

Abstract
Background
Cardiomyopathy is a major determinant of overall Fabry disease (FD) prognosis, with the worst outcomes in patients with myocardial fibrosis. Late gadolinium enhancement is currently the gold standard for evaluation of replacement myocardial fibrosis; however, this event is irreversible, thus identification of biomarkers of earlier diffuse fibrosis is paramount.
Methods and Results
Type I collagen synthesis and degradation biomarkers (PICP [carboxyterminal propeptide of procollagen type I], ICTP [carboxyterminal telopeptide of type I collagen], and MMP1 [matrix metalloproteinase 1] and MMP2) and markers of bone synthesis and degradation were evaluated (to adjust type I collagen metabolism to bone turnover) in FD patients and controls. FD patients were grouped by cardiomyopathy severity, according to echocardiogram: (1) normal, (2) tissue Doppler abnormalities, (3) left ventricular hypertrophy. A significant increase in PICP and a significant decrease in matrix metalloproteinases were observed in FD patients; even the group with normal echocardiogram had a significant increase in PICP. We also found a significant correlation between left ventricular mass and PICP (ρ=0.378, P=0.003) and MMP1 (ρ=−0.484, P<0.001). PICP (adjusted for bone turnover) was the better predictor of left ventricular mass in multivariable regression, and its diagnostic accuracy to predict late gadolinium enhancement was also significant.
Conclusions
Collagen type I synthesis is increased in FD cardiomyopathy, even in the earlier stages of the disease, and this profibrotic state has good predictive value for and is likely to be critical to the development of overt left ventricular hypertrophy. Moreover, inhibition of enzymes involved in collagen type I cleavage also seems crucial to myocardial collagen deposition.
Keywords: biomarkers, carboxyterminal propeptide of procollagen type I, cardiac fibrosis, Fabry disease cardiomyopathy, matrix metalloproteinases
Subject Categories: Biomarkers, Fibrosis, Cardiomyopathy
Clinical Perspective
What Is New?
Collagen type I synthesis is increased in Fabry disease cardiomyopathy, even in the early preclinical stages.
Inhibition of enzymes involved in collagen type I degradation also seems crucial for myocardial collagen type I deposition.
What Are the Clinical Implications?
Serum biomarkers of collagen type I metabolism may identify ongoing fibrosis in the early stages of Fabry disease cardiomyopathy and might predict the development of left ventricular hypertrophy.
Introduction
Fabry disease (FD) is an X‐linked lysosomal storage disorder caused by mutations in the GLA (galactosidase α) gene that encodes the enzyme α‐galactosidase A, resulting in diffuse lysosomal accumulation of neutral glycosphingolipids (mainly Gb3 [globotriaosylceramide]). Both classical and attenuated phenotypes are associated with significant cardiac involvement. In adulthood, involvement of the heart, kidney, and brain causes significant morbidity and premature death.1, 2 Most recent studies have shown cardiovascular disease as the main cause of death in FD and a major determinant of overall disease prognosis.3, 4 Arrhythmias are the most frequent cardiac event in FD,4, 5 with a recent study reporting the annual increase in cardiac fibrosis as the sole independent predictor of malignant ventricular arrhythmias.6
Late gadolinium enhancement (LGE) imaging techniques using cardiac magnetic resonance imaging (MRI) is the gold standard for noninvasive detection of focal replacement fibrosis in the myocardium. More than 50% of FD patients present with LGE, with characteristic midmyocardial distribution in the inferolateral basal or midbasal segments of the left ventricle wall that seems to be specific of FD cardiomyopathy.7, 8 Nevertheless, LGE has several limitations as an imaging biomarker: (1) It detects only irreversible tissue damage with focal replacement fibrosis and has limited resolution of ≈0.2g 9; (2) it may not detect early, potentially reversible, diffuse interstitial fibrosis10; (3) there is no universally accepted technique to quantify fibrosis volume.11 A novel technique, T1 mapping, has been studied for assessment of diffuse interstitial fibrosis.10 In FD, however, it has been studied more extensively as an imaging biomarker for early detection of cardiac involvement (due to lipid storage, yielding low native T1) than for evaluation of diffuse interstitial fibrosis.12, 13
Enzyme replacement therapy (ERT) is the standard of care in the treatment of FD; however, the benefits of ERT may be limited in patients with cardiac fibrosis14, 15 and are probably greater when administered early in the course of the disease. A study using tissue Doppler imaging to detect early cardiac involvement reported that ERT prevented the appearance of tissue Doppler abnormalities in FD patients with no left ventricular (LV) hypertrophy or tissue Doppler abnormalities at baseline.16
The presence of LGE on cardiac MRI is a late event and predicts a worse prognosis. Consequently, identifying early predictors of overt disease is clinically relevant. Echocardiography is currently the gold standard to assess early signs of cardiomyopathy, which is associated with diastolic dysfunction. Several studies have demonstrated that tissue Doppler abnormalities precede and correlate with LV hypertrophy progression.16, 17 Newer tools like speckle tracking, which allows measurement of myocardial systolic and diastolic strains, seem to be superior to conventional echocardiographic measurements (including tissue Doppler) for the identification of myocardial contraction and relaxation abnormalities.18, 19
These manifestations may be preceded by histologic changes or deregulated gene expression, as demonstrated previously in hypertrophic cardiomyopathy (HCM) due to sarcomere protein gene mutations.20, 21 These results indicate that a profibrotic milieu, with extracellular matrix expansion and collagen deposition, is present early in the pathogenesis of the disease, even when cardiac function and histology are normal.
The identification of biomarkers of collagen synthesis and degradation could represent an advance in the identification of preclinical involvement of the heart in FD, with possible therapeutic implications. Type I collagen is the main collagen type of the myocardium. During its synthesis from its precursor, procollagen type 1, PICP (carboxyterminal propeptide of procollagen type I) is released into the bloodstream with a stoichiometric ratio of 1:1, and its serum level reliably reflects myocardial type I collagen synthesis.22, 23 Collagen turnover biomarkers have been studied in HCM, hypertension, heart failure, and myocardial infarction.24, 25, 26 In the HCM model, an increase in serum PICP was reported even in mutation carriers without LV hypertrophy or visible fibrosis in cardiac MRI.26 In FD cardiomyopathy, to our knowledge, there are only 2 publications of a limited evaluation of extracellular matrix turnover, reporting increased levels of MMP9 (matrix metalloproteinase 9), PICP, ICTP (carboxyterminal telopeptide of type I collagen), and PIIINP (procollagen type III aminoterminal propeptide).6, 27
In this study, we investigated several biomarkers of collagen type I turnover in a large cohort of FD patients (within the entire spectrum of FD cardiomyopathy severity), with an emphasis on the usefulness of these markers in early and prehypertrophic stages of FD cardiomyopathy.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. See Data S1 for extended methods.
Study Design and Population
In this multicenter, cross‐sectional, and prospective study, a cohort of 60 consecutive FD patients was recruited from 3 centers (Centro Hospitalar Lisboa Norte, Lisbon, Portugal; Hospital Senhora da Oliveira, Guimarães, Portugal; Royal Free Hospital, London, United Kingdom) between February 2013 and June 2014. Twenty healthy controls were also recruited.
For FD patients, the only inclusion criteria were diagnosis of FD and age ≥18 years. FD was defined in male patients as low α‐galactosidase A activity and/or identification of a proven pathogenic mutation in the GLA gene and in female patients as the presence of a proven pathogenic mutation in the GLA gene.
FD patients with conditions, other than the usual manifestations of FD cardiomyopathy, that possibly affected cardiac collagen turnover were excluded, namely, HCM due to sarcomere protein gene mutations or other cardiomyopathies, previous myocardial infarction, moderate or severe valvular heart disease, previous heart surgery, a cardiac device implanted in the previous 6 months, surgery or major trauma within the previous 6 months, inflammatory or fibrotic diseases, and active cancer.
To enroll patients who were representative of the entire spectrum of FD cardiomyopathy severity, recruitment was done in accordance with subgroups of increasing severity of FD cardiomyopathy (aiming at a specific number of patients), defined by echocardiogram. Subgroup 1 had no evidence of cardiac involvement: no LV hypertrophy or tissue Doppler abnormalities (20 patients). Subgroup 2 had tissue Doppler abnormalities (defined as at least 1 of the following: systolic tissue Doppler velocities <6 cm/s; early diastolic tissue Doppler velocities <10, <8, or <6 cm/s at the septal corner of the mitral annulus in patients aged <40, between 41 and 60, and >60 years, respectively; early diastolic tissue Doppler velocities <14, <12, or <6 cm/s at the lateral corner of the mitral annulus in patients aged <40, between 41 and 60, and >60 years, respectively) and no LV hypertrophy (20 patients). Subgroup 3 had LV hypertrophy, defined as diastolic interventricular septum or posterior wall thickness ≥12 mm (20 patients).
The control group included healthy individuals with normal echocardiograms who were age and sex matched with the less severe FD subgroup (subgroup 1) and who did not have conditions influencing cardiac collagen turnover, including not only those previously listed for FD patients but also systemic arterial hypertension (defined as systolic blood pressure ≥140 mm Hg, diastolic blood pressure ≥90 mm Hg, or use of antihypertensive medication), LV hypertrophy from any cause, coronary artery disease, pacemaker placement (regardless of time since implantation), and atrial fibrillation.
The study protocol was approved by the local or national ethics committees of each participating center, and the study was conducted in accordance with this protocol and the ethics principles of the Declaration of Helsinki. Written informed consent was obtained from all participants before enrollment.
Clinical Assessment
For each patient recruited, routine follow‐up data were collected, namely sex, age, age at diagnosis, plasma α‐galactosidase A activity, GLA gene mutation, current medication (angiotensin‐converting enzyme inhibitors, angiotensin II receptor blockers, aldosterone antagonists, and β‐blockers), data about ERT (product, dose, and duration), clinical manifestations (to calculate the disease severity indexes: Mainz Score Severity Index [MSSI]28 and Fabry International Prognostic Index [FIPI]29), echocardiographic measurements, presence of LGE on cardiac MRI (if available), and laboratory results (NT‐proBNP [amino‐terminal fragment of the pro‐hormone of brain natriuretic peptide], kidney function tests [glomerular filtration rate, creatinine, and albuminuria] and plasma lyso‐Gb3 [globotriaosylsphingosine]).
Cardiomyopathy assessment/reference test and outcomes
Cardiac function and structure were evaluated by echocardiogram and cardiac MRI. Echocardiogram (LV mass and tissue Doppler abnormalities) was used as the reference test for comparison with index tests because it is considered the gold standard for evaluation of early cardiac dysfunction in FD.
Data from the M‐mode, 2‐dimensional, and Doppler transthoracic echocardiographic study were collected for each patient and control. FD patients’ echocardiograms were done using a Vivid 7 (General Electric) ultrasonographic system at all recruitment sites. Evaluation of control participants was performed using a Vivid 7 or Toshiba Xario ultrasonographic systems, with a protocol identical to that for FD patients for imaging acquisition. Echocardiograms were undertaken in routine clinical practice, and there was no core reading of the results.
LV dimensions (interventricular septum, posterior wall, and LV end‐diastolic diameter) were assessed from the long‐axis view, and ventricular mass was calculated according to the Devereux formula and normalized for height (g/m2.7). LV hypertrophy was defined as LV mass ≥50 g/m2.7. Left atrium area was obtained in the apical 4‐chamber view and expressed in square centimeters.
Mitral inflow pattern (rapid filling [E wave] and atrial contraction [A wave] peak velocities, E‐wave decelerating time, A‐wave duration, and isovolumic relaxation time) was obtained at the mitral level by Doppler echocardiography in the apical 4‐chamber view. Retrograde atrial flow velocity and duration were acquired from the pulmonary veins also by Doppler echocardiography in the same view.
Myocardial velocities during systole and early diastole were collected from tissue Doppler imaging at lateral and septal corner of the mitral annulus in the apical 4‐chamber view. Tissue Doppler abnormalities were defined as aforementioned. The ratio between LV rapid filling and average early diastole (septal and lateral) was used to estimate LV end‐diastolic pressure.
Echocardiographic studies were performed and reported by people who were blinded to the measurement of collagen turnover biomarkers (index tests) and clinical data.
Data about cardiac MRI were collected if it was performed as part of the follow‐up protocol and there was no core reading of the images. Cardiac MRI studies were performed in a 3.0‐T system (Philips Intera). LGE is the standard for detection of focal myocardial replacement fibrosis and was defined in the study as 2 SD above the mean signal intensity of the distant myocardium.
The specified outcomes were to compare the index tests with tissue Doppler abnormalities in the identification of incipient Fabry cardiomyopathy (comparing patients in subgroup 1 with controls) and to correlate index tests with LV mass and their diagnostic accuracy to identify LGE in cardiac MRI.
Measurement of Collagen Turnover Biomarkers and Index Tests
To assess type I collagen turnover, we measured levels of peptides released during collagen synthesis and degradation and enzymes involved in collagen degradation. During collagen synthesis, PICP is cleaved from procollagen I and released into blood. Collagen is degraded by matrix metalloproteinases (MMPs) as follows: MMP1 cleaves collagen I and releases ICTP into the blood; further degradation is performed by gelatinases MMP2 and MMP9.
Type I collagen is also a major component of bone; so the measured pro‐ and telopeptides may reflect bone formation and resorption. To minimize such confounding factors, we also measured markers of bone synthesis (B‐AP [bone‐specific alkaline phosphatase]) and resorption (TRAP5b [tartrate‐resistant acid phosphatase type 5b]) and determined the ratios of PICP:B‐AP and ICTP:TRAP5b.
Peripheral venous blood samples were obtained <6 months apart from the echocardiographic study, spun within 120 minutes after phlebotomy (489×g, 10 minutes), immediately stored at −20°C, transported to the laboratory in dry ice, and thawed and mixed thoroughly just before the assay.
TRAP5b was evaluated by an ELISA method, according to manufacturer instructions (Immunodiagnostic Systems), and measured at 405 nm; MMP1 and MMP2 were evaluated by an ELISA method, according to manufacturer instructions (SunRed), and measured at 450 nm; B‐AP and PICP were evaluated by an ELISA method, according to manufacturer instructions (Quidel Corp), and measured at 405 nm; ICTP was evaluated by an electrochemiluminescence immunoassay in a COBAS e411 instrument (Roche). Duplicate determinations were made for each individual, and the average result was considered. Laboratory researchers were blinded to clinical data and cardiac assessments (reference test).
Because there are no validated reference values for the tested biomarkers, the upper limit of the 95% confidence interval for the mean of the control group was assumed to be the upper limit of the reference value.
Statistical Analysis
Statistical analysis was performed with SPSS (version 21; IBM Corp) software. Categorical variables were expressed as number and percentage and continuous variables as median and interquartile range.
For categorical variables, the χ2 or Fisher exact tests were used to compare the variable distributions between the groups. Normal distribution of continuous variables was tested using the Shapiro–Wilk test. For continuous variables, comparison of means or medians was performed using the Student t test for variables that followed a normal distribution and the Mann–Whitney test or related‐samples Wilcoxon signed rank test for variables that did not. If the qualitative variable had >2 categories, ANOVA (post hoc analysis with Bonferroni correction) was used for variables with normal distribution, and a Kruskal–Wallis test was used for those without.
Because of the skewed distribution of all studied biomarkers (index and reference tests), the Spearman correlation coefficient was determined to evaluate the correlation between the biomarkers and the quantitative variables.
To construct a regression model to quantify LV mass, all potentially related variables were correlated, using the Spearman rank correlation coefficient, with LV mass (in univariate analysis). Those variables showing significant correlation (P<0.05) were entered into the multivariable analysis. For multivariable analysis, stepwise regression combining both forward selection and backward elimination was used. A cutoff limit to remain in the model was set at an F‐statistic P value of <0.05.
We evaluated the diagnostic accuracy of the studied biomarkers to detect cardiac fibrosis (defined by presence of LGE in cardiac MRI) by calculating the sensitivity, specificity, and area under the curve from receiver operating characteristic curves (we determined the confidence intervals of the area under the curve [nonparametric method], with values between 1 [perfect test] and 0.5 [useless test]).
For all comparisons and correlations, P<0.05 was considered significant.
Results
Population Characteristics
From February 2013 to June 2014, we recruited 60 FD patients (20 in each subgroup) and 20 controls (age and sex matched with subgroup 1; Figure 1). Population characteristics are given in Table 1. Only 1 FD patient was taking a mineralocorticoid receptor antagonist (in subgroup 3). Overall, 28 different GLA pathogenic mutations were identified (Table S1), with p.N215S and p.F113L—2 mutations associated with attenuated or late‐onset phenotypes with predominant cardiac involvement—accounting for 31.7% of the patients; the remaining mutations are usually associated with a classical phenotype. As expected, no adverse events resulted from collecting blood samples to determine index tests.
Figure 1.
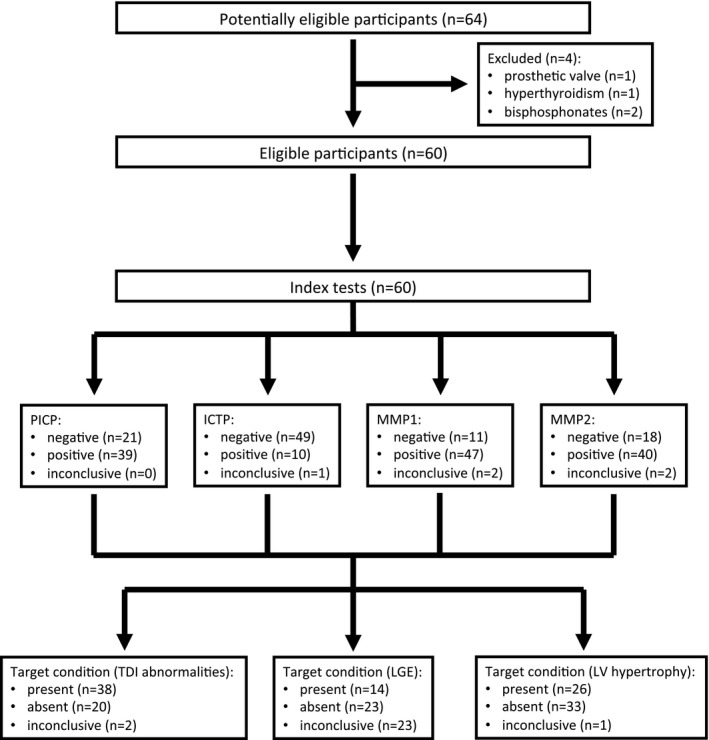
Study flow diagram. ICTP indicates carboxyterminal telopeptide of type I collagen; LGE, late gadolinium enhancement; LV, left ventricular; MMP, matrix metalloproteinase; PICP, carboxyterminal propeptide of procollagen type I; TDI, tissue Doppler imaging.
Table 1.
Clinical Characteristics of Study Population
| Controls (n=20) | FD Cohort (n=60) | FD Subgroups | |||
|---|---|---|---|---|---|
| 1 (n=20) | 2 (n=20) | 3 (n=20) | |||
| Categorical variables, n (%) | |||||
| Sex (female)a | 14 (70.0) | 37 (61.7) | 14 (70.0) | 18 (90.0) | 5 (25.0) |
| HBP (yes) | 0 (0.0) | 25 (41.7) | 8 (40.0) | 10 (50.0) | 7 (35.0) |
| HF class (0/I/II) | ··· | 39/6/14 | 15/3/2 | 14/2/4 | 10/2/8 |
| ERT (yes) | ··· | 39 (65.0) | 14 (70.0) | 8 (40.0) | 17 (85.0) |
| ACEI/ARB (yes) | 0 (0.0) | 29 (48.3) | 9 (45.0) | 10 (50.0) | 10 (50.0) |
| β‐Blockers (yes) | 0 (0.0) | 11 (18.3) | 3 (15.0) | 3 (15.0) | 5 (25.0) |
| Continuous variable, median (IQR) | |||||
| Age, yb | 41.0 (23.0) | 44.0 (23.0) | 40.5 (21.0) | 48.5 (18.0) | 59.5 (13.0) |
| Age at diagnosis, yc | ··· | 41.5 (22.0) | 34.5 (24.0) | 44.0 (15.0) | 50.5 (35.0) |
| Age at ERT initiation, yd | ··· | 49.0 (17.1) | 38.1 (25.4) | 52.4 (17.0) | 49.4 (19.9) |
| Time in ERT, y | ··· | 6.3 (8.4) | 4.8 (8.0) | 5.9 (7.6) | 8.2 (9.1) |
| MSSIe | ··· | 18.5 (21.0) | 11.0 (11.0) | 12.5 (18.0) | 31.0 (16.0) |
| FIPIe | ··· | 2.0 (3.0) | 1.0 (2.0) | 1.0 (3.0) | 4.0 (2.0) |
| Plasma lyso‐Gb3, nmol/Lf | ··· | 9.1 (16.6) | 3.6 (27.3) | 8.3 (9.9) | 18.8 (45.1) |
| Plasma α‐gal A (female), nmol/h/mL | ··· | 4.3 (4.1) | 5.1 (2.8) | 3.5 (4.5) | 3.8 (.) |
| Plasma α‐gal A (male), nmol/h/mL | ··· | 0.11 (0.3) | 0.14 (0.50) | 0.06 (.) | 0.20 (0.30) |
| NT‐proBNP, ng/mLb | ··· | 142.0 (334.0) | 40.0 (122.0) | 129.5 (166.0) | 940.7 (1602) |
| eGFR, mL/min/1.73 m2 b | 108.5 (20.0) | 93.0 (51.0) | 99.5 (53.0) | 101.0 (91.0) | 67.5 (51.0) |
ACEI indicates angiotensin‐converting enzyme inhibitors; α‐gal A, α‐galactosidase A; ARB, angiotensin II receptor blockers; eGFR, estimated glomerular filtration rate; ERT, enzyme replacement therapy; FD, Fabry disease; FIPI, Fabry International Prognostic Index; HBP, high blood pressure (arterial hypertension); HF, heart failure; IQR, interquartile range; lyso‐Gb3, globotriaosylsphingosine; MSSI, Mainz Severity Score Index; NT‐proBNP, N‐terminal probrain natriuretic peptide.
P<0.01 for difference in the distribution of categorical variables between subgroups.
P<0.01 for difference between subgroups 1 and 3.
P<0.05 for difference between subgroups 1 and 2.
P<0.05 for difference between subgroups 1 and 3.
P<0.01 for difference between group 3 and all other groups.
P<0.05 for difference between subgroups 2 and 3.
No significant echocardiographic differences were noted between the control group and FD subgroup 1 (Table 2). A gadolinium‐based contrast cardiac MRI result was available for 37 FD patients (60%; 9, 13, and 15 in subgroups 1, 2, and 3, respectively); LGE was present in 0%, 15.4%, and 80.0% of patients in FD subgroups 1, 2, and 3, respectively.
Table 2.
Echocardiographic Characteristics of Study Population
| Controls | FD Subgroups | |||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| dIVS, mma | 9.0 (2.0) | 9.0 (2.0) | 9.0 (2.2) | 14.5 (4.0) |
| dLVPW, mma | 9.0 (1.0) | 9.0 (3.0) | 8.9 (2.8) | 12.1 (4.5) |
| LVMi, g/m2.7 a | 39.3 (10.9) | 35.2 (9.7) | 39.5 (19.6) | 63.3 (10.6) |
| LA area, cm2 a | ··· | 17.0 (4.6) | 18.9 (5.6) | 23.3 (10.2) |
| E/A ratio | ··· | 1.47 (0.85) | 1.25 (0.87) | 0.98 (0.95) |
| DT, msb | ··· | 184.0 (73.0) | 230.0 (62.0) | 249.5 (25.0) |
| S′(s), cm/sc | 8.0 (2.0) | 8.0 (2.0) | 6.0 (2.0) | 6.0 (2.0) |
| S′(l), cm/sc | 9.5 (5.0) | 11.0 (3.0) | 7.0 (3.0) | 6.5 (3.0) |
| E′(s), cm/sd | 11.5 (5.0) | 11.5 (3.0) | 7.0 (4.0) | 5.0 (2.0) |
| E′(l), cm/sc | 15.5 (6.0) | 14.5 (4.0) | 9.0 (4.0) | 8.0 (4.0) |
| E/E′c | 6.3 (2.1) | 6.0 (2.3) | 9.4 (5.0) | 11.8 (6.5) |
Data are shown as median (interquartile range). dIVS indicates diastolic interventricular septal thickness; dLVPW, diastolic left ventricular posterior wall thickness; DT, E‐wave decelerating time; E/A, left ventricular rapid filling (E wave)/atrial contraction (A wave) peak velocities; E/E′(a), left ventricular rapid filling velocity/average (septal and lateral) early diastolic myocardial velocity measured at the mitral annulus; E′(l), early diastolic myocardial velocity measured at the lateral corner of the mitral annulus; E′(s), early diastolic myocardial velocity measured at the septal corner of the mitral annulus; LA, left atrium; LVMi, left ventricular mass indexed for height; S′(l), systolic myocardial velocity measured at the lateral corner of the mitral annulus; S′(s), systolic myocardial velocity measured at the septal corner of the mitral annulus.
P<0.01 for difference between subgroup 3 and all other FD groups.
P<0.01 for difference between subgroups 1 and 3.
P<0.01 for difference between subgroup 1 and all other FD groups.
P<0.01 for difference between all FD groups.
Collagen Type I Turnover Biomarkers (Index Tests)
Collagen type I synthesis (PICP) was increased in FD patients (Table 3). Compared with PICP levels in controls, PICP levels in FD subgroup 1 were significantly elevated, a 61% increase (P=0.006). Comparing FD subgroups, PICP levels were significantly higher in FD subgroup 3 (P=0.001). The significant results comparing FD subgroup 1 and controls and FD subgroups remained with the PICP:B‐AP ratio.
Table 3.
Collagen Type I Turnover Biomarkers in Controls and FD Subgroups
| Controls (n=20) | FD Subgroups | P Valuea | P Valueb | |||
|---|---|---|---|---|---|---|
| 1 (n=20) | 2 (n=20) | 3 (n=20) | ||||
| PICP, ng/mL | 107.3 (39.3) | 148.5 (129.0) | 115.7 (88.8) | 219.0 (98.4) | 0.006 | 0.001 |
| PICP/B‐AP | 10.1 (4.3) | 16.2 (10.3) | 12.7 (9.1) | 19.4 (24.1) | 0.003 | 0.001 |
| ICTP, ng/mL | 0.34 (0.20) | 0.25 (0.27) | 0.20 (0.19) | 0.29 (0.23) | 0.687 | 0.236 |
| ICTP/TRAP5b | 0.11 (0.07) | 0.16 (0.10) | 0.11 (0.07) | 0.13 (0.06) | 0.011 | 0.186 |
| MMP1, ng/mL | 16.8 (26.3) | 13.5 (19.3) | 14.1 (4.3) | 9.6 (4.6) | 0.159 | 0.002 |
| MMP2, ng/mL | 440.3 (541.0) | 341.8 (849.5) | 356.5 (187.0) | 318.7 (101.9) | 0.872 | 0.108 |
Data are shown as median (interquartile range). B‐AP indicates bone‐specific alkaline phosphatase; FD, Fabry disease; ICTP, carboxyterminal telopeptide of type I collagen; MMP, matrix metalloproteinase; PICP, carboxyterminal propeptide of procollagen type I; TRAP5b, tartrate‐resistant acid phosphatase type 5b.
Controls vs FD disease subgroup 1.
Between FD subgroups.
Collagen type I degradation (ICTP) and levels of the enzymes involved in its degradation (MMP1 and MMP2) were similar between the control group and FD subgroup 1 (Table 3). The difference between these 2 groups attained statistical significance only after adjustment to bone degradation with the ICTP:TRAP5b ratio (P=0.011). Moreover, there was a significant trend of decreased activity of MMP1 as severity increased.
The PICP:ICTP ratio is considered to reflect the balance between type I collagen synthesis and degradation. PICP:ICTP evaluation reveals a clear trend (significant only after adjustment for bone turnover [P=0.012]) of prevalent type I collagen synthesis over degradation within the subgroups of FD patients with greater severity (Figure S1). Correlation between PICP:ICTP and MMP1 reflects the influence of higher collagenase activity balancing PICP:ICTP toward type I collagen degradation and lesser collagen type I deposition. Accordingly, we identified a significant inverse correlation between PICP:ICTP and MMP1 activity before (ρ=−0.348; P=0.008) and after (ρ=−0.322; P=0.015) adjustment to bone turnover.
Correlation With Cardiac Imaging and LGE (Reference Standards)
For the entire FD cohort, a significant direct correlation between the biomarker of collagen type I synthesis (PICP) and LV wall thickness and mass (Table 4) was found (PICP with LV mass: ρ=0.378, P=0.003). This significant correlation remained after adjustment for bone turnover (PICP:B‐AP ratio with LV mass: ρ=0.313, P=0.016). For biomarkers of collagen type I degradation, there was a significant inverse correlation between the enzyme MMP1, involved collagen type I cleavage, and LV thickness and mass parameters (MMP1 with LV mass: ρ=−0.484, P<0.001). A significant direct correlation was found for the MMP1 and echocardiographic parameters of diastolic dysfunction, namely, early diastolic mitral velocities measured at the septal (ρ=0.354, P=0.016) and lateral (ρ=0.280, P=0.042) corners of the mitral annulus.
Table 4.
Correlation Between Type I Collagen Turnover Biomarkers and Cardiac Imaging Parameters
| PICP (ng/mL) | PICP/B‐AP | ICTP (ng/mL) | ICTP/TRAP5b | MMP1 (ng/mL) | MMP2 (ng/mL) | |
|---|---|---|---|---|---|---|
| Echocardiogram | ||||||
| dIVS, mm | 0.438a | 0.378a | 0.178 | −0.017 | −0.519a | −0.300a |
| dLVPW, mm | 0.322a | 0.234 | 0.204 | 0.043 | −0.438a | −0.294a |
| LVMi, g/m2.7 | 0.378a | 0.313a | 0.197 | 0.013 | −0.484a | −0.235 |
| LA area, cm2 | 0.173 | 0.066 | 0.092 | 0.080 | −0.129 | −0.239 |
| DT, ms | 0.103 | 0.059 | −0.024 | −0.187 | −0.294a | −0.145 |
| S′(s), cm/s | −0.023 | 0.071 | 0.020 | −0.046 | 0.354a | 0.188 |
| S′(l), cm/s | −0.029 | −0.017 | −0.063 | 0.083 | 0.230 | 0.127 |
| E′(s), cm/s | −0.125 | −0.113 | 0.130 | 0.139 | 0.354a | 0.288 |
| E′(l), cm/s | −0.037 | 0.000 | 0.127 | 0.234 | 0.280a | 0.200 |
| E/E′(a) | 0.105 | 0.005 | 0.027 | −0.082 | −0.097 | 0.087 |
| Cardiac MRI | ||||||
| LGE (yes) | 88.8a | 12.5a | 0.066 | −0.024 | −5.27 | −239.3 |
Data are shown as mean difference for categorical variables and as ρ for continuous variables. B‐AP indicates bone‐specific alkaline phosphatase; dIVS, diastolic interventricular septal thickness; dLVPW, diastolic left ventricular posterior wall thickness; DT, E‐wave decelerating time; E/E′(a), left ventricular rapid filling velocity/average (septal and lateral) early diastolic myocardial velocity measured at the mitral annulus; E′(l), early diastolic myocardial velocity measured at the lateral corner of the mitral annulus; E′(s), early diastolic myocardial velocity measured at the septal corner of the mitral annulus; ICTP, carboxyterminal telopeptide of type I collagen; LA, left atrium; LGE, late gadolinium enhancement; LVMi, left ventricular mass indexed for height; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; PICP, carboxyterminal propeptide of procollagen type I; S′(l), systolic myocardial velocity measured at the lateral corner of the mitral annulus; S′(s), systolic myocardial velocity measured at the septal corner of the mitral annulus; TRAP5b, tartrate‐resistant acid phosphatase type 5b.
P<0.05.
There was a clear trend for higher values of collagen type I synthesis and decreased activity of the enzymes involved in collagen type I cleavage in patients with LGE in cardiac MRI (Table 4), but the difference between patients with and without LGE was significant only for PICP, even after adjustment for bone turnover (for PICP:B‐AP ratio: mean difference 12.5, a 74% increase for LGE positive group; P=0.01).
After adjustment for bone turnover, there was a significant correlation between PICP:ICTP and LV mass and higher values of this ratio in LGE positive patients (Figure S2).
Correlation With ERT, Severity Indexes, and Other Variables
For the rest of the variables (including demographic, diagnostic, and severity‐ and treatment‐related variables), not directly related to cardiac imaging, consistent and significant correlations were found among the biomarker of collagen type I synthesis or MMP1 and the disease severity indexes (MSSI and FIPI) and NT‐pro BNP (Table 5). Correlations among other biomarkers and these variables seemed less reliable, except for the correlations between age or estimated glomerular filtration rate and MMPs; however, the significant correlation with estimated glomerular filtration rate did not remain after adjustment for LV mass.
Table 5.
Influence of Other Variables in Biomarkers of Collagen Type I Turnover
| PICP (ng/mL) | PICP/B‐AP | ICTP (ng/mL) | ICTP/TRAP5b | MMP1 (ng/mL) | MMP2 (ng/mL) | |
|---|---|---|---|---|---|---|
| Demographic | ||||||
| Sex (female) | −48.9a | −7.5a | −0.11a | −0.02 | 5.7 | −55.6 |
| Age, y | 0.077 | 0.071 | −0.153 | −0.175 | −0.334a | −0.277a |
| Diagnosis | ||||||
| Age at diagnosis, y | −0.059 | 0.009 | −0.142 | −0.100 | −0.308a | −0.309a |
| Plasma α‐gal A (female), nmol/h/mL | −0.318 | −0.444a | 0.028 | 0.029 | 0.071 | −0.017 |
| Plasma α‐gal A (male), nmol/h/mL | 0.120 | 0.145 | 0.012 | 0.035 | −0.121 | −0.055 |
| Disease severity | ||||||
| MSSI | 0.394a | 0.287a | 0.194 | 0.189 | −0.392a | −0.222 |
| FIPI | 0.258a | 0.141 | 0.158 | 0.043 | −0.374a | −0.211 |
| Plasma lyso‐Gb3 (female), nmol/L | 0.402a | 0.463a | 0.225 | 0.255 | −0.123 | 0.094 |
| Plasma lyso‐Gb3 (male), nmol/L | 0.240 | 0.095 | 0.171 | 0.308 | 0.037 | 0.266 |
| NT‐proBNP, ng/mL | 0.289 | 0.489a | −0.163 | −0.084 | −0.533a | −0.262 |
| eGFR, mL/min/1.73 m2 | −0.122 | −0.081 | −0.060 | −0.059 | 0.289a | 0.262a |
| Treatment | ||||||
| ERT (yes) | 13.6 | 3.8 | 0.03 | −0.01 | −1.0 | −56.3 |
| Age at ERT initiation, y | −0.187 | −0.139 | −0.217 | −0.218 | −0.217 | −0.220 |
| Time in ERT, y | 0.204 | 0.110 | −0.213 | −0.288 | −0.014 | 0.146 |
| ACEI/ARB (yes) | −27.0 | −1.8 | −0.03 | −0.04 | −8.2 | −173.4 |
| β‐Blockers (yes) | 6.3 | 0.2 | −0.03 | −0.00 | 4.7 | 67.5 |
Data are shown as mean difference for categorical variables and as ρ for continuous variables. ACEI indicates angiotensin‐converting enzyme inhibitors; α‐gal A, α‐galactosidase A; ARB, angiotensin II receptor blockers; B‐AP, bone‐specific alkaline phosphatase; eGFR, estimated glomerular filtration rate; ERT, enzyme replacement therapy; FIPI, Fabry International Prognostic Index; ICTP, carboxyterminal telopeptide of type I collagen; lyso‐Gb3, globotriaosylsphingosine; MMP, matrix metalloproteinase; MSSI, Mainz Severity Score Index; NT‐proBNP, N‐terminal probrain natriuretic peptide; PICP, carboxyterminal propeptide of procollagen type I; TRAP5b, tartrate‐resistant acid phosphatase type 5b.
P<0.05.
The serum concentration of PICP and PICP adjusted for bone turnover was significantly higher in male patients, and there was a nonsignificant trend of lower serum MMP1 concentration in the same sex. This is in agreement with the unbalanced sex distribution across FD patient subgroups and the significantly higher mean LV mass in male patients (57.6 g/m2.7 versus 41.4 g/m2.7; P<0.001). Nonetheless, the significant direct correlation between LV mass and PICP and the inverse correlation between LV mass and MMP1 remained significant for both sexes in the subanalysis by sex.
Association Between Predictive Variables and Clinical End Points (LV Mass and LGE)
Variables with significant correlation with LV mass (in univariate analysis) were included in a multivariable regression model (Table 6). Within this model only 2 variables retained statistical significance: PICP:B‐AP ratio and age, with the former as the better predictor of LV mass (β=0.919; SE=0.095; P<0.001).
Table 6.
Predictive Model of LV Mass by Univariate and Multivariable Regression Analysis
| LV Mass (Univariate) | LV Mass (Multivariable) | |||
|---|---|---|---|---|
| R | P Value | β | P Value | |
| PICP | 0.413 | 0.001 | 0.081 | 0.418 |
| PICP/B‐AP | 0.510 | <0.001 | 0.919 | <0.001 |
| ICTP | 0.170 | 0.202 | ||
| ICTP/TRAP5b | 0.016 | 0.906 | ||
| MMP1 | −0.339 | 0.010 | −0.066 | 0.646 |
| MMP2 | −0.285 | 0.032 | 0.016 | 0.910 |
| PICP/ICTP | 0.217 | 0.102 | ||
| PICP/ICTP adjusted | 0.379 | 0.004 | 0.035 | 0.751 |
| Age | 0.486 | <0.001 | 0.392 | 0.010 |
| Age at diagnosis | 0.274 | 0.035 | 0.066 | 0.644 |
| Age at ERT initiation | 0.310 | 0.058 | ||
| Time in ERT | 0.320 | 0.050 | −0.094 | 0.288 |
| Plasma α‐gal A (female) | −0.027 | 0.906 | ||
| Plasma α‐gal A (male) | −0.004 | 0.987 | ||
| MSSI | 0.631 | <0.001 | 0.041 | 0.686 |
| FIPI | 0.658 | <0.001 | −0.149 | 0.417 |
| Plasma lyso‐Gb3 | 0.409 | 0.009 | −0.012 | 0.898 |
| NT‐proBNP | 0.414 | 0.040 | 0.028 | 0.759 |
| eGFR | −0.388 | 0.003 | −0.057 | 0.645 |
α‐gal A indicates α‐galactosidase A; B‐AP, bone‐specific alkaline phosphatase; eGFR, estimated glomerular filtration rate; FIPI, Fabry International Prognostic Index; ICTP, carboxyterminal telopeptide of type I collagen; lyso‐Gb3, globotriaosylsphingosine; LV, left ventricular; MMP, matrix metalloproteinase; MSSI, Mainz Severity Score Index; NT‐proBNP, N‐terminal probrain natriuretic peptide; PICP, carboxyterminal propeptide of procollagen type I; TRAP5b, tartrate‐resistant acid phosphatase type 5b.
The global diagnostic accuracy of several biomarkers to detect patients with LGE in cardiac MRI was significant for LV mass, PICP, PICP:B‐AP ratio, PICP:ICTP ratio adjusted to bone turnover, and NT‐proBNP (Figure 2), achieving diagnostic accuracy of 75.9% for the PICP:ICTP adjusted ratio. Moreover, lower estimated glomerular filtration rate, systolic average, and early diastolic average had significant predictive value for presence of LGE in cardiac MRI.
Figure 2.
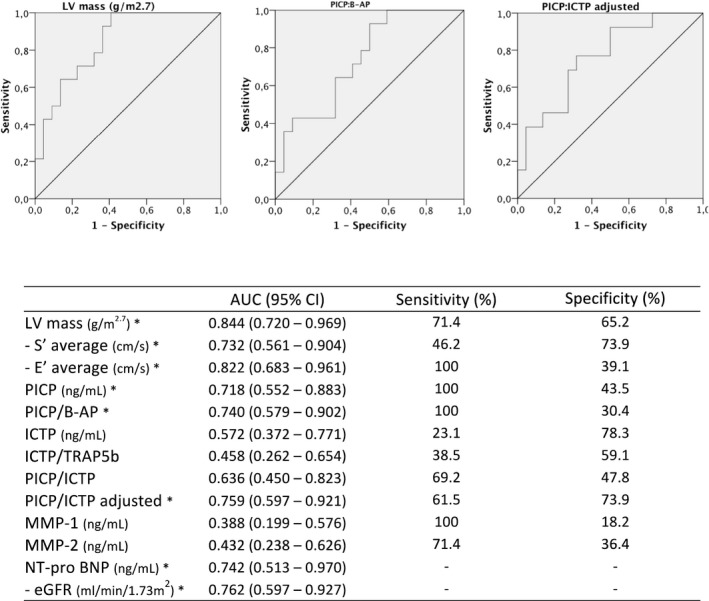
Diagnostic accuracy of several biomarkers for presence of LGE in cardiac MRI. Minus S′ average, E′ average, and eGFR have been used to identify the presence of LGE; lower values of these variables are associated with the presence of LGE. *P<0.05 for true area=0.5. B‐AP indicates bone‐specific alkaline phosphatase; E′, early diastolic myocardial velocity measured at the mitral annulus; eGFR, estimated glomerular filtration rate; ICTP, carboxyterminal telopeptide of type I collagen; LV, left ventricular; MMP, matrix metalloproteinase; NT‐proBNP, N‐terminal probrain natriuretic peptide; PICP, carboxyterminal propeptide of procollagen type I; S′, systolic myocardial velocity measured at the mitral annulus; TRAP5b, tartrate‐resistant acid phosphatase type 5b.
Using a model of binary logistic regression for the diagnosis of LGE, a model with LV mass and NT‐proBNP correctly classified 73.9% of patients regarding LGE status; this percentage increased to 87.0% after addition of the PICP:ICTP adjusted ratio to the model.
Discussion
Increased Collagen Type I Synthesis
The results of our study suggest increased myocardial collagen type I synthesis in FD patients with established LV hypertrophy and, to a lesser extent, in FD patients with normal echocardiograms or with isolated tissue Doppler abnormalities. PICP increase remained after adjustment for bone synthesis (with the ratio to B‐AP), confirming the myocardial origin of the excessive collagen type I synthesis (rather than higher bone metabolic activity). This correction for bone activity is important mainly in postmenopausal women because the concentration of collagen type I synthesis and degradation biomarkers is increased in this population.30
Serum PICP concentration was slightly, but nonsignificantly, lower in patients with tissue Doppler abnormalities (subgroup 2) compared with patients with normal echocardiograms (subgroup 1). We attributed this to the similar LV masses in these subgroups because there was a significant direct correlation between LV mass and serum PICP.
Elevation of PICP levels in FD patients with normal echocardiograms suggests that it may be used as a serologic marker of risk, detectable before the earlier echocardiographic signs of cardiac dysfunction. The ongoing longitudinal evaluation of these biomarkers will help clarify this issue. Moreover, it hints that the stimulus for myocardial fibrosis is an early event in FD cardiomyopathy (probably directly related to the disease pathophysiology) and not a secondary event caused by mechanical stress or ischemia at microcirculatory level. Similar to our study, in HCM it has been shown that serum levels of PICP are elevated even in mutation carriers without LV hypertrophy, supporting a profibrotic state preceding development of overt cardiomyopathy.26 Moreover, a histologic study in cats with preclinical HCM showed an increase in myocardial collagen deposition.31 Furthermore, studies in murine models of HCM have shown upregulation of several extracellular matrix proteins (CTGF [connective tissue growth factor] and POSTN [periostin]) in the very early stage of the disease (prehypertrophic stage, with normal histologic findings).20, 21
In FD, one of the accumulating substrates is lyso‐Gb3, a product of Gb3 deacylation. Lyso‐Gb3 promotes proliferation of vascular smooth muscle cells in culture32 and increases the expression of TGF‐β (transforming growth factor β) in human cultured podocytes, with subsequent increase in extracellular matrix synthesis.33 These findings support our observations that upregulation of cardiac fibrogenesis is an early event and directly related to the disease pathophysiology.
Type I Collagen Degradation: MMP Downregulation
In contrast, we found no increase in collagen type I degradation biomarker (ICTP), even after adjustment for bone turnover. Nonetheless, we found a significant decrease in the activity of MMP1 across the subgroups of increasing severity of FD cardiomyopathy. As an index of degree of coupling between the synthesis and degradation of collagen type I, the PICP:ICTP ratio, after adjustment to bone turnover, was significantly higher in the subgroup of patients with established LV hypertrophy and inversely correlated with MMP1 activity. Thus, as reported in HCM,26, 34, 35 we can hypothesize that in incipient FD cardiomyopathy, increased collagen synthesis is balanced by degradation (limiting fibrogenesis), but when collagen type I synthesis exceeds the degradation, there is deposition of collagen in the myocardium and LV hypertrophy; the suppression of MMPs may be another mechanism of myocardial collagen type I buildup.
The suppression of MMPs as a pathophysiological mechanism for cardiac fibrosis has been described previously in HCM.34, 35 The multiple pathways involved in MMP suppression are not well understood, but recent studies imply aldosterone‐induced expression of TIMP1 (tissue inhibitor of metallopeptidase 1), acting mainly through MMP1 inhibition increasing net cardiac collagen content.36 Furthermore, microRNA miR‐214 (an antifibrotic microRNA) may play an important role in MMP1 regulation, as shown by Dong et al,37 demonstrating that suppression of MMP1 in cardiac fibroblasts, induced by angiotensin II, could be reversed after administration of pre–miR‐214 (this also decreased TIMP1 and TGF‐β).
Correlations Between Collagen Metabolism Biomarkers and Cardiac Imaging
The biomarker of type I collagen synthesis (PICP), before and after adjustment for bone turnover, had a significant correlation with LV mass, and after multivariable adjustment, PICP:B‐AP ratio remained the better predictor of LV mass. In addition, there was a significant inverse correlation between MMP‐1 and LV mass. These 2 findings might indicate that increased collagen synthesis and inhibition of collagenolysis are syntropic in the process of LV hypertrophy.
Moreover, although female patients presented significantly lower serum PICP concentration and LV mass, subanalysis by sex showed that PICP or MMP1 remained predictors of LV mass in both sexes.
Nevertheless, the correlation between biomarkers of collagen type I metabolism and LV thickness and mass is not a universal finding in previous studies in HCM.26, 34, 35 Studies in HCM elucidating the relative contribution of fibrosis to the magnitude of hypertrophy and heart weight found clear but weak correlations.38 In FD, the magnitude of the increase of fibrotic tissue in endomyocardial tissue was not as high as the cardiomyocyte area and glycosphingolipids vacuoles.39 Despite the possibility of greater contribution of cardiomyocytes hypertrophy than fibrosis to the degree LV hypertrophy, the predictive value of the collagen type I synthesis biomarker to LV mass is evident.
In addition, there was significant correlation between MMP1 and tissue Doppler systolic and early diastolic myocardial velocities, further supporting the role of MMP inhibition in diastolic function impairment, as described previously for HCM.34
As expected, PICP was the sole biomarker with significant difference between LGE‐positive and ‐negative FD patients; however, we cannot rule out that these findings reflect only the significant correlation between PICP and LV mass, as LV mass was significantly higher in patients with LGE. Furthermore, PICP was already elevated in FD patients with normal echocardiograms and without LGE. Nonetheless, the diagnostic accuracy of PICP and the PICP:ICTP ratio (reflecting the balance between synthesis and degradation) to detect LGE was high, ≈75% after adjustment for bone metabolism, and significant. Moreover, the addition of the PICP:ICTP ratio to routine clinical predictors (LV mass and NT‐proBNP) seems to increase the ability to correctly classify a patient regarding LGE status.
Previous results in HCM found no significant correlation between collagen type I metabolism biomarkers and LGE,26 and these results are certainly related to the limited resolution of LGE to detect focal replacement fibrosis9 and the inability to detect early diffuse interstitial fibrosis.10 Nevertheless, sensitive ELISAs may detect very small amounts of circulating PICP; they provide a more sensitive index of fibrosis and reflect subtle changes in myocardial composition that are not detectable by cardiac MRI.
A novel cardiac MRI technique, T1 mapping (assessing T1 relaxation times), may overcome the inability of LGE to detect diffuse interstitial fibrosis. High T1 relaxation times are observed in diffuse fibrosis, protein deposition, and water in edema. Low T1 values are seen in iron or lipid deposition; however, in FD cardiomyopathy, cardiac lipid storage is a hallmark, and T1 mapping has been studied as an imaging biomarker for early detection of cardiac involvement (with low native T1).12, 13 Consequently, the capacity of T1 mapping to detect early diffuse fibrosis may be impaired because of low native T1 (fibrosis may be detected only in the “pseudonormalization” stage, when the amount of fibrotic tissue is enough to overcome the low native characteristic FD cardiomyopathy) and needs further evaluation.
Collagen Type I Metabolism Biomarkers: Previous Findings in FD Cardiomyopathy
To our knowledge, only 2 studies have evaluated cardiac extracellular matrix turnover in FD. Shah et al27 reported increased levels of MMP9, with significant correlation with MSSI and inverse correlation with midwall fractional shortening of the left ventricle. No difference was found in levels of TIMP1 and TIMP2 between FD patients and controls. In contrast to the identified decrease in MMP1 and MMP2 with increasing cardiomyopathy severity, MMP9 has been identified in HCM as a profibrotic marker.35
Krämer et al6 reported an increase in 3 biomarkers of collagen metabolism (PICP, ICTP, and PIIINP) compared with healthy historical controls; however, no difference in these markers was observed between patients with and without fibrosis in the cardiac MRI. The authors explained this finding assuming a systemic fibrotic state in FD, involving the heart, the kidneys, and other organs. Nevertheless, a subanalysis by sex and adjustment to bone turnover was not performed and could be crucial; usually female patients have a milder phenotype, but the rise of the collagen markers due to bone turnover can misleadingly overestimate myocardial fibrosis. Moreover, the previously described limitations of LGE techniques to detect cardiac fibrosis certainly influenced the results.
Limitations
This study has several limitations. First, histologic correlation is absent, although it could be very useful for biomarker validation, but endomiocardial biopsy is an invasive and potentially harmful technique. Second, the cross‐sectional design hinders the prognostic value of these biomarkers and the influence of ERT and angiotensin‐converting enzyme inhibitors or angiotensin II receptor blockers on them (we are currently performing a longitudinal evaluation). Third, laboratory standardization of these biomarker assays is an urgent need to improve their clinical application. Fourth, no core reading of the echocardiograms and cardiac MRIs was performed as part of the standardized research protocol. Fifth, there is no correlation with speckle‐tracking analysis of echocardiograms because this was not available in the majority of the cohort, and there is no quantification of the LGE area, although no correlation was identified in previous studies. Sixth, there is no correlation with T1 mapping cardiac MRI techniques.
Moreover, in this study, we recruited a relatively small cohort of FD patients that does not represent the entire spectrum of FD cardiomyopathy phenotypes. Further research is needed to address the value of the studied biomarkers in a larger and more heterogeneous cohort of FD patients.
Conclusion
This study provides new, relevant data to understand the natural history of fibrogenesis in FD cardiomyopathy. It shows, for the first time, that serum biomarkers of collagen type I metabolism identify ongoing fibrosis in the early stages of FD cardiomyopathy and possibly predict the development of LV hypertrophy, highlighting the importance of developing therapies to mitigate fibrosis and change the natural history of FD cardiomyopathy.
Author Contributions
Research idea and study design: Aguiar, Azevedo, Ducla Soares and Hughes; data acquisition and laboratorial measurements: Aguiar, Pinto, Marino, Cardoso; data analysis/interpretation: Aguiar, Pinto; statistical analysis: Aguiar; supervision: Sousa, Cunha, Ducla Soares, Hughes. Each author contributed important intellectual content during article drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. Aguiar takes responsibility that this study has been reported honestly, accurately and transparently; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned have been explained.
Sources of Funding
This study has been supported by an investigator‐initiated research grant from Shire Pharmaceuticals. The funder did not have a role in study design; collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication.
Disclosures
Aguiar has received speaker honoraria from Genzyme, Shire and Biomarin Corporations and research grants from Shire Corporation. Hughes has received: speaker honoraria from Genzyme, Shire, Protalix and Amicus Corporations; consulting fees from Genzyme, Shire, Amicus, Protalix and Actelion Corporations; research grants from Genzyme and Shire. The remaining authors have no disclosures to report.
Supporting information
Data S1. Supplemental methods.
Table S1. Mutation Frequency
Figure S1. Box plot of PICP:ICTP ratio (before and after adjustment for bone collagen turnover) in controls and Fabry disease subgroups. ICTP indicates carboxyterminal telopeptide of type I collagen; PICP, carboxyterminal propeptide of procollagen type I.
Figure S2. Correlation between PICP:ICTP ratio (after adjustment for bone collagen turnover) and left ventricular mass (upper image); box plot of PICP:ICTP ratio (after adjustment for bone collagen turnover) in LGE‐negative and ‐positive patients (lower image). ICTP indicates carboxyterminal telopeptide of type I collagen; LGE, late gadolinium enhancement; PICP, carboxyterminal propeptide of procollagen type I.
Acknowledgments
Clinical support by Atul Metha, J.M. Braz Nogueira, Francisco Araújo, and Anabela Oliveira is gratefully acknowledged. We are also grateful to Hatim Ibrahim, André Domingues, and Rita Fernandes for laboratory support.
(J Am Heart Assoc. 2018;7:e007124 DOI: 10.1161/JAHA.117.007124.)29535138
References
- 1. MacDermot KD, Holmes A, Miners AH. Anderson‐Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacDermot KD, Holmes A, Miners AH. Anderson‐Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carriers females. J Med Genet. 2001;38:769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mehta A, Clarke JTR, Giugliani R, Elliott P, Linhart A, Beck M, Sunder‐Plassmann G; on behalf of the FOS Investigators . Natural course of Fabry disease: changing pattern of cause of death in FOS—Fabry Outcome Survey. J Med Genet. 2009;46:548–552. [DOI] [PubMed] [Google Scholar]
- 4. Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11:790–796. [DOI] [PubMed] [Google Scholar]
- 5. Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24:2102–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krämer J, Niemann M, Störk S, Frantz S, Beer M, Ertl G, Wanner C, Weidemann F. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am J Cardiol. 2014;114:895–900. [DOI] [PubMed] [Google Scholar]
- 7. Moon JCC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennella DJ, Leed PJ, Elliott PM. Gadolinium enhanced cardiovascular magnetic resonance in Anderson‐Fabry disease: evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003;24:2151–2155. [DOI] [PubMed] [Google Scholar]
- 8. De Cobelli F, Esposito A, Belloni E, Pieroni M, Perseghin G, Chimenti C, Frustaci A, Del Maschio A. Delayed‐enhanced cardiac MRI for differentiation of Fabry's disease from symmetric hypertrophic cardiomyopathy. AJR Am J Roentgenol. 2009;192:97–102. [DOI] [PubMed] [Google Scholar]
- 9. Wu E, Judd RM, Vargas JD, Klocke FJ, Bonow RO, Kim RJ. Visualization of presence, location and transmural extent of healed Q‐wave and non‐Q‐wave myocardial infarction. Lancet. 2001;357:21–28. [DOI] [PubMed] [Google Scholar]
- 10. Iles LM, Ellims AH, Llewellyn H, Hare JL, Kaye DM, McLean CA, Taylor AJ. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur Heart J Cardiovasc Imaging. 2015;16:14–22. [DOI] [PubMed] [Google Scholar]
- 11. Flett AS, Hasleton J, Cook C, Hausenloy D, Quarta G, Ariti C, Muthurangu V, Moon JC. Evaluation of techniques for the quantification of myocardial scar of differing etiology using cardiac magnetic resonance. JACC Cardiovasc Imaging. 2011;4:150–156. [DOI] [PubMed] [Google Scholar]
- 12. Sado DM, White SK, Piechnik SK, Banypersad SN, Treibel T, Captur G, Fontana M, Maestrini V, Flett AS, Robson MD, Lachmann RH, Murphy E, Mehta A, Hughes D, Neubauer S, Elliott PM, Moon JC. Identification and assessment of Anderson‐Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. 2013;6:392–398. [DOI] [PubMed] [Google Scholar]
- 13. Pica S, Sado DM, Maestrini V, Fontana M, White SK, Treibel T, Captur G, Anderson S, Piechnik SK, Robson MD, Lachmann RH, Murphy E, Mehta A, Hughes D, Kellman P, Elliott PM, Herrey AS, Moon JC. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2014;16:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, Voelker W, Ertl G, Wanner C, Strotmann J. Long‐term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119:524–529. [DOI] [PubMed] [Google Scholar]
- 15. Beer M, Weidemann F, Breunig F, Knoll A, Koeppe S, Machann W, Hahn D, Wanner C, Strotmann J, Sandstede J. Impact of enzyme replacement therapy on cardiac morphology and function and late enhancement in Fabry's cardiomyopathy. Am J Cardiol. 2006;97:1515–1518. [DOI] [PubMed] [Google Scholar]
- 16. Zamorano J, Serra V, Isla LP, Feltes G, Calli A, Barbado FJ, Torras J, Hernandez S, Herrera J, Herrero JA, Pintos G. Usefulness of tissue Doppler in early detection of cardiac disease in Fabry patients and potential role of enzyme replacement therapy (ERT) for avoiding progression of disease. Eur J Echocardiogr. 2011;12:671–677. [DOI] [PubMed] [Google Scholar]
- 17. Toro R, Perez‐Isla L, Doxastaquis G, Barba MA, Gallego AR, Pintos G, Barbados FJ, Mangas A, Zamorano JL. Clinical usefulness of tissue Doppler imaging in predicting preclinical Fabry cardiomyopathy. Int J Cardiol. 2009;132:38–44. [DOI] [PubMed] [Google Scholar]
- 18. Saccheri MC, Cianciulli TF, Lax JA, Gagliardi JA, Cáceres GL, Quarin AE, Kisinovsky I, Rozenfeld PA, Reisin RC; AADELFA . Two‐dimensional speckle tracking echocardiography for early detection of myocardial damage in young patients with Fabry disease. Echocardiography. 2013;30:1069–1077. [DOI] [PubMed] [Google Scholar]
- 19. Morris DA, Blaschke D, Canaan‐Kühl S, Krebs A, Knobloch G, Walter TC, Haverkamp W. Global cardiac alterations detected by speckle‐tracking echocardiography in Fabry disease: left ventricular, right ventricular, and left atrial dysfunction are common and linked to worse symptomatic status. Int J Cardiovasc Imaging. 2015;31:301–313. [DOI] [PubMed] [Google Scholar]
- 20. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires Tgf‐β. J Clin Invest. 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sasagawa S, Nishimura Y, Okabe S, Murakami S, Ashikawa Y, Yuge M, Kawaguchi K, Kawase R, Okamoto R, Ito M, Tanaka T. Downregulation of GSTK1 is a common mechanism underlying hypertrophic cardiomyopathy. Front Pharmacol. 2016;7:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pearlman ES, Weber KT, Janicki JS, Pietra GG, Fishman AP. Muscle fiber orientation and connective tissue content in the hypertrophied human heart. Lab Invest. 1982;46:158–164. [PubMed] [Google Scholar]
- 23. Querejeta R, López B, González A, Sánchez E, Larman M, Martínez Ubago JL, Díez J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. 2004;110:1263–1268. [DOI] [PubMed] [Google Scholar]
- 24. Poulsen SH, Host NB, Egstrup K. Long‐term changes in collagen formation expressed by serum carboxyterminal propeptide of type I procollagen and relation to left ventricular function after acute myocardial infarction. Cardiology. 2001;96:45–50. [DOI] [PubMed] [Google Scholar]
- 25. Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the Randomized Aldactone Evaluation Study (RALES). Circulation. 2000;102:2700–2706. [DOI] [PubMed] [Google Scholar]
- 26. Ho CY, López B, Coelho‐Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, González A, Colan SD, Seidman JG, Díez J, Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shah JS, Hughes DA, Tayebjee MH, MacFadyen RJ, Mehta AB, Elliott PM. Extracellular matrix turnover and disease severity in Anderson‐Fabry disease. J Inherit Metab Dis. 2007;39:88–95. [DOI] [PubMed] [Google Scholar]
- 28. Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E, Baehner F, Kim K, Bajbouj M, Schwarting A, Gal A, Beck M. The Mainz Severity Score Index: a new instrument for quantifying the Anderson‐Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004;65:299–307. [DOI] [PubMed] [Google Scholar]
- 29. Hughes D, Malmenas M, Deegan P, Elliott PM, Ginsberg L, Hajioff D, Ioannidis AS, Orteu CH, Ramaswami U, West M, Pastores GM, Jenkinson C. Fabry International Prognostic Index: a predictive severity score for Anderson‐Fabry disease. J Med Genet. 2012;49:212–220. [DOI] [PubMed] [Google Scholar]
- 30. Michelsen J, Wallaschofski H, Friedrich N, Spielhagen C, Rettig R, Ittermann T, Nauck M, Hannemann A. Reference intervals for serum concentrations of three bone turnover markers for men and women. Bone. 2013;57:399–404. [DOI] [PubMed] [Google Scholar]
- 31. Khor KH, Campbell FE, Owen H, Shiels IA, Mills PC. Myocardial collagen deposition and inflammatory cell infiltration in cats with pre‐clinical hypertrophic cardiomyopathy. Vet J. 2015;203:161–168. [DOI] [PubMed] [Google Scholar]
- 32. Aerts JM, Groner JE, Kuiper S, Donker‐Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox‐Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA. 2008;105:2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanchez‐Niño MD, Sanz AB, Carrasco S, Saleem MA, Mathieson PW, Valdivielso JM, Ruiz‐Ortega M, Egido J, Ortiz A. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant. 2011;26:1797–1802. [DOI] [PubMed] [Google Scholar]
- 34. Lombardi R, Betocchi S, Losi MA, Tocchetti CG, Aversa M, Miranda M, D'Alessandro G, Cacace A, Ciampi Q, Chiariello M. Myocardial collagen turnover in hypertrophic cardiomyopathy. Circulation. 2003;108:1455–1460. [DOI] [PubMed] [Google Scholar]
- 35. Münch J, Avanesov M, Bannas P, Säring D, Krämer E, Mearini G, Carrier L, Suling A, Lund G, Patten M. Serum matrix metalloproteinases as quantitative biomarkers for myocardial fibrosis and sudden cardiac death risk stratification in patients with hypertrophic cardiomyopathy. J Card Fail. 2016;22:845–850. [DOI] [PubMed] [Google Scholar]
- 36. Hung CS, Chou CH, Liao CW, Lin YT, Wu XM, Chang YY, Chen YH, Wu VC, Su MJ, Ho YL, Chen MF, Wu KD, Lin YH; TAIPAI Study Group . Aldosterone induces tissue inhibitor of metalloproteinases‐1 expression and further contributes to collagen accumulation: from clinical to bench studies. Hypertension. 2016;67:1309–1320. [DOI] [PubMed] [Google Scholar]
- 37. Dong H, Dong S, Zhang L, Gao X, Lv G, Chen W, Shao S. MicroRNA‐214 exerts a cardio‐protective effect by inhibition of fibrosis. Anat Rec (Hoboken). 2016;299:1348–1357. [DOI] [PubMed] [Google Scholar]
- 38. Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. [DOI] [PubMed] [Google Scholar]
- 39. Chimenti C, Hamdani N, Boontje NM, DeCobelli F, Esposito A, Bronzwaer JG, Stienen GJ, Russo MA, Paulus WJ, Frustaci A, Velden J. Myofilament degradation and dysfunction of human cardiomyocytes in Fabry disease. Am J Pathol. 2008;172:1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Mutation Frequency
Figure S1. Box plot of PICP:ICTP ratio (before and after adjustment for bone collagen turnover) in controls and Fabry disease subgroups. ICTP indicates carboxyterminal telopeptide of type I collagen; PICP, carboxyterminal propeptide of procollagen type I.
Figure S2. Correlation between PICP:ICTP ratio (after adjustment for bone collagen turnover) and left ventricular mass (upper image); box plot of PICP:ICTP ratio (after adjustment for bone collagen turnover) in LGE‐negative and ‐positive patients (lower image). ICTP indicates carboxyterminal telopeptide of type I collagen; LGE, late gadolinium enhancement; PICP, carboxyterminal propeptide of procollagen type I.