

Abstract
Background
Epinephrine administered during cardiopulmonary resuscitation (CPR) is associated with severe post‐resuscitation myocardial dysfunction. We previously demonstrated that therapeutic hypothermia reduced the severity of post‐resuscitation myocardial dysfunction caused by epinephrine; however, the relationship between myocardial adrenoceptor expression and myocardial protective effects by hypothermia remains unclear.
Methods and Results
Rats weighing between 450 and 550 g were randomized into 5 groups: (1) normothermic placebo, (2) normothermic epinephrine, (3) hypothermic placebo, (4) hypothermic epinephrine, and (5) sham (not subject to cardiac arrest and resuscitation). Ventricular fibrillation was induced and untreated for 8 minutes for all other groups. Hypothermia was initiated coincident with the start of CPR and maintained at 33±0.2°C for 4 hours. Placebo or epinephrine was administered 5 minutes after the start of CPR and 3 minutes before defibrillation. Post‐resuscitation ejection fraction was measured hourly for 4 hours then hearts were harvested. Epinephrine increased coronary perfusion pressure during CPR (27±6 mm Hg versus 21±2 mm Hg P<0.05). Post‐resuscitation myocardial function was impaired in the normothermic epinephrine group compared with other groups. The concentration of myocardial cAMP doubled in the normothermic epinephrine group (655.06±447.63 μmol/L) compared with the hypothermic epinephrine group (302.51±97.98 μmol/L; P<0.05). Myocardial β1‐adrenoceptor expression decreased with normothermia cardiac arrest but not with hypothermia regardless of epinephrine.
Conclusions
Epinephrine, administered during normothermic CPR, increased the severity of post‐resuscitation myocardial dysfunction. This adverse effect was inhibited by intra‐arrest hypothermia resuscitation. Declined cAMP with more preserved β1‐adrenoceptors in hypothermia‐resuscitated myocardium is associated with improved post‐resuscitated myocardial function in vivo.
Keywords: adrenoceptor, CPR, epinephrine, hypothermia, post‐resuscitation
Subject Categories: Cardiopulmonary Resuscitation and Emergency Cardiac Care
Clinical Perspective
What Is New?
The effect of protecting post‐resuscitation myocardial contractility by intra‐arrest hypothermia may prompt early clinical utilization of hypothermia ahead of resuscitation rather than delay to post‐resuscitation.
It is novel that there are more preserved β1‐adrenoceptors in hypothermia‐resuscitated myocardium.
What Are the Clinical Implications?
This indicates more cautious administration of β1‐adrenoceptor agonist clinically for the hypothermic post‐resuscitation duration.
Introduction
Epinephrine has been the preferred vasopressor agent for CPR.1 Its efficacy is attributable to its α‐adrenergic vasopressor effect, which increases coronary and cerebral perfusion during resuscitation. The potential adverse effects of epinephrine, including ventricular dysrhythmias and post‐resuscitation myocardial dysfunction, have been attributed primarily to its inotropic and chronotropic actions of β1‐adrenergic actions,2 resulting in increased myocardial oxygen consumption and severity of myocardial ischemic injury. When epinephrine is combined with a β1‐adrenergic–blocking agent during CPR, the severity of post‐resuscitation myocardial dysfunction is significantly reduced.2 We have recently demonstrated that hypothermia attenuates the detrimental effects of epinephrine and exerts myocardial protection after restoration of spontaneous circulation (ROSC).3 Myocardial protection provided by hypothermia was considered to be attributable to the reduction in myocardial metabolism and oxygen consumption.4 We further demonstrated that the β‐adrenergic effects of epinephrine diminished during hypothermia, whereas α‐adrenergic effects were not altered, suggesting the need to further explore the impact of hypothermia on myocardial adrenergic receptors (ARs) during and after resuscitation.3
Studies have demonstrated abnormalities at several levels of the β‐AR pathways in heart failure that are characterized by left ventricular dysfunction, decreased myocardial β1‐AR, and increased Gαi.5 Chronic increases in catecholamines following activation of the sympathetic nervous system leads to both β‐AR desensitization and downregulation.5, 6 The declined β1‐AR expression may lead to cardiac dysfunction. Along with the effects of β1‐AR downregulation on left ventricular contractile function, the augmented activity of β‐AR signaling also accounts for heart failure, which is similar to the phenotype of post‐resuscitation cardiac dysfunction. β‐AR blockers dramatically improve the outcome of both heart failure7, 8 and cardiac arrest (CA)2, 9 and may further indicate the pathological toxicity caused by excess β‐AR stimulation.
The present study investigated post‐resuscitation myocardial function, adrenoceptor expression, and second messenger levels in a rat heart that underwent different resuscitation themes in vivo. We hypothesized that β1‐AR would be sustained by intra‐arrest hypothermia CPR, while α1‐AR expression may not be affected.
Materials and Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author upon reasonable request to other researchers for purposes of reproducing the results or replicating the procedure. This study was approved by the Animal Care and Use Committee at Virginia Commonwealth University. All procedures were in accordance with institutional guidelines. Animals received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the National Institute of Health publication on the Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources.
Animal Preparation
Male Sprague‐Dawley rats weighing 450 to 550 g were fasted overnight except for free access to water. After inhalation of CO2 for 10 seconds, the animals were anesthetized by an intraperitoneal injection of pentobarbital (45 mg/kg). The trachea was orally intubated with a 14‐G cannula mounted on a blunt needle (Abbocath‐T; Abbott Hospital Products Division) with a 145‐degree angled tip. End‐tidal CO2 (ETCO2) was continuously monitored with a side‐stream infrared CO2 analyzer (model 200; Instrument Laboratory) interposed between the tracheal cannula and the ventilator. A conventional lead II ECG was continuously monitored. The core temperature (aortic blood temperature) was maintained at 37°C by adjusting the distance between animals and the heating lamp.
A PE‐50 catheter (Becton Dickinson) was advanced into the descending aorta for measurement of arterial pressure. A thermocouple microprobe (9030‐12‐D‐34; Columbus Instruments) was advanced to the descending aorta for measurement of blood temperature. Through the left external jugular vein, another PE‐50 catheter was advanced into the right atrium for measurement of right atrial pressures. Aortic and right atrial pressures were measured with high‐sensitivity transducers (model 42584‐01; Abbott Critical Care Systems). A 3F PE catheter (model C‐PMS‐301J; Cook Critical Care) was inserted through the right external jugular vein into the right atrium. A precurved guide wire was then advanced through the catheter into the right ventricle confirmed by transthoracic ECG. All catheters were flushed intermittently with saline containing 2.5 IU/mL of crystalline bovine heparin.
Experimental Procedures
Fifteen minutes before inducing ventricular fibrillation (VF), animals were randomly assigned into 1 of 5 groups (n=8 in each group): normothermic placebo, normothermic epinephrine, hypothermic control, hypothermic epinephrine, and sham. For the sham group, animals were euthanized at baseline without VF and CPR; then heart tissue was collected for testing β1‐AR and cAMP. VF was then induced through a guide wire advanced from the right jugular vein into the right ventricle. A progressive increase in 60‐Hz current up to a maximum of 3.5 mA was then delivered to the right ventricular endocardium. The current flow was continued for 3 minutes to prevent spontaneous defibrillation. Precordial compression, together with mechanical ventilation (tidal volume 0.6 mL/100 g body weight, frequency 100 breaths per min, fraction of inspired oxygen 1.0), was initiated after 8 minutes of untreated VF with a pneumatically driven mechanical chest compressor. Precordial chest compressions were maintained at a rate of 200 per minute and synchronized to provide a compression/ventilation ratio of 2:1 with equal compression‐relaxation for 8 minutes. Epinephrine (20 μg/kg) or saline placebo was injected into the right atrium 5 minutes after the start of precordial compression. Mechanical ventilation was discontinued after onset of VF. Resuscitation was attempted with up to three 2‐J countershocks after 8 minutes of CPR. ROSC was defined as the return of supraventricular rhythm with a mean aortic pressure >50 mm Hg for 5 minutes. After ROSC, a fraction of inspired oxygen of 1.0 was continued for 1 hour, adjusted to 0.5 for the second hour and 0.21 thereafter.
In animals assigned to therapeutic hypothermia, body cooling was initiated coincident with the start of CPR (Figure 1). Blood temperature was reduced to 34±0.2°C when epinephrine was injected and further reduced to 33±0.2°C when defibrillation was attempted. Body cooling was induced with the aid of ice packs and an electrical fan. Once reached, target temperature was maintained with the aid of a Blanketrol II (Cincinnati Sub Zero) and continued for 4 hours after resuscitation. For the animals not subjected to cooling, core temperature was maintained at 37±0.2°C with a heating lamp. Room temperature was kept at 23 to 25°C.
Figure 1.
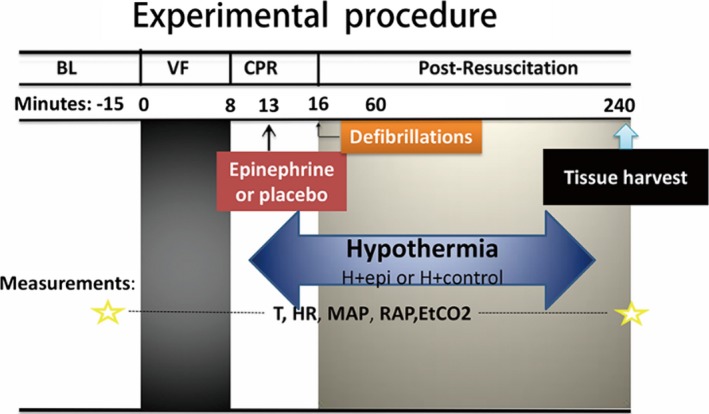
Experimental procedure. −15 indicates 15 minutes before ventricular fibrillation was induced; BL, baseline; DF, defibrillation; EtCO2, end‐tidal CO2; H+control, hypothermic control group; H+epi, hypothermic epinephrine group; HR, heart rate; MAP, mean arterial pressure; RAP, right atrium pressure; T, temperature; VF, ventricular fibrillation.
At the end of 4 hours after ROSC, animals were euthanized with an intravenous injection of 150 mg/kg pentobarbital. The heart was then rapidly excised and frozen in liquid nitrogen for further testing. A necropsy was performed to inspect for gross abnormalities, including evidence of traumatic injuries consequent to cannulation, airway management, or precordial compression.
Measurements
Heart rate, aortic and right atrial pressures, ECG, body temperature, and ETCO2 values were continuously recorded on a personal computer–based data‐acquisition system supported by WINDAQ software (DATAQ). Coronary perfusion pressure (CPP) was calculated as the difference between aortic and time‐coincident right atrial pressures measured at the end of each minute of precordial compression.
Ejection fraction (EF), cardiac output, and myocardial performance index (MPI) were measured by echocardiography (HD11XE; Philips Medical Systems) with a 12.5‐HZ transducer at baseline and hourly after ROSC. All echo measurements were reviewed and confirmed separately by 2 investigators. The EF was used to assess myocardial contractility. MPI, the indicator of both systolic and diastolic function, was calculated using the formula (a−b)/b, where a=mitral closure‐to‐opening interval (time interval from cessation to onset of mitral inflow) and b=ET (aortic flow ejection time, obtained at the left ventricular outflow tract).3, 10
Myocardial concentrations of cAMP and inositol trisphosphate were measured with commercial ELISA kits according to the manufacturer's instructions (category No. STA‐500, Cell Biolabs; category No. ABIN457242, Antibodies), respectively. Total proteins were extracted in a Triton 1% buffer with anti‐phosphatase/protease inhibitor (Sigma‐Aldrich). All protein concentrations were determined using Bradford reagent (BioRad). After denaturation in Laemmli buffer, a fixed amount of mixed proteins was loaded in each lane of a sodium dodecyl sulfate polyacrylaminde gel electrophoresis (9–15%). Proteins were separated by electrophoresis in a migration buffer and then transferred to a nitrocellulose membrane (Hybond; GE Healthcare). After saturation in milk, each membrane was incubated overnight at 4°C with primary antibodies: anti–β1‐adrenoceptor (1:200, Santa Cruz Biotechnology), anti–α1‐adrenoceptor (1:1000, Abcam), and β‐actin (1:5000, Sigma‐Aldrich). The following day, membranes were washed with Tris‐buffered saline Tween and incubated with appropriate secondary antibody. The protein was detected (β1‐adrenoceptor at 65 kDa and α1‐adrenoceptor at 60 kDa). All Western blot experiments were quantified using ImageJ software (National Institutes of Health) and β‐actin (42 kDa), ensuring no variation in protein gel loading.
Statistical Analysis
Data were analyzed using SPSS version 19.0 (IBM). Measurements were reported as mean±SD. The added effects of either epinephrine or hypothermia on post‐resuscitation myocardial function (EF, cardiac output, and MPI) over time were examined using a 2‐factor (groups×time) repeated‐measures ANOVA. Both comparison between time‐based measurements within each group and the total response over time among groups were examined in the model. For measurements among groups, 1‐way ANOVA and Scheffe's multiple comparison were used. The outcomes including second messengers, ARs, defibrillation numbers, arrhythmia start time, and arrhythmia episode differences were analyzed with Kruskal–Wallis test. A value of P<0.05 was regarded as significant.
Results
Forty rats were used and 39 were analyzed in the present experiment. There was no difference among the 5 groups at baseline (Table 1). The number of defibrillations required during CPR did not differ among groups (Table 2). CPP was augmented with epinephrine in both hypothermia and normothermia groups. There was no difference in CPP in both placebo control groups regardless of temperature (Figure 2). Except for 1 rat in the normothermic placebo group, all animals that underwent CA were successfully resuscitated. There was a trend towards shorter arrhythmia duration and the incidence of refractory and/or recurrent VF in the hypothermia groups, but the differences were not statistically significant (Table 2).
Table 1.
Baseline Characteristics
| Group | Control | H+Control | H+epi | Epinephrine |
|---|---|---|---|---|
| Body weight, g | 503.3±18.4 | 506.6±13.4 | 503±17.7 | 505±20.7 |
| Heart rate, beats per min | 370±22.04 | 374±32.25 | 365±14.24 | 363±33.45 |
| MAP, mm Hg | 142.71±13.26 | 144.43±13.11 | 148.63±9.49 | 141.88±12.17 |
| ETCO2, mm Hg | 37.96±2.56 | 38.93±2.86 | 38.44±4.27 | 39.96±3.10 |
| RA, mm Hg | 1.2±0.15 | 1.3±0.11 | 1.2±0.21 | 1.2±0.18 |
| Temperature, °C | 37.1±0.29 | 36.8±0.66 | 36.8±0.25 | 36.9±0.27 |
| CO, mL/min | 102.88±9.37 | 101.75±6.84 | 103.125±11.27 | 109.875±11.48 |
| EF, % | 73.75±2.82 | 73.5±3.82 | 71.5±2.45 | 73.25±3.49 |
| MPI | 0.63±0.088 | 0.67±0.086 | 0.59±0.080 | 0.61±0.092 |
| Lactate, mmol/L | 0.79±0.11 | 0.80±0.33 | 0.89±0.38 | 0.70±0.15 |
Values are presented as mean±SD. CO indicates cardiac output; EF, ejection fraction; ETCO2, end‐tidal CO2; H+Control, hypothermic control group; H+epi, hypothermic epinephrine group; MAP, mean aortic pressure; MPI, myocardial performance index; RA, right atrial pressure.
Table 2.
Defibrillation Numbers, 15‐Minute Post‐Resuscitation Ventricular Arrhythmias, and Outcome of Survival
| Group | Control | H+Control | H+epi | Epinephrine |
|---|---|---|---|---|
| Defibrillation numbers | 2.14±1.21 | 1±0.53 | 1.125±0.64 | 1.88±1.36 |
| Arrhythmia start time, s | 3.29±2.63 | 3.38±3.2 | 6.13±9.06 | 4.63±3.07 |
| Arrhythmia episode, min | 7.26±7.98 | 5.94±6.52 | 6.88±6.94 | 10.41±7.65 |
| Resuscitation | 7/8 | 8/8 | 8/8 | 8/8 |
| 4‐h Survival | 7/7 | 8/8 | 8/8 | 7/8 |
H+Control indicates hypothermic control group; H+epi, hypothermic epinephrine group.
Figure 2.
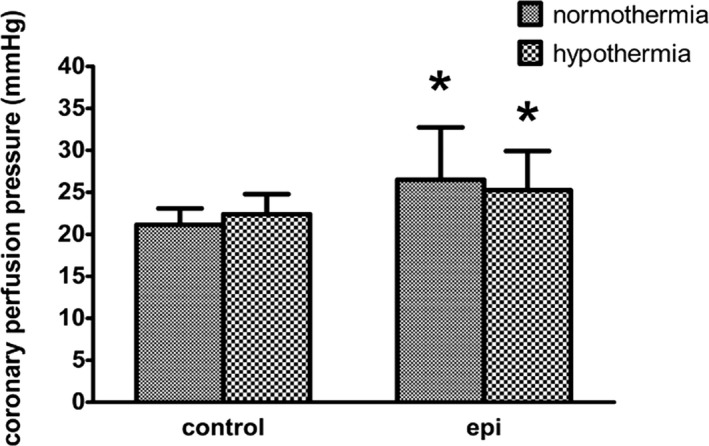
Coronary perfusion pressure after epinephrine administered during CPR in 4 groups (each group contains 8 rats). *P<0.05 vs the normothermic control group.
There was less impairment in post‐resuscitation myocardial systolic and diastolic function and slower heart rates in rats treated with hypothermia (Figures 3, 4, 5 through 6). The combination of epinephrine and hypothermia improved post‐resuscitation myocardial contractility when compared with normothermic rats treated with epinephrine, which led to the most severe post‐resuscitation myocardial dysfunction (Figures 3, 4 through 5).
Figure 3.
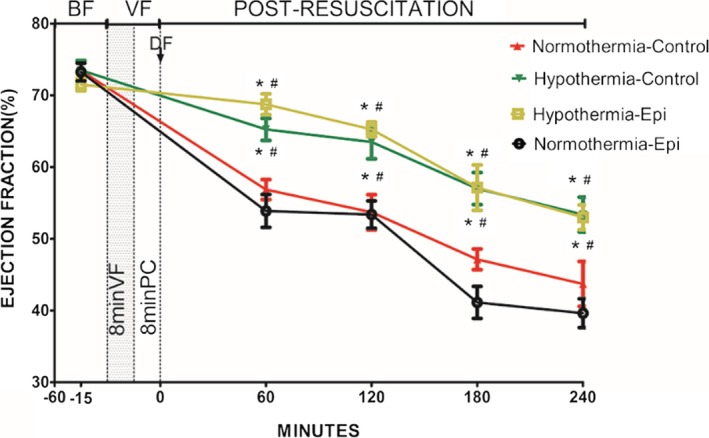
Ejection fraction in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each) (percentage). *P<0.05 vs the normothermic epinephrine group. # P<0.05 vs the normothermic control group. Data points are mean±SEM. −15 indicates 15 minutes before ventricular fibrillation was induced; −60, 60 minutes before ventricular fibrillation was induced; BF, before fibrillation; DF, defibrillation; Epi, epinephrine; PC, precordial compression; VF, ventricular fibrillation.
Figure 4.
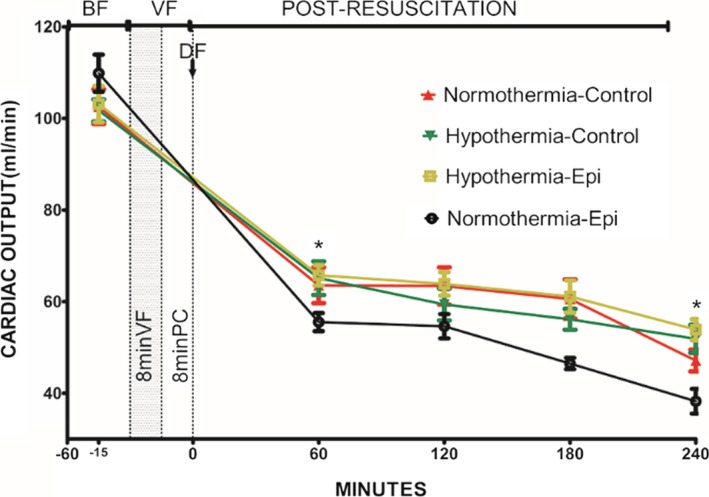
Cardiac output in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each) (mL/min). *P<0.05 vs the normothermic epinephrine group. Data points are mean±SEM. −15 indicates 15 minutes before ventricular fibrillation was induced; −60, 60 minutes before ventricular fibrillation was induced; BF, before fibrillation; DF, defibrillation; Epi, epinephrine; PC, precordial compression; VF, ventricular fibrillation.
Figure 5.
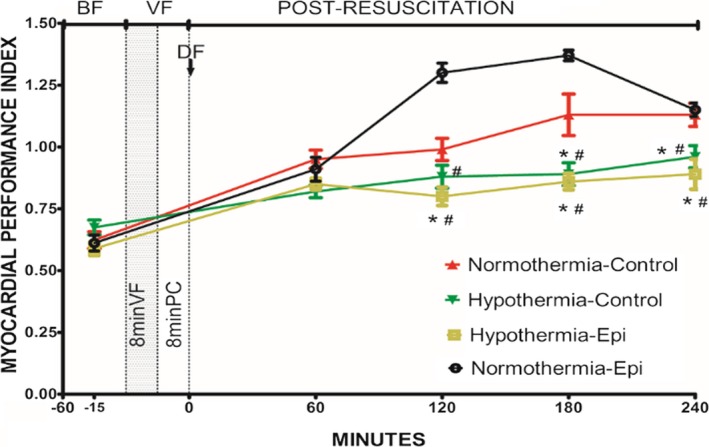
Myocardial performance index in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each). *P<0.05 vs the normothermic control group. # P<0.05 vs the normothermic epinephrine group. Data points are mean±SEM. −15 indicates 15 minutes before ventricular fibrillation was induced; −60, 60 minutes before ventricular fibrillation was induced; BF, before fibrillation; DF, defibrillation; Epi, epinephrine; PC, precordial compression; VF, ventricular fibrillation.
Figure 6.
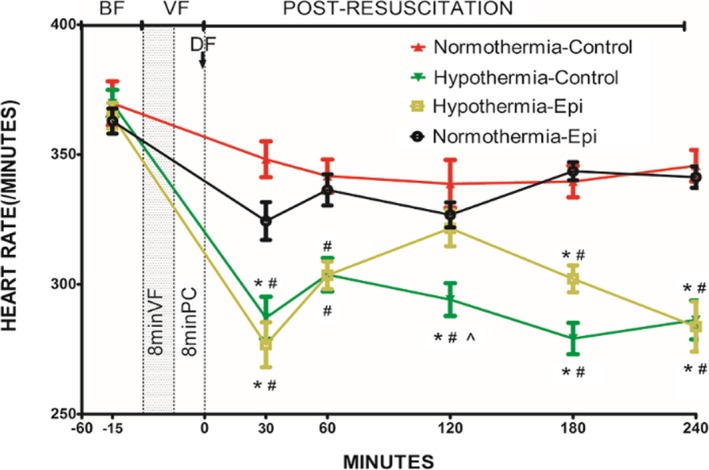
Heart rate in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each) (beats per minute). *P<0.05 vs the normothermic epinephrine group. # P<0.05 vs the normothermic control group. ^ P<0.05 vs the hypothermic epinephrine group. Data points are mean±SEM. −15 indicates 15 minutes before ventricular fibrillation was induced; −60, 60 minutes before ventricular fibrillation was induced; BF, before fibrillation; DF, defibrillation; Epi, epinephrine; PC, precordial compression; VF, ventricular fibrillation.
cAMP within the sham group was at the lowest level compared with the other groups. A significant enhancement of cAMP was observed in the hypothermia‐treated group regardless of the use of epinephrine (Figure 7). There was no difference between the normothermia control and normothermia epinephrine groups, although the mean value of the former was larger than the latter. There was no difference in the concentration of inositol trisphosphate among the 4 groups (Table 3).
Figure 7.
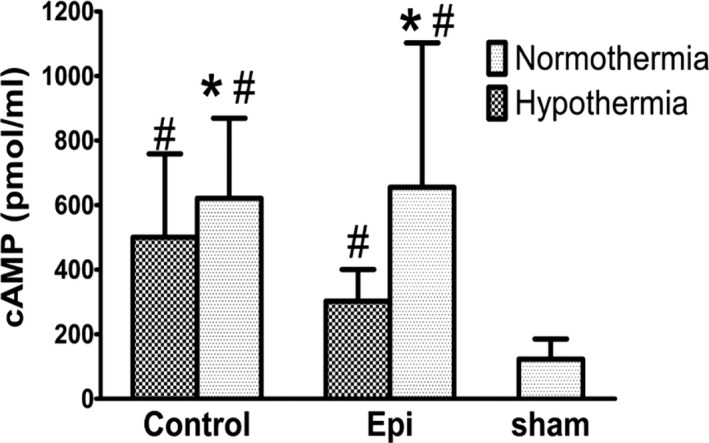
Myocardial cAMP levels *P<0.05 vs the hypothermic epinephrine group. # P<0.05 vs the sham group.
Table 3.
Myocardial Inositol Trisphosphate Concentration
| Group | Control | H+Control | H+epi | Epinephrine |
|---|---|---|---|---|
| Inositol trisphosphate, μmol/L | 5±4.05 | 3.16±4.09 | 1.28±0.97 | 2.31±1.72 |
H+Control indicates hypothermic control group; H+epi, hypothermic epinephrine group.
Myocardial β1‐AR expression decreased sharply in the normothermia groups regardless of the use of epinephrine. However, there was no significant difference in myocardial β1‐AR expression between sham‐ and hypothermia‐treated groups (Figure 8). No difference was found between the normothermia control and normothermia epinephrine groups. There was also no difference in the expression of α1‐AR among the 4 groups (Figure 9).
Figure 8.
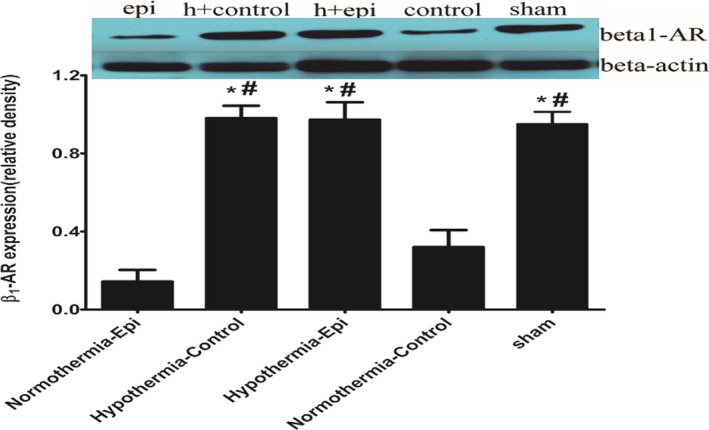
Myocardial β1‐adrenergic receptor (AR) expression at 4 hours after return of spontaneous circulation in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each). *P<0.05 vs the normothermic epinephrine group. # P<0.05 vs the normothermic control group. ^ P<0.05 vs the sham group.
Figure 9.
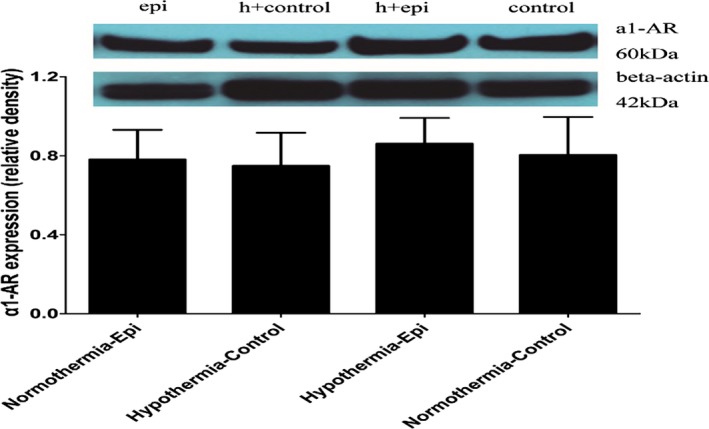
Myocardial α1‐adrenergic receptor (AR) expression at 4 hours after return of spontaneous circulation in 4 groups (note: except the normothermic control group containing 7 rats, the other groups have 8 rats each).
Discussion
The present study demonstrated elevated post‐resuscitation myocardial EF concurrent with decreased MPI when animals were treated with hypothermia regardless of epinephrine. Furthermore, CA reduced β1‐adrenoceptors expression compared with sham, which was prevented by hypothermia‐initiated intra‐arrest and maintained post‐resuscitation. In contrast, post‐resuscitation myocardial α1‐adrenoceptor expression was unchanged by hypothermia or epinephrine use and hypothermia did not affect the α1‐adrenoceptor effect that elevated CPP during CPR.
Therapeutic hypothermia provides myocardial protection after resuscitation in both patients with clinical CA and animal models.11, 12, 13, 14 In animal models of CA, enhanced post‐resuscitation myocardial systolic and diastolic function is observed with the use of mild therapeutic hypothermia.3, 10 Hsu et al15 demonstrated that hypothermia (30°C) maintained left ventricular function and reduced myocardial damage after CA and resuscitation in an asphyxia‐induced CA animal model. The results, which showed lower MPI but higher EF in resuscitated rats given hypothermia, are in accordance with the previous study. It is reported that reduced metabolic rate and oxygen consumption are possible mechanisms of myocardial protection.4, 16, 17 When epinephrine is administered during moderate hypothermia, oxygen consumption increases and myocardial efficiency (calculated by the ratio between energy output and energy input) is impaired in the isolated beating rat heart.17 Thus, except for reduced oxygen consumption, preservation of myocardial β1‐AR expression may account for hypothermia‐induced improved post‐resuscitation myocardial contractility whenever epinephrine is given, as demonstrated in the present study.
The β‐AR agonists, such as epinephrine, mediate inotropic effects via the sarcolemmal G‐protein–protein kinase A signaling pathway by increasing intracellular cAMP levels with normal core temperatures. During hypothermia, the cardiac β‐adrenergic response to β‐AR agonists remains controversial. Williams and Broadley18 reported that hypothermia induces supersensitivity of the β‐adrenoceptor in guinea pig atria, which correlates with the in vivo 4‐fold increase of cardiac tissue cAMP observed at 15°C following epinephrine infusion. Dietrichs19 described elevated cardiac β‐adrenoceptor sensitivity and significantly greater cAMP levels in rat heart with the same conditions. The possible explanation for hypersensitivity is that hypothermia reduces catechol‐O‐methyltransferase activity, increasing epinephrine's half‐life and reducing phosphodiesterase 3 function.20 However, Tveita and Sieck21 demonstrated diminished inotropic effects of β‐AR agonists during hypothermia. Hypothermia induced a reduction of β‐adrenoceptor sensitivity and transition from β‐ to α‐adrenoceptor in 17°C in vitro rat hearts, which was first reported by Kunos and Nickerson,22 who also indicated that the conversion may begin at a higher temperature. Han et al23 further demonstrated that the positive inotropic and chronotropic effects mediated by β‐adrenoceptors diminished at core temperature levels <33°C. In the present study, post‐resuscitation EF declined relative to baseline, and hypothermia reduced this decline compared with normothermia, which indicates that moderate hypothermia might enhance the positive inotropic effect primarily mediated by myocardial β‐adrenoceptor in the context of CA following resuscitation. The decline of post‐resuscitation cardiac output relative to baseline is consistent with EF; however, it cannot be changed by hypothermia. A possible reason might be the slower heart rate, which reduces cardiac output despite a stronger heart beat in hypothermia. It seems that slower heart rate by hypothermia is not consistent with higher β1‐AR expression. That may lead to a stronger positive chronotropic effect. Actually, a factor that primarily affects heart rate in hypothermia is impaired myocardial oxidative metabolism, as suggested by Hannon.24 The activation of α2‐AR in mild hypothermia also could result in bradycardia.25
It is well known that β1‐adrenoceptors comprise ≈70% to 80% of the β‐AR population expressed in cardiomyocytes and lead to cAMP synthesis when coupled with Gαs.5 cAMP binds to and regulates the function of ion channels such as L‐type Ca2+ channels and leads to the alteration of intracellular Ca2+ concentration. The present study demonstrated that the cAMP level in the sham group was significantly lower compared with the other 4 groups subjected to VF followed by CPR. In summary, myocardial cAMP concentration was lower in the hypothermic resuscitated animals regardless of epinephrine when compared with those with normothermia. Although, there was no significant statistical difference in cAMP concentration comparing normothermia with hypothermia and saline‐treated animals. Moreover, lower myocardial cAMP concentration was associated with higher post‐resuscitation EF and lower MPI in hypothermia. Thus, we infer that decreasing cAMP levels may result in a low influx of calcium from the extracellular space, thus preventing cytosolic calcium overload17 and myofilament hypercontracture, and therefore preserving myocardial compliance. Although the weakened phosphodiesterase function by hypothermia3 may reduce cAMP decomposition, the adenylate cyclase that facilitates cAMP synthesis could also be inhibited by anandamide that can induce hypothermia.26 Thus, the understanding of why hypothermia reduces post‐resuscitation myocardial cAMP concentration is still uncertain and needs further exploration.
The present study further demonstrated a significantly reduced density of myocardial β1‐AR in normothermic post‐resuscitation animals, while the density of myocardial β1‐AR in hypothermic post‐resuscitation animals was equal to the sham group. Thus, we believe that post‐resuscitation hypothermia may preserve cardiac β1‐AR, which will be the basis for subsequent endogenic catecholamine stimulation. To the best of our knowledge, this is the first study to suggest that mild hypothermia may preserve β1‐AR in a rat heart in vivo after ROSC. Either a higher sustained β1‐AR expression or decreased myocardial cAMP might play a respective role in myocardial protection demonstrated with intra‐arrest and post‐resuscitation hypothermia. In terms of the relationship between declined cAMP levels and elevated β1‐AR in hypothermia‐treated resuscitated rat hearts, we surmise that hypothermia CPR reduced heterologous adrenoceptor desensitization through cAMP‐dependent protein kinase A activation,27 which may result in increased β1‐AR expression. However, this hypothesis will need further investigation.
At a normal core temperature, epinephrine activates α‐AR and causes vasopressor effects through the sarcolemmal G‐protein–phospholipase C signaling pathway by increasing inositol trisphosphate/diacylglycerol levels. The α‐adrenergic vasopressor effects increase CPP during CPR and thus improve the likelihood of successful resuscitation. Our study demonstrated that epinephrine increases CPP at a low core temperature, which indicates that there is an adequate vascular response to epinephrine during hypothermia.28, 29 We further demonstrated that the rat heart inositol trisphosphate concentration remained unchanged after hypothermia resuscitation regardless of epinephrine treatment. This result is similar to the finding from Kohlhauer et al that hypothermia (32°C) did not affect the vascular response to adrenaline during CA.30 However, profound hypothermia with lower temperatures of 10°C may alter the response of isolated carotid arteries to noradrenaline and induce vasodilation.31
Limitations
The current study has several limitations. First, the animals utilized were free from heart disease and anesthetized by pentobarbital. Second, the present study did not determine catecholamine levels after ROSC, which may affect myocardial cAMP levels. Third, hypothermia was provided and achieved rapidly during resuscitation, as opposed to the slow induction of therapeutic hypothermia after ROSC that is typically seen in clinical practice. Fourth, as a pilot study, there is paucity of direct experimentation either in vivo or in vitro to test the adrenoceptor function or signaling effectors (eg, phosphorylation of phospholamban, L‐type Ca2+ channels, troponin I, and the cardiac ryanodine receptor). Therefore, the results of the present study may not articulate the mechanistic hypothesis. Further research is needed to explore the precise mechanism of the results found in the present study.
Conclusions
CA following normothermic CPR regardless of epinephrine impairs post‐resuscitation myocardial contractile function. This adverse effect could be inhibited by hypothermia‐initiated intra‐arrest and maintained post‐resuscitation. Declined cAMP with preservation of β1‐adrenoceptors in hypothermia‐resuscitated myocardium is associated with improved post‐resuscitated myocardial function in vivo. In contrast, hypothermia resuscitation does not affect α1‐AR expression in the post‐resuscitation heart regardless of epinephrine administration during CPR. Further studies are needed to elucidate the mechanism of β1‐AR preservation induced by hypothermia resuscitation.
Sources of Funding
This study was funded in part by the project of Leading Talents in Pearl River Talent Plan of Guangdong Province (No. 81000‐42020004) and by the Weil Family Foundation.
Disclosures
None.
Acknowledgments
This work was performed at Weil Institute of Emergency and Critical Care Research, Virginia Commonwealth University, Richmond, VA.
(J Am Heart Assoc. 2018;7:e006573 DOI: 10.1161/JAHA.117.006573.)29572320
References
- 1. Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, Gabrielli A, Silvers SM, Zaritsky AL, Merchant R, Vanden Hoek TL, Kronick SL; Association American Heart . Part 9: post‐cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S78–S786. [DOI] [PubMed] [Google Scholar]
- 2. Tang W, Weil MH, Sun S, Noc M, Yang L, Gazmuri RJ. Epinephrine increases the severity of postresuscitation myocardial dysfunction. Circulation. 1995;92:3089–3093. [DOI] [PubMed] [Google Scholar]
- 3. Sun S, Tang W, Song F, Yu T, Ristagno G, Shan Y, Weng YL, Weil MH. The effects of epinephrine on outcomes of normothermic and therapeutic hypothermic cardiopulmonary resuscitation. Crit Care Med. 2010;38:2175–2180. [DOI] [PubMed] [Google Scholar]
- 4. Ning XH, Chi EY, Buroker NE, Chen SH, Xu CS, Tien YT, Hyyti OM, Ge M, Portman MA. Moderate hypothermia (30 degrees C) maintains myocardial integrity and modifies response of cell survival proteins after reperfusion. Am J Physiol Heart Circ Physiol. 2007;293:H2119–H2128. [DOI] [PubMed] [Google Scholar]
- 5. Rockman HA, Koch WJ, Lefkowitz RJ. Seven‐transmembrane‐spanning receptors and heart function. Nature. 2002;415:206–212. [DOI] [PubMed] [Google Scholar]
- 6. Bristow MR. Why does the myocardium fail? Insights from basic science. Lancet. 1998;352(suppl 1):Si8–Si14. [DOI] [PubMed] [Google Scholar]
- 7. Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334:1349–1355. [DOI] [PubMed] [Google Scholar]
- 8. Packer M, Coats AJ, Fowler MB, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Castaigne A, Roecker EB, Schultz MK, DeMets DL; Carvedilol Prospective Randomized Cumulative Survival Study Group . Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658. [DOI] [PubMed] [Google Scholar]
- 9. Yang M, Hu X, Lu X, Wu X, Xu J, Yang Z, Qian J, Sun S, Cahoon J, Tang W. The effects of alpha‐ and beta‐adrenergic blocking agents on postresuscitation myocardial dysfunction and myocardial tissue injury in a rat model of cardiac arrest. Transl Res. 2015;165:589–598. [DOI] [PubMed] [Google Scholar]
- 10. Ye S, Weng Y, Sun S, Chen W, Wu X, Li Z, Weil MH, Tang W. Comparison of the durations of mild therapeutic hypothermia on outcome after cardiopulmonary resuscitation in the rat. Circulation. 2012;125:123–129. [DOI] [PubMed] [Google Scholar]
- 11. Neumar RW, Eigel B, Callaway CW, Estes NA III, Jollis JG, Kleinman ME, Morrison LJ, Peberdy MA, Rabinstein A, Rea TD, Sendelbach S; American Heart Association . American Heart Association response to the 2015 Institute of Medicine report on strategies to improve cardiac arrest survival. Circulation. 2015;132:1049–1070. [DOI] [PubMed] [Google Scholar]
- 12. Drury PP, Gunn ER, Bennet L, Gunn AJ. Mechanisms of hypothermic neuroprotection. Clin Perinatol. 2014;41:161–175. [DOI] [PubMed] [Google Scholar]
- 13. Scirica BM. Therapeutic hypothermia after cardiac arrest. Circulation. 2013;127:244–250. [DOI] [PubMed] [Google Scholar]
- 14. Moore EM, Nichol AD, Bernard SA, Bellomo R. Therapeutic hypothermia: benefits, mechanisms and potential clinical applications in neurological, cardiac and kidney injury. Injury. 2011;42:843–854. [DOI] [PubMed] [Google Scholar]
- 15. Hsu CY, Huang CH, Chang WT, Chen HW, Cheng HJ, Tsai MS, Wang TD, Yen ZS, Lee CC, Chen SC, Chen WJ. Cardioprotective effect of therapeutic hypothermia for postresuscitation myocardial dysfunction. Shock. 2009;32:210–216. [DOI] [PubMed] [Google Scholar]
- 16. Wood T, Thoresen M. Physiological responses to hypothermia. Semin Fetal Neonatal Med. 2015;20:87–96. [DOI] [PubMed] [Google Scholar]
- 17. Schiffmann H, Gleiss J, von Hirscheydt A, Schröder T, Kahles H, Hellige G. Effects of epinephrine on the myocardial performance and haemodynamics of the isolated rat heart during moderate hypothermia—importance of calcium homeostasis. Resuscitation. 2001;50:309–317. [DOI] [PubMed] [Google Scholar]
- 18. Williams RG, Broadley KJ. Responses mediated via beta 1, but not beta 2‐adrenoceptors, exhibit hypothermia‐induced supersensitivity. Life Sci. 1982;31:2977–2983. [DOI] [PubMed] [Google Scholar]
- 19. Dietrichs ES, Schanche T, Kondratiev T, Gaustad SE, Sager G, Tveita T. Negative inotropic effects of epinephrine in the presence of increased beta‐adrenoceptor sensitivity during hypothermia in a rat model. Cryobiology. 2015;70:9–16. [DOI] [PubMed] [Google Scholar]
- 20. Zhou J, Poloyac SM. The effect of therapeutic hypothermia on drug metabolism and response: cellular mechanisms to organ function. Expert Opin Drug Metab Toxicol. 2011;7:803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tveita T, Sieck GC. The physiologic responses to epinephrine during cooling and after rewarming in vivo. Crit Care. 2011;15:R225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kunos G, Nickerson M. Effects of sympathetic innervation and temperature on the properties of rat heart adrenoceptors. Br J Pharmacol. 1977;59:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han YS, Tveita T, Kondratiev TV, Prakash YS, Sieck GC. Changes in cardiovascular beta‐adrenoceptor responses during hypothermia. Cryobiology. 2008;57:246–250. [DOI] [PubMed] [Google Scholar]
- 24. Hannon JP. Effect of temperature on the heart rate, electrocardiogram and certain myocardial oxidations of the rat. Circ Res. 1958;6:771–778. [DOI] [PubMed] [Google Scholar]
- 25. Kim SY, Raikoff K, Wülfert E, Hanin I. Bradycardia induced by mivazerol, a selective alpha 2‐adrenoceptor agonist, is amplified during mild hypothermia in the pentobarbital‐anesthetized rat. Cardiovasc Drugs Ther. 1997;11:751–755. [DOI] [PubMed] [Google Scholar]
- 26. Vogel Z, Barg J, Levy R, Saya D, Heldman E, Mechoulam R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem. 1993;61:352–355. [DOI] [PubMed] [Google Scholar]
- 27. Rapacciuolo A, Suvarna S, Barki‐Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. Protein kinase A and G protein‐coupled receptor kinase phosphorylation mediates beta‐1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–35411. [DOI] [PubMed] [Google Scholar]
- 28. Krismer AC, Lindner KH, Kornberger R, Wenzel V, Mueller G, Hund W, Oroszy S, Lurie KG, Mair P. Cardiopulmonary resuscitation during severe hypothermia in pigs: does epinephrine or vasopressin increase coronary perfusion pressure? Anesth Analg. 2000;90:69–73. [DOI] [PubMed] [Google Scholar]
- 29. Wira CR, Becker JU, Martin G, Donnino MW. Anti‐arrhythmic and vasopressor medications for the treatment of ventricular fibrillation in severe hypothermia: a systematic review of the literature. Resuscitation. 2008;78:21–29. [DOI] [PubMed] [Google Scholar]
- 30. Kohlhauer M, Darbera L, Lidouren F, Chenoune M, Ghaleh B, Vivien B, Carli P, Dabire H, Berdeaux A, Tissier R. Comparative effect of hypothermia and adrenaline during cardiopulmonary resuscitation in rabbits. Shock. 2014;41:154–158. [DOI] [PubMed] [Google Scholar]
- 31. Mustafa S, Thulesius O. Cooling‐induced carotid artery dilatation: an experimental study in isolated vessels. Stroke. 2002;33:256–260. [DOI] [PubMed] [Google Scholar]