

Abstract
Pompe disease is a metabolic myopathy with a wide spectrum of clinical presentation. The gold-standard diagnostic test is acid alpha-glucosidase assay on skin fibroblasts, muscle or blood. Identification of two GAA pathogenic variants in-trans is confirmatory. Optimal effectiveness of enzyme replacement therapy hinges on early diagnosis, which is challenging in late-onset form of the disease due to non-specific presentation. Next-generation sequencing-based panels effectively facilitate diagnosis, but the sensitivity of whole-exome sequencing (WES) in detecting pathogenic GAA variants remains unknown.
We analyzed WES data from 93 patients with confirmed Pompe disease and GAA genotypes based on PCR/Sanger sequencing. After ensuring that the common intronic variant c.-32-13T > G is not filtered out, whole-exome sequencing identified both GAA pathogenic variants in 77/93 (83%) patients. However, one variant was missed in 14/93 (15%), and both variants were missed in 2/93 (2%). One complex indel leading to a severe phenotype was incorrectly called a nonsynonymous substitution c.-32-13T > C due to misalignment.
These results demonstrate that WES may fail to diagnose Pompe disease. Clinicians need to be aware of limitations of WES, and consider tests specific to Pompe disease when WES does not provide a diagnosis in patients with proximal myopathy, progressive respiratory failure or other subtle symptoms.
Keywords: Whole-exome sequencing, Pompe disease, Glycogen storage disease type II, Limb girdle muscular dystrophy, Acid-alpha-1, 4-glucosidase deficiency, GAA
1. Introduction
Pompe disease (glycogen storage disease type II) is an autosomal recessive myopathy primarily affecting cardiac, skeletal, and smooth muscles that encompasses a spectrum of clinical presentations and severity. Biallelic pathogenic variants in the GAA gene, encoding the lysosomal enzyme acid-alpha-1,4-glucosidase (GAA), result in deficiency of the enzyme, causing accumulation of glycogen in the lysosomes of all tissue types, and subsequent progressive muscle destruction. Pompe disease is broadly categorized as infantile-onset and late-onset Pompe disease (IOPD and LOPD), based on age of onset and presenting symptoms. Classical IOPD is characterized by prominent cardiomegaly, hepatomegaly, hypotonia, and, without treatment, death due to cardiorespiratory failure within the first year of life. Atypical IOPD is characterized by presentation in the first year of life with less severe or no cardiac involvement. LOPD presents as a slowly progressive proximal myopathy that involves mainly skeletal and respiratory muscles, and may present in early childhood or even as late as the sixth decade of life [1]. The prevalence of IOPD and LOPD combined in the general population has been estimated to be between 1 in 40,000 to 1 in 60,000 [2], but findings from newborn screening programs in Taiwan and the United States indicate that the prevalence is underestimated [3,4]. Early and timely diagnosis is vital, because treatment with alglucosidase alfa (human recombinant GAA) results in best outcomes when initiated before severe muscle damage has occurred [5,6]. A definitive diagnosis of Pompe disease is made by showing a deficiency of GAA enzyme activity in skin fibroblasts, muscle, or blood-based assays, along with identification of two pathogenic in-trans variants in the GAA gene.
Diagnosis of LOPD in adults can be especially challenging because of the non-specific and insidious nature of disease onset and progression. The disease involves multiple organs and its clinical presentation overlaps with many other neuromuscular conditions. Subtle symptoms such as lingual weakness or dysphagia may be overlooked [7,8]. Studies from the Pompe registry showed that LOPD may not be diagnosed until after symptoms have progressed significantly [9], and is probably often underdiagnosed [10] or misdiagnosed [11,12]. The diagnostic delay for Pompe disease across the spectrum is approximately 7 years [9]. A patient with LOPD may be asymptomatic except for elevation of serum transaminases and creatine kinase (CK), which indicates muscle injury [12,13]. Differential diagnosis of LOPD includes muscle glycogen storage diseases, other metabolic and non-metabolic myopathies, myositis, and limb-girdle muscular dystrophy (LGMD). In many LOPD cases, histological analysis of muscle biopsy can result in a negative diagnosis if the biopsied tissue lacks diseased muscle fiber, or if appropriate staining or specific enzyme testing on the muscle are not performed. Muscle biopsy histology is known to be normal in 25% of adults with LOPD [14].
Recently, next generation sequencing (NGS)-based panels have been shown to facilitate the diagnosis of LOPD [15,16]. Whole-exome sequencing (WES) is now routinely ordered in clinic for many conditions including undiagnosed causes of myopathy and neurological indications [16–18], and has successfully diagnosed patients with Pompe disease [19]. WES offers a high diagnostic yield when differential diagnosis cannot be narrowed [17]. However, the sensitivity of WES in detecting pathogenic GAA variants has never been explored or published. Several studies have compared the results of NGS and Sanger sequencing in different genes, but their focus was on the Sanger-confirmation of variants found by NGS technology. These studies did not assess the standard WES bioinformatics pipelines that automatically analyze WES data to detect known pathogenic variants [20,21].
WES is an application of NGS, in which only coding regions of the genome (exome), which make up about 1% of the human genome, are captured for sequencing [22]. NGS is performed by breaking down DNA extracted from a patient’s specimen into small fragments, amplifying the fragments by PCR, and simultaneously determining sequences of the fragments (primary analysis). A series of computer software tools are used to align the sequence reads to specific locations in the human reference genome to identify sequence variants that do not match the reference (secondary analysis), and filter out variants that are unlikely to cause the disease of interest (tertiary analysis) [23,24]. WES was developed to efficiently sequence regions most likely to include pathogenic variants. However, with the dramatical drop in price of sequencing per base pair, whole-genome sequencing (WGS) will likely become available for clinical use in the near future. While secondary and tertiary analysis on WGS will continue to focus on the exome, WGS is not affected by biases introduced by capture and amplification process.
The GAA gene is a mid-size gene spanning 18,175 base pairs (bp) and 20 exons, with 2859 bp coding DNA sequence, encoding 952 amino acids. The first exon is non-coding with the start codon in exon 2 [25,26]. The gene harbors rare pathogenic variants of diverse types and recurrent pathogenic variants such as: intronic c.-32-13T > G affecting splicing of exon 2 seen in 68–90% of Caucasians with LOPD; a large deletion encompassing exon18 c.2481 + 102_2646 + 31del (p.Gly828_Asn882del); a frameshift c.525delT (p.Glu176Argfs*45); and a nonsense c.2560C > T (p.Arg854*) seen in African Americans [27]. A European newborn screening study showed that the carrier frequency of c.-32-13T > G in the European general population is 1 in 154; the exon 18 deletion, 1 in 187; and c.525delT, 1 in 284 [28]. Currently, > 400 pathogenic variants have been reported in the GAA gene (http://cluster15.erasmusmc.nl/klgn/pompe/mutations.html?lang=en accessed May 22, 2017) [29,30]. The c.-32-13T > G variant, seen in 68–90% of Caucasian patients with LOPD in a heterozygous or homozygous state [31–36], results in alternatively spliced transcripts with deletion of exon 2 and leakage of some normal transcripts [32,37]. This variant has never been observed in classic IOPD, though it has been observed in atypical IOPD. Phenotypic diversity seen in patients with this variant, even when the other GAA genotype is the same, is likely due to genomic and environmental modifiers [27,30]. The GAA gene has a high (> 60%) overall GC (guanine-cytosine) content (Fig. S1), which may undermine PCR amplification during the NGS process and lead to low read depth coverage and inaccurate variant calling in certain cases. These interesting characteristics of the GAA gene make it a good candidate on which to test the validity of WES technology to detect all pathogenic variants in this gene.
We performed WES on 93 patients with confirmed Pompe disease who had a spectrum of phenotypes ranging from IOPD to LOPD. In this cohort, GAA genotypes had been confirmed by Sanger sequencing in a clinical molecular diagnostics laboratory. The Sanger sequencing and the preceding PCR amplification were carefully designed to capture the common intronic pathogenic variant c.-32-13T > G and the deletion variant of exon 18. Thus, we had a unique opportunity to assess the sensitivity of WES in diagnosing Pompe disease by comparing the results of WES with PCR/Sanger sequencing of GAA in patients who had a confirmed diagnosis of Pompe disease.
2. Methods
2.1. Subjects
A waiver of written consent was obtained by the Duke institutional review board. The study included 93 patients with a confirmed diagnosis of Pompe disease and known GAA genotypes. One patient had a heterozygous pathogenic GAA variant and a second had a heterozygous variant of unknown significance, but both had a confirmed diagnosis of Pompe disease based on reduced GAA enzyme activity on skin fibroblasts and a clinical phenotype consistent with the disease.
2.2. Sanger sequencing
Sanger sequencing for molecular diagnosis of Pompe disease was performed in the College of American Pathologist (CAP)-accredited and Clinical Laboratory Improvement Amendments (CLIA)-certified molecular diagnostics laboratory at Duke University or at other similarly qualified molecular laboratories. The coding regions of the GAA gene and surrounding exon/intron boundaries (minimum of 20 base pairs) were sequenced following PCR amplification, amplicon purification, and loading onto an ABI 3130×1 Genetic Analyzer (Perkin Elmer, Santa Clara, CA). The primers used for PCR contained M13 universal primer “tails” at their 5′ ends, and had 3′ ends that were homologous to their genomic target sequence. PCR was performed using primers flanking the known breakpoints. This amplification product was sized on a gel, and sequenced to confirm the breakpoints. Sequences were compared to the GAA reference DNA sequence (GenBank Accession: NM_000152.3) to identify genetic variants.
2.3. GAA artifacts in genome-in-a-bottle project
Given the high GC content of the GAA gene, we investigated potential systematic artifacts in the GAA region by checking its depth and breadth of sequence read coverage in the ExAC database [38], as well as checking its inclusion in the regions in which highly accurate genotype calls were achieved in the Genome-in-a-Bottle project [39]. A recent study defined sites with systematic errors that can be caused by PCR amplification, particular sequence contexts, local alignment errors, and/or global mapping errors [40]. We examined if any GAA position was included in these error prone positions.
2.4. Whole exome sequencing
DNA was extracted from peripheral blood or skin fibroblast samples. Sequencing was performed with Illumina HiSeq2500 sequencers. Nimblegen SeqCap EZ V3.0 Exome Enrichment kit was used to target 64 Mb of the genome, which includes 98% of RefSeq, 98% of Vega, 97% of Gencode, 99% of Ensembl, 99% of consensus coding sequence (CCDS) [41] and 98% of miRBase sequences. Across the exome, an average of 93.0% (median 91.6%) covered CCDS. The paired-end reads were aligned to a Genome Reference Consortium Human Genome Build 37 (GRCh37)-derived alignment set including decoy sequences using the Burrows-Wheeler Aligner (BWA-0.5.10) [42]. PCR duplicates were removed with Picard (http://picard.sourceforge.net), and single nucleotide variants (SNV’s) and small insertions and deletions (indels) were called with GATK UnifiedGenotyper following the GATK Best Practices [24]. The resulting variant data for all samples underwent standard quality control checks, including validation of the reported sex, relatedness, and ethnicity. Variants were annotated by snpEff and the resulting genotypes were stored in a centralized database. Columbia University in-house software package Analysis Tool for Annotated Variants (ATAV; https://redmine.igm.cumc.columbia.edu/projects/atav/wiki) was used for filtration and for read depth coverage calculation. Transcript ENST00000302262 was used for GAA variant notations. Standard filtering is Coverage > 10×, VQSLOD score ≤ 99–99.9%, Quality By Depth > 2, Mapping Quality > 40, Genotype Quality > 20 in the CCDS region; SNV’s also require VQSLOD score ≤ 99.9%, and indels require strand bias < 200 and read positive rank sum > −20.
2.5. Concordance
Substitution and indel variant positions were converted to genomic positions using LUMC Mutalyzer Position Converter on a GAA transcript NM_000152.3 [43]. Confirmation of indels were performed manually [44]. When an expected variant was not in the filtered variant list, VCF files were inspected to see if it had been called by GATK UnifiedGenotyper and later filtered out. CODEX package [45] was used to detect exon-level large deletions based on indexed BAM files.
3. Results
3.1. Subjects
Phenotypes of the 93 patients are presented in Table 1. There were 51 specimens from patients with IOPD and 42 specimens from patients with LOPD.
Table 1.
Genotype and phenotype of samples and WES genotype detection.
| Pathogenic variant 1 | Pathogenic variant 2 | Phenotype | Onset for LOPD |
|---|---|---|---|
| c.1075G > T(p.Gly359*) | c.1075G > T(p.Gly359*) | IOPD | |
| c.1195-19_2190-17del(p.Asp399fs*6)U | c.1195-19_2190-17del (p.Asp399fs*6)U | IOPD | |
| c.1209delC(p.Asp531Glyfs*37)U | c.1209delC(p.Asp531Glyfs*37) | IOPD | |
| c.148_859-11del(p.Glu50Hisfs*37; exons2–4deletion) | c.686insCGGC (p.Arg229fsProfs*102) | IOPD | |
| c.546+2_5deltggg | c.1650dupG(p.Thr551Aspfs*85) | IOPD§1 | |
| c.546+2_5deltggg | c.1650dupG(p.Thr551Aspfs*85) | IOPD§1 | |
| c.546+2_5deltggg | c.2501_2502delCA (p.Thr834Argfs*49) | IOPD | |
| c.1754+1G > A | c.722_723delTT (p.Phe241Cysfs*88) | IOPD | |
| c.1826dupA(p.Tyr609*) | c.2238G > A(p.Trp746*) | IOPD | |
| c.1978C > T(p.Arg660Cys) | c.2221G > A(p.Asp741Asn) | IOPD | |
| c.2237G > A(p.Trp746*) | c.437delT(p.Met146Argfs*7) | IOPD | |
| c.2560C > T(p.Arg854*) | c.1654delC(p.Leu552Serfs*26) | IOPD | |
| c.2560C > T(p.Arg854*) | c.1292_1295dupTGCA (p.Gln433Alafs*74) | IOPD | |
| c.2560C > T(p.Arg845*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2560C > T(p.Arg854*) | IOPD | |
| c.2560C > T(p.Arg854*) | c.1129G > C(p.Gly377Arg) | IOPD | |
| c.2560C > T(p.Arg854*) | c.1710C > G(p.Asn570Lys) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2236T > C(p.Trp746Arg) | IOPD | |
| c.2560C > T(p.Arg854*) | c.2459_2461delCTG(p.Ala820del) | IOPD | |
| c.722_723delTT(p.Phe241Cysfs*88) | c.1687C > T(p.Gln563*) | IOPD | |
| [del Exons 15–20]U | c.2012T > G(p.Met671Arg) | IOPD | |
| c.1411_1414del4(p.Glu471fs*5) | c.460_465del6(Arg154_Thre155del)1 | IOPD | |
| c.1099T > C(p.Trp367Arg) | c.1802C > T(p.Ser601Leu) | IOPD | |
| c.1293_1312del20(p.Gln433Aspfs*66) | c.1716C > G(p.His572Gln) | IOPD | |
| c.1327-2A > C | c.1327-2A > C | IOPD | |
| c.1438-1G > C | c.1655T > C(p.Leu552Pro) | IOPD | |
| c.1933G > A(p.Asp645Asn) | c.1933G > A(p.Asp645Asn) | IOPD | |
|
| |||
| Mutation1 | Mutation2 | Phenotype | Onset for LOPD |
|
| |||
| c.1933G > A(p.Asp645Asn) | c.1933G > A(p.Asp645Asn) | IOPD | |
| c.1942G > A(p.Gly648Ser) | c.1942G > A(p.Gly648Ser) | IOPD | |
| c.2238G > A(p.Trp746*) | c.1843G > A(p.Gly615Arg) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.437delT(p.Met146Argfs*7) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.1210G > A(p.Asp404Asn) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.1912G > T(p.Gly638Trp) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.525delT(p.Glu176Argfs*45) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.525delT(p.Glu176Argfs*45) | IOPD | |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.2481+102_2646+31del (p.Gly828_Asn882del)U | IOPD | |
| c.2512C > T(p.Gln838*) | c.2105G > T(p.Arg702Leu) | IOPD | |
| c.2815_2816delGT(p.Val939fs*78) | c.1935C > A(p.Asp645Glu) | IOPD | |
| c.525delT(p.Glu176Argfs*45) | c.1799G > T(p.Arg600Leu) | IOPD | |
| c.525delT(p.Glu176Argfs*45) | c.1880C > T(p.Ser627Phe) | IOPD | |
| c.525delT(p.Glu176Argfs*45) | c.1655T > C(p.Leu552Pro) | IOPD | |
| c.655G > A(p.Gly219Arg) | c.1979G > A(p.Arg660His) | IOPD | |
| c.655G > A(p.Gly219Arg) | c.655G > A(p.Gly219Arg) | IOPD | |
| c.-32-17_-32-10delins TCCCTGCTGAGCCTCCTACAGGCCTCCCGCW | c.1447G > A(p.Gly483Arg) | IOPD | |
| c.1210G > A(p.Asp404Asn) | c.1924G > T(p.Val642Phe) | IOPD | |
| c.525delT(p.Glu176Argfs*45) | c.-32-13T > GF | IOPD | |
| c.2015G > T(p.Arg672Leu)) | c.2783A > G(p.Tyr928Cys) | LOPD | Juvenile |
| c.1655T > C(p.Leu552Pro) | c.1655T > C(p.Leu552Pro) | LOPD | Juvenile |
| c.1437+2T > C | c.-32-13T > GF | LOPD | Juvenile |
| c.953T > C(p.Met318Thr) | c.-32-13T > GF | LOPD | Juvenile |
| c.1796C > A(p.Ser599Tyr) | c.-32-13T > GF | LOPD | Juvenile |
| c.1396_1397insG(p.Val466fs*39) | c.-32-13T > GF | LOPD | Adult |
| c.1143delC(p.Ala382Leufs*10) | c.-32-13T > GF | LOPD | Adult |
| c.1441T > C(p.Trp481Arg) | c.-32-13T > GF | LOPD | Adult |
| c.1445C > G(p.Pro482Arg) | c.-32-13T > GF | LOPD§2 | Adult |
| c.1445C > G(p.Pro482Arg) | c.-32-13T > GF | LOPD§2 | Adult |
| c.1548G > A(p.Trp516*) | c.-32-13T > GF | LOPD | Adult |
| c.1548G > A(p.Trp516*) | c.-32-13T > GF | LOPD | Adult |
| c.1655T > C(p.Leu552Pro) | c.-32-13T > GF | LOPD | Adult |
| c.1798C > T(p.Arg600Cys) | c.-32-13T > GF | LOPD | Adult |
| c.1827delC(p.Tyr609*) | c.-32-13T > GF | LOPD | Adult |
| c.1835A > C(p.His612Pro)2 | c.-32-13T > GF | LOPD | Adult |
| c.1880C > T(p.Ser627Phe) | c.-32-13T > GF | LOPD | Adult |
| c.2238G > A(p.Trp746*) | c.-32-13T > GF | LOPD | Adult |
| c.2238G > A(p.Trp746*) | c.-32-13T > GF | LOPD | Juvenile |
| c.2242dupG (p.Glu748Glyfs*48) | c.-32-13T > GF | LOPD | Adult |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.-32-13T > GF | LOPD | Adult |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.-32-13T > GF | LOPD | Adult |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.-32-13T > GF | LOPD | Adult |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.-32-13T > GF | LOPD | Adult |
| c.2481+102_2646+31del (p.Gly828_Asn882del)U | c.-32-13T > GF | LOPD | Adult |
| c.2560C > T(p.Arg854*) | c.-32-13T > GF | LOPD | Adult |
| c.2608C > T(p.Arg870*) | c.-32-13T > GF | LOPD | Adult |
| c.525delT(p.Glu176Argfs*45) | c.-32-13T > GF | LOPD | Adult |
| c.525delT(p.Glu176Argfs*45) | c.-32-13T > GF | LOPD | Adult |
| c.525delT(p.Glu176Argfs*45) | c.-32-13T > GF | LOPD | Adult |
| c.525delT(p.Glu176Argfs*45) | c.-32-13T > GF | LOPD | Adult |
| c.742delC(p.Leu248Profs*20) | c.-32-13T > GF | LOPD | Adult |
| c.743T > C(p.Leu248Pro) | c.-32-13T > GF | LOPD | Adult |
| c.784G > A(p.Glu262Lys) | c.-32-13T > GF | LOPD | Adult |
| c.784G > A(p.Glu262Lys) | c.-32-13T > GF | LOPD§3 | Adult |
| c.784G > A(p.Glu262Lys)3 | c.-32-13T > G3F | LOPD§3 | Adult |
| c.836G > A(p.Trp279*) | c.-32-13T > GF | LOPD | Adult |
| c.877G > A(p.Gly293Arg) | c.-32-13T > GF | LOPD | Adult |
| c.925G > A(p.Gly309Arg)F | c.-32-13T > GF | LOPD | Adult |
| c.925G > A(p.Gly309Arg)U | c.-32-13T > GF | LOPD | Adult |
| c.1143delC(p.Ala382Leufs*10) | c.-32-13T > GF | LOPD§4 | Adult |
| c.1143delC(p.Ala382Leufs*10) | c.-32-13T > GF | LOPD§4 | Adult |
IOPD infantile Pompe disease.
LOPD late-onset Pompe disease.
The significance of this variant is unknown. This patient also had c.752C > T(p.Ser251Leu) and c.761C > T(p.Ser254Leu) variants, both of which were detected by WES.
The significance of this variant is unknown.
Genotype for this patient was assumed based on the genotype of her sib.
Sib pairs.
The variant was called but filtered out by the tertiary analysis pipeline.
The variant was detected but was called as a wrong genotype by the variant caller.
The variant was not called by the variant caller.
3.2. Sanger sequencing result
GAA Genotypes by Sanger sequencing are listed in Table 1.
3.3. Evaluation for systematic artifacts in the GAA coding region
Despite the relatively high GC content, all coding regions in the GAA gene are included in the regions in which highly accurate genotype calls were achieved in the Genome-in-a-Bottle project (Fig. S2) [39]. The gene regions were also well covered in the ExAC database (Fig. S3). Only one intronic position in the GAA gene, chr17:g.78084704 (NM_ 000152.3:c.1552-36del; a position not reported to be pathogenic) was identified to be subject to systematic errors [40]. Thus except for that one intronic position, we found no evidence of systematic artifacts due to GC bias, poor alignment, tandem repeats, or availability of reference sequence.
3.4. Read depth coverage in WES
The median overall coverage for captured regions was 84× with a minimum overall coverage of 34×. All samples had at least 5× read depth in > 97% of the exome. However, analysis of regions covered at minimum of 20× read depth in the GAA gene revealed some GAA regions with insufficient read depth; 14 samples without large deletions in the GAA gene had regions in exons 4, 5, 15, 18 or 19 with < 20 read depth, resulting in overall ≥20× coverage of 83%–94% for the GAA gene. These large deletions negatively affected the percentage of regions that had ≥20× coverage. For example, a sample with a homozygous deletion of exon 18 [c.2481 + 102_2646 + 31del (p.Gly828_Asn882del)] of the GAA gene had ≥20× read depth coverage for 83% of the GAA gene. Similarly, a sample with a homozygous deletion of exons 8–15 had ≥20× read depth coverage in 65% of the GAA gene. A sample reported to have a heterozygous c.148_859-11del (p.Glu50Hisfs*37(deletion of exons 2–4) along with an insertion c.686insCGGC(p.Arg229fsProfs*102) in the other allele showed < 20 read depth in exons 2 and 4, resulting in ≥20× read depth coverage in 96% of the GAA gene. A sample reported to have a heterozygous deletion of exons 15–20 along with a missense pathogenic variant c.2012T > G(p.Met671Arg) in the other allele showed < 20 read depth in exons 17–19, resulting in ≥20× read depth coverage in 92% of the GAA gene. Eight of the 10 samples with a heterozygous deletion overlapping the exon 18 along with a SNV in the other allele showed < 20× read depth in regions within exon 18. As a result, the ≥20× read depth coverage of the 8 samples ranged between 83% and 99% for the GAA gene. The coverage of each exon across the 93 samples is summarized in Table S1. The position chr17:78078341-T-G corresponding to c.-32-13T > G was covered at least 20× read depth in all 93 samples.
3.5. Concordance
Both of the pathogenic GAA variants were detected in 42/93 (45%) samples by WES using the standard bioinformatics pipeline of the research laboratory; one of the pathogenic GAA variants was missed in 42/93 (45%) samples by WES; both of the pathogenic GAA variants were missed in 9/93 (10%) samples. The concordance between the WES and Sanger sequencing results for each variant in each sample is summarized in Table 1 and Fig. 1. Fig. 2 shows the concordance for each unique pathogenic variant. The common intron 1 variant c.-32-13T > G was called correctly by the GATK UnifiedGenotyper, as well as passed quality control criteria in all of the 41 samples that harbor this pathogenic variant. However, it had been filtered out by the tertiary analysis pipeline because it was not in the CCDS region. As expected, none of the large deletions, including the common exon 18 deletion, were detected in any of the samples harboring the deletion variant. One missense pathogenic variant, c.925G > A (p.Gly309Arg) in exon 5, was not called as a heterozygous variant as only two out of 12 reads at this position had the change (Table 2). This sample had a poor coverage in exon 5; it had < 20× read depth at all base pair positions in exon 5 and low read depth of 12× at the variant position. In another sample, the same variant was also called but was filtered out due to low genotype quality score and read depth (Table 2). This sample also had poor coverage in exon 5; < 20× read depth at all base pair positions in exon 5 and low read depth of 7× at the variant position. A complicated deletion/insertion c.-32-17_-32-10delins TCCCTGCTGAGCCTCCTACAGGCCTCCCGC in intron 1, beginning seventeen nucleotides upstream of the beginning of exon 2 (which is 32 nucleotides upstream of the translation initiation site) and replacing eight nucleotides with thirty, was incorrectly called as c.-32-13T > C due to incorrect alignment (Fig. S5). The Sanger sequencing report interpreted the indel as pathogenic as it overlaps the common pathogenic c.32-13T > G variant and is predicted to affect exon 2 splicing [46]. Variant IDs with genomic positions confirmed for each pathogenic variant are provided in the supplementary material to avoid ambiguity (Table S2). The CODEX software detected the homozygous large deletion c.1195-19_2190-17del (p.Asp399fs*6), among numerous other deletion/duplications of unknown significance, but did not detect other large deletions, including the common exon 18 deletion, either in a heterozygous or homozygous state (data not shown).
Fig. 1.
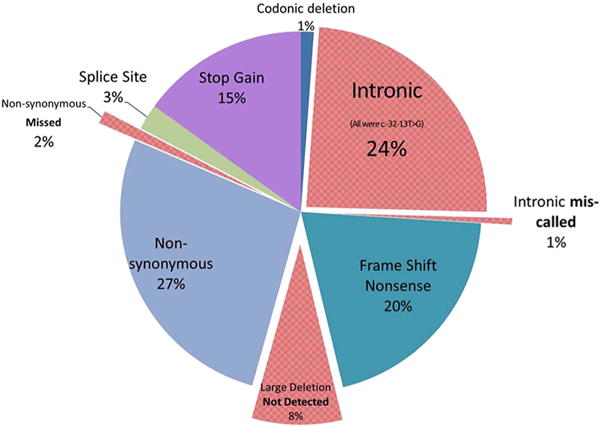
Concordance data between Sanger sequencing based molecular analysis of GAA gene and WES detected mutation type; total 173 mutations (n = 93 patients). Mutations in red were either missed or miscalled by WES. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
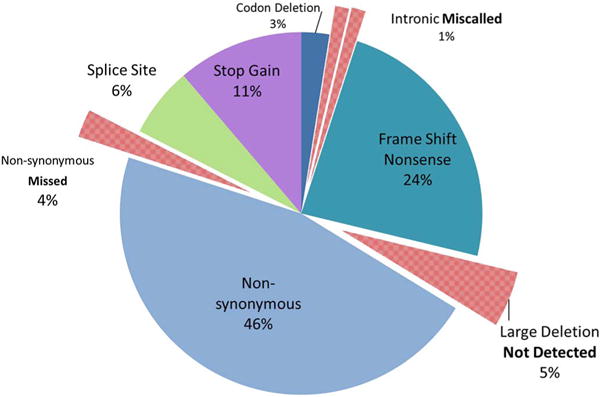
Concordance data between mutation type through Sanger molecular sequence analysis of GAA and WES detected mutant alleles; 79 Unique Mutations (n = 93 patients).
Table 2.
Mutations missed by standard WES pipeline and mechanism.
| Sanger call | WES call | Ref/Alt | Genotype quality (> 20) | Quality By Depth (> 2) | Mapping quality (> 40) | Fisher strand | Nature of failure |
|---|---|---|---|---|---|---|---|
| c.925G > A (p.Gly309Arg) | chr17:78081665G > A | 4/3 | 72.74 | 8.74 | 60 | 0 | Missense, filtered out |
| c.925G > A (p.Gly309Arg) | Not called out by GATK | 10/2 | – | – | – | – | Missense, not called |
| c.-32-17_-32-10delins30 | chr17:g.78078341T > C | 9/3 | 99 | 4.05 | 54.44 | 0 | Insertion not called. VQSLOD 3.26 |
| c.1195-19_2190-17del (p.Asp399fs*6) 8273 bp deletion in intron7-exon15 seen in Hispanic population | Not called out by GATK UnifiedGenotyper | – | – | – | – | – | Very large deletion |
| c.2481 + 102_2646 + 31del common 537 bp deletion from intron17-intron18 | Not called out by GATK UnifiedGenotyper | – | – | – | – | – | Large deletion |
| c.148_859del exons 2–4 | Not called out by GATK UnifiedGenotyper | – | – | – | – | – | Large deletion |
| Exons 15–20 deletion breakpoint not localized | Not called out by GATK UnifiedGenotyper | – | – | – | – | – | Very large deletion |
Many clinical laboratories would evaluate intronic pathogenic variants in variant databases such as ClinVar and the Human Gene Mutation Database (HGMD). If the pathogenic c.-32-13T > G variant is detected in a clinical WES laboratory, then both pathogenic GAA variants would be detectable in 77/93 (83%) samples. One pathogenic GAA variant would have been missed in 14/93 (15%) patients and both pathogenic GAA variants would have been missed in 2/93 (2%) patients. Interestingly, these patients had homozygous large deletions, one with c.1195-19_2190-17del (p.Asp399fs*6) and the other with the common exon 18 deletion (Figs. S6–7).
4. Discussion
WES technology is limited by its ability to detect certain types of pathogenic variants. For example, WES does not reliably detect structural variants such as large deletions or duplications. Recent studies have shown that even the latest variant-calling algorithms do not reliably call SNV’s and small insertions/deletions [40,47,48]. In addition, standard WES pipelines typically filter out intronic variants. The analytical validity of homozygous or heterozygous SNV’s can be compromised when the read depth is poor (< 20×), the primary reason for a false negative result in WES [40]. The poor read coverage in WES is attributable to non-uniform read depth across the exome compared to other NGS-based test such as targeted gene panels [49]. High GC content (> 75%) and repeat sequence can also lead to a false negative SNV call [40]. In contrast, NGS-based gene panels offered by clinical laboratories ensure good read coverage over the targeted genes, and typically supplemented with Sanger sequencing if adequate coverage is not achieved. Many of these gene panels are offered along with a matching exon-array gene panel to detect large deletions and duplications in the targeted genes. Though Sanger sequencing has previously been used to detect pathogenic variants in the GAA gene to confirm a diagnosis of Pompe disease, WES is increasing being utilized. However, the GAA gene harbors a number of deletions and intronic pathogenic variants, which may be not be detected through WES.
This study demonstrates various mechanisms by which WES technology and variant analysis pipelines can fail to detect pathogenic variants. Unlike WGS, WES data is very limited in revealing large deletions that span exon(s) because it lacks intergenic sequence data and suffers from enrichment bias [50]. Several tools designed to detect structural variants, including large deletions based on WES read depth data, have been developed [51,52]. CODEX is one of the more recent tools with relatively superior sensitivity and specificity compared to others [45]. Using the default setting, the tool had very limited use in detecting large deletions in our patient cohort, demonstrating the difficulty in calling large deletions even with specialized algorithms.
WES reports should clearly state that large deletions and duplications are often not detectable by WES, particularly in genes in which pathogenic large deletions are common such as in the GAA gene. The position at the common pathogenic variant c.-32-13T > G had a good read depth in all samples and was called by the GATK UnifiedGenotyper, the standard variant caller. The position had good read depth despite being in the intron because this variant is located 13 base pairs from the noncoding part of exon 2, still within the parameters of capture regions for the exome sequencing. However, the standard filtration protocol filtered out this variant as it was not in the CCDS region. We recommend that laboratories performing WES implement a system to evaluate known intronic pathogenic variants for all genes to ensure that they are not filtered out. Tertiary WES analysis generally is focused on exons and canonical splice sites, and the inclusion of intronic variants may require manual alteration of the filtering pipeline. As our result show, WES will inevitably end up with some regions with low read depth, possibly resulting in false negative findings or inaccurate calls of SNV and indel variants. Thus, WES can miss a diagnosis of other single gene diseases especially those caused by a gene in which deletions or intronic mutations are common, or systematic artifacts exist. Accordingly, clinicians should be aware of these limitations of WES.
The inability of our research WES to detect the common exon 18 deletion (allele frequency 6% among IOPD + LOPD in the 93 samples) and c.-32-13T > G variant alleles (allele frequency 48% among LOPD; not observed in IOPD), which are common GAA pathogenic variants, could have resulted in missed diagnosis in patients with Pompe disease. Incorrect variant calls of indels as SNV’s are also concerning as clinical laboratories may not perform confirmatory Sanger sequencing for SNV’s [20,21]. Our WES pipeline incorrectly identified c.-32-17_-32-10delins30 as an SNV c.-32-13T > C, and it may have been incorrectly interpreted it as consistent with an LOPD phenotype because an SNV occurring at the same position c.-32-13T > G is seen in LOPD. In fact, in this situation, the c.-32-17_-32-10delins30 is a likely severe pathogenic variant based on the phenotype of IOPD and the fact that the other pathogenic variant c.1447G > A(p.Gly483Arg) is predicted to be less severe according to the Pompe Disease Mutation Database [29]. The miscall of an indel as an SNV and resultant genotype prediction would be even more critical in prognostication of asymptomatic patients detected by newborn screening, or by persistent elevation of CK, compared to diagnostic testing in patients with a clear phenotype of Pompe disease. Many clinical laboratories are moving away from Sanger-confirmation of SNV’s [21]. This example underscores the importance of visual inspection of BAM files for reportable variants, and Sanger-confirmation when needed.
Since the application of WES was first proposed as a diagnostic test for human Mendelian disorders in 2009 [53], this method has become routine for both research and clinical applications [17]. In a work-up of patients presenting with progressive muscle weakness, respiratory failure, a LGMD phenotype, polymyositis or persistently elevated CK, it is important not to miss a treatable disease. Clinicians should be aware that WES can fail to provide a complete molecular diagnosis of Pompe disease. Particularly in cases where only one pathogenic variant is identified by WES, the possibility of a deletion on the other allele should be considered. A NGS-based gene panel specifically designed to detect all clinically relevant pathogenic GAA variants including intronic variants and small and large deletions for neuromuscular disease/LGMD may be a better initial test when the differential diagnosis cannot be narrowed. Clinical laboratories that offer such panels typically implement methods to ensure that common deletion pathogenic variants or intronic pathogenic variants are detectable.
However, it should be noted that some of these commercially available gene panels are not necessarily designed to detect all deletion variants [15] or intronic pathogenic variants in the GAA gene, and thus may fail to detect them.
Correlation between genotype and phenotype in the cohort of 93 patients gave us an opportunity to confirm prior findings that having c.-32-13T > G on one allele, and a pathogenic variant on the other allele, almost always leads to the adult onset LOPD phenotype [34] but that genotype-phenotype correlation is not consistent [27]. In our cohort, five patients had the c.-32-13T > G/c.525delT GAA genotype. Four of them had the adult LOPD phenotype as expected. However, one of them had an early presentation in the first year of life with no cardiac involvement. We further investigated to determine whether this patient had pathogenic variants in other genes that can cause neuromuscular phenotypes but no other pathogenic variants were identified.
A limitation of our study is that the WES was performed as a research methodology and did not necessarily meet the CLIA standards or proposed best clinical practice [23]. A base pair coverage of 20× is now recommended for detection of SNVs [54], and clinical laboratories offering WES may move towards improving the coverage at 20× read depth. WES capturing technology and analytical bioinformatics adopted in clinical laboratories are constantly improving. WGS will soon be available for clinical use. However, the problems discussed here, such as insufficient read depth, tertiary analysis of intronic variants, and alignment problems could still all occur, although to a lesser degree. Our sample size may be considered limited for a research study but a cohort size of 93 patients with Pomple disease is substantial for a rare autosomal recessive disease. Moreover, we were able to illustrate molecular examples in which WES did not correctly identify pathogenic variants.
5. Conclusion
Patients with Pompe disease frequently harbor pathogenic variants that are not reliably detected by WES. As treatment is available for this metabolic myopathy, clinicians should be aware of the limitations of the test and rule out the disorder by specific GAA gene and enzyme testing. The limitations of WES should also be recognized for other genetic conditions caused by genes in which deletion or intronic pathogenic variants are common, or when coverage is poor because of systematic artifacts or non-uniform coverage. If WES is ordered and does not result in a diagnosis, a specific diagnostic test for Pompe disease should be considered if clinical suspicion for Pompe disease remains. Acid alpha-glucosidase (GAA) enzyme activity assay on dried blood spot is a less-invasive and cost-effective test with a quick turnaround time [55]. PCR/Sanger sequencing of the GAA gene, which is specifically designed to detect the common intronic pathogenic variant and exon 18 deletion, is helpful if the enzyme testing is not available or indicates an indeterminate result. Clinicians are also advised to be aware of other limitations of WES that are not discussed in this paper, such as its inability to detect repeat expansion variants that may underlie disorders such as oculopharyngeal muscular dystrophy, or methylation variants that may underlie conditions such as Prader-Willi syndrome.
Supplementary Material
Acknowledgments
We appreciate Mr. Cheng Lu’s work for maintenance of biorepository and specimen handling, and Dr. Constance D. Baldwin and Ms. Heidi Cope for editorial assistance. We would like to acknowledge the following individuals or groups for the contributions of control samples:
D. Daskalakis; R Buckley; J. Milner; M.Hauser; A. Need; J. McEvoy; R. Ottman; D. Levy; C. Chen; J.Hoover-Fong, N. L. Sobreira and D. Valle; A. Poduri; T. Young and K. Whisenhunt; Z. Farfel, D. Lancet, and E. Pras; R. Gbadegesin and M. Winn; K. Schmader, S. McDonald, H. K. White and M. Yanamadala; S. Palmer; G. Cavalleri; N. Delanty; G. Nestadt; V. Shashi; M. Carrington; The Murdock Study Community Registry and Biorepository; the Carol Woods and Crosdaile Retirement Communities; National Institute of Allergy and Infectious Diseases Center for HIV/AIDS Vaccine Immunology (CHAVI); National Institute of Allergy and Infectious Diseases Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery; K. Welsh-Bomer, C. Hulette, J. Burke; S. Schuman, E. Nading; Epi4K Consortium and Epilepsy Phenome/Genome Project; and DUHS (Duke University Health System) Nonalcoholic Fatty Liver Disease Research Database and Specimen Repository.
The collection of control samples and data was funded in part by: The Duke Chancellor’s Discovery Program Research Fund 2014; Bill and Melinda Gates Foundation (50957); B57 SAIC-Fredrick Inc. M11-074; the Division of Intramural Research; The Ellison Medical Foundation New Scholar award AG-NS-0441-08; National Institute of Mental Health (K01MH098126, R01MH097993; RC2MH089915); National Institute of Allergy and Infectious Diseases (1R56AI098588-01A1); National Human Genome Research Institute (U01HG007672); National Institute of Neurological Disorders and Stroke (U01-NS077303, U01-NS053998; RC2NS070344); National Institutes of Health (U01MH105670); and National Institute of Allergy and Infectious Diseases Center (U19-AI067854, UM1-AI100645); and the National Institute on Aging (P30AG028377).
The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about).
Funding:
This research was supported by a grant from the Genzyme Corporation, a Sanofi Company (Cambridge, MA), and in part the Lysosomal Disease Network (U54NS065768), a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS and the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Abbreviations
- ATAV
Analysis Tool for Annotated Variants
- BWA
Burrows-Wheeler Aligner
- CAP
College of American Pathologist
- CCDS
Consensus Coding Sequence
- CK
Creatine Kinase
- CLIA
Clinical Laboratory Improvement Amendments
- GAA
Acid-alpha-14-glucosidase
- GC
Guanine-Cytosine
- GRCh
Genome Reference Consortium Human Genome Build
- HGMD
Human Gene Mutation Database
- IOPD
Infantile-onset Pompe Disease
- LGMD
Limb-girdle muscular dystrophy
- LOPD
Late-onset Pompe disease
- NGS
Next-generation sequencing
- SNV
Single Nucleotide Variant
- WES
Whole-exome sequencing
- WGS
Whole-genome sequencing
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgme.2017.10.008.
Footnotes
Ethics approval and consent to participate
A waiver of written consent was obtained by the Duke institutional review board.
Conflict of interest disclosures
Priya S. Kishnani has received research/grant support from Genzyme Corporation (Sanofi), Valerion Therapeutics, Shire Pharmaceuticals, Roche, Pfizer, Alexion, NIH, FDA, and PCORI. She has also received consulting fees and honoraria from Genzyme Corporation (Sanofi), Shire Pharmaceuticals, Alexion Pharmaceuticals, Amicus Therapeutics and Roche. She served as a member of the Pompe and Gaucher Disease Registry Advisory Board for Genzyme Corporation (Sanofi); Scientific Advisory Board for Shire Pharmaceuticals; and Registry Board Member for Alexion Pharmaceuticals. She also serves on the Data Safety Monitoring Board for PTC Therapeutics.
Author contributions
MM conceived and designed the study, collected clinical data, performed data analysis and wrote the manuscript. GH conceived the study, aided in data interpretation, and critically reviewed the manuscript. JG, CR, and DB collected data, aided in data interpretation, and critically reviewed the manuscript. XZ and EC generated variant data using ATAV, advised data analysis, aided in data interpretation, and critically reviewed the manuscript. ZK and SD collected specimens, phenotyped the patients, and critical reviewed the manuscript. PSK conceived the study, collected clinical data, and critically reviewed the manuscript. All authors read and approved the final manuscript.
References
- 1.Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. http://dx.doi.org/10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leslie N, Tinkle BT. In: Glycogen Storage Disease Type II (Pompe Disease) Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews®, Seattle (WA): University of Washington, Seattle; 1993. [Google Scholar]
- 3.Kemper AR, Comeau AM, Prosser LA, Green NS, Tanksley S, Goldenberg A, et al. Evidence report: newborn screening for Pompe disease. Book evidence report: newborn screening for Pompe disease. http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/nominatecondition/reviews/pompereport2013.pdf Accessed 2015 Apr; 23.
- 4.Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ, Hwu WL. Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr. 2011;158:1023–1027.e1. doi: 10.1016/j.jpeds.2010.11.053. http://dx.doi.org/10.1016/j.jpeds.2010.11.053. [DOI] [PubMed] [Google Scholar]
- 5.Prater SN, Banugaria SG, DeArmey SM, Botha EG, Stege EM, Case LE, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med Off J Am Coll Med Genet. 2012;14:800–810. doi: 10.1038/gim.2012.44. http://dx.doi.org/10.1038/gim.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid α-glucosidase major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. http://dx.doi.org/10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 7.Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord. 2013;23:319–323. doi: 10.1016/j.nmd.2012.12.003. http://dx.doi.org/10.1016/j.nmd.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Dubrovsky A, Corderi J, Lin M, Kishnani PS, Jones HN. Expanding the phenotype of late-onset pompe disease: tongue weakness: a new clinical observation. Muscle Nerve. 2011;44:897–901. doi: 10.1002/mus.22202. http://dx.doi.org/10.1002/mus.22202. [DOI] [PubMed] [Google Scholar]
- 9.Byrne BJ, Kishnani PS, Case LE, Merlini L, Müller-Felber W, Prasad S, et al. Pompe disease: design, methodology, and early findings from the Pompe registry. Mol Genet Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. http://dx.doi.org/10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Preisler N, Lukacs Z, Vinge L, Madsen KL, Husu E, Hansen RS, et al. Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab. 2013;110:287–289. doi: 10.1016/j.ymgme.2013.08.005. http://dx.doi.org/10.1016/j.ymgme.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Dubrovsky A, Corderi J, Karasarides T, Taratuto AL. Pompe disease, the must-not-miss diagnosis: a report of 3 patients. Muscle Nerve. 2013;47:594–600. doi: 10.1002/mus.23643. http://dx.doi.org/10.1002/mus.23643. [DOI] [PubMed] [Google Scholar]
- 12.Pérez-López J, Selva-O’Callaghan A, Grau-Junyent JM, Gallego-Galindo L, Coll MJ, García-Morillo S, et al. Delayed diagnosis of late-onset Pompe disease in patients with myopathies of unknown origin and/or hyperCKemia. Mol Genet Metab. 2015;114:580–583. doi: 10.1016/j.ymgme.2015.02.004. http://dx.doi.org/10.1016/j.ymgme.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Gutiérrez-Rivas E, Bautista J, Vílchez JJ, Muelas N, Díaz-Manera J, Illa I, et al. Targeted screening for the detection of Pompe disease in patients with unclassified limb-girdle muscular dystrophy or asymptomatic hyperCKemia using dried blood: a Spanish cohort. Neuromuscul Disord. 2015;25:548–553. doi: 10.1016/j.nmd.2015.04.008. http://dx.doi.org/10.1016/j.nmd.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Vissing J, Lukacs Z, Straub V. Diagnosis of pompe disease: muscle biopsy vs blood-based assays. JAMA Neurol. 2013;70(7):923. doi: 10.1001/2013.jamaneurol.486. http://dx.doi.org/10.1001/2013.jamaneurol.486. [DOI] [PubMed] [Google Scholar]
- 15.Lévesque S, Auray-Blais C, Gravel E, Boutin M, Dempsey-Nunez L, Jacques PE, et al. Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing, Orphanet. J Rare Dis. 2016;11 doi: 10.1186/s13023-016-0390-6. http://dx.doi.org/10.1186/s13023-016-0390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angelini C, Savarese M, Fanin M, Nigro V. Next generation sequencing detection of late onset Pompe disease. Muscle Nerve. 2016:n/a–n/a. doi: 10.1002/mus.25042. https://doi.org/10.1002/mus.25042. [DOI] [PubMed]
- 17.Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. 2014;16:922–931. doi: 10.1038/gim.2014.58. http://dx.doi.org/10.1038/gim.2014.58. [DOI] [PubMed] [Google Scholar]
- 18.Ghaoui R, Cooper ST, Lek M, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. 2015;72:1424–1432. doi: 10.1001/jamaneurol.2015.2274. http://dx.doi.org/10.1001/jamaneurol.2015.2274. [DOI] [PubMed] [Google Scholar]
- 19.Schabhüttl M, Wieland T, Senderek J, Baets J, Timmerman V, Jonghe PD, et al. Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol. 2014;261:970–982. doi: 10.1007/s00415-014-7289-8. http://dx.doi.org/10.1007/s00415-014-7289-8. [DOI] [PubMed] [Google Scholar]
- 20.Strom SP, Lee H, Das K, Vilain E, Nelson SF, Grody WW, et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16:510–515. doi: 10.1038/gim.2013.183. http://dx.doi.org/10.1038/gim.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck TF, Mullikin JC. Program on behalf of the NCS, Biesecker LG. Systematic evaluation of sanger validation of next-generation sequencing variants. Clin Chem. 2016;62:647–654. doi: 10.1373/clinchem.2015.249623. http://dx.doi.org/10.1373/clinchem.2015.249623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altmüller J, Budde BS, Nürnberg P. Enrichment of target sequences for next-generation sequencing applications in research and diagnostics. Biol Chem. 2014;395 doi: 10.1515/hsz-2013-0199. http://dx.doi.org/10.1515/hsz-2013-0199. [DOI] [PubMed] [Google Scholar]
- 23.Gargis AS, Kalman L, Bick DP, da Silva C, Dimmock DP, Funke BH, et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat Biotechnol. 2015;33:689–693. doi: 10.1038/nbt.3237. http://dx.doi.org/10.1038/nbt.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. http://dx.doi.org/10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martiniuk F, Bodkin M, Tzall S, Hirschhorn R. Isolation and partial characterization of the structural gene for human acid alpha glucosidase. DNA Cell Biol. 1991;10:283–292. doi: 10.1089/dna.1991.10.283. http://dx.doi.org/10.1089/dna.1991.10.283. [DOI] [PubMed] [Google Scholar]
- 26.Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA. Characterization of the human lysosomal alpha-glucosidase gene. Biochem J. 1990;272:493–497. doi: 10.1042/bj2720493. http://dx.doi.org/10.1042/bj2720493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJJ. The genotype–phenotype correlation in Pompe disease. Am J Med Genet C: Semin Med Genet. 2012;160C:59–68. doi: 10.1002/ajmg.c.31318. http://dx.doi.org/10.1002/ajmg.c.31318. [DOI] [PubMed] [Google Scholar]
- 28.Ausems MGEM, Verbiest J, Hermans MMP, Kroos MA, Beemer FA, Wokke JHJ, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713. doi: 10.1038/sj.ejhg.5200367. http://dx.doi.org/10.1038/sj.ejhg.5200367. [DOI] [PubMed] [Google Scholar]
- 29.Kroos M, Hoogeveen-Westerveld M, Michelakakis H, Pomponio R, Van der Ploeg A, Halley D, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat. 2012;33:1161–1165. doi: 10.1002/humu.22108. http://dx.doi.org/10.1002/humu.22108. [DOI] [PubMed] [Google Scholar]
- 30.De Filippi P, Saeidi K, Ravaglia S, Dardis A, Angelini C, Mongini T, et al. Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J Rare Dis. 2014;9:102. doi: 10.1186/s13023-014-0102-z. http://dx.doi.org/10.1186/s13023-014-0102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montalvo ALE, Bembi B, Donnarumma M, Filocamo M, Parenti G, Rossi M, et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II, Hum. Mutat. 2006;27:999. doi: 10.1002/humu.20374. http://dx.doi.org/10.1002/humu.20374. [DOI] [PubMed] [Google Scholar]
- 32.Musumeci O, Thieme A, Claeys KG, Wenninger S, Kley RA, Kuhn M, et al. Homozygosity for the common GAA gene splice site mutation c.-32-13T > G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul Disord NMD. 2015;25:719–724. doi: 10.1016/j.nmd.2015.07.002. http://dx.doi.org/10.1016/j.nmd.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Bali DS, Tolun AA, Goldstein JL, Dai J, Kishnani PS. Molecular analysis and protein processing in late-onset pompe disease patients with low levels of acid α-glucosidase activity. Muscle Nerve. 2011;43:665–670. doi: 10.1002/mus.21933. http://dx.doi.org/10.1002/mus.21933. [DOI] [PubMed] [Google Scholar]
- 34.Kroos MA, Pomponio RJ, Hagemans ML, Keulemans JLM, Phipps M, DeRiso M, et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T→G haplotype. Neurology. 2007;68:110–115. doi: 10.1212/01.wnl.0000252798.25690.76. http://dx.doi.org/10.1212/01.wnl.0000252798.25690.76. [DOI] [PubMed] [Google Scholar]
- 35.Laforêt P, Laloui K, Granger B, Hamroun D, Taouagh N, Hogrel JY, et al. The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease. Rev Neurol (Paris) 2013;169:595–602. doi: 10.1016/j.neurol.2013.07.002. http://dx.doi.org/10.1016/j.neurol.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 36.van Capelle CI, van der Meijden JC, van den Hout JMP, Jaeken J, Baethmann M, Voit T, et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J Rare Dis. 2016;11:65. doi: 10.1186/s13023-016-0442-y. http://dx.doi.org/10.1186/s13023-016-0442-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boerkoel CF, Exelbert R, Nicastri C, Nichols RC, Miller FW, Plotz PH, et al. Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am J Hum Genet. 1995;56:887–897. [PMC free article] [PubMed] [Google Scholar]
- 38.Exome Aggregation Consortium. Lek M, Karczewski K, Minikel E, Samocha K, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. BioRxiv. 2015:030338. doi: 10.1038/nature19057. http://dx.doi.org/10.1101/030338. [DOI] [PMC free article] [PubMed]
- 39.Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32:246–251. doi: 10.1038/nbt.2835. http://dx.doi.org/10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
- 40.Goldfeder RL, Priest JR, Zook JM, Grove EM, Waggott D, Wheeler MT, et al. Medical implications of technical accuracy in genome sequencing. Genome Med. 2016;8:24. doi: 10.1186/s13073-016-0269-0. http://dx.doi.org/10.1186/s13073-016-0269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farrell CM, O’Leary NA, Harte RA, Loveland JE, Wilming LG, Wallin C, et al. Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res. 2013:gkt1059. doi: 10.1093/nar/gkt1059. https://doi.org/10.1093/nar/gkt1059. [DOI] [PMC free article] [PubMed]
- 42.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. http://dx.doi.org/10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PEM. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. http://dx.doi.org/10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- 44.Oetting WS, Brenner SE, Brookes AJ, Greenblatt MS, Hart RK, Karchin R, et al. Pathogenicity interpretation in the age of precision medicine: the 2015 annual scientific meeting of the human genome variation society: human mutation. Hum Mutat. 2016;37:406–411. doi: 10.1002/humu.22958. http://dx.doi.org/10.1002/humu.22958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y, Oldridge DA, Diskin SJ, Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015;43:e39. doi: 10.1093/nar/gku1363. http://dx.doi.org/10.1093/nar/gku1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feeney EJ, Austin S, Chien YH, Mandel H, Schoser B, Prater S, et al. The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile-and adult-onset patients. Acta Neuropathol Commun. 2014;2:2. doi: 10.1186/2051-5960-2-2. http://dx.doi.org/10.1186/2051-5960-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics. 2014;15:247. doi: 10.1186/1471-2105-15-247. http://dx.doi.org/10.1186/1471-2105-15-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang H, Wu Y, Narzisi G, ORawe JA, Barrón LTJ, Rosenbaum J, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. http://dx.doi.org/10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747. doi: 10.1038/gim.2013.92. http://dx.doi.org/10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. http://dx.doi.org/10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tattini L, D’Aurizio R, Magi A. Detection of genomic structural variants from next-generation sequencing data. Bioinforma Comput Biol. 2015;92 doi: 10.3389/fbioe.2015.00092. http://dx.doi.org/10.3389/fbioe.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu X, Guo H, et al. An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum Mutat. 2014;35:899–907. doi: 10.1002/humu.22537. http://dx.doi.org/10.1002/humu.22537. [DOI] [PubMed] [Google Scholar]
- 53.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. http://dx.doi.org/10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meynert AM, Bicknell LS, Hurles ME, Jackson AP, Taylor MS. Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinformatics. 2013;14:195. doi: 10.1186/1471-2105-14-195. http://dx.doi.org/10.1186/1471-2105-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of α-glucosidase activities in dried blood spots. Mol Genet Metab. 2007;90:449–452. doi: 10.1016/j.ymgme.2006.12.006. http://dx.doi.org/10.1016/j.ymgme.2006.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.