

Abstract
Background
Platelet‐derived growth factor BB, a potent mitogen of pulmonary artery smooth muscle cells (PASMCs), has been implicated in pulmonary arterial remodeling, which is a key pathogenic feature of pulmonary arterial hypertension. Previous microRNA profiling in platelet‐derived growth factor BB–treated PASMCs found a significantly downregulated microRNA, miR‐1281, but it has not been associated with any cellular function, and we investigated the possibility.
Methods and Results
Real‐time quantitative reverse transcription–polymerase chain reaction assay proved that downregulation of miR‐1281 was a conserved phenomenon in human and rat PASMCs. Overexpression and inhibition of miR‐1281 in PASMCs promoted and suppressed, respectively, the cell proliferation and migration. Bioinformatic prediction and 3′‐untranslated region reporter assay identified histone deacetylase 4 to be a direct target of miR‐1281. Supporting this, proliferation and migration assay demonstrated the cellular function of histone deacetylase 4 is inversely correlated with that of miR‐1281. Mechanistically, it is found that platelet‐derived growth factor BB activates the phosphatidylinositol 3‐kinase pathway, which then induces the expression of DNA methyltransferase 1, leading to enhanced methylation of a flanking CpG island and repressed miR‐1281 expression. Finally, a reduced miR‐1281 level was consistently identified in hypoxic PASMCs in vitro, in pulmonary arteries of rats with monocrotaline‐induced pulmonary arterial hypertension, and in serum of patients with coronary heart disease–pulmonary arterial hypertension. These data suggest that there may be a diagnostic and therapeutic use for miR‐1281.
Conclusions
Herein, we report a novel regulatory axis, phosphatidylinositol 3‐kinase–DNA methyltransferase 1–miR‐1281–histone deacetylase 4, integrating multiple epigenetic regulators that participate in platelet‐derived growth factor BB–stimulated PASMC proliferation and migration and pulmonary vascular remodeling.
Keywords: DNA methyltransferase 1, epigenetics, histone deacetylase 4, hypertension, miRNA, platelet‐derived growth factor, pulmonary, pulmonary arterial smooth muscle cells, vascular remodeling, vascular smooth muscle
Subject Categories: Vascular Disease, Pulmonary Hypertension, Epigenetics, Growth Factors/Cytokines, Smooth Muscle Proliferation and Differentiation
Clinical Perspective
What Is New?
Platelet‐derived growth factor BB–induced repression of miR‐1281 is a conserved phenomenon in human and rat pulmonary artery smooth muscle cells.
miR‐1281 inhibits pulmonary artery smooth muscle cell proliferation and migration, at least in part through targeting histone deacetylase 4.
Phosphatidylinositol 3‐kinase–DNA methyltransferase 1–miR‐1281–histone deacetylase 4 is an integrated regulatory axis mediating platelet‐derived growth factor BB function.
Expression of miR‐1281 is consistently reduced in hypoxic pulmonary artery smooth muscle cells, rats with monocrotaline‐induced pulmonary arterial hypertension, and patients with coronary heart disease–pulmonary arterial hypertension.
What Are the Clinical Implications?
Inhibiting the activity of platelet‐derived growth factor receptor is an effective approach reversing pulmonary arterial hypertension–related pulmonary artery smooth muscle cell proliferation and vascular remodeling; identifying the molecular agents mediating platelet‐derived growth factor function is critical for seeking novel therapeutic targets.
DNA methyltransferases, microRNA, and histone deacetylases are important epigenetic agents regulating the pulmonary arterial hypertension–proliferative phenotype; their interactions and integrated biological roles are barely understood.
Introduction
Pulmonary arterial hypertension (PAH), manifested by a maladaptive elevation in pulmonary arterial pressure, is a progressive lung vascular disorder that causes occlusion of the pulmonary vascular lumen, increased pulmonary vascular resistance, right‐sided heart failure, and eventual death.1, 2 Because of deficiencies in the present knowledge of PAH pathogenesis, effective therapies are limited and the mortality rate in the patient population is still high.3, 4, 5 Hence, there is a need for a better understanding of the molecular pathogenesis of this disease.
Pulmonary arterial remodeling is an important pathologic hallmark of PAH,6, 7 and excessive proliferation and migration of pulmonary artery smooth muscle cells (PASMCs) is thought to be a major cause of the vascular remodeling.8, 9 Platelet‐derived growth factor (PDGF), a potent mitogen of PASMCs,2, 10, 11 with PDGFBB being the key homodimer isoform, plays a critical role in vascular remodeling.12, 13 It is known that, in the pulmonary artery, PDGFBB is synthesized by both endothelial cells and fibroblasts and it can act in a paracrine manner on PASMCs in the medial layer of the vessel wall. It induces PASMCs to switch from a contractile phenotype to a proliferative and migratory phenotype, which then leads to medial vessel wall remodeling.14, 15, 16 It is also known that in settings of experimental PAH, expression of PDGFBB and phosphorylation of PDGF receptor are increased in lungs with PAH and lung vascular cells2, 17, 18; the administration of the PDGF receptor inhibitor, imatinib mesylate, has been shown to significantly reverse pulmonary vascular remodeling in animal models of PAH.19, 20 Consistent with these findings, high levels of PDGFBB in lung tissues of patients with PAH have correlated with the severity of PAH.21, 22 Hence, the role of PDGF in PAH pathogenesis deserves greater attention, and this study elucidating the downstream regulatory mechanisms of PDGF is highly relevant.
MicroRNAs (miRNAs) are noncoding RNAs of ≈19 to 25 nucleotides that negatively regulate gene expression at the posttranscriptional and/or translational level.23 Downregulation of specific miRNAs is a common feature of proliferative diseases, such as cancer, and identification of miRNA targets with critical functions supports the role of corresponding miRNAs as tumor suppressors.24, 25, 26 At this interface, miRNAs are emerging as a class of therapeutic agents that can control disease in a propathogenic or antipathogenic manner.27 Recently, using miRNA profiling techniques, many miRNAs have been identified to have altered expression in patients with PAH28, 29, 30 and in PAH animal models,31, 32, 33, 34 but the great variation in these screening results requires cautious analysis of the function and mechanisms involved of each identified miRNA candidate. To date, the roles of a few miRNAs in mediating differentiation, migration, and/or proliferation of PASMCs have been reported28, 35, 36, 37, 38; however, more complex studies are required to explore the potential pathogenic or therapeutic roles of PAH‐dysregulated miRNAs.
In a recent study, we focused on the identification of PDGFBB‐regulated miRNAs and their functions related to PASMC proliferation and migration. By profiling miRNA expression in PDGFBB‐stimulated human PASMCs, we found 15 miRNAs that were significantly dysregulated, among which miR‐1281 was 1 of the 3 most downregulated miRNAs.39 miR‐1281 is a relatively less conserved miRNA annotated only in 3 placental mammals (ie, humans, chimpanzees, and cattle; http://www.ncbi.nlm.nih.gov/gene/?term=miR-1281) and is only associated with a few diseases as a putative biomarker.40, 41, 42 In this study, we found that the position and sequence of miR‐1281 are conserved between humans and rodents and, therefore, took advantage of this to closely investigate the function and regulatory mechanism of miR‐1281 action in rat PASMCs. As a result, the epigenetic regulators DNA methyltransferase 1 (DNMT1) and histone deacetylase 4 (HDAC4) were identified as agents that form a regulatory axis with miR‐1281 (ie, phosphatidylinositol 3‐kinase [PI3K]–DNMT1–miR‐1281–HDAC4) and that function downstream of PDGFBB to control PASMC proliferation and migration.
Methods
The data, analytic methods, and study materials will be available, on request, from Shenzhen University (Shenzhen, China).
Monocrotaline‐Induced PAH Model in Rats
Sprague‐Dawley rats were obtained from the Medical and Experimental Animal Center of Gongdong Province (China). Adult male rats (weight, 250–300 g; age, 10–12 weeks) were used for experiments. All animal procedures conformed to the Guide for Care and Use of Laboratory Animals, published by the US National Institutes of Health (8th edition, 2011), and were approved by the Animal Care and Use Committee of Shenzhen University.
PAH was induced in Sprague‐Dawley rats (10 per experimental group) by a single IP injection with monocrotaline (60 mg/kg; Sigma‐Aldrich, St Louis, MO). Pulmonary arteries collected from rats with confirmed monocrotaline‐induced PAH and healthy control rats were used for experiments (Figure S12).
Human Subjects
Serum samples were collected from healthy infants and infants with coronary heart disease–PAH (including ventricular septal defect or atrial septal defect) at Fuwai Hospital (Beijing, China; refer to Table S1 for detailed participant information). The blood samples were centrifuged at 3000g for 10 minutes at 4°C and stored at −80°C until use. This study was approved by the ethics committee of Fuwai Hospital, with the requirement for informed consent waived.
Cell Culture Experiments
Human embryonic kidney 293T and 293A cells were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% fetal bovine serum. Human pulmonary arterial smooth muscle cells (PASMCs) were purchased from Sciencell, San Diego, CA and cultured in smooth muscle cell medium, as previously described.43 Rat PASMCs were isolated and cultured in DMEM supplemented with 10% fetal bovine serum, as previously described.34 The purity of rat PASMCs was checked by immunofluorescence staining with α‐smooth muscle actin (Sigma‐Aldrich, Burlington, MA). PASMCs at passage 3 or 4 were used for experiments.
For treatment with growth factors, PASMCs were prestarved for 12 hours (0.2% fetal bovine serum) and then treated with growth factors (angiotensin 2 [100 ng/mL], endothelin 1 [25 ng/mL], β‐fibroblast growth factor [20 ng/mL], insulin‐like growth factor‐1 [20 ng/mL], PDGFAA [20 ng/mL], PDGFBB [20 ng/mL], transforming growth factor‐β [20 ng/mL], and vascular endothelial growth factor [VEGF; 20 ng/mL]) (R&D Systems, Minneapolis, MN) for different periods of time, as indicated. For kinase or DNMT inhibition, starved cells were pretreated with respective inhibitors (imatinib [5 μmol/L], SP600125 [10 μmol/L], SCH772984 [20 μmol/L], enzastaurin [5 μmol/L], saracatinib [0.1 μmol/L], and pictilisib [1 μmol/L]) (Selleck Chemical, Houston, TX) for 1 hour or with 5‐aza‐2′‐deoxycytidine (Sigma‐Aldrich, Burlington, MA) for 24 hours, and then stimulated with PDGFBB (30 ng/mL).
Vectors, Transfection, and Infection
For construction of lentiviral short hairpin RNA vector, each short hairpin RNA was prepared by 2 primers annealed and cloned into a pLVX‐Puro backbone (Clontech, Mountain View, CA; refer to Kang et al37). For construction of luciferase vectors, a 3′‐untranslated region (UTR) fragment of protein tyrosine phosphate, reporter type, D (PTPRD), HDAC4, and wingless‐type MMTV integration site family member 3A (WNT3A) was amplified from human genomic DNA and cloned into pmirGLO dual‐luciferase vector (Promega, Madison, WI). For corresponding mutational 3′‐UTR reporter vectors, the region that base paired with miR‐1281 seeding sequences was mutated by site‐directed mutagenesis. Sequences of primers used for vector construction are summarized in Table S2. All constructs were confirmed by DNA sequencing.
Transfection of DNA plasmids into 293T or 293A cells was performed with polyethylenimine (Geneups, China), as previously reported.44 High‐titer lentivirus was generated through a Lenti‐X HT packaging system in 293T cells, according to manufacturer's instruction (Clontech, Mountain View, CA).
Transfection of miRNA Mimic, Inhibitor, and Small Interfering RNA Oligonucleotides
miR‐1281 mimic, inhibitor, and their corresponding negative controls, small interfering (si)‐EGR1, si‐HDAC4, si‐DNMT1, and the negative control si‐negative control were chemically synthesized by Ribobio (China). The siRNA sense sequences designed were as follows: EGR1, 5′‐CCAACAGTGGCAACACTTT‐3′; si‐HDAC4, 5′‐GGATGAGCCCTACCTAGAT‐3′; and si‐DNMT1, 5′‐TATTGGTGCATACTCTGGGCT‐3′.
Cell Proliferation and Migration Assays
Cell proliferation was assessed by 5‐Ethynyl‐2′‐deoxyuridine (EdU) incorporation. 5‐Ethynyl‐2′‐deoxyuridine labeling was performed using an EdU Assay Kit (Ribobio), according to manufacturer's instruction. The stained cells were examined using a fluorescent‐inverted microscope (Nikon, Tokyo, Japan). Cell proliferation rate was calculated as the number of EdU‐stained cells/the number of 4′,6‐diamidino‐2‐phenylindole–stained cells.
Cell migration was determined by the wound‐healing assay, as reported previously.44 In brief, cells were maintained in starvation condition for 12 hours, then a line scratch was made in the cell layer and photographed immediately as initial image (0 hour). The second image (24 hours) of the same line scratch was taken after 24 hours of culturing, and cell migration was determined by measuring the decreased width of corresponding scratch from 0 to 24 hours.
RNA Isolation and Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was extracted with RNAiso Plus (Takara, China) and quantified using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). For quantification of mRNA expression, 500 ng total RNA was reverse transcribed using M‐MLV Reverse Transcriptase (Takara) with oligo(dT)18 plus random hexamer primers (Promega, Madison, WI). Real‐time quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) was performed with gene‐specific primers (Table S2) and SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA) on an ABI StepOne real‐time PCR System (Applied Biosystems, Waltham, MA). The expression level of each gene was normalized to internal control β‐ACTIN gene and calculated using the 2(−ΔΔCT) method. For quantification of miR‐1281 expression, S‐Poly (T) Plus real‐time PCR method was applied with the protocol, as detailed in our previous work.45, 46 Small nucleolar RNA‐44 and small nucleolar RNA‐202 were used as reference genes for human and rat PASMC miRNAs, respectively. Primer sequences for miRNA retrotranscription and qRT‐PCR were summarized in Table S2.
Protein Preparation and Western Blot
Total proteins were extracted using ice‐cold radioimmunoprecipitation assay buffer (50 mmol/L Tris‐HCl, pH 7.5; 150 mmol/L NaCl; 1% NP‐40; 0.25% sodium deoxycholate; and 1 mmol/L EDTA), supplemented with a protease inhibitor cocktail (Sigma‐Aldrich, Burlington, MA). The samples were homogenized and centrifuged for 10 minutes at 12 000g at 4°C. The resulting supernatant was collected and used for protein concentration determination by bicinchoninic acid assay protein kit (Thermo Scientific, Waltham, MA). Equal amounts of proteins (10–20 μg) were electrophoresed on an SDS polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Merck Millipore, Bedford, MA). The membranes were blocked for 2 hours at room temperature in Tris‐buffered saline with 0.1% Tween 20, containing 5% nonfatted dry milk (blocking solution). After that, the membrane was incubated with antibodies against α‐smooth muscle actin (Sigma‐Aldrich, Burlington, MA), β‐tubulin and Smooth Muscle (SM)‐22α (Abcam, Cambridge, MA), HDAC4 and DNMT1 (Proteintech, China), and β‐actin and Smoothelin (Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C. For histone acetylation analysis, acetyl‐histone antibody sampler kits (No. 9927, 8346, and 9933; Cell Signaling Technology, Beverly, MA) provided the antibodies against H3, H3K9ac, H3K18ac, H3K54ac, H4, H4K5ac, H4K8ac, H4K12ac, H2A, H2AK5ac, H2B, and H2BK5ac. After 3×5 minute washes in Tris‐buffered saline with 0.1% Tween 20 solution, the membrane was incubated for 1 hour at room temperature with horseradish peroxidase–conjugated secondary antibody (Jackson Immuno‐Research, PA). The protein bands were visualized by the SuperSignal chemiluminescent detection module (Pierce, TX) under x‐ray film.
Luciferase Assay
Approximately 1×104 293A cells were seeded in quadruplicate in 48‐well plates, and they were cotransfected with dual‐luciferase reporter plasmids together with either miR‐1281 or control mimics. The cells were lysed for luciferase activity measurement using a Lumat LB9508 luminometer (Berthold, Wildbad, Germany) after 48 hours of transfection. The firefly luciferase activity expressed in the same vector was used as an internal control for normalization.
DNA Methylation Assay
Genomic DNA was isolated from rat PASMCs treated with PDGFBB or dimethyl sulfoxide for 72 hours. Approximately 1 μg of DNA from each sample was bisulfite converted and cleaned up using EpiTect Bisulfite kit (Qiagen, Hilden, Germany), following the manufacturer's instruction. Nested PCR was used to amplify the CpG island from bisulfite‐treated DNA samples (primer sequences are summarized in Table S2), with the PCR setup described by Wang et al.47 The PCR product was gel purified and sequenced directly with BigDye 3.1 using primer CpG‐F2. For cytosine methylation analysis, a simple method verified and recommended by Finn et al48 was adopted (namely, BioEdit software was used to view sequencing results and obtain the sequencing trace file). The ratios of trace values (cytosine [C] verses thymine [T] plus cytosine [C/(T+C)]) were calculated as the percentage of cytosine being methylated in every CG site.
Statistical Analysis
Statistical analyses were performed using the SPSS version 13.0 software 261 package (SPSS) for Windows. All data are presented as mean values±SD of 3 experiments (for proliferation and migration assays, data were extracted from 5 and 6 batches of experiments, respectively). When only 2 groups were compared, the statistical differences were evaluated with the double‐sided Student t test. Significant differences between groups were analyzed by 1‐way ANOVA, taking P<0.05 as a significant difference.
Results
miR‐1281 Is Downregulated by PDGFBB in PASMCs
Previously, we identified that miR‐1281 is downregulated by PDGFBB treatment in human PASMCs.39 According to the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov/gene/100302237), miR‐1281 originates from the upstream region of EP300 5′‐UTR in the human genome (EP300: E1A binding protein p300, a lysine acetyltransferase). Through a sequence alignment (Figure 1A), the location and sequence of the MIR‐1281 gene were highly conserved among human, rat, and mouse genomes, with them differing only at 1 nucleotide outside the mature miR‐1281 sequence. Hence, all experiments have been done with rat PASMCs.
Figure 1.
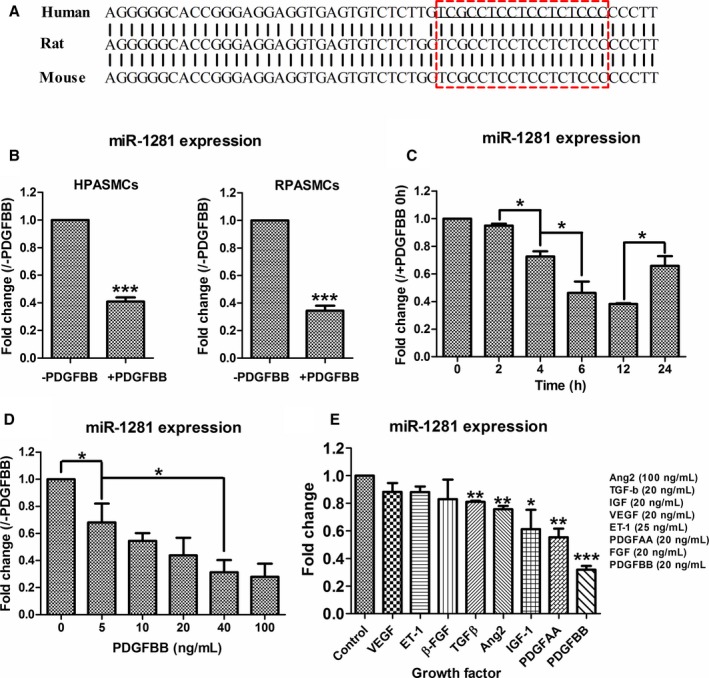
miR‐1281 is downregulated in platelet‐derived growth factor (PDGF) BB–stimulated pulmonary artery smooth muscle cells (PASMCs). A, Sequence of human miR‐1281 precursor was aligned with corresponding sequences 5′ upstream of gene EP300 in rat and mouse genomes. Red box indicates the region generating mature miR‐1281 (underlined). B, Real‐time quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) measurements of miR‐1281 level in human and rat PASMCs (HPASMCs and RPASMCs, respectively) treated with dimethyl sulfoxide (DMSO; ‐PDGFBB) or PDGFBB. C, Time‐course assay of PDGFBB effect on miR‐1281 level in rat PASMCs. Significant difference was analyzed for each of 2 neighboring time points and marked by asterisks and broken lines. D, Dosage‐course assay of PDGFBB effect on miR‐1281 level in rat PASMCs. Significant differences were analyzed for each of 2 neighboring dosage points and as broken line indicated. E, Effects of different growth factors on PASMC miR‐1281 expression were evaluated by qRT‐PCR. DMSO treatment was used as negative control. Angiotensin 2 (Ang2), transforming growth factor‐β (TGF‐β), insulin‐like growth factor (IGF), vascular endothelial growth factor (VEGF), endothelin 1 (ET‐1), PDGFAA, and fibroblast growth factor (FGF) were applied at concentrations as indicated. For this and all subsequent qRT‐PCR data: miRNA levels are normalized with small nucleolar 202 or small nucleolar 44 for rat or human samples, respectively, and mRNA levels are normalized with β‐actin. Fold changes are calculated by comparing each normalized measurement with normalized corresponding control. Data are expressed as means±SD, verified by at least 3 biological replicates. *P<0.05, **P<0.01, ***P<0.001 compared with control or as specified by broken lines in respective figures. Exact P values in consecutive order are: 0.000599, 0.000926, 0.016403, 0.044714, 0.030503,0.040582, 0.047031, 0.001946, 0.009166, 0.033907, 0.002544, 0.000013.
The repressing effect of PDGFBB on miR‐1281 in PASMCs was revalidated in both human and rat PASMCs (downregulated by ≈60%; Figure 1B). Further sets of qRT‐PCR revealed that the expression level of miR‐1281 was inversely correlated with time (within 12 hours; Figure 1C) and concentration (<100 ng/mL; Figure 1D) of PDGFBB treatment. When other growth factors (ie, VEGF, endothelin 1, β‐fibroblast growth factor, transforming growth factor‐β, angiotensin 2, insulin‐like growth factor‐1, PDGFAA, and PDGFBB) were applied to PASMCs, most of them did not significantly alter miR‐1281 expression; however, PDGF (especially PDGFBB) significantly repressed miR‐1281 expression (Figure 1E), indicating a major role for miR‐1281 in PASMCs, acting downstream of PDGF.
Expression of miR‐1281 Suppresses the Proliferative and Migratory Phenotype of PASMCs
To examine if miR‐1281 modulates PASMC phenotype, miR‐1281 mimic was transfected into PASMCs, which led to an increase of miR‐1281 steady‐state level by almost 200‐fold (Figure 2A). Using an EdU incorporation assay, the miR‐1281 mimic repressed PASMC proliferation by ≈30%, with or without added PDGFBB (Figure 2B), indicating that miR‐1281 can directly suppress PASMC proliferation and attenuate PDGFBB‐stimulated proliferation. To confirm these data, miR‐1281 inhibitor was used to inhibit endogenous miR‐1281, which decreased the steady‐state level of miR‐1281 by 75% (Figure 2C); this was greater than the maximum extent of repression achieved by treatment with PDGFBB (Figure 1D). Transfection of miR‐1281 inhibitor significantly increased PASMC proliferation (Figure 2D), supporting the role of miR‐1281 as an inhibitor of PASMC proliferation.
Figure 2.
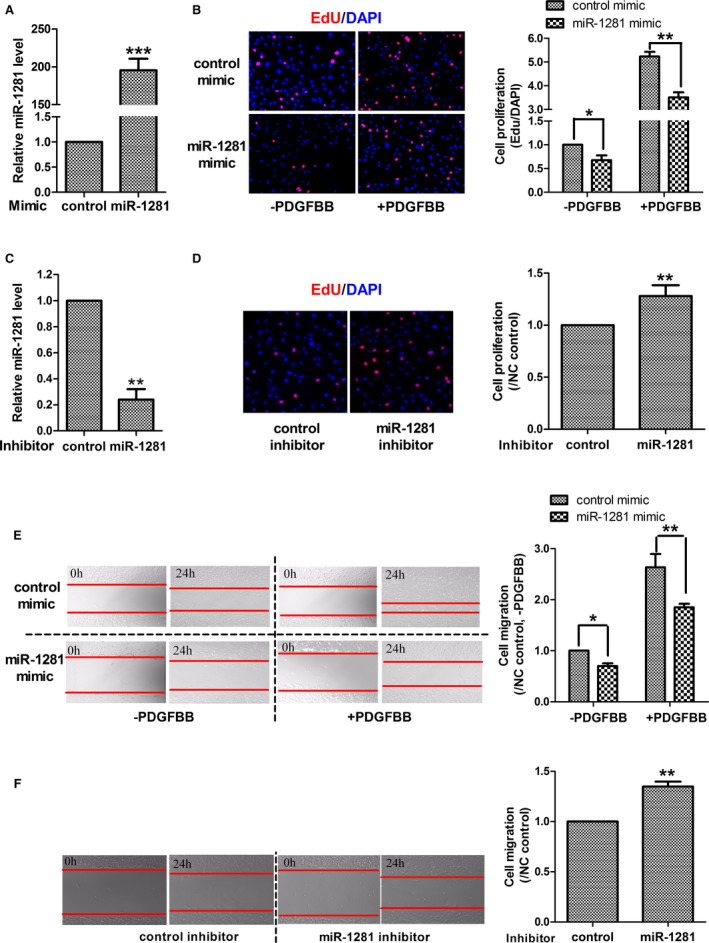
miR‐1281 suppresses pulmonary artery smooth muscle cell (PASMC) proliferation and migration. A, Real‐time quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) analysis of overexpression level of miR‐1281 in PASMCs achieved by transfection of miR‐1281 mimic. B, Representative figures of EdU assay and statistical analysis showing that miR‐1281 mimic inhibited PASMC proliferation under culturing conditions without and with platelet‐derived growth factor (PDGF) BB treatments. C, qRT‐PCR analysis of inhibitory efficiency of miR‐1281 inhibitor in PASMCs. D, Representative figures of EdU assay and statistical analysis showing that miR‐1281 inhibitor promoted PASMC proliferation. E, Representative figures of wound‐healing assay and statistical analysis showing that miR‐1281 mimic inhibited PASMC migration under culturing conditions without and with PDGFBB treatments. F, Representative figures of wound‐healing assay and statistical analysis showing that miR‐1281 inhibitor promoted PASMC migration. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; and NC, negative control. *P<0.05, **P<0.01, ***P<0.001 compared with control or as specified by broken lines in respective figures. Exact P values in consecutive order are: 0.00045, 0.012642, 0.001155, 0.001081, 0.009504,0.03544, 0.003835, 0.002613.
Next, the wound‐healing assay was performed to assess the effect of miR‐1281 on cell migration. Transfection of miR‐1281 mimic significantly suppressed PASMC migration both with and without PDGFBB treatments (Figure 2E). Complementing these findings are the results with an miR‐1281 inhibitor, in which the inhibition of miR‐1281 promoted PASMC migration (Figure 2F). Taken together, these data indicate that miR‐1281 represses PASMC proliferation and migration and that this is one mechanism by which PDGFBB induces PASMC proliferation and migration and vessel wall remodeling.
To see if miR‐1281 influences other PASMC phenotypes, we analyzed the expression of cell contractile marker proteins (ie, Smoothelin, α‐smooth muscle actin, and SM‐22α) via Western blotting; we did not find a significant change in miR‐1281 mimic‐transfected PASMCs in comparison with control mimic‐transfected PASMCs (Figure S1). This suggests that miR‐1281 has no influence on the contractile phenotype of PASMCs.
miR‐1281 Targets HDAC4 in PASMCs
According to the targetscan webserver (release 6.2; http://www.targetscan.org/cgi-bin/targetscan/vert_61/targetscan.cgi?species=Human&gid=&mir_sc=&mir_c=&mir_nc=miR-1281&mirg=), 12 transcript targets were predicted to carry conserved binding sites for miR‐1281, where 3 are annotated to play roles in diverse cellular processes, including cell proliferation and migration (namely HDAC4,49 PTPRD,50 and WNT3A).51 Hence, we determined if HDAC4, PTPRD, and WNT3A are directly targeted by miR‐1281. First, each 3′‐UTR fragment of these genes carrying miR‐1281 binding site (Figure 3A) was cloned into a luciferase reporter plasmid, and 3′‐UTR luciferase assays were conducted. As shown in Figure 3B, compared with control mimic, cotransfection of miR‐1281 mimic with either PTPRD–3′‐UTR or HDAC4–3′‐UTR reporter plasmids displayed significantly reduced luciferase activity, which was not observed for the cotransfection event of miR‐1281 mimic and WNT3A–3′‐UTR plasmids. This suggests that only HDAC4 and PTPRD are direct targets of miR‐1281, but WNT3A is not. Supporting this, it was demonstrated that the miR‐1281–induced repression could be abolished by mutating the miR‐1281 binding sites in HDAC4 3′‐UTR and PTPRD 3′‐UTR (Figure 3C and Figure S2A), suggesting that 3′‐UTR fragments of these 2 genes contain functional binding sites of miR‐1281 seed region.
Figure 3.
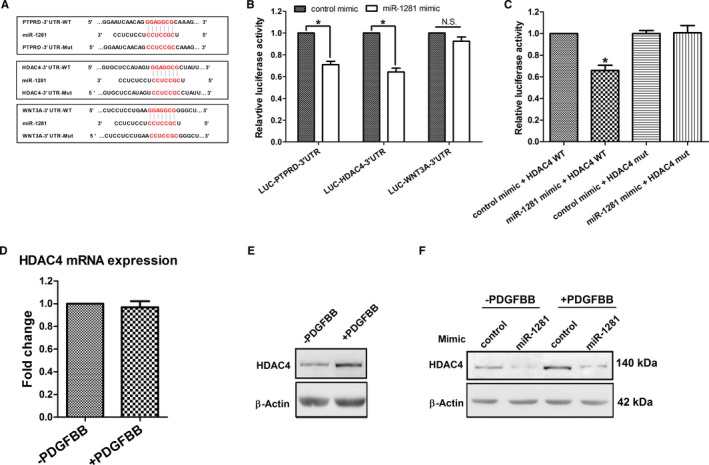
Histone deacetylase 4 (HDAC4) is a direct target gene of miR‐1281 in pulmonary artery smooth muscle cells (PASMCs). A, Alignment of miR‐1281 with its putative binding sites in 3′‐untranslated regions (UTRs) of protein tyrosine phosphate, recepter type, D (PTPRD), HDAC4, and wingless‐type MMTV integration site family member 3A (WNT3A) (3′‐UTR–wild type [WT]), and with corresponding mutated 3′‐UTR binding sites (3′‐UTR Mut). B, Respective 3′‐UTR–WT construct was cotransfected with either miR‐1281 mimic or control mimic into human embryonic kidney 293A cells, and luciferase reporter assay was performed to assess interactions between miR‐1281 and 3′‐UTRs of PTPRD, HDAC4, and WNT3A. C, Abolishing the interaction by cotransfecting miR‐1281 mimic with HDAC4 3′‐UTR Mut recovered miR‐1281 inhibition of luciferase expression. D, mRNA level of HDAC4 stayed unchanged in platelet‐derived growth factor (PDGF) BB–treated PASMCs. E, Protein level of HDAC4 increased in PDGFBB‐treated PASMCs. (F) Protein level of HDAC4 was downregulated by miR‐1281 mimic in PASMCs in the absence or presence of PDGFBB. β‐Actin was used as loading control. N.S. indicates not significant. *P<0.05 compared with control mimic. Exact P values in consecutive order are: 0.011313, 0.010948, 0.011313.
Next, to check if expression of HDAC4 and PTPRD are regulated by miR‐1281 in PDGFBB‐stimulated PASMCs, the mRNA and/or protein levels of HDAC4 and PTPRD in response to PDGFBB were analyzed. As shown in Figure S2B, mRNA level of PTPRD was reduced in response to PDGFBB, which disagreed with an expectation of target deregulation caused by PDGFBB‐repressed miR‐1281. This finding indicates that in PASMCs, PTPRD may not be regulated primarily by miR‐1281, but some other PDGF‐related regulatory mechanism may be involved. In contrast, mRNA level of HDAC4 stayed unchanged, whereas the protein level increased significantly (Figure 3D and 3E), which is consistent with the PDGFBB‐induced repression of miR‐1281. Further verification was performed by transfecting miR‐1281 mimic into PASMCs, and the reduction of HDAC4 protein level was observed both with and without PDGFBB treatment (Figure 3F). Therefore, these data demonstrate that, in PASMCs, miR‐1281 regulates HDAC4 expression at the translational level; they also show that PDGFBB‐induced suppression of miR‐1281 would result in deregulated expression of HDAC4 protein.
Cellular Function of HDAC4 Inversely Correlates With Expression of miR‐1281
We investigated if HDAC4 mediates the effects of miR‐1281. To address this, shHDAC4 was constructed and transducted into PASMCs via lentiviral infection, which achieved ≈80% inhibition of HDAC4 protein level (Figure S3A). An EdU incorporation assay showed that shHDAC4 infection suppressed PASMC proliferation either with or without PDGFBB treatment (Figure S3B); a wound‐healing assay proved that shHDAC4 infection also suppressed PASMC migration (Figure S3C). On the basis of these observations, we can infer that expression of HDAC4 promotes proliferation and migration of PASMCs and contributes to PDGFBB‐induced cell dysfunction, which is inversely correlated with the cellular function of miR‐1281.
miR‐1281/HDAC4 Module Acts by Influencing Activities of Related Transcription Factors
To determine the mode of action of HDAC4, 2 possible regulatory pathways of miR‐1281/HDAC4 pathway were analyzed. First, by transfecting mimic miR‐1281 into PASMCs, we tested if the expression of miR‐1281 influenced the histone acetylation status of PASMCs. However, we found that overexpression of miR‐1281 failed to significantly enhance the acetylation status of histone H3, H4, H2A, or H2B (Figure S4); therefore, we do not expect miR‐1281 to exert its cellular effects by influencing the PASMC transcriptome globally.
Second, because HDAC4 can act as a transcriptional coregulator that may interact with myocyte enhancer factor 2,52 activating transcription factor 4,53 transforming growth factor‐β,54 and hypoxia‐inducible factor (HIF)‐1α55, 56 in PASMCs, we investigated if miR‐1281/HDAC4 pathway influenced the transcription of corresponding downstream genes kruppel‐like factor 2 (KLF2), KLF4, CCAAT/enhancer‐binding protein‐homologous protein (CHOP), tribbles homolog 3 (TRB3), drosophila mothers against decapentaplegic protein 3 (SMAD3), vascular endothelial growth factor (VEGF), and glucose transporter‐1 (GLUT‐1). Our results demonstrate that inhibition of HDAC4 by siHDAC4 led to decreased mRNA levels of CHOP, SMAD3, VEGF, and GLUT‐1, but increased mRNA level of TRB3 (Figure 4A); agreeing with this, overexpression of miR‐1281 by miR‐1281 mimic also led to decreased mRNA levels of SMAD3, VEGF, and GLUT‐1 (Figure 4B). Hence, it appears that genes functioning downstream of HDAC4 can be, in part, regulated by miR‐1281. Notably, miR‐1281 overexpression also induced downregulation of KLF2 and TRB3. This is supported by miR‐1281 inhibitor‐induced upregulation of these 2 genes (Figure 4C), reminding us of the difference between miR‐1281 and HDAC4 downstream regulatory networks.
Figure 4.
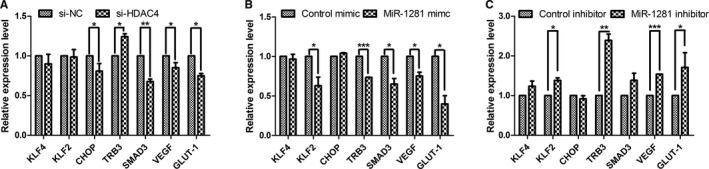
miR‐1281/histone deacetylase 4 (HDAC4) pathway regulates the transcription of multiple related transcription factors. A through C, A set of real‐time quantitative reverse transcription–polymerase chain reaction analyses exploring the possible regulatory network downstream of HDAC4 and miR‐1281. RNA samples were obtained from pulmonary artery smooth muscle cells transfected with small interfering (si)‐HDAC4, miR‐1281 mimic, miR‐1281 inhibitor, and their corresponding negative controls (NCs). CHOP indicates CCAAT/enhancer‐binding protein‐homologous protein; GLUT‐1, glucose transporter‐1; KLF, kruppel‐like factor; SMAD3, drosophila mothers against decapentaplegic protein 3; TRB3, tribbles homolog 3; and VEGF, vascular endothelial growth factor. *P<0.05, **P<0.01, ***P<0.001 compared with si‐NC or mimic or inhibitor control as broken lines indicated in each figure. Exact P values in consecutive order are: 0.045223, 0.021835, 0.009286, 0.042967, 0.020433, 0.019755, 0.000044, 0.037415, 0.034118, 0.029028, 0.024684, 0.001313, 0.000403, 0.045261.
EGR1 Regulation of EP300 Is Not the Key Mechanism Leading to Repression of miR‐1281 by PDGFBB
Next, the regulatory mechanism underlying PDGFBB repression of miR‐1281 was investigated. As mentioned, MIR‐1281 originates from an upstream region of EP300 5′ end, and the latter carries 6 EGR1 binding sites and can respond to EGR1 activation.57 In rat EP300 5′ end, 6 EGR1 binding sites were identified to scatter around the MIR‐1281 flanking region (Figure S5A); hence, the effect of EGR1 on miR‐1281 expression was analyzed. First, in PDGFBB‐treated PASMCs, the mRNA levels of EGR1 increased by >4‐fold within 4 hours of PDGFBB treatment, and then decreased gradually and returned to the original level after 12 hours (Figure S5B). This revealed an earlier response of EGR1 than that of miR‐1281 (at 12 hours; Figure 1C) to PDGFBB treatment. Accordingly, an siRNA against EGR1 was transfected into PASMCs, which knocked down the mRNA expression of EGR1 by 70% (Figure S5C). Under normal culture conditions, transfection of an siRNA against EGR1 induced the expression of miR‐1281 by >60% in PASMCs (Figure S5D); however, under PDGFBB treatment, the siRNA against EGR1 failed to significantly alter miR‐1281 expression (at 12 hours; Figure S5E), suggesting the possibility that other regulatory mechanism(s) exist to mediate the repressing effect of PDGFBB on miR‐1281 expression.
PI3K‐Induced DNMT1 Expression Results in Repression of miR‐1281
Via promoter prediction webserver (FirstEF, http://rulai.cshl.org/tools/FirstEF), we identified a CpG island located in the vicinity of 5′ upstream region of MIR‐1281 and EP300 (Figure S6A). By sequence alignment, we found that this CpG island is highly conserved between human and rat genomes (Figure S6B). Then, using DNA samples isolated from rat PASMCs treated with either PDGFBB or dimethyl sulfoxide (control), the methylation status of CG dinucleotide sites within this CpG island was analyzed, and we found that PDGFBB treatment enhanced methylation status of most cytosine residues in these CG dinucleotides (Figure S6D). Moreover, addition of DNMT inhibitor (ie, 5‐aza‐2′‐deoxycytidine) reversed the PDGFBB‐induced repression of miR‐1281, from <40% to ≈80% (Figure S6E). Hence, these data emphasize the importance of PDGFBB‐induced CpG island methylation on miR‐1281 expression, and the member(s) of DNMT is(are) assumed to act as the important repressor(s).
Previously, PDGFBB treatment was induced significant upregulation of DNMT1, but not DNMT3A or DNMT3B39; therefore, DNMT1 is assumed to be the DNMT responsible for repressed miR‐1281 expression. Herein, we found that the protein level of DNMT1 correlated with PDGFBB treatment in a dose‐dependent manner (Figure 5A), which is in line with the PDGFBB dose‐dependent repression of miR‐1281 (Figure 1D). Then, we demonstrated that silencing DNMT1 expression by transfecting si‐DNMT1 in PASMCs partially recovers PDGFBB‐induced miR‐1281 repression (Figure 5B and 5C). Thus, these data strongly support the notion that increased expression of DNMT1 underlies PDGFBB‐induced repression of miR‐1281.
Figure 5.
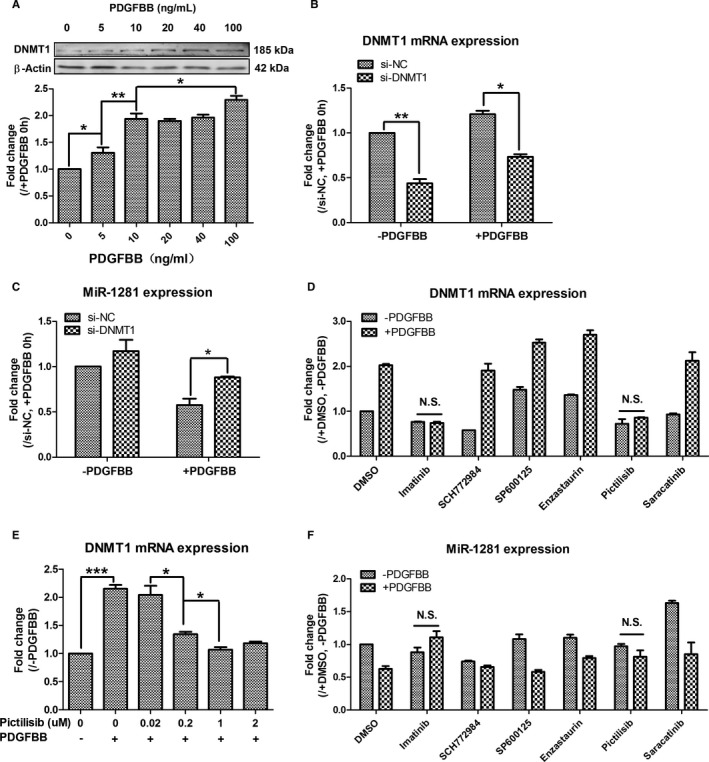
Platelet‐derived growth factor (PDGF) BB–phosphatidylinositol 3‐kinase–DNA methyltransferase 1 (DNMT1) activation cassette mediates PDGFBB repression of miR‐1281. A, Expression of DNMT1 protein increased along with the increase of PDGFBB use. B, Real‐time quantitative reverse transcription–polymerase chain reaction analysis of small interfering (si)‐DNMT1 silencing efficiency in pulmonary artery smooth muscle cells (PASMCs) without and with PDGFBB treatments. C, Transfection of si‐DNMT1 recovered PDGFBB repression of miR‐1281 in PASMCs. D, Effects of different pathway inhibitors on DNMT1 expression were analyzed, and pictilisib was identified to inhibit PDGFBB upregulated mRNA level of DNMT1. E, Pictilisib inhibited PDGFBB upregulation of DNMT1 in a dose‐dependent manner. F, Pictilisib also inhibited PDGFBB‐repressed expression of miR‐1281. DMSO indicates dimethyl sulfoxide; NC, negative control; and N.S., not significant. *P<0.05, **P<0.01, ***P<0.001 compared with control or as specified by broken lines in respective figures. Exact P values in consecutive order are: 0.034235, 0.009507, 0.043504, 0.003449, 0.010124, 0.049599, 0.000249, 0.010004, 0.036281.
To investigate which PDGFBB‐activated pathway(s) upregulate(s) DNMT1 in PASMCs, we used specific inhibitors (SCH772948, SP600125, enzastaurin, pictilisib, and saracatinib) to block PDGF‐related signaling pathways (extracellular signal–regulated kinase [ERK] 1/2, c‐Jun N‐terminal Kinase, protein kinase Cβ, PI3K, and sarcoma (Src), respectively) in PDGFBB‐treated PASMCs. The inhibitor of PDGF receptor (ie, imatinib) was used as a positive control to illustrate complete inhibition of PDGFBB effect on DNMT1. As shown in Figure 5D, pretreatment with either imatinib (positive control) or pictilisib fully inhibited PDGFBB‐induced increase in DNMT1 mRNA, suggesting that the upregulation of DNMT1 was attributable to activation of the PI3K pathway by PDGF receptor. After this, different concentrations of pictilisib were applied to PASMCs, which were then treated with PDGFBB. The qRT‐PCR analysis showed that pictilisib attenuates the effect of PDGFBB on DNMT1 mRNA expression at 0.02 to 1 μmol/L, but it did not induce any further reduction in DNMT1 mRNA levels when higher concentrations of pictilisib were applied (Figure 5E). Finally, miR‐1281 level was also measured in the previously described RNA samples. As expected, pretreatment with pictilisib attenuated PDGFBB repression of miR‐1281 level (Figure 5F). Overall, the data indicate that PDGFBB‐activated PI3K pathway leads to upregulation of DNMT1, which, in turn, acts as the repressor of miR‐1281 expression. Besides, the inhibitor of ERK1/2 (ie, SCH772984) also attenuated the inhibitory effect of PDGFBB on miR‐1281 expression (Figure 5F), but it did not influence the mRNA level of DNMT1 (Figure 5D); these findings indicated that the expression of miR‐1281 can be controlled by another PDGF‐related pathway, and this would be independent of DNMT1 regulation.
Reduced miR‐1281 Levels Were Found in Hypoxic PASMCs in Vitro, in Pulmonary Arteries of Rats With Monocrotaline‐Induced PAH, and in Serum of Patients With Coronary Heart Disease–PAH
To correlate downregulated miR‐1281 levels with the presence of PAH, we compared miR‐1281 level between hypoxic and normoxic PASMCs in vitro, because hypoxia does induce pulmonary arterial remodeling and PAH.58 The results revealed a significant downregulation of miR‐1281 in PASMCs after 12 and 24 hours of hypoxia (Figure 6A). Then, we measured miR‐1281 level in pulmonary arteries isolated from rats with monocrotaline‐induced PAH and found that miR‐1281 levels were significantly decreased compared with those of control rats (Figure 6B). Finally, we measured miR‐1281 level in serum samples collected individually from 13 healthy donors and 29 patients with coronary heart disease–PAH (refer to Table S1 for participant information). The miR‐1281 level was significantly lower in serum of patients with PAH compared with healthy donors (Figure 6C). In summary, these data consistently suggest a close relationship between miR‐1281 and PAH, and they suggest that a reduction in the level of miR‐1281 could be a potential biomarker of PAH.
Figure 6.
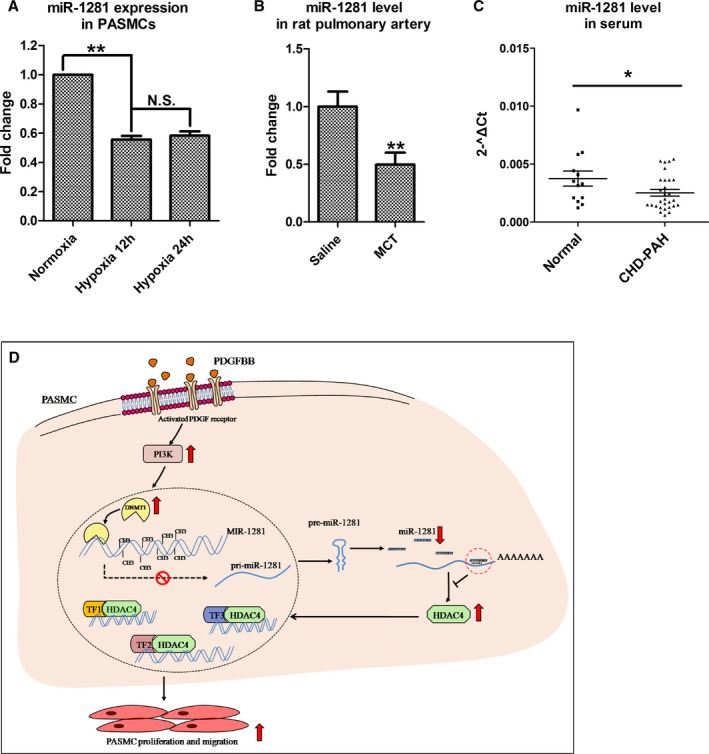
Reduced miR‐1281 level is identified in hypoxic pulmonary artery smooth muscle cells (PASMCs), rats with pulmonary arterial hypertension (PAH), and patients with PAH. The relative miR‐1281 levels were estimated by real‐time quantitative reverse transcription–polymerase chain reaction in PASMCs exposed to normoxia (21% O2) or hypoxia (3% O2) for 12 and 24 hours (A), pulmonary arteries individually collected from 4 rats with monocrotaline (MCT)–induced PAH and 4 controls (rats received intraperitoneal injections of normal saline; B), and serum individually collected from 13 healthy participants and 29 patients with coronary heart disease (CHD)–PAH (C). D, Proposed regulatory model of phosphatidylinositol 3‐kinase (PI3K)–DNA methyltransferase 1 (DNMT1)–miR‐1281–histone deacetylase 4 (HDAC4) axis. Upward arrow indicates upregulation or activation. Downward arrow indicates downregulation. N.S. indicates not significant; PDGF, platelet‐derived growth factor; and TF, transcription factor. *P<0.05, **P<0.01 compared with normoxia, saline or normal control. Exact P values in consecutive order are: 0.00332, 0.001079, 0.024024.
Discussion
Mature quiescent PASMCs retain a significant capacity to become proproliferative, promigratory, and antiapoptotic in response to a multitude of stimuli,16, 59, 60 and previous studies have revealed that HDAC members are involved in vasculature remodeling of PAH through their regulations of this process. Boucherat et al61 reported the robust increase of HDAC6 protein expression in PAH‐PASMCs and distal pulmonary arteries and demonstrated that inhibition of HDAC6 not only rescues PASMC proliferation and antiapoptotic phenotype, but also reverses PAH symptoms in monocrotaline rat model. Usui et al62 found that HDAC4 mediates the development of hypertension in spontaneously hypertensive rats, by controlling proliferation and migration of vascular SMCs. Hence, revealing the mechanism regulating each HDAC expression would be important for selective inhibition of HDAC members and evaluating their individual therapeutic potential. In this study, we used PDGFBB treatment as the trigger for PASMC proliferation and migration and found that significant downregulation of miR‐1281, coupled with a significant upregulation of HDAC4 (Figures 1B and 3E) (the latter being a direct target of miR‐1281) (Figure 3B and 3C). miR‐1281 inhibits PASMC proliferation and migration (Figure 2), whereas HDAC4 has an opposite effect (Figure S3). These data indicate that the miR‐1281/HDAC4 complex and signaling pathway regulates PASMC function and that HDAC4 is downstream from miR‐1281. Because the cellular effects of this miRNA pathway were evident both with and without PDGFBB treatment, it is reasonable to assume that the miR‐1281/HDAC4 module would also come into play if the expression of miR‐1281 was downregulated by other stimuli. Supporting this, we found reduced miR‐1281 levels in hypoxia‐treated PASMCs and pulmonary arteries isolated from monocrotaline‐treated rats (Figure 6A and 6B). Therefore, further investigation may be needed to analyze the broader biological role of the miR‐1281/HDAC4 pathway. Further experimentation would help clarify the findings of Zhao et al, who identified the increased expression of HDAC1/4/5 in lungs from human idiopathic PAH, but only focused on the role of HDAC1/5 in remodeled pulmonary vessels.63 Nozik‐Grayck and colleagues found that HDAC3 induced the suppression of superoxide dismutase (ie, superoxide dismutase 3) in PASMCs and suggested that the HDAC pan‐inhibitor Trichostatin A (TSA) may protect against idiopathic PAH, in part, by blocking HDAC3 activity and increasing PASMC superoxide dismutase 3 expression.64
To elucidate the regulatory mechanisms of the miR‐1281/HDAC4 pathway, 2 possible actions of HDAC4 were considered: it may be globally influencing the PASMC transcriptome by altering the acetylation status of histone(s), or it may be specifically interacting with related commanding transcription factors. The data from our study support the latter case. Namely, overexpression of miR‐1281 did not significantly influence the acetylation status of any PASMC histone (Figure S4), but only altered the transcription of specific transcription factors that may also function downstream of HDAC4 (eg, VEGF and GLUT‐1; Figure 4). The reduced mRNA level of VEGF and GLUT‐1 in si‐HDAC4– or miR‐1281 mimic–transfected PASMCs indicates a reduced transcriptional activity of HIF‐1α, which is in line with the reduced mRNA level of HIF‐1α identified in si‐HDAC4– and miR‐1281 mimic–transfected PASMCs (Figure S7). Hence, it is hypothesized that the miR‐1281/HDAC4 pathway could change the HIF‐1α expression and transcriptional activity. More interestingly, the association of VEGF and HIF‐1α with PAH development65 may indicate that they participated in the functional regulatory network downstream of miR‐1281/HDAC4. However, the increased TRB3 mRNA level identified in both miR‐1281 mimic–treated and si‐HDAC4–transfected cells illustrates the difference between HDAC4 and miR‐1281 regulation of PASMCs (Figure 4A and 4B), which is reasonable considering that miRNAs usually serve as fine tuners of target genes and exert their influence by targeting several genes. Hence, to fully understand the regulatory network and elucidate the difference between HDAC4 and miR‐1281 regulation of PASMCs, relative RNA‐sequencing studies may be required.
Recently, altered miR‐1281 level has been identified in some diseases: it is upregulated in serum samples of patients with abdominal aortic aneurysm40 and in patients with malignant pleural mesothelioma,41 whereas it is downregulated in plasma samples of patients with immune thrombocytopenic purpura.42 Hence, miR‐1281 has been known for its potential to be a biomarker in a few disease diagnoses, but no reported data have associated miR‐1281 with any specific cellular function. This study connected miR‐1281 with PAH by revealing a downregulated miR‐1281 level in serum of patients with coronary heart disease–PAH; moreover, this study provides the first evidence for miR‐1281 to act as a downstream mediator of PDGF effects in PASMCs. We found that not only the sequence and location of MIR‐1281 gene were conserved between human and murine genomes (Figure 1A), but also the downregulation of miR‐1281 is a conserved phenomenon between PDGFBB‐treated human and rat PASMCs (Figure 1B; data obtained from human PASMCs are not shown). The expression of rat PASMC miR‐1281 is regulated by PDGFBB treatment in a dose‐ and time‐dependent manner (Figure 1C and 1D), indicating the tight correlation between PASMC miR‐1281 levels and the effect of PDGFBB. Of the many growth factors that were tested (ie, VEGF, endothelin 1, β‐fibroblast growth factor, transforming growth factor‐β, angiotensin 2, and insulin‐like growth factor‐1), because PDGFBB induced the greatest decrease in miR‐1281 level in PASMCs (Figure 1E) and this appeared to be a specific phenomenon in PASMCs (Figure S8), we suggest that miR‐1281 may be a good indicator of PDGFBB effect on PASMCs, which could be used as a reference miRNA in related studies in the future.
The annotated location of MIR‐1281 gene flanks the 5′ ends of EP300 first exon, where we identified 6 EGR1 binding sites scattered around. Because EGR1 has been reported to activate or repress EP300 promoter, depending on the stimulus,57 the identified PDGFBB upregulation of EGR1 (Figure S5B) is supposed to influence the transcription of miR‐1281. However, our experiment did not support this because inhibition of EGR1 by an siRNA against EGR1 did not affect PDGFBB‐induced repression of miR‐1281 (Figure S5E). Instead, PDGFBB induced a dose‐dependent upregulation of DNMT1 activity, and the inhibition of DNMT1 activity by either DNMT pan‐inhibitor 5‐aza‐2′‐deoxycytidine or si‐DNMT1 was found to greatly recover PDGFBB repression of miR‐1281 (Figure S6E and Figure 5C). This indicates that DNMT1 is a major inhibitor of miR‐1281 in PDGFBB‐treated PASMCs, and it may surpass the effects of EGR1 regulation of miR‐1281 by maintaining a hypermethylation state of the CpG islands around miR‐1281 promoter. On the basis of these findings, we hypothesize a scenario in which in normal PASMCs, the methylation status of miR‐1281 5′ upstream is low, such that EGR1 can bind to the miR‐1281 promoter and/or flanking region and regulate the expression of miR‐1281. However, in PDGFBB‐stimulated PASMCs, DNMT1 expression is up‐regulated, leading to enhanced methylation of miR‐1281 5′ upstream region (especially the CpG island). This may block the binding of EGR1 to the miR‐1281 promoter, so that even though the expression of EGR1 is increased by PDGF, it would not be able to alter miR‐1281 expression, and the expression of miR‐1281 remains low.
PI3K–protein kinase B and mitrogen‐activated protein kinase (MEK) 1/2‐ERK1/2 are 2 canonical signaling pathways that regulate cell proliferation, migration, and apoptosis.66 We found that inhibition of both PI3K and ERK1/2 attenuated PDGFBB‐induced repression of miR‐1281, with PI3K's effect being mediated by upregulation of DNMT1 expression (Figure 5D and 5E). These data complement the recent study that identified PI3K–protein kinase B as the key pathway that increased DNMT1 expression by protecting DNMT1 protein from degradation in lung fibroblasts.67 Our data also reveal that the PI3K–protein kinase B pathway transcriptionally regulates DNMT1 expression in other cell types, such as PASMCs. PI3K–protein kinase B and MEK1/2‐ERK1/2 pathways are known to maintain multiple cross‐talk points in a context‐dependent way, and their coordinated signaling determines cell fate.68, 69 Via analyzing the phosphorylation status of ERK in PASMCs treated with pictilisib (PI3K inhibitor) before PDGFBB, it is demonstrated that inhibition of PI3K activation negatively influences PDGFBB‐induced ERK signaling (Figure S9), suggesting the existence of PI3K and ERK cross talk in PASMCs. Hence, it is likely that PI3K and ERK signaling pathways synergistically regulate miR‐1281 expression, at least in part, via this cross talk and miR‐1281 serves as a downstream effector for both of them to regulate proliferation and migration of PASMCs. Notably, the PDGF is known to induce activation of Src and subsequent phosphorylation of STAT3.70, 71 STAT3 interacted with DNMT1 and facilitated its binding to CpG island of specific promoters72, 73 or enhanced DNMT1 expression in some cell lines.74, 75 However, this study did not find any correlation of Src activation with miR‐1281 expression, suggesting the DNMT1 expression may not be majorly regulated by Src/STAT3 pathway in PDGFBB‐treated PASMCs. Also, the upregulated HDACs and downregulated EP300 expression (data unpublished) in PDGFBB‐treated PASMCs may maintain STAT3 in a low acetylated status, which deprived its interaction with DNMT1.72
In conclusion, we propose that the regulatory axis of PI3K–DNMT1–miR‐1281–HDAC4 is a novel PDGFBB‐responsive pathway that plays an important role in PASMC proliferation and migration (Figure 6D). The hierarchical relationship between DNMT1 and HDAC4 may provide a new explanation for a recent report that lung DNA dysmethylation underlies pulmonary vascular dysfunction in the offspring of pregnant mice with a restrictive diet, whereas administration of TSA to offspring of pregnant mice with a restrictive diet normalized their vascular function.76 Overall, our data point to a promising role for miR‐1281 as a therapeutic target in the treatment of pulmonary hypertension. miR‐328 is another functional miRNA being identified that is downregulated in PDGFBB‐treated PASMCs.39 We transfected miR‐1281 inhibitor and miR‐328 inhibitor individually or together into PASMCs, and found no influence of miR‐1281 inhibition on miR‐328 expression and vice versa (Figures S10 and S11). Functional assays demonstrated that simultaneous inhibition of both miR‐1281 and miR‐328 did not produce greater effect on PASMC proliferation or migration than individual inhibition. Hence, it is suggested that PDGFBB can be downregulated by both miR‐1281 and miR‐328; they function independently to ensure an accelerated cell proliferation and migration outcome.
Sources of Funding
This project is supported by National Natural Science Foundation of China (91739109, 81570046, 81500044, and 81700054); the Transgenic New Species Breeding Program of China (2014ZX08009‐051B); Shenzhen Basic Research Program (JCYJ20150729104027220, JCYJ20160520174032607); Interdisciplinary Innovation Team Project of Shenzhen University; China Postdoctoral Science Foundation (2016M602524); and National Institutes of Health Grant HL123804.
Disclosures
None.
Supporting information
Table S1. Information of Healthy Donors and CHD‐PAH Patients Providing Serum for This Project
Table S2. List of Primers for Vector Construction, mRNA/miRNA Retro‐Transcription and qRT‐PCR
Figure S1. MiR‐1281 does not influence the expression of contractile maker proteins in PASMCs.
Figure S2. PTPRD is a direct target of miR‐1281 but its expression is not primarily regulated by miR‐1281 in PDGFBB‐treated PASMCs.
Figure S3. HDAC4 promotes PASMC proliferation and migration.
Figure S4. Over‐expression of miR‐1281 does not significantly alter histone acetylation status of PASMCs.
Figure S5. EGR1 does not influence the expression of miR‐1281 in PDGFBB‐treated PASMCs.
Figure S6. PDGFBB induces DNA methylation of CpG island 5′ upstream of MIR‐1281/EP300 gene.
Figure S7. miR‐1281/HDAC4 pathway influences the mRNA expression of HIF‐1α in PASMCs.
Figure S8. PDGFBB‐induced downregulation of miR‐1281 is a specific phenomenon in PASMCs.
Figure S9. Inhibition of PI3K kinase negatively influences the ERK phosphorylation.
Figure S10. Expression of miR‐1281 does not influence the expression of miR‐328 in rat PASMCs.
Figure S11. miR‐1281 and miR‐328 are independent miRNAs in terms of regulating human PASMC proliferation and migration.
Figure S12. Validation of MCT‐induced PAH rat model.
(J Am Heart Assoc. 2018;7:e007572 DOI: 10.1161/JAHA.117.007572.)29514810
References
- 1. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ten Freyhaus H, Berghausen EM, Janssen W, Leuchs M, Zierden M, Murmann K, Klinke A, Vantler M, Caglayan E, Kramer T, Baldus S, Schermuly RT, Tallquist MD, Rosenkranz S. Genetic ablation of PDGF‐dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2015;35:1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoeper MM, Huscher D, Ghofrani HA, Delcroix M, Distler O, Schweiger C, Grunig E, Staehler G, Rosenkranz S, Halank M. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol. 2013;168:871–880. [DOI] [PubMed] [Google Scholar]
- 4. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier J‐F, Chabot F. Survival in patients with idiopathic, familial, and anorexigen‐associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. [DOI] [PubMed] [Google Scholar]
- 5. Benza RL, Miller DP, Gomberg‐Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG. Predicting survival in pulmonary arterial hypertension insights from the registry to evaluate early and long‐term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122:164–172. [DOI] [PubMed] [Google Scholar]
- 6. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. [DOI] [PubMed] [Google Scholar]
- 7. Sakao S, Tatsumi K, Voelkel NF. Reversible or irreversible remodeling in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2010;43:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stenmark KR, Fagan KA, Frid MG. Hypoxia‐induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. [DOI] [PubMed] [Google Scholar]
- 9. Cantoni S, Galletti M, Zambelli F, Valente S, Ponti F, Tassinari R, Pasquinelli G, Galie N, Ventura C. Sodium butyrate inhibits platelet‐derived growth factor‐induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition. FEBS J. 2013;280:2042–2055. [DOI] [PubMed] [Google Scholar]
- 10. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet‐derived growth factor. Physiol Rev. 1999;79:1283–1316. [DOI] [PubMed] [Google Scholar]
- 11. Chen CN, Li YS, Yeh YT, Lee PL, Usami S, Chien S, Chiu JJ. Synergistic roles of platelet‐derived growth factor‐BB and interleukin‐1beta in phenotypic modulation of human aortic smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:2665–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leppanen O, Janjic N, Carlsson MA, Pietras K, Levin M, Vargeese C, Green LS, Bergqvist D, Ostman A, Heldin CH. Intimal hyperplasia recurs after removal of PDGF‐AB and ‐BB inhibition in the rat carotid artery injury model. Arterioscler Thromb Vasc Biol. 2000;20:E89–E95. [DOI] [PubMed] [Google Scholar]
- 13. Noiseux N, Boucher CH, Cartier R, Sirois MG. Bolus endovascular PDGFR‐beta antisense treatment suppressed intimal hyperplasia in a rat carotid injury model. Circulation. 2000;102:1330–1336. [DOI] [PubMed] [Google Scholar]
- 14. Kawai‐Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292:C59–C69. [DOI] [PubMed] [Google Scholar]
- 15. Hughes A, Clunn G, Refson J, Demoliou‐Mason C. Platelet‐derived growth factor (PDGF): actions and mechanisms in vascular smooth muscle. Gen Pharmacol. 1996;27:1079–1089. [DOI] [PubMed] [Google Scholar]
- 16. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 17. Li L, Xu M, Li X, Lv C, Zhang X, Yu H, Zhang M, Fu Y, Meng H, Zhou J. Platelet‐derived growth factor‐B (PDGF‐B) induced by hypoxia promotes the survival of pulmonary arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell Physiol Biochem. 2015;35:441–451. [DOI] [PubMed] [Google Scholar]
- 18. Katayose D, Ohe M, Yamauchi K, Ogata M, Shirato K, Fujita H, Shibahara S, Takishima T. Increased expression of PDGF A‐ and B‐chain genes in rat lungs with hypoxic pulmonary hypertension. Am J Physiol. 1993;264:L100–L106. [DOI] [PubMed] [Google Scholar]
- 19. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. [DOI] [PubMed] [Google Scholar]
- 20. Souza R, Sitbon O, Parent F, Simonneau G, Humbert M. Long term imatinib treatment in pulmonary arterial hypertension. Thorax. 2006;61:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perros F, Montani D, Dorfmüller P, Durand‐Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O. Platelet‐derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. [DOI] [PubMed] [Google Scholar]
- 22. Ogawa A, Nakamura K, Matsubara H, Fujio H, Ikeda T, Kobayashi K, Miyazaki I, Asanuma M, Miyaji K, Miura D. Prednisolone inhibits proliferation of cultured pulmonary artery smooth muscle cells of patients with idiopathic pulmonary arterial hypertension. Circulation. 2005;112:1806–1812. [DOI] [PubMed] [Google Scholar]
- 23. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. [DOI] [PubMed] [Google Scholar]
- 24. Lu J, Getz G, Miska EA, Alvarez‐Saavedra E, Lamb J, Peck D, Sweet‐Cordero A, Ebert BL, Mak RH, Ferrando AA. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. [DOI] [PubMed] [Google Scholar]
- 25. Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA‐127 with downregulation of the proto‐oncogene BCL6 by chromatin‐modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. [DOI] [PubMed] [Google Scholar]
- 26. Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setién F, Casado S, Suarez‐Gauthier A, Sanchez‐Cespedes M, Gitt A. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. [DOI] [PubMed] [Google Scholar]
- 27. Shah MY, Calin GA. MicroRNAs as therapeutic targets in human cancers. Wiley Interdiscip Rev RNA. 2014;5:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J. Role for miR‐204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu D, Talbot CC Jr, Liu Q, Jing ZC, Damico RL, Tuder R, Barnes KC, Hassoun PM, Gao L. Identifying microRNAs targeting Wnt/β‐catenin pathway in end‐stage idiopathic pulmonary arterial hypertension. J Mol Med. 2016;94:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen R, Guth‐Gundel S, Southwood M, Morrell NW, Thomas M. MicroRNA‐140‐5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest. 2016;126:2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30:716–723. [DOI] [PubMed] [Google Scholar]
- 32. Sharma S, Umar S, Centala A, Eghbali M. Role of miR206 in genistein‐induced rescue of pulmonary hypertension in monocrotaline model. J Appl Physiol. 2015;119:1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joshi SR, Dhagia V, Gairhe S, Edwards JG, McMurtry IF, Gupte SA. MicroRNA‐140 is elevated and mitofusin‐1 is downregulated in the right ventricle of the Sugen5416/hypoxia/normoxia model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2016;311:H689–H698. DOI: 10.1152/ajpheart.00264.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeng Y, Liu H, Kang K, Wang Z, Hui G, Zhang X, Zhong J, Peng W, Ramchandran R, Raj JU, Gou D. Hypoxia inducible factor‐1 mediates expression of miR‐322: potential role in proliferation and migration of pulmonary arterial smooth muscle cells. Sci Rep. 2015;5:12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cushing L, Costinean S, Xu W, Jiang Z, Madden L, Kuang P, Huang J, Weisman A, Hata A, Croce CM. Disruption of miR‐29 leads to aberrant differentiation of smooth muscle cells selectively associated with distal lung vasculature. PLoS Genet. 2015;11:e1005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grant JS, White K, MacLean MR, Baker AH. Micrornas in pulmonary arterial remodeling. Cell Mol Life Sci. 2013;70:4479–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L, Weng T, Zhang H, Ramchandran R, Raj JU, Gou D, Liu L. MicroRNA‐124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem. 2013;288:25414–25427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeng Y, Pan Y, Liu H, Kang K, Wu Y, Hui G, Peng W, Ramchandran R, Raj JU, Gou D. MiR‐20a regulates the PRKG1 gene by targeting its coding region in pulmonary arterial smooth muscle cells. FEBS Lett. 2014;588:4677–4685. [DOI] [PubMed] [Google Scholar]
- 39. Qian Z, Zhang L, Chen J, Li Y, Kang K, Qu J, Wang Z, Zhai Y, Li L, Gou D. MiR‐328 targeting PIM‐1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget. 2016;7:54998–55011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang W, Shang T, Huang C, Yu T, Liu C, Qiao T, Huang D, Liu Z, Liu C. Plasma microRNAs serve as potential biomarkers for abdominal aortic aneurysm. Clin Biochem. 2015;48:988–992. [DOI] [PubMed] [Google Scholar]
- 41. Bononi I, Comar M, Puozzo A, Stendardo M, Boschetto P, Orecchia S, Libener R, Guaschino R, Pietrobon S, Ferracin M, Negrini M, Martini F, Bovenzi M, Tognon M. Circulating microRNAs found dysregulated in ex‐exposed asbestos workers and pleural mesothelioma patients as potential new biomarkers. Oncotarget. 2016;7:82700–82711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuo B, Zhai J, You L, Zhao Y, Yang J, Weng Z, Dai L, Wu Q, Ruan C, He Y. Plasma microRNAs characterising patients with immune thrombocytopenic purpura. Thromb Haemost. 2017;117:1420–1431. [DOI] [PubMed] [Google Scholar]
- 43. Qian ZJ, Li YJ, Chen JD, Li X, Gou DM. miR‐4632 mediates PDGFBB‐induced proliferation and anti‐apoptosis of human pulmonary artery smooth muscle cells via targeting cJUN. Am J Physiol Cell Physiol. 2017;313:C380–C391. [DOI] [PubMed] [Google Scholar]
- 44. Zeng Y, Zhang X, Kang K, Chen J, Wu Z, Huang J, Lu W, Chen Y, Zhang J, Wang Z, Zhai Y, Qu J, Ramchandran R, Raj JU, Wang J, Gou D. MicroRNA‐223 attenuates hypoxia‐induced vascular remodeling by targeting RhoB/MLC2 in pulmonary arterial smooth muscle cells. Sci Rep. 2016;6:24900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niu Y, Zhang L, Qiu H, Wu Y, Wang Z, Zai Y, Liu L, Qu J, Kang K, Gou D. An improved method for detecting circulating microRNAs with S‐Poly(T) plus real‐time PCR. Sci Rep. 2015;5:15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang K, Zhang X, Liu H, Wang Z, Zhong J, Huang Z, Peng X, Zeng Y, Wang Y, Yang Y, Luo J, Gou D. A novel real‐time PCR assay of microRNAs using S‐Poly(T), a specific oligo(dT) reverse transcription primer with excellent sensitivity and specificity. PLoS One. 2012;7:e48536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang MB, Wesley SV, Finnegan EJ, Smith NA, Waterhouse PM. Replicating satellite RNA induces sequence‐specific DNA methylation and truncated transcripts in plants. RNA. 2001;7:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Finn TE, Wang L, Smolilo D, Smith NA, White R, Chaudhury A, Dennis ES, Wang MB. Transgene expression and transgene‐induced silencing in diploid and autotetraploid Arabidopsis. Genetics. 2011;187:409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics. 2014;6:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walia V, Prickett TD, Kim JS, Gartner JJ, Lin JC, Zhou M, Rosenberg SA, Elble RC, Solomon DA, Waldman T. Mutational and functional analysis of the tumor‐suppressor PTPRD in human melanoma. Hum Mutat. 2014;35:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. He S, Lu Y, Liu X, Huang X, Keller ET, Qian C‐N, Zhang J. Wnt3a: functions and implications in cancer. Chin J Cancer. 2015;34:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, McLean DL, Ju H, Comhair SA. Restoration of impaired endothelial MEF2 function rescues pulmonary arterial hypertension. Circulation. 2015;131:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang P, Sun Q, Zhao C, Ling S, Li Q, Chang Y‐Z, Li Y. HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell Signal. 2014;26:556–563. [DOI] [PubMed] [Google Scholar]
- 54. Yu S‐L, Lee DC, Son JW, Park CG, Lee HY, Kang J. Histone deacetylase 4 mediates SMAD family member 4 deacetylation and induces 5‐fluorouracil resistance in breast cancer cells. Oncol Rep. 2013;30:1293–1300. [DOI] [PubMed] [Google Scholar]
- 55. Kato H, Tamamizu‐Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia‐inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–41974. [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y, Ren YJ, Guo LC, Ji C, Hu J, Zhang HH, Xu QH, Zhu WD, Ming ZJ, Yuan YS, Ren X, Song J, Yang JM. Nucleus accumbens‐associated protein‐1 promotes glycolysis and survival of hypoxic tumor cells via the HDAC4‐HIF‐1alpha axis. Oncogene. 2017;36:4171–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu J, de Belle I, Liang H, Adamson ED. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell. 2004;15:83–94. [DOI] [PubMed] [Google Scholar]
- 58. Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high‐altitude long‐term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1001–L1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138:1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 61. Boucherat O, Chabot S, Paulin R, Trinh I, Bourgeois A, Potus F, Lampron MC, Lambert C, Breuils‐Bonnet S, Nadeau V, Paradis R, Goncharova EA, Provencher S, Bonnet S. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci Rep. 2017;7:4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Usui T, Morita T, Okada M, Yamawaki H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension. 2014;63:397–403. [DOI] [PubMed] [Google Scholar]
- 63. Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nozik‐Grayck E, Woods C, Stearman RS, Venkataraman S, Ferguson BS, Swain K, Bowler RP, Geraci MW, Ihida‐Stansbury K, Stenmark KR, McKinsey TA, Domann FE. Histone deacetylation contributes to low extracellular superoxide dismutase expression in human idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;311:L124–L134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. [DOI] [PubMed] [Google Scholar]
- 66. Choudhury GG, Karamitsos C, Hernandez J, Gentilini A, Bardgette J, Abboud HE. PI‐3‐kinase and MAPK regulate mesangial cell proliferation and migration in response to PDGF. Am J Physiol. 1997;273:F931–F938. [DOI] [PubMed] [Google Scholar]
- 67. Koh HB, Scruggs AM, Huang SK. Transforming growth factor‐1 increases DNA methyltransferase 1 and 3a expression through distinct post‐transcriptional mechanisms in lung fibroblasts. J Biol Chem. 2016;291:19287–19298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross‐talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans. 2012;40:139–146. [DOI] [PubMed] [Google Scholar]
- 69. Mendoza MC, Er EE, Blenis J. The Ras‐ERK and PI3K‐mTOR pathways: cross‐talk and compensation. Trends Biochem Sci. 2011;36:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gao P, Niu N, Wei T, Tozawa H, Chen X, Zhang C, Zhang J, Wada Y, Kapron CM, Liu J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget. 2017;8:69139–69161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Takikita‐Suzuki M, Haneda M, Sasahara M, Owada MK, Nakagawa T, Isono M, Takikita S, Koya D, Ogasawara K, Kikkawa R. Activation of Src kinase in platelet‐derived growth factor‐B‐dependent tubular regeneration after acute ischemic renal injury. Am J Pathol. 2003;163:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, Yu H. Acetylated STAT3 is crucial for methylation of tumor‐suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A. 2012;109:7765–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3‐ and DNA methyltransferase 1‐mediated epigenetic silencing of SHP‐1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu J, Tang Q, Yang L, Chen Y, Zheng F, Hann SS. Interplay of DNA methyltransferase 1 and EZH2 through inactivation of Stat3 contributes to beta‐elemene‐inhibited growth of nasopharyngeal carcinoma cells. Sci Rep. 2017;7:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Quan Z, He Y, Luo C, Xia Y, Zhao Y, Liu N, Wu X. Interleukin 6 induces cell proliferation of clear cell renal cell carcinoma by suppressing hepaCAM via the STAT3‐dependent up‐regulation of DNMT1 or DNMT3b. Cell Signal. 2017;32:48–58. [DOI] [PubMed] [Google Scholar]
- 76. Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol. 2011;301:H247–H252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information of Healthy Donors and CHD‐PAH Patients Providing Serum for This Project
Table S2. List of Primers for Vector Construction, mRNA/miRNA Retro‐Transcription and qRT‐PCR
Figure S1. MiR‐1281 does not influence the expression of contractile maker proteins in PASMCs.
Figure S2. PTPRD is a direct target of miR‐1281 but its expression is not primarily regulated by miR‐1281 in PDGFBB‐treated PASMCs.
Figure S3. HDAC4 promotes PASMC proliferation and migration.
Figure S4. Over‐expression of miR‐1281 does not significantly alter histone acetylation status of PASMCs.
Figure S5. EGR1 does not influence the expression of miR‐1281 in PDGFBB‐treated PASMCs.
Figure S6. PDGFBB induces DNA methylation of CpG island 5′ upstream of MIR‐1281/EP300 gene.
Figure S7. miR‐1281/HDAC4 pathway influences the mRNA expression of HIF‐1α in PASMCs.
Figure S8. PDGFBB‐induced downregulation of miR‐1281 is a specific phenomenon in PASMCs.
Figure S9. Inhibition of PI3K kinase negatively influences the ERK phosphorylation.
Figure S10. Expression of miR‐1281 does not influence the expression of miR‐328 in rat PASMCs.
Figure S11. miR‐1281 and miR‐328 are independent miRNAs in terms of regulating human PASMC proliferation and migration.
Figure S12. Validation of MCT‐induced PAH rat model.