11.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuzuki S, Mikami M, Yamada S. Origin of attraction, magnitude, and directionality of interactions in benzene complexes with pyridinium cations. J Am Chem Soc. 2007;129:8656–8662. doi: 10.1021/ja071372b. [DOI] [PubMed] [Google Scholar]
- 50.Zhong W, et al. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proc Natl Acad Sci USA. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. Classic physical organic study demonstrating relevance of cation–π interactions in biological systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiu X, Puskar NL, Shanata JAP, Lester HA, Dougherty DA. Nicotine binding to brain receptors requires a strong cation-π interaction. Nature. 2009;458:534–537. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc Natl Acad Sci USA. 2007;104:11184–11188. doi: 10.1073/pnas.0610850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 54.Morikubo N, et al. Cation-π interaction in the polyolefin cyclization cascade uncovered by incorporating unnatural amino acids into the catalytic sites of squalene cyclase. J Am Chem Soc. 2006;128:13184–13194. doi: 10.1021/ja063358p. [DOI] [PubMed] [Google Scholar]
- 55.Wendt KU, Poralla K, Schulz GE. Structure and function of a squalene cyclase. Science. 1997;277:1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 56.Knowles RR, Lin S, Jacobsen EN. Enantioselective thiourea-catalyzed cationic polycyclizations. J Am Chem Soc. 2010;132:5030–5032. doi: 10.1021/ja101256v. Landmark example of rigorous quantification of an attractive NCI in asymmetric catalysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uyeda C, Jacobsen EN. Transition-state charge stabilization through multiple non-covalent interactions in the guanidinium-catalyzed enantioselective Claisen rearrangement. J Am Chem Soc. 2011;133:5062–5075. doi: 10.1021/ja110842s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin S, Jacobsen EN. Thiourea-catalysed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nat Chem. 2012;4:817–824. doi: 10.1038/nchem.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gamez P, Mooibroek TJ, Teat SJ, Reedijk J. Anion binding involving π-acidic heteroaromatic rings. Acc Chem Res. 2007;40:435–444. doi: 10.1021/ar7000099. [DOI] [PubMed] [Google Scholar]
- 60.Frontera A, Gamez P, Mascal M, Mooibroek TJ, Reedijk J. Putting anion-π interactions into perspective. Angew Chem Int Ed. 2011;50:9564–9583. doi: 10.1002/anie.201100208. [DOI] [PubMed] [Google Scholar]
- 61.Giese M, Albrecht M, Rissanen K. Experimental investigation of anion-π interactions—applications and biochemical relevance. Chem Commun. 2016;52:1778–1795. doi: 10.1039/c5cc09072e. [DOI] [PubMed] [Google Scholar]
- 62.Lucas X, Bauzá A, Frontera A, Quiñonero D. A thorough anion–π interaction study in biomolecules: on the importance of cooperativity effects. Chem Sci. 2016;7:1038–1050. doi: 10.1039/c5sc01386k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alkorta I, Rozas I, Elguero J. Interaction of anions with perfluoro aromatic compounds. J Am Chem Soc. 2002;124:8593–8598. doi: 10.1021/ja025693t. [DOI] [PubMed] [Google Scholar]
- 64.Mascal M, Armstrong A, Bartberger MD. Anion-aromatic bonding: a case for anion recognition by π-acidic rings. J Am Chem Soc. 2002;124:6274–6276. doi: 10.1021/ja017449s. [DOI] [PubMed] [Google Scholar]
- 65.Quiñonero D, et al. Counterintuitive interaction of anions with benzene derivatives. Chem Phys Lett. 2002;359:486–492. [Google Scholar]
- 66.Estarellas C, Bauzá A, Frontera A, Quiñonero D, Deyà PM. On the directionality of anion-π interactions. Phys Chem Chem Phys. 2011;13:5696–5702. doi: 10.1039/c0cp01894e. [DOI] [PubMed] [Google Scholar]
- 67.Hay BP, Custelcean R. Anion–π interactions in crystal structures: commonplace or extraordinary? Cryst Growth Des. 2009;9:2539–2545. [Google Scholar]
- 68.Frontera A, et al. Anion-π interactions in cyanuric acids: a combined crystallographic and computational study. Chemistry. 2005;11:6560–6567. doi: 10.1002/chem.200500783. [DOI] [PubMed] [Google Scholar]
- 69.Lu T, Wheeler SE. Quantifying the role of anion-π interactions in anion-π catalysis. Org Lett. 2014;16:3268–3271. doi: 10.1021/ol501283u. [DOI] [PubMed] [Google Scholar]
- 70.Wheeler SE, Bloom JWG. Anion-π interactions and positive electrostatic potentials of N-heterocycles arise from the positions of the nuclei, not changes in the π-electron distribution. Chem Commun. 2014;50:11118–11121. doi: 10.1039/c4cc05304d. [DOI] [PubMed] [Google Scholar]
- 71.Estarellas C, Frontera A, Quiñonero D, Deyà PM. Relevant anion-π interactions in biological systems: the case of urate oxidase. Angew Chem Int Ed. 2011;50:415–418. doi: 10.1002/anie.201005635. [DOI] [PubMed] [Google Scholar]
- 72.Berryman OB, Sather AC, Hay BP, Meisner JS, Johnson DW. Solution phase measurement of both weak σ and C–H…X− hydrogen bonding interactions in synthetic anion receptors. J Am Chem Soc. 2008;130:10895–10897. doi: 10.1021/ja8035652. [DOI] [PubMed] [Google Scholar]
- 73.Dawson RE, et al. Experimental evidence for the functional relevance of anion-π interactions. Nat Chem. 2010;2:533–538. doi: 10.1038/nchem.657. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Y, et al. Catalysis with anion-π interactions. Angew Chem Int Ed. 2013;52:9940–9943. doi: 10.1002/anie.201305356. Seminal example demonstrating the possibility of exploiting anion–π interactions for catalysis. [DOI] [PubMed] [Google Scholar]
- 75.Zhao Y, Sakai N, Matile S. Enolate chemistry with anion-π interactions. Nat Commun. 2014;5:3911. doi: 10.1038/ncomms4911. [DOI] [PubMed] [Google Scholar]
- 76.Zhao Y, Benz S, Sakai N, Matile S. Selective acceleration of disfavored enolate addition reactions by anion–π interactions. Chem Sci. 2015;6:6219–6223. doi: 10.1039/c5sc02563j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao Y, Cotelle Y, Avestro AJ, Sakai N, Matile S. Asymmetric anion-π catalysis: enamine addition to nitroolefins on π-acidic surfaces. J Am Chem Soc. 2015;137:11582–11585. doi: 10.1021/jacs.5b07382. [DOI] [PubMed] [Google Scholar]
- 78.Miros FN, et al. Enolate stabilization by anion-π interactions: deuterium exchange in malonate dilactones on π-acidic surfaces. Chemistry. 2016;22:2648–2657. doi: 10.1002/chem.201504008. [DOI] [PubMed] [Google Scholar]
- 79.Cotelle Y, et al. Anion-π catalysis of enolate chemistry: rigidified Leonard turns as a general motif to run reactions on aromatic surfaces. Angew Chem Int Ed. 2016;55:4275–4279. doi: 10.1002/anie.201600831. [DOI] [PubMed] [Google Scholar]
- 80.Singh SK, Das A. The n → π * interaction: a rapidly emerging non-covalent interaction. Phys Chem Chem Phys. 2015;17:9596–9612. doi: 10.1039/c4cp05536e. Thorough review of various manifestations of ground-state lp–π interactions. [DOI] [PubMed] [Google Scholar]
- 81.Gallivan JP, Dougherty DA. Can lone pairs bind to a π system? The water…hexafluorobenzene interaction. Org Lett. 1999;1:103–106. doi: 10.1021/ol990577p. [DOI] [PubMed] [Google Scholar]
- 82.Alkorta I, Rozas I, Elguero J. An attractive interaction between the π-cloud of C6F6 and electron-donor atoms. J Org Chem. 1997;62:4687–4691. [Google Scholar]
- 83.Amicangelo JC, Gung BW, Irwin DG, Romano NC. Ab initio study of substituent effects in the interactions of dimethyl ether with aromatic rings. Phys Chem Chem Phys. 2008;10:2695–2705. doi: 10.1039/b718722j. [DOI] [PubMed] [Google Scholar]
- 84.Ran J, Hobza P. On the nature of bonding in lone pair…π-electron complexes: CCSD(T)/complete basis set limit calculations. J Chem Theory Comput. 2009;5:1180–1185. doi: 10.1021/ct900036y. [DOI] [PubMed] [Google Scholar]
- 85.Badri Z, Foroutan-Nejad C, Kozelka J, Marek R. On the non-classical contribution in lone-pair-π interaction: IQA perspective. Phys Chem Chem Phys. 2015;17:26183–26190. doi: 10.1039/c5cp04489h. [DOI] [PubMed] [Google Scholar]
- 86.Gung BW, et al. Quantitative study of interactions between oxygen lone pair and aromatic rings: substituent effect and the importance of closeness of contact. J Org Chem. 2008;73:689–693. doi: 10.1021/jo702170j. [DOI] [PubMed] [Google Scholar]
- 87.Singh SK, Kumar S, Das A. Competition between n → π (Ar)* and conventional hydrogen bonding (N–H···N) interactions: an ab initio study of the complexes of 7-azaindole and fluorosubstituted pyridines. Phys Chem Chem Phys. 2014;16:8819–8827. doi: 10.1039/c3cp54169j. [DOI] [PubMed] [Google Scholar]
- 88.Ao M-Z, Tao Z-q, Liu H-X, Wu D-Y, Wang X. A theoretical investigation of the competition between hydrogen bonding and lone pair…π interaction in complexes of TNT with NH3. Comput Theor Chem. 2015;1064:25–34. [Google Scholar]
- 89.Egli M, Gessner RV. Stereoelectronic effects of deoxyribose O4′ on DNA conformation. Proc Natl Acad Sci USA. 1995;92:180–184. doi: 10.1073/pnas.92.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Egli M, Sarkhel S. Lone pair-aromatic interactions: to stabilize or not to stabilize. Acc Chem Res. 2007;40:197–205. doi: 10.1021/ar068174u. [DOI] [PubMed] [Google Scholar]
- 91.Mooibroek TJ, Gamez P, Reedijk J. Lone pair–π interactions: a new supramolecular bond? CrystEngComm. 2008;10:1501–1515. [Google Scholar]
- 92.Korenaga T, Tanaka H, Ema T, Sakai T. Intermolecular oxygen atom…π interaction in the crystal packing of chiral amino alcohol bearing a pentafluorophenyl group. J Fluor Chem. 2003;122:201–205. [Google Scholar]
- 93.Korenaga T, Shoji T, Onoue K, Sakai T. Demonstration of the existence of intermolecular lone pair…π interaction between alcoholic oxygen and the C6F5 group in organic solvent. Chem Commun. 2009:4678–4680. doi: 10.1039/b908752d. Rare experimental evidence of intermolecular lp–π interaction. [DOI] [PubMed] [Google Scholar]
- 94.Gung BW, Xue X, Reich HJ. Off-center oxygen-arene interactions in solution: a quantitative study. J Org Chem. 2005;70:7232–7237. doi: 10.1021/jo050874+. [DOI] [PubMed] [Google Scholar]
- 95.Pavlakos I, et al. Noncovalent lone pair···(no-π !)-heteroarene interactions: the Janus-faced hydroxy group. Angew Chem Int Ed. 2015;54:8169–8174. doi: 10.1002/anie.201502103. [DOI] [PubMed] [Google Scholar]
- 96.Neel AJ, Milo A, Sigman MS, Toste FD. Enantiodivergent fluorination of allylic alcohols: data set design reveals structural interplay between achiral directing group and chiral anion. J Am Chem Soc. 2016;138:3863–3875. doi: 10.1021/jacs.6b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seguin TJ, Wheeler SE. Competing noncovalent interactions control the stereoselectivity of chiral phosphoric acid catalyzed ring openings of 3-substituted oxetanes. ACS Catal. 2016;6:7222–7228. [Google Scholar]
- 98.Raynal M, Ballester P, Vidal-Ferran A, van Leeuwen PW. Supramolecular catalysis. Part 1: non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem Soc Rev. 2014;43:1660–1733. doi: 10.1039/c3cs60027k. [DOI] [PubMed] [Google Scholar]
- 99.Milo A, Neel AJ, Toste FD, Sigman MS. A data-intensive approach to mechanistic elucidation applied to chiral anion catalysis. Science. 2015;347:737–743. doi: 10.1126/science.1261043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buitrago Santanilla A, et al. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science. 2015;347:49–53. doi: 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- 101.Sigman MS, Harper KC, Bess EN, Milo A. The development of multidimensional analysis tools for asymmetric catalysis and beyond. Acc Chem Res. 2016;49:1292–1301. doi: 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
- 102.Wheeler SE, Seguin TJ, Guan Y, Doney AC. Noncovalent interactions in organocatalysis and the prospect of computational catalyst design. Acc Chem Res. 2016;49:1061–1069. doi: 10.1021/acs.accounts.6b00096. [DOI] [PubMed] [Google Scholar]