

Abstract
Background
Thiazide and thiazide‐like diuretics are first‐line medications for treating uncomplicated hypertension. However, their use has been associated with adverse metabolic events, including hyperglycemia and incident diabetes mellitus, with incompletely understood mechanisms. Our goal was to identify genomic variants associated with thiazide‐like diuretic/chlorthalidone‐induced glucose change.
Methods and Results
Genome‐wide analysis of glucose change after treatment with chlorthalidone was performed by race among the white (n=175) and black (n=135) participants from the PEAR‐2 (Pharmacogenomic Evaluation of Antihypertensive Responses‐2). Single‐nucleotide polymorphisms with P<5×10−8 were further prioritized using in silico analysis based on their expression quantitative trait loci function. Among blacks, an intronic single‐nucleotide polymorphism (rs9943291) in the HMGCS2 was associated with increase in glucose levels following chlorthalidone treatment (ß=12.5; P=4.17×10−8). G‐allele carriers of HMGCS2 had higher glucose levels (glucose change=+16.29 mg/dL) post chlorthalidone treatment compared with noncarriers of G allele (glucose change=+2.80 mg/dL). This association was successfully replicated in an independent replication cohort of hydrochlorothiazide‐treated participants from the PEAR study (ß=5.54; P=0.023). A meta‐analysis of the 2 studies was performed by race in Meta‐Analysis Helper, where this single‐nucleotide polymorphism, rs9943291, was genome‐wide significant with a meta‐analysis P value of 3.71×10−8. HMGCS2, a part of the HMG‐CoA synthase family, is important for ketogenesis and cholesterol synthesis pathways that are essential in glucose homeostasis.
Conclusions
These results suggest that HMGCS2 is a promising candidate gene involved in chlorthalidone and Hydrochlorothiazide (HCTZ)‐induced glucose change. This may provide insights into the mechanisms involved in thiazide‐induced hyperglycemia that may ultimately facilitate personalized approaches to antihypertensive selection for hypertension treatment.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifiers: NCT00246519 and NCT01203852.
Keywords: chlorthalidone, diabetes mellitus, genome‐wide association study, glucose, hydrochlorothiazide, hyperglycemia, pharmacogenomics
Subject Categories: Genetics; Hypertension; Genetic, Association Studies; Clinical Studies
Clinical Perspective
What Is New?
In spite of studies showing that chlorthalidone is a superior diuretic compared with hydrochlorothiazide in terms of potency and decreasing cardiovascular events, development of hyperglycemia, and new‐onset diabetes mellitus post chlorthalidone treatment plagues the utility of this drug.
Using genome‐wide association study, we discovered a novel signal rs9943291 in the HMGCS2 gene associated with increased chlorthalidone‐induced glucose change and successfully replicated this signal in the independent hydrochlorothiazide treated cohort.
Variants carriers of rs9943291 (G allele) had significantly higher glucose increase post chlorthalidone treatment compared with noncarriers.
What Are the Clinical Implications?
These findings suggest that HMGCS2 is involved in chlorthalidone‐induced glucose change, and further understanding of this signal can provide insights into the mechanisms involved in thiazide‐induced increased glucose.
Replication of these signals in other larger, independent studies may aid clinicians to use this information for a priori identification of patients at increased risk for thiazide‐induced hyperglycemia and can prescribe alternate antihypertensive medications.
Introduction
In the United States and worldwide, hypertension is a common condition that strongly influences risk for coronary heart disease, stroke, peripheral vascular disease, heart failure, renal insufficiency, retinal bleeding, and visual impairment.1, 2 Of the treatment options available, thiazide diuretics are among the most commonly prescribed antihypertensive medications and remain a first‐line treatment option in the current Joint National Committee recommendations for treatment of uncomplicated hypertension.3 Their use, however, has been associated with several adverse metabolic side effects, including hyperglycemia and increased risk for new‐onset diabetes mellitus.4, 5, 6, 7, 8, 9 Findings from the ALLHAT (antihypertensive and lipid lowering treatment to prevent heart attack trial) report that among chlorthalidone‐treated patients, the 4‐year incidence of new‐onset diabetes mellitus was significantly higher (11.6%) compared with the other treatment groups (9.8% for amlodipine and 8.1% for lisinopril).10 Similar results, with a higher incidence of new‐onset diabetes mellitus in patients treated with a beta‐blocker and/or thiazide diuretic, have also been observed in other large hypertension outcomes clinical trials.11, 12 Mechanistically, some studies attribute the observed rise in glucose following thiazide diuretics to the associated hypokalemia, which, in turn, affects potassium‐dependent insulin secretion and/or increased insulin resistance.13, 14, 15, 16, 17, 18 However, the exact mechanism is unknown, and considering the cross‐talk between the pathways related to hypertension and glucose homeostasis, it is highly unlikely that hypokalemia is the only mechanism involved in thiazide‐induced glucose dysregulation.
Studies support that chlorthalidone is a more‐potent diuretic compared with hydrochlorothiazide in treating hypertension.19 Like other thiazides, chlorthalidone impairs glucose tolerance, leading to hyperglycemia in many patients. There is, however, a void in the literature regarding the understanding of the genetic basis of chlorthalidone‐induced glucose change. Furthermore, the benefits of reduction in blood pressure (BP) with antihypertensive medications could possibly be offset by the risk for hyperglycemia and new‐onset diabetes mellitus. Identifying and understanding genetic variation that may predispose treated individuals to this adverse metabolic effect could ultimately optimize selection of antihypertensive agents. Therefore, we conducted a genome‐wide association study (GWAS) of change in glucose following treatment of chlorthalidone among a population of essential hypertensive patients to identify genomic variants underlying the variability observed in the blood glucose change post chlorthalidone treatment.
Methods
All the genotype and phenotype data for the PEAR (Pharmacogenomic Evaluation of Antihypertensive Responses) study have been made publically available at the dbGaP database under dbGaP Accession: phs000649.v1.p1 and can be accessed at (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000649.v1.p1).20 The PEAR‐2 genotype and phenotype data are currently in the process of being uploaded to dbGaP and will soon be available to other researchers under dbGaP Accession: phs000649.v2.p1.
PEAR‐2 and PEAR Study Design
The PEAR studies were prospective, multicenter, clinical trials conducted to assess the genomic variability attributed to BP and adverse metabolic effects following sequential monotherapy treatment with metoprolol and chlorthalidone in PEAR‐2 (clinicaltrials.gov identifier: NCT01203852) and randomized monotherapy treatment with hydrochlorothiazide and atenolol in PEAR (clinicaltrials.gov identifier NCT00246519). Details of both of these clinical trials have been described previously.21, 22 Briefly, in both studies, males and females of any race and ethnicity with mild‐to‐moderate uncomplicated hypertension were recruited, and participants with secondary hypertension and a known history of cardiovascular disease or diabetes mellitus were excluded. Eligible participants with uncomplicated hypertension underwent an antihypertensive medication washout period of 3 to 4 weeks. Following confirmation of BP eligibility based on both home and office BP measurements, participants were treated with protocol‐specified antihypertensive drugs and doses.
Both PEAR‐2 and PEAR included beta‐blocker monotherapy treatment, but, for brevity, details are not provided here and we focus on the thiazide diuretic components of the trials, because these are the data included in this report. In PEAR‐2, participants were treated with chlorthalidone 15 mg daily for 2 weeks (and later in the study, when sale of the 15‐mg tablet was discontinued, 25 mg daily 4 times per week), followed by uptitration to 25 mg daily for 6 weeks. In PEAR, participants randomized to hydrochlorothiazide received 12.5 mg once‐daily followed by uptitration to 25 mg once‐daily for a total 9 weeks' treatment. In both the PEAR‐2 and PEAR studies, all patients with a BP of >120/70 mm Hg had their dose titrated.
At the end of washout (baseline) and following the 8‐ to 9‐week treatment period in both studies, blood samples were collected for DNA, RNA, plasma, and serum, along with measurement of BP.
Both PEAR studies were reviewed and approved by the institutional review boards at the participating sites (University of Florida in Gainesville, FL; Mayo Clinic in Rochester, MN; and Emory University in Atlanta, GA). All participants provided voluntary, written informed consent, and the studies were conducted in accord with the principles outlined in the Declaration of Helsinki.
Phenotype
Fasting plasma glucose measurements were made at Mayo Clinic using a Hitachi 911 Chemistry Analyzer (Roche Diagnostic, Indianapolis, IN). For both PEAR‐2 and PEAR, change in glucose was defined as the difference in glucose measurement from baseline to the end of the drug treatment.
Genotyping and Imputation
DNA samples from PEAR‐2 participants were genotyped for ≈2.5 million single‐nucleotide polymorphism (SNPs) using the Illumina Human Omni2.5S Beadchip (Illumina, San Diego, CA), whereas DNA samples from PEAR participants were genotyped for ≈1 million SNPs using the Illumina Human Omni1MQuad BeadChip (Illumina). Quality‐control procedures applied to the genetic data for both studies have been previously published.23 A principal component analysis was used to determine PEAR‐2 and PEAR participants' genetic ancestry and confirm self‐identified race/ethnicity using the EIGENSTRAT method,24 and genetic data from both studies were imputed to the 1000 genomes phase I reference panel using high‐quality SNPs that passed quality control, using MaCH (version 1.0.16)25 for prephasing followed by imputation by Minimac.26 Post imputation quality control involved filtering and excluding SNPs with imputation quality <0.3 and minor allele frequency <5%.
Statistical Analysis
For the purpose of this article, our analyses were focused on identifying the genetic predictors of chlorthalidone‐induced glucose change in black and white hypertensive participants. Chlorthalidone‐treated hypertensive participants from PEAR‐2 served as the discovery cohort. Because there was no available replication cohort for chlorthalidone effects, hydrochlorothiazide‐treated participants from PEAR comprised the replication cohort, under the assumption that the mechanisms of the adverse effect on glucose are similar or the same for hydrochlorothiazide and chlorthalidone. For both studies, the primary outcome variable was defined as change in glucose from baseline to the end of the drug treatment. Participants with a standardized residual greater than 3 SDs were excluded from the analysis for both discovery and replication cohorts (PEAR‐2 blacks [n=3], PEAR‐2 whites [n=4], PEAR blacks [n=0], PEAR whites [n=4]; Figure S1). We exclude these because the most likely explanation for the extreme response is that the study participants were not fasting at the time of sample collection. For both the studies, the final participant's clinical characteristics of the participants are presented as mean±SD for continuous variables and numbers and percentages for categorical variables.
GWAS Analysis
The genome‐wide association analysis was conducted separately by race using a staged approach with PEAR‐2 serving as the discovery cohort and PEAR as the replication cohort. Multiple linear regression analysis was conducted using ProbABEL,27 assuming an additive mode of inheritance. The analysis was adjusted for age, sex, and baselines glucose because these were associated with the change in glucose post chlorthalidone treatment in our study. Also, studies have shown baselines glucose to be a significant predictor of thiazide‐associated glucose change.7, 28 Furthermore, even though we did not find any population substructure in our cohort, principal components 1 and 2 were included in the model to account for ancestry. SNPs with a P<5×10−8 were considered to have met genome‐wide significance. To focus on the regulatory and functional variants that affect the change in expression, an SNP prioritization approach was undertaken to filter the genome‐wide significant SNPs to be tested for replication. Studies have shown the utility of expression quantitative trait loci (eQTL) signal in elucidating true associations and understanding the mechanistic underpinnings of these associations in GWAS.29, 30 The genome‐wide significant SNPs were hence prioritized based on their eQTL annotation as listed in the Haploreg Database.31 After prioritizing SNPs based on their eQTL annotation, linkage disequilibrium (LD) pruning was performed to prune multiple SNPs in LD (r 2>0.08) that were present in the same gene locus, using LDlink3.0.32 Thus, a single SNP with an eQTL annotation representing an independent signal in that locus was selected and moved forward for replication. SNPs that passed the prioritization filter were further validated, by testing for replication in an independent cohort of hydrochlorothiazide‐treated participants from PEAR. Because our hypothesis for replication was 1‐sided, SNPs with a 1‐sided P≤0.05 with association in the same direction were considered replicated. Bonferroni correction was used to adjust for multiple testing during replication (0.05/number of association tested). To reduce the risk for bias, we had multiple steps built into our analysis plan. These consisted of (1) eQTL prioritization for the significant SNPs identified in the discovery analysis, (2) use of Bonferroni correction for multiple testing during replication, and (3) use of a cohort with the same ancestry for replication as the discovery cohort.
The results of PEAR and PEAR‐2 were further combined by doing a meta‐analysis within each race for blacks and whites using Meta‐Analysis Helper using a fixed‐effect model and inverse variance weighing33 Locus Zoom was used to display the association results using AFR population for blacks and CEU population for whites.34
Results
Demographics and baseline characteristics of the black and white study participants treated with chlorthalidone in PEAR‐2 and HCTZ in PEAR are represented in Table 1. Black and white cohorts in both PEAR and PEAR‐2 study had a similar mean age of ≈50 years. The other clinical characteristics were similar between the 2 studies.
Table 1.
Baseline Characteristics of PEAR‐2 and PEAR Participants
| Baseline Characteristics | PEAR‐2 (Chlorthalidone) | PEAR (Hydrochlorothiazide) | ||
|---|---|---|---|---|
| Blacks (n=135) | Whites (n=175) | Blacks (n=140) | Whites (n=222) | |
| Age, y | 50.30±8.75 | 51.22±8.82 | 48.91±9.38 | 50.16±9.46 |
| Female, N (%) | 65 (48.14) | 75 (42.85) | 86 (61.26) | 89 (40.09) |
| Body mass index, kg/m2 | 31.25±5.4 | 30.76±5.02 | 31.8±6.2 | 30.31±5.20 |
| Waist circumference, cm | 97.51±11.6 | 100.46±13.24 | 94.72±9.2 | 98.21±13.15 |
| Systolic blood pressure | 146.49±11.06 | 147.48±10.34 | 150.16±12.96 | 151.06±13.41 |
| Diastolic blood pressure | 94.88±5.99 | 94.31±5.04 | 97.34±5.68 | 97.05±5.24 |
Values are presented as mean±SD unless otherwise noted. PEAR indicates Pharmacogenomic Evaluation of Antihypertensive Response.
Glucose response to chlorthalidone in PEAR‐2 and hydrochlorothiazide for PEAR are listed in Table 2. Following 8 weeks of treatment with chlorthalidone, mean glucose change was a 4.30±9.83 mg/dL increase for blacks and 4.39±7.51 mg/dL increase in whites for the PEAR‐2 participants. Among the PEAR participants, mean glucose change post‐treatment with hydrochlorothiazide was 2.38±10.82 mg/dL for blacks and 0.97±10.13 mg/dL for whites, suggesting that, similarly to the BP effect, chlorthalidone also has a more‐potent effect than hydrochlorothiazide on adverse glucose response. The change in glucose from baseline to post‐treatment was greater for chlorthalidone compared with hydrochlorothiazide in both races. This change in glucose post‐treatment between chlorthalidone and hydrochlorothiazide was statistically significant only in whites (P=0.0002) and not in blacks. However, there was substantial interindividual variability in the blood glucose change post chlorthalidone treatment in PEAR‐2 and post hydrochlorothiazide treatment in PEAR, as shown in Figure 1.
Table 2.
Glucose Characteristics of PEAR‐2 and PEAR Participants
| Glucose Characteristics | PEAR‐2 (Chlorthalidone) | PEAR (Hydrochlorothiazide) | ||
|---|---|---|---|---|
| Blacks (n=135) | Whites (n=175) | Blacks (n=139) | Whites (n=222) | |
| Baseline glucose, mg/dL | 94.46±11.37 | 94.18±9.39 | 90.65±12.78 | 92.85±12.28 |
| Glucose post‐treatment, mg/dL | 99.77±6.23 | 99.10±16.23 | 93.1±14.84 | 93.82±11.93 |
| Glucose change, mg/dL | 4.30±9.83 | 4.39±7.51 | 2.38±10.82 | 0.97±10.13a |
Values are presented as mean±SD unless otherwise noted. PEAR indicates Pharmacogenomic Evaluation of Antihypertensive Response.
Indicates P≤0.05 for chlorthalidone (PEAR‐2) vs Hydrochlorothiazide (PEAR) within race.
Figure 1.
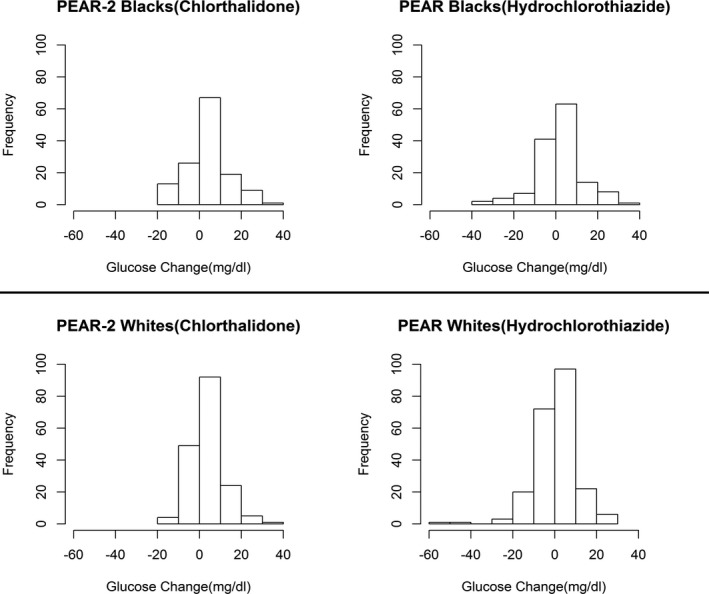
Distribution of glucose change post‐treatment showing the interindividual variability of the response among PEAR‐2 and PEAR participants. PEAR indicates Pharmacogenomic Evaluation of Antihypertensive Response.
Genome‐Wide Analysis of Chlorthalidone‐Induced Glucose Change in PEAR‐2
Among blacks, genome‐wide association analyses of glucose change revealed 10 SNPs representing 4 independent signals that reached genome‐wide significance (Table 3). All the variants in high LD (r 2>0.8) with one another were considered as representing a single independent signal. The Manhattan plot corresponding to the association of chlorthalidone‐induced glucose change in blacks is shown in Figure 2. The red line is the strict genome‐wide significance level with P=5×10−8. The 4 independent genome‐wide significant signals represented 4 loci: SLC4A2 (1 SNP); intergenic region of C1orf98 (4 SNPs); HMGCS2 (4 SNPs); and SNX29 (1 SNP).
Table 3.
Genome‐wide Significant SNPs From Genome‐wide Association Results for Glucose Change Among PEAR‐2 Blacks
| SNP | CHR | BP | Nearest Gene | Minor Allele | MAF | Imputation Quality (Rsq) | PEAR‐2 Chlorthalidone Response | |
|---|---|---|---|---|---|---|---|---|
| β | P Value | |||||||
| rs201505549 | 9 | 19 743 120 | SLC24A2 | D | 0.0556 | 0.94 | 15.41 | 2.11×10−09 |
| rs61824877 | 1 | 200 242 632 | 69 kb 3′ of C1orf98 | A | 0.0606 | 0.98 | 12.74 | 4.82×10−09 |
| rs9927344 | 16 | 12 388 109 | SNX29 | T | 0.11819 | 0.99 | 9.61 | 3.86×10−08 |
| rs9943291 | 1 | 120 292 290 | HMGCS2 | G | 0.0637 | 0.97 | 12.51 | 4.17×10−08 |
BP indicates base‐pair position: hg19 position; CHR, chromosome; D, deletion; MAF, minor allele frequency; PEAR‐2, Pharmacogenomic Evaluation of Antihypertensive Response‐2; SE, standard error of the beta coefficient; SNP, single‐nucleotide polymorphism; β, regression coefficient for minor allele.
Figure 2.
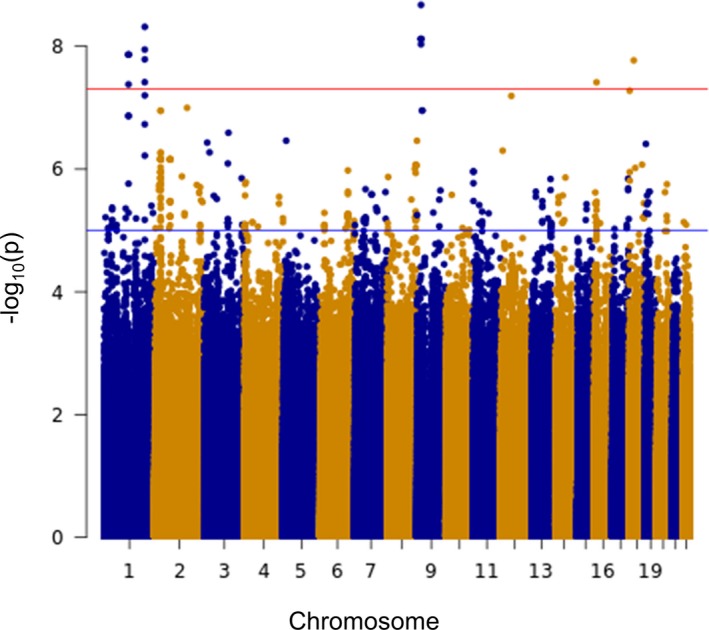
Manhattan plots of glucose change post chlorthalidone treatment among PEAR‐2 black participants. Genome‐wide significance threshold (red line): P<5×10−8. PEAR‐2 indicates Pharmacogenomic Evaluation of Antihypertensive Response‐2.
Of these, an intronic SNP rs201505549 in SLC24A2, present on chromosome 9q21.13, had the strongest association for glucose change in blacks with the lowest P value of 2.11×10−09 and was associated with increased glucose (β=15.41) post chlorthalidone treatment (Table 3). Another independent signal rs61827877 was present in chromosome 1q12 in the intergenic region near C1orf98 and also achieved genome‐wide significance for association with increased blood glucose post chlorthalidone treatment (Table 3). Rs61824877 is also a cis‐eQTL signal for ZNF218 in whole blood.35 Another strong association was present in the locus of HMGCS2 that reached genome‐wide significance (Table 3). SNP rs9943291 in this gene was associated with a significant increase in blood glucose levels at the end of the chlorthalidone treatment. This SNP is also a cis‐eQTL for another gene, PHGDH, in whole blood.35 PHGDH is present very close to the HMGCS2 (Figure S2). Last, one other SNP rs9927344 in SNX29 was also genome‐wide significant (Table 3) and was associated with an increase in blood glucose levels post chlorthalidone treatment. All the genome‐wide significant SNPs before LD pruning are reported in Table S1.
Among whites, no SNPs reached genome‐wide significance. The Manhattan plot corresponding to the association of chlorthalidone‐induced glucose change in whites is presented in Figure S3. Four SNPs that met the suggestive level of significance (P<1×10−6) are listed in Table S2. Furthermore, none of the genome‐wide significant SNPs of the black ancestry group were associated with glucose response in the white cohort (Table S3), and none of the SNPs that met the suggestive level of significance in the white cohort were associated with glucose response in the black cohort (Table S4).
Replication in Hydrochlorothiazide‐Induced Glucose Change in PEAR
Genome‐wide significant SNPs from the PEAR‐2 discovery cohort were further prioritized based on their eQTL annotation to screen for functional and regulatory SNPs to be tested for replication. Among the blacks, 2 of the 4 genome‐wide significant loci had eQTL annotations. The SNP in the intergenic region of C1orf98 is an eQTL for ZNF281, and the SNP in the HMGCS2 region is an eQTL for PHGDH. Based on this, these 2 independent signals (loci) were taken forward for replication. Replication for association of change in blood glucose levels post chlorthalidone treatment was tested in an independent cohort of 140 hydrochlorothiazide‐treated hypertensive blacks from PEAR. SNP rs9943291 from the HMGCS2 signal met the Bonferroni significance (0.05/2=0.025) of replication (1‐sided P=0.023) in the same direction (β=5.54) and was associated with increased blood glucose post hydrochlorothiazide treatment (Table 4). The signal from the intergenic region of C1orf98 did not replicate in PEAR (P=0.135; β=3.87).
Table 4.
PEAR‐2 Association Tested for Replication in PEAR for Blacks
| SNP | CHR | BP | Nearest Gene | Minor Allele | MAF | PEAR‐2 Chlorthalidone Response | PEAR Hydrochlorothiazide Response | Meta‐Analysis | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β | P Value | β | One‐Sided P Value | β | P Value | ||||||
| rs9943291 | 1 | 120 292 290 | HMGCS2 | G | 0.0637 | 12.51 | 4.17×10−08 | 5.54 | 0.023 | 10.01 | 3.71×10−08 |
| rs61824877 | 1 | 200 242 632 | 69 kb 3′ of C1orf98 | A | 0.0606 | 12.74 | 4.82×10−09 | 3.87 | 0.135 | 9.08 | 5.20×10−08 |
BP indicates base‐pair position: hg19 position; CHR, chromosome; D, deletion; MAF, minor allele frequency; PEAR, Pharmacogenomic Evaluation of Antihypertensive Response; SE, standard error of the beta coefficient; SNP, single‐nucleotide polymorphism; β, regression coefficient for minor allele.
Meta‐Analysis PEAR‐2 and PEAR
Association results for all the SNPs from the 2 independent cohorts of PEAR‐2 and PEAR were further combined by race to perform the meta‐analysis. Among the black cohort, 2 variants representing 2 independent signals met genome‐wide significance upon meta‐analysis. Rs202033909, an intronic SNP in the FAT3, was the top signal and was associated with increased glucose change post chlorthalidone treatment (meta‐analysis, P=1.87×10−8; Table S5). Also, rs9943291, in the HMGCS2, which was genome‐wide significant in PEAR‐2 and successfully replicated in PEAR, reached genome‐wide significance with meta‐analysis as expected (meta‐analysis P=3.71×10−8; Table 4). rs9943291 was associated with higher glucose levels at the end of the thiazide treatment in both PEAR‐2 as well as PEAR, as was assessed using additive model in GWAS. However, because we had only 1 homozygote for the minor allele (GG genotype) in PEAR‐2 and no homozygotes for the minor allele in PEAR for rs9943291, we also performed the association analysis using a dominant model by grouping the carriers of the minor allele (GG+GT) versus noncarriers (TT) and tested whether the variant would be significantly associated with glucose change under a dominant model as well. rs9943291 was significantly associated with higher glucose change in PEAR‐2 with least squares adjusted mean change of 16.29±2.4 mg/dL and 2.80±0.81 mg/dL for the G‐allele carriers and T/T genotype carriers, respectively. Similarly in PEAR, carriers of the G allele of this variant had a least squares adjusted mean glucose change of 7.31±2.61 mg/dL and participants with the T/T genotype had a mean glucose change of 1.79±0.88 mg/dL (Figure 3).
Figure 3.
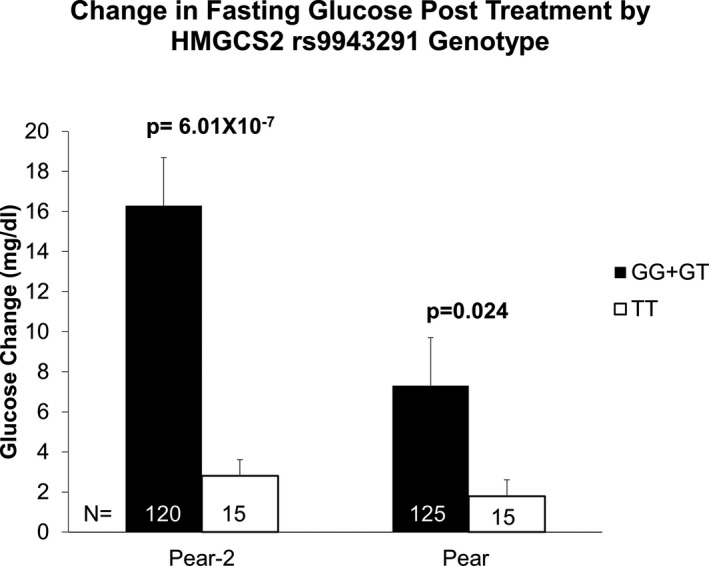
Glucose change post chlorthalidone treatment among PEAR‐2 blacks (N=135) and PEAR blacks (N=140) post hydrochlorothiazide by HMGCS2 rs9943291 genotype. Glucose change for both studies is adjusted for pretreatment glucose levels, age, sex, and principal components 1 and 2. P values are for contrast of least square adjusted means between genotype groups. PEAR indicates Pharmacogenomic Evaluation of Antihypertensive Response.
Among the whites, none of the SNPs met genome‐wide significance in the meta‐analysis. However, 4 SNPs that met the suggestive level of significance and are listed in Table S6.
Discussion
Hyperglycemia or increased risk of new‐onset diabetes mellitus is one of the most common adverse metabolic side effects of thiazide therapy. A previous PEAR study has shown that thiazide diuretic and beta‐blocker treatment causes the advent of adverse metabolic events, including glucose impairment and new‐onset diabetes mellitus, as early as within 9 weeks of starting treatment.36 This increases the risk of developing diabetes mellitus long term given that thiazides are usually lifelong therapeutics for hypertensives. In this study, we performed a GWAS and identified SNPs associated with change in blood glucose levels after treatment with chlorthalidone. Among blacks, our strongest association was found in HMGCS2. An intronic SNP in this gene was associated at the genome‐wide significant level with change in blood glucose levels following chlorthalidone treatment in blacks in PEAR‐2 and successfully replicated in the independent cohort of hydrochlorothiazide‐treated blacks in the PEAR study. This variant also achieved genome‐wide significance in the PEAR‐2/PEAR meta‐analysis (P=3.17×10−8). Individuals who carried the variant allele at this SNP had greater chlorthalidone‐induced glucose increase. The stringent checks utilized during the analysis to reduce bias, including eQTL prioritization, Bonferroni correction for multiple testing, as well as using the same ancestry for replication, provide confidence in the association and its successful replication. Also, additional evidence from power analyses indicate that our study was powered to successfully detect an association of this effect size in both our discovery and replication cohorts, further strengthening the confidence that this is not a spurious or false‐positive association, but a true signal.
HMGCS2 encodes the mitochondrial enzyme, 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2, that is part of the HMG‐CoA family. The HMGCS2 enzyme is an integral part of hepatic metabolism and is important for ketogenesis by multifaceted regulation. Hepatic metabolism and ketogenesis is an important regulator of glucose and lipid metabolism and has an important implication on the pathogenesis of obesity, metabolic syndrome, and also increased risk for type 2 diabetes mellitus (reviewed in an earlier work37). Studies have shown increased expression of lipid‐metabolism–related genes, including HMGSC2, in individuals with nonalcoholic fatty liver disease.38, 39 HMCSC2 is transcriptionally coregulated by insulin, which can suppress its transcription and glucagon, which induces its transcription.40, 41, 42, 43 HMGCS2 also indirectly interacts with PPAR‐alpha, wherein fatty acids can induce expression of HMGCS2 in a PPAR‐alpha dependent manner, and, moreover, HMGCS2 can self‐regulate its own transcription by directly interacting with PPAR‐alpha upon translocation to the nucleus.44 This is an important interaction given that studies have shown that PPAR‐alpha activation improves insulin resistance and prevents the development of diabetes mellitus.45 Studies in mice have shown that absence of HMGCS2 impairs the process of ketogenesis because of insufficient derivation of ketone bodies from fatty acids. Mice lacking HMGCS2 exhibited a mild increase in blood glucose levels with no change in insulin concentrations.46 This further strengthens the evidence for involvement of HMGCS2 in metabolism and glucose and insulin homeostasis. Furthermore, rs9943291 in HMGCS2, the genome‐wide significant SNP, is also an eQTL for PHGDH according to the Haploregv4.1 database and is also present in high LD with some other variants of the PHGDH. PHGDH is adjacent to HMGCS2 on chromosome 1 and encodes for the enzyme, phosphoglycerate dehydrogenase, that is important for L‐serine synthesis, which is essential for amino acid synthesis.47 It is possible that chlorthalidone treatment affects one of these interactions related to HMGCS2, causing chlorthalidone‐induced glucose increase. Further studies are needed to elucidate the exact mechanism of this gene and its cross‐talk with chlorthalidone‐induced effects that will further provide insights into the underlying mechanism driving this association.
Some of our other top signals in the GWAS of chlorthalidone‐induced glucose change in PEAR‐2 blacks also had literature support for involvement with mechanisms related to our phenotype of interest. Variants in SLC24A2 were genome‐wide significant for their association with chlorthalidone‐induced glucose change. SLC24A2 encodes for solute carrier family 24 member 2 also known as sodium‐calcium‐potassium exchanger.48 It is involved in exchanging four Na+ ions for one Ca+ and one K+ ion and decreases Ca+ concentration in response to light in the retinal rod for light adaptation.49 Its role, if any, in potassium‐dependent glucose regulation in response to thiazide treatment needs to be explored further. Another interesting region was C1orf98 intergenic region. rs61824877 was genome‐wide significant as well as an eQTL signal for ZNF281 according to Haploregv4.1, which encodes for zinc finger protein 281. ZNF281 is a potential target for microRNA‐33 that has been shown to be involved in cholesterol regulation and glucose metabolism.
We acknowledge that our study had limitations, the biggest being that we did not have a distinct chlorthalidone replication cohort. Even though chlorthalidone belongs to the thiazide‐like diuretics class and hydrochlorothiazide belongs to the thiazide diuretics class, which are often considered to be alike and similar classes of diuretics,3 there are distinct differences in their potency, chemical structure, and effectiveness.50 Chlorthalidone is a much more‐potent diuretic compared with hydrochlorothiazide. Moreover, the mean glucose change induced by chlorthalidone in PEAR‐2 was higher compared with that induced by hydrochlorothiazide in PEAR. This difference in effect sizes might be one of the underlying reasons for the failure to replicate certain signals from the chlorthalidone‐treated cohorts in PEAR‐2 to the hydrochlorothiazide‐treated cohort in PEAR. We performed a power analysis using the effect size f2 of 0.14 that was observed with a change in glucose response in our discovery cohort, using linear multiple regression (α=0.05, 2‐sided hypothesis). Based on these inputs, we had 98% power to detect an effect size of 0.14 in 135 chlorthalidone‐treated participants. However, to acknowledge the winners curse phenomenon, when the power analysis was performed using the effect size f2=0.034 observed in the replication cohort, we only had 58% power to identify the association between rs9943291 and the hydrochlorothiazide glucose response under the same assumptions (α=0.05, 2‐sided hypothesis). Considering this, even though we were underpowered to detect small effect sizes, we had sufficient power for moderate‐to‐high effect sizes, which are common in pharmacogenomic studies.51 Moreover, the successful replication of this variant in an independent cohort reduces the likelihood of the discovered association being spurious. Last, the same data sets—PEAR‐2 and PEAR—that were used to find the associations were also used to assess the extent of the effect sizes based on genotype. Further replication of these associations in an independent chlorthalidone‐treated cohort will aid in not only testing the association, but also assessing the magnitude of the effect sizes.
In conclusion, by using the chlorthalidone and hydrochlorothiazide‐treated cohorts and conducting a GWAS analysis, we were able to identify several key signals and genes, which may further our understanding of the mechanisms of thiazide‐induced hyperglycemia. A SNP, s9943291, in HMGCS2 met the genome‐wide significance level in the chlorthalidone‐treated discovery cohort, cleared the prioritization filter, and was successfully replicated in the hydrochlorothiazide‐treated replication cohort. Furthermore, it was also genome‐wide significant in the meta‐analysis between the 2 cohorts. This SNP was also an eQTL for the PHGDH gene, which makes both of these genes promising candidates for further studies. Further elucidation and functional validation of these loci is required to completely understand the underlying mechanisms and the role of genetic variation in thiazide‐induced glucose impairment. This can further help improve individualized approaches for effective management of hypertension by identifying individuals at higher risk of glucose impairment attributed to thiazides, thus reducing the incidences of adverse events that are associated with thiazide diuretics.
Sources of Funding
Both PEAR studies were supported by the National Institute of Health (NIH) Pharmacogenetics Research Network grant U01‐GM074492 and the National Center for Advancing Translational Sciences. Award numbers UL1 TR000064 (University of Florida); UL1 TR000454 (Emory University); and UL1 TR000135 (Mayo Clinic). PEAR was also supported by funds from the Mayo Foundation.
Disclosures
None.
Supporting information
Table S1. Genome‐wide Significant SNPs From Genome‐wide Association Results for Glucose Change Among PEAR‐2 Blacks
Table S2. SNPs Which Met Suggestive Significance Level 1×10−6 in the Genome‐wide Association With Respect to Chlorthalidone‐Induced Glucose Change in PEAR‐2 White Participants
Table S3. Details of the Top Associations for Blacks in Whites
Table S4. Details of the Top Associations for Whites in Blacks
Table S5. Genome‐wide Significant Signals From the Meta‐Analysis of Chlorthalidone‐Induced Glucose Change in PEAR and HCTZ Induced Glucose Change in PEAR‐2 Black Participants
Table S6. SNPs Which Met Suggestive Significance Level 1×10−6 in the Meta‐Analysis of Chlorthalidone‐Induced Glucose Change in PEAR‐2 and HCTZ‐Induced Glucose Change in PEAR White Participants
Figure S1. Overall analyses flowchart of the study depicting the total number of participants that were excluded from the analysis, the final number of participants included in the analysis, and the various steps of the analysis framework.
Figure S2. Regional plot for rs9943291 (HMGCS2 gene) the GWAS of glucose change postchlorthalidone treatment.
Figure S3. Manhattan plots of glucose change postchlorthalidone treatment among PEAR‐2 White participants.
Acknowledgments
We thank the participants, staff, and study physicians of PEAR and PEAR‐2 study for their contributions.
(J Am Heart Assoc. 2018;7:e007339 DOI: 10.1161/JAHA.117.007339.)29523524
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 2. NCD Risk Factor Collaboration (NCD‐RisC) . Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population‐based measurement studies with 19.1 million participants. Lancet. 2016;389:37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. James PA, Oparil S, Carter BL, Cushman WC, Dennison‐Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC, Svetkey LP, Taler SJ, Townsend RR, Wright JT, Narva AS, Ortiz E. 2014 evidence‐based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507–520. [DOI] [PubMed] [Google Scholar]
- 4. Karnes JH, McDonough CW, Gong Y, Vo TT, Langaee TY, Chapman AB, Gums JG, Beitelshees AL, Bailey KR, Del‐Aguila JL, Boerwinkle EA, Pepine CJ, Turner ST, Johnson JA, Cooper‐DeHoff RM. Association of KCNJ1 variation with change in fasting glucose and new onset diabetes during HCTZ treatment. Pharmacogenomics J. 2013;13:430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karnes JH, Gong Y, Pacanowski MA, McDonough CW, Arwood MJ, Langaee TY, Pepine CJ, Johnson JA, Cooper‐Dehoff RM. Impact of TCF7L2 single nucleotide polymorphisms on hydrochlorothiazide‐induced diabetes. Pharmacogenet Genomics. 2013;23:697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gong Y, McDonough CW, Beitelshees AL, Karnes JH, O'Connell JR, Turner ST, Chapman AB, Gums JG, Bailey KR, Boerwinkle E, Johnson JA, Cooper‐DeHoff RM. PROX1 gene variant is associated with fasting glucose change after antihypertensive treatment. Pharmacotherapy. 2014;34:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karnes JH, Gong Y, Arwood MJ, Gums JG, Hall KL, Limacher MC, Johnson JA, Cooper‐DeHoff RM. Alteration in fasting glucose after prolonged treatment with a thiazide diuretic. Diabetes Res Clin Pract. 2014;104:363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cooper‐DeHoff RM. Thiazide‐induced dysglycemia: it's time to take notice. Expert Rev Cardiovasc Ther. 2008;6:1291–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carter BL, Einhorn PT, Brands M, He J, Cutler JA, Whelton PK, Bakris GL, Brancati FL, Cushman WC, Oparil S, Wright JT; Working Group from the National Heart, Lung, and Blood Institute . Thiazide‐induced dysglycemia: call for research from a working group from the National Heart, Lung, and Blood Institute. Hypertension. 2008;52:30–36. [DOI] [PubMed] [Google Scholar]
- 10. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial . Major outcomes in high‐risk hypertensive patients randomized to angiotensin‐converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288:2981–2997. [DOI] [PubMed] [Google Scholar]
- 11. Verdecchia P, Reboldi G, Angeli F, Borgioni C, Gattobigio R, Filippucci L, Norgiolini S, Bracco C, Porcellati C. Adverse prognostic significance of new diabetes in treated hypertensive subjects. Hypertension. 2004;43:963–969. [DOI] [PubMed] [Google Scholar]
- 12. Pepine CJ, Handberg EM, Cooper‐DeHoff RM, Marks RG, Kowey P, Messerli FH, Mancia G, Cangiano JL, Garcia‐Barreto D, Keltai M, Erdine S, Bristol HA, Kolb HR, Bakris GL, Cohen JD, Parmley WW; INVEST Investigators . A calcium antagonist vs a non‐calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil‐Trandolapril Study (INVEST): a randomized controlled trial. JAMA. 2003;290:2805–2816. [DOI] [PubMed] [Google Scholar]
- 13. Gorden P. Glucose intolerance with hypokalemia. Failure of short‐term potassium depletion in normal subjects to reproduce the glucose and insulin abnormalities of clinical hypokalemia. Diabetes. 1973;22:544–551. [DOI] [PubMed] [Google Scholar]
- 14. Rowe JW, Tobin JD, Rosa RM, Andres R. Effect of experimental potassium deficiency on glucose and insulin metabolism. Metabolism. 1980;29:498–502. [DOI] [PubMed] [Google Scholar]
- 15. Sica DA. Diuretic‐related side effects: development and treatment. J Clin Hypertens (Greenwich). 2004;6:532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zillich AJ, Garg J, Basu S, Bakris GL, Carter BL. Thiazide diuretics, potassium, and the development of diabetes: a quantitative review. Hypertension. 2006;48:219–224. [DOI] [PubMed] [Google Scholar]
- 17. Eriksson JW, Jansson PA, Carlberg B, Hägg A, Kurland L, Svensson MK, Ahlström H, Ström C, Lönn L, Ojbrandt K, Johansson L, Lind L. Hydrochlorothiazide, but not Candesartan, aggravates insulin resistance and causes visceral and hepatic fat accumulation: the mechanisms for the diabetes preventing effect of Candesartan (MEDICA) Study. Hypertension. 2008;52:1030–1037. [DOI] [PubMed] [Google Scholar]
- 18. Perez‐Stable E, Caralis PV. Thiazide‐induced disturbances in carbohydrate, lipid, and potassium metabolism. Am Heart J. 1983;106(1 Pt 2):245–251. [DOI] [PubMed] [Google Scholar]
- 19. Rosendorff C. Why are we still using hydrochlorothiazide? J Clin Hypertens (Greenwich). 2011;13:867–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson JA. Pharmacogenomic evaluation of antihypertensive responses (PEAR). NCBI's Database of Genotypes and Phenotypes: dbGaP. 2016. Accession phs000649.v1.p1.
- 21. Hamadeh IS, Langaee TY, Dwivedi R, Garcia S, Burkley BM, Skaar TC, Chapman AB, Gums JG, Turner ST, Gong Y, Cooper‐DeHoff RM, Johnson JA. Impact of CYP2D6 polymorphisms on clinical efficacy and tolerability of metoprolol tartrate. Clin Pharmacol Ther. 2014;96:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson JA, Boerwinkle E, Zineh I, Chapman AB, Bailey K, Cooper‐DeHoff RM, Gums J, Curry RW, Gong Y, Beitelshees AL, Schwartz G, Turner ST. Pharmacogenomics of antihypertensive drugs: rationale and design of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study. Am Heart J. 2009;157:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gong Y, Wang Z, Beitelshees AL, McDonough CW, Langaee TY, Hall K, Schmidt SO, Curry RW, Gums JG, Bailey KR, Boerwinkle E, Chapman AB, Turner ST, Cooper‐DeHoff RM, Johnson JA. Pharmacogenomic genome‐wide meta‐analysis of blood pressure response to β‐blockers in hypertensive African Americans. Hypertension. 2016;67:556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 25. Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome‐wide association studies through pre‐phasing. Nat Genet. 2012;44:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genome‐wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore MJ, Gong Y, Hou W, Hall K, Schmidt SO, Curry RW, Beitelshees AL, Chapman A, Turner ST, Schwartz GL, Bailey K, Boerwinkle E, Gums JG, Cooper‐DeHoff RM, Johnson JA. Predictors for glucose change in hypertensive participants following short‐term treatment with atenolol or hydrochlorothiazide. Pharmacotherapy. 2014;34:1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait‐associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Machiela MJ, Chanock SJ. LDlink: a web‐based application for exploring population‐specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics. 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, Zhernakova A, Zhernakova DV, Veldink JH, Van den Berg LH, Karjalainen J, Withoff S, Uitterlinden AG, Hofman A, Rivadeneira F, Hoen PA, Reinmaa E, Fischer K, Nelis M, Milani L, Melzer D, Ferrucci L, Singleton AB, Hernandez DG, Nalls MA, Homuth G, Nauck M, Radke D, Völker U, Perola M, Salomaa V, Brody J, Suchy‐Dicey A, Gharib SA, Enquobahrie DA, Lumley T, Montgomery GW, Makino S, Prokisch H, Herder C, Roden M, Grallert H, Meitinger T, Strauch K, Li Y, Jansen RC, Visscher PM, Knight JC, Psaty BM, Ripatti S, Teumer A, Frayling TM, Metspalu A, van Meurs JB, Franke L. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cooper‐DeHoff RM, Wen S, Beitelshees AL, Zineh I, Gums JG, Turner ST, Gong Y, Hall K, Parekh V, Chapman AB, Boerwinkle E, Johnson JA. Impact of abdominal obesity on incidence of adverse metabolic effects associated with antihypertensive medications. Hypertension. 2010;55:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sales V, Patti ME. The ups and downs of insulin resistance and type 2 diabetes: lessons from genomic analyses in humans. Curr Cardiovasc Risk Rep. 2013;7:46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Younossi ZM, Baranova A, Ziegler K, Del Giacco L, Schlauch K, Born TL, Elariny H, Gorreta F, VanMeter A, Younoszai A, Ong JP, Goodman Z, Chandhoke V. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology. 2005;42:665–674. [DOI] [PubMed] [Google Scholar]
- 39. Younossi ZM, Gorreta F, Ong JP, Schlauch K, Del Giacco L, Elariny H, Van Meter A, Younoszai A, Goodman Z, Baranova A, Christensen A, Grant G, Chandhoke V. Hepatic gene expression in patients with obesity‐related non‐alcoholic steatohepatitis. Liver Int. 2005;25:760–771. [DOI] [PubMed] [Google Scholar]
- 40. Arias G, Asins G, Hegardt FG, Serra D. The effect of fasting/refeeding and insulin treatment on the expression of the regulatory genes of ketogenesis in intestine and liver of suckling rats. Arch Biochem Biophys. 1997;340:287–298. [DOI] [PubMed] [Google Scholar]
- 41. Arias G, Matas R, Asins G, Hegardt FG, Serra D. The effect of fasting and insulin treatment on carnitine palmitoyl transferase I and mitochondrial 3‐hydroxy‐3‐methylglutaryl coenzyme A synthase mRNA levels in liver from suckling rats. Biochem Soc Trans. 1995;23:493S. [DOI] [PubMed] [Google Scholar]
- 42. Quant PA, Tubbs PK, Brand MD. Glucagon activates mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur J Biochem. 1990;187:169–174. [DOI] [PubMed] [Google Scholar]
- 43. Thumelin S, Forestier M, Girard J, Pegorier JP. Developmental changes in mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase gene expression in rat liver, intestine and kidney. Biochem J. 1993;292:493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodríguez JC, Gil‐Gómez G, Hegardt FG, Haro D. Peroxisome proliferator‐activated receptor mediates induction of the mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase gene by fatty acids. J Biol Chem. 1994;269:18767–18772. [PubMed] [Google Scholar]
- 45. Koh EH, Kim MS, Park JY, Kim HS, Youn JY, Park HS, Youn JH, Lee KU. Peroxisome proliferator‐activated receptor (PPAR)‐alpha activation prevents diabetes in OLETF rats: comparison with PPAR‐gamma activation. Diabetes. 2003;52:2331–2337. [DOI] [PubMed] [Google Scholar]
- 46. Cotter DG, Ercal B, Huang X, Leid JM, d'Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA. Ketogenesis prevents diet‐induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124:5175–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson JR, Bell JK, Bratt J, Grant GA, Banaszak LJ. Vmax regulation through domain and subunit changes. The active form of phosphoglycerate dehydrogenase. Biochemistry. 2005;44:5763–5773. [DOI] [PubMed] [Google Scholar]
- 48. Ginger RS, Askew SE, Ogborne RM, Wilson S, Ferdinando D, Dadd T, Smith AM, Kazi S, Szerencsei RT, Winkfein RJ, Schnetkamp PP, Green MR. SLC24A5 encodes a trans‐Golgi network protein with potassium‐dependent sodium‐calcium exchange activity that regulates human epidermal melanogenesis. J Biol Chem. 2008;283:5486–5495. [DOI] [PubMed] [Google Scholar]
- 49. Sharon D, Yamamoto H, McGee TL, Rabe V, Szerencsei RT, Winkfein RJ, Prinsen CF, Barnes CS, Andreasson S, Fishman GA, Schnetkamp PP, Berson EL, Dryja TP. Mutated alleles of the rod and cone Na‐Ca+K‐exchanger genes in patients with retinal diseases. Invest Ophthalmol Vis Sci. 2002;43:1971–1979. [PubMed] [Google Scholar]
- 50. Hughes AD. How do thiazide and thiazide‐like diuretics lower blood pressure? J Renin Angiotensin Aldosterone Syst. 2004;5:155–160. [DOI] [PubMed] [Google Scholar]
- 51. Maranville JC, Cox NJ. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J. 2016;16:388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome‐wide Significant SNPs From Genome‐wide Association Results for Glucose Change Among PEAR‐2 Blacks
Table S2. SNPs Which Met Suggestive Significance Level 1×10−6 in the Genome‐wide Association With Respect to Chlorthalidone‐Induced Glucose Change in PEAR‐2 White Participants
Table S3. Details of the Top Associations for Blacks in Whites
Table S4. Details of the Top Associations for Whites in Blacks
Table S5. Genome‐wide Significant Signals From the Meta‐Analysis of Chlorthalidone‐Induced Glucose Change in PEAR and HCTZ Induced Glucose Change in PEAR‐2 Black Participants
Table S6. SNPs Which Met Suggestive Significance Level 1×10−6 in the Meta‐Analysis of Chlorthalidone‐Induced Glucose Change in PEAR‐2 and HCTZ‐Induced Glucose Change in PEAR White Participants
Figure S1. Overall analyses flowchart of the study depicting the total number of participants that were excluded from the analysis, the final number of participants included in the analysis, and the various steps of the analysis framework.
Figure S2. Regional plot for rs9943291 (HMGCS2 gene) the GWAS of glucose change postchlorthalidone treatment.
Figure S3. Manhattan plots of glucose change postchlorthalidone treatment among PEAR‐2 White participants.