

Abstract
Background
Abdominal aortic aneurysm (AAA) is a potentially life‐threatening disease that is common in older individuals. Currently, therapeutic options are limited to surgical interventions. Although it has long been known that AAA tissue is enriched in B cells and immunoglobulins, their involvement in AAA pathogenesis remains controversial.
Methods and Results
We investigated the role of B cells and immunoglobulins in a murine model of AAA, induced with a periaortic application of CaCl2, and in human AAA. Both human and mouse AAA tissue showed B‐cell infiltration. Mouse AAA tissue showed deposition of IgG and activation of Syk, a key molecule in B‐cell activation and immunoglobulin function, which were localized to infiltrating cells including B cells and macrophages. B‐cell–deficient muMT mice showed suppression of AAA development that was associated with reduced activation of Syk and less expression of matrix metalloproteinase‐9. Administration of exogenous immunoglobulins restored the blunted Syk activation and AAA development in muMT mice. Additionally, exogenous immunoglobulins induced interleukin‐6 and metalloproteinase‐9 secretions in human AAA tissue cultures. Furthermore, administration of R788, a specific Syk inhibitor, suppressed AAA expansion, reduced inflammatory response, and reduced immunoglobulin deposition in AAA tissue.
Conclusions
From these results, we concluded that B cells and immunoglobulins participated in AAA pathogenesis by promoting inflammatory and tissue‐destructive activities. Finally, we identified Syk as a potential therapeutic target.
Keywords: abdominal aortic aneurysm, B cells, immune system, immunoglobulins, inflammation, Syk
Subject Categories: Animal Models of Human Disease, Inflammation, Mechanisms, Vascular Biology, Cell Signalling/Signal Transduction
Clinical Perspective
What Is New?
Immunoglobulins, the major effector molecules of B cells, promoted chronic inflammation both in mouse model of abdominal aortic aneurysm (AAA) and in human AAA tissue in culture.
Syk, a tyrosine kinase that is essential both for B‐cell activation and for the function of immunoglobulins, was activated in mouse AAA tissue.
Genetic deletion of mature B cells or pharmacological inhibition of Syk ameliorated AAA development in mice.
What Are the Clinical Implications?
Activation of B cells, pathogenic effect of immunoglobulins, and activation of Syk represent potential therapeutic targets for AAA.
Monitoring their activities may provide a clue for the disease activity of AAA.
Abdominal aortic aneurysm (AAA) is a common aortic disease in older individuals (over 70 years old).1 AAA is caused by the local weakening of aortic walls. Although patients with AAA seldom show clinical symptoms or signs for years, the AAA diameter slowly expands and leads to the catastrophic events of aortic rupture and sudden death. Currently, no therapy is available to halt the expansion of AAA. Thus, the only therapeutic option is watchful observation and, before rupture, replacing the diseased aorta with an artificial graft, either through open surgery or endovascular aneurysm repair (EVAR) with a self‐expandable stent‐graft.2 Although EVAR is less invasive than open repair, long‐term aortic complications or stent‐graft–associated complications occur more commonly in EVAR, possibly due to the fragility of the aortic wall.3, 4 To develop strategies for slowing AAA expansion and to provide better clinical outcomes with EVAR, it is essential to understand the pathogenesis of AAA.5
Decades of research have revealed the molecular pathogenesis of AAA.6, 7, 8 We know that chronic inflammation plays a central role in promoting tissue destruction and in suppressing tissue repair. Suppressing inflammatory signaling was proven to be effective in preventing the progression and promoting the healing of AAA in animal models.6, 8, 9 Human AAA tissue is characterized by an early infiltration of inflammatory cells, including neutrophils, macrophages, T cells, and B cells, which occurs before detectable extracellular matrix (ECM) destruction and aortic diameter expansion.1 Although both AAA and aortic occlusive disease show prominent cellular infiltration, AAA is more enriched in B cells and immunoglobulins, which suggested that these factors might be involved in AAA pathogenesis.10, 11, 12 Indeed, a series of studies have demonstrated that B cells promoted AAA by producing immunoglobulins against fibrinogen, which then activated the complement pathway, in an elastase‐induced mouse model.13, 14 Consistently, it has been reported that the depletion of B cells with an anti‐CD20 antibody protected the aorta from AAA development in the elastase‐induced model and in an angiotensin II–induced model in Apoe‐deficient mice.15 However, the involvement of B cells in AAA pathogenesis is controversial. Another study reported that, with the same elastase‐induced AAA model, B cell–deficient mice developed AAA equivalent to wild‐type (WT) mice, and B2‐cell administration ameliorated AAA development.16 Those findings indicated that further evaluations should be performed to determine the significance of B cells in AAA pathogenesis, preferably in a different model of AAA, because currently used animal models of AAA are significantly different from each other, and no single model recapitulates all aspects of human AAA.6 Moreover, evaluating the role of B cells and immunoglobulins in human AAA tissue is preferable because no single animal model has recapitulated all aspects of human AAA.6 In addition, if B cells are involved in AAA pathogenesis, therapeutic targets that can modulate B‐cell function should be explored.
The goal of the current study was to evaluate the role of B cells/immunoglobulins in a widely used murine AAA model, which is induced with a periaortic application of CaCl2,17 and in human AAA tissue cultures. We also explored the therapeutic potential of inhibiting Syk, a tyrosine kinase that plays a central role in B‐cell activation and in immunoglobulin effector functions.18, 19, 20, 21
Methods
The data, analytic methods, and study materials will be available to other researchers for purposes of reproducing the results or replicating the procedure, as long as the situation allows. The analytic methods and data are described in this article except for the transcriptome data set, which has been deposited to the Gene Expression Omnibus of the National Center for Biotechnology Information (accession #GSE109640).22 The study materials are commercially available except for human AAA tissue samples, which will not be publicly available.
Animal Experiments
All animal experimental protocols were approved by the Animal Experiments Review Boards of Kurume University. To create a mouse model of AAA, we placed a periaortic application of 0.5 mol/L CaCl2 onto the infrarenal aorta for 20 minutes. This treatment caused chronic local inflammation, as described previously.17 We previously confirmed that the local inflammation was not elicited by a sham operation in which physiological saline had been used instead of CaCl2.23 Mice were killed with a pentobarbital overdose, and blood and tissue samples were collected at 2 time points: at 7 days after the CaCl2 treatment and at 42 days after the CaCl2 treatment. Aortic tissue was excised either immediately, for protein and mRNA expression analyses, or after perfusion and fixation with 4% paraformaldehyde in phosphate‐buffered saline at physiological pressure, for histological analysis. Aortic enlargement was defined as a diameter equal to or exceeding 1.5‐fold greater than the control aorta diameter. For protein and mRNA expression analyses, aortic tissue was rapidly frozen in liquid nitrogen and stored at −80°C until sample extraction.
To assess the role of B cells in AAA pathogenesis, we used muMT mice, which are deficient in membrane‐bound IgM, on a background of C57BL/6J (Jackson Laboratory #002288, Bar Harbor, ME). WT mice with the same genetic background served as controls. To evaluate the role of Syk in AAA pathogenesis, we used R788 (fostamatinib, MedChemeExpress, Monmouth Junction, NJ, USA), an orally available prodrug of the selective Syk inhibitor, R406. R788 was given ad libitum for 7 days before the CaCl2 treatment and afterwards, during the observational period. The concentration of R788 was 3 mg/mL in drinking water,24 with 0.1% carboxymethyl cellulose, 0.1% methylparaben, 0.02% propylparaben, and 1% sucrose. The control group received drinking water with 0.1% carboxymethyl cellulose, 0.1% methylparaben, 0.02% propylparaben, and 1% sucrose, without R788 (vehicle).
Human AAA Tissue
All protocols that involved human specimens were approved by the Institutional Review Board at Kurume University Hospital, and all samples were obtained with informed consent from the patients. Human AAA tissue was obtained from patients during open surgery performed to repair AAA. Tissues were acquired from the anterior wall of the aneurysm, at the junction of a section with a normal diameter on 1 side and the dilated lesion on the other side. For histological analyses, AAA tissues were fixed in paraformaldehyde, paraffin embedded, and sliced into tissue sections 5 μm thick. For ex vivo cultures, we obtained fresh AAA samples from 2 patients, acquired during surgery as stated above, that were cleaned of thrombi and loose connective tissue. The aortic tissue was cut horizontally into 3 parts. Each part of the aortic wall was minced into pieces ≈2×2 mm, which maintained the full thickness of the aortic wall. Minced tissues were transferred to 12‐well plates with serum‐free DMEM (Invitrogen, Carlsbad, CA). Into each well we placed an equal number of pieces from all 3 parts of the AAA sample to minimize the effect of AAA tissue heterogeneity. At 24 hours after starting the culture, culture media were exchanged with fresh DMEM, and samples were incubated for 48 hours (the first 48 hours) to obtain conditioned media for measuring basal secretions of interleukin‐6 (IL‐6) and matrix metalloproteinase‐9 (MMP9). Then, culture media were exchanged with fresh DMEM, with or without 0.5 mg/mL human polyclonal IgG (Jackson ImmunoResearch, West Grove, PA; #767‐71594), and cultures were incubated for another 48 hours (the second 48 hours) to obtain conditioned media after stimulation. Secreted IL‐6 and MMP9 were measured with an IL‐6 ELISA kit (R&D Systems, Minneapolis, MN; #S6050) and gelatin zymography (ThermoFisher Scientific, Waltham, MA, USA; #EC61755BOX), respectively. To adjust for tissue heterogeneity, all measured values were normalized to the basal secretion of a given molecule by dividing the measurement of the second 48 hours by the measurement of the first 48 hours from the same well. IL‐6 and MMP9 levels were evaluated as the fold change in normalized values relative to the normalized levels observed in unstimulated cultures, (designated as 1).
Expression Analysis
For protein expression analysis, aortic samples were pulverized with an SK mill (TOKKEN, Kashiwa, Japan), and proteins were extracted with RIPA buffer. The protein concentrations were determined by bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA) to adjust the protein loading on a NuPAGE system (Invitrogen, Carlsbad, CA). After resolving the proteins on the NuPAGE system, we performed immunoblotting with antibodies specific for Stat3 (Cell Signaling Technology, Danvers, MA; #4904), phospho‐Stat3 (Tyr705, Cell Signaling Technology; #9145), Jnk (Abcam, Cambridge, UK; #ab112501), phospho‐Jnk (Thr183/Tyr185, Cell Signaling Technology; #4671), Syk (Cell Signaling Technology; #2712), phospho‐Syk (Tyr323, Cell Signaling Technology; #2715), and lysyl oxidase (Abcam; #ab31238). MMP9 expression was evaluated with gelatin zymography (ThermoFisher Scientific, Waltham, MA, USA; #EC61755BOX). Expression levels of α‐tubulin were determined using a specific antibody (Abcam; #ab40742) and used as an internal control to normalize the protein expressions. For mRNA expression analyses, we used aortic tissue from the same part of the aorta that was used in the protein expression analysis. We homogenized the tissues in TRIzol (Invitrogen) and isolated total RNA with RNeasy (Qiagen, Hilden, Germany). We performed transcriptome analyses with the SurePrint G3 Mouse Gene Expression 8x60K Microarray Kit (Agilent Technologies, Santa Clara, CA).
Histological Analysis
We stained paraffin‐embedded sections of aortic tissue with elastica van Gieson or hematoxylin and eosin. We performed immunohistochemical staining of mouse aortic tissue with antibodies specific for phospho‐Syk (Tyr323, Cell Signaling Technology; #2715), B cells (B220, BD #553086), and macrophages (Iba‐1, Millipore, Billerica, MA; #MABN92). Deposition of immunoglobulins was detected by fluorescently labeled antibodies for mouse IgM (Abcam; #ab150121) and IgG (Abcam; #ab150117). We performed immunohistochemical staining of human AAA tissue with antibodies specific for B cells (CD19, Dako, Agilent Technologies; #M729629), smooth muscle cells (smooth muscle α‐actin, Sigma‐Aldrich, St. Louis, MO; #A5228), T cells (CD3, Abcam; #ab16669), neutrophils (neutrophil elastase, Dako, Agilent Technologies; #M075201), and macrophages (CD68, Dako, Agilent Technologies; #M0876).
Statistical Analysis
Quantitative data are presented by box‐and‐whisker plots to show medians and interquartile ranges. Statistical analyses were performed with GraphPad PRISM 5 (GraphPad Software, San Diego, CA). When the data passed the D'Agostino and Pearson normality test and Bartlett test for equal variances, we performed 1‐way ANOVAs to compare 3 or more groups, followed by Bonferroni multiple comparison test. For nonnormal data distributions, we performed the Kruskal‐Wallis test, followed by a Dunn multiple comparison test. Statistical significance was indicated by a P<0.05.
Results
Presence of B Cells in AAA Tissue
We examined the presence of B cells and other inflammatory cells in AAA tissue samples obtained from patients who underwent open surgery to repair AAA (Figure 1A). Immunostaining with antibodies specific for different cell types, including B cells, indicated that B cells formed clusters with other inflammatory cells, including T cells and macrophages, which surrounded small vessels.10, 11 We next examined whether B cells were present in mouse aorta (Figure 1B). In the control, untreated mouse aorta, we found no B cells. We used a well‐established method to create a mouse model of AAA with a periaortic application of 0.5 mol/L CaCl2 for 20 minutes.17 This procedure caused inflammation, followed by chronic inflammation and destructive changes in the treated aortic walls. At 6 weeks after CaCl2 challenge, AAA had developed. When aortic tissues from these AAA mice were examined with immunostaining after the CaCl2 challenge, we observed a few B cells that had infiltrated the adventitia and had formed small clusters (Figure 1B). During the 6‐week time course of AAA development, B cells were observed throughout the study period, at 3, 7, 21, and 42 days after a CaCl2 challenge. The number of infiltrating B cells did not significantly change during this time course. We examined the presence of immunoglobulins, the main effector molecules of B cells, in the aortic walls during the development of AAA (Figure 1C). Depositions of IgM and IgG were barely detectable by Western blot before CaCl2 challenge but were readily detected in AAA tissue throughout the 6‐week observation period, and they apparently peaked at 1 week after the CaCl2 challenge. On the other hand, IgA and IgE were undetectable in our hands.
Figure 1.
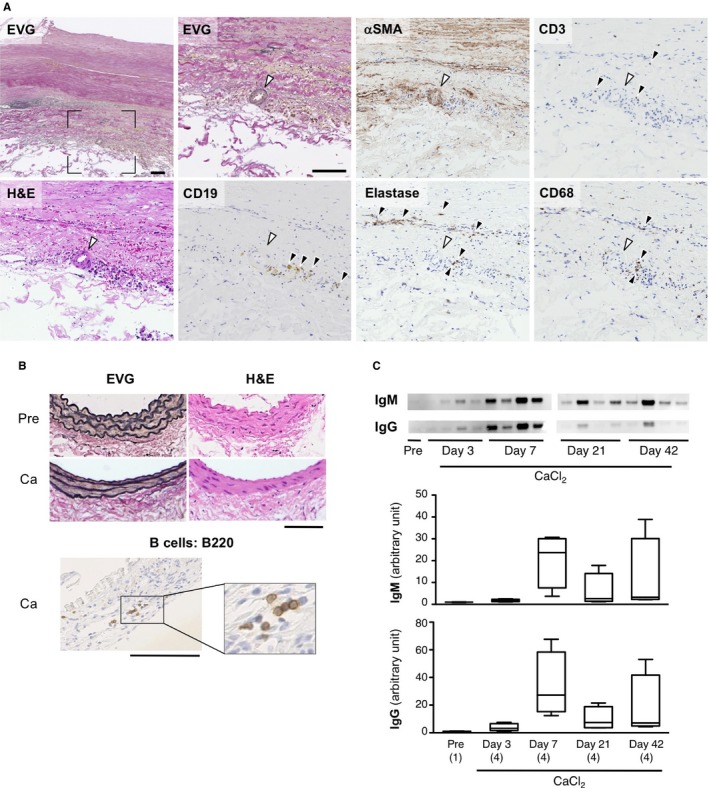
Histology of human and mouse AAA. A, Representative photomicrographs of human AAA tissues. Sections were stained with elastica van Gieson (EVG), with hematoxylin and eosin (H&E), and with antibodies specific for B cells (CD19), smooth muscle cells (αSMA), T cells (CD3), neutrophils (Elastase), and macrophages (CD68). Black arrowheads in each panel indicate positive staining of the corresponding antigen. White arrowheads indicate small blood vessels. The section marked with black rectangle corners on the low‐magnification image (top left) is enlarged in the adjacent image. Scale bars: 100 μm. B, Mouse AAA model was created by treating with a periaortic application of 0.5 mol/L CaCl2. Upper, Representative aortic sections, histologically stained with EVG and H&E, are shown before (Pre) and 6 weeks after CaCl2 treatment (Ca). Lower, B‐cell infiltration is indicated with B220 immunostaining of aortic tissue 6 weeks after CaCl2 treatment. Black rectangle indicates the area enlarged in the inset. Scale bars: 50 μm. All images are shown with the luminal side up and adventitial side down. C, Immunoblotting (top) and quantification (bottom) of IgM and IgG in mouse aorta before and after CaCl2 treatment. Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plots. AAA indicates abdominal aortic aneurysm.
We also performed immunostaining of IgM and IgG in aortic sections before and 1 week after a CaCl2 challenge (Figure 2). In aortic tissue without CaCl2 challenge, IgM and IgG were barely detectable. After CaCl2 challenge, IgM was detected in intima and adventitia, whereas IgG was diffusely stained in the adventitia of the aortic wall (Figure 2B). Immunofluorescence staining of B cells showed that part of IgM staining was in close proximity of B220‐positive B cells (Figure 2C). On the other hand, IgG was overlapping with most of the cells in the adventitia. These results indicated that inflammation of AAA tissue was accompanied by B‐cell infiltration in both human and mouse. The development of AAA in mice was also accompanied by the deposition of IgM and IgG.
Figure 2.
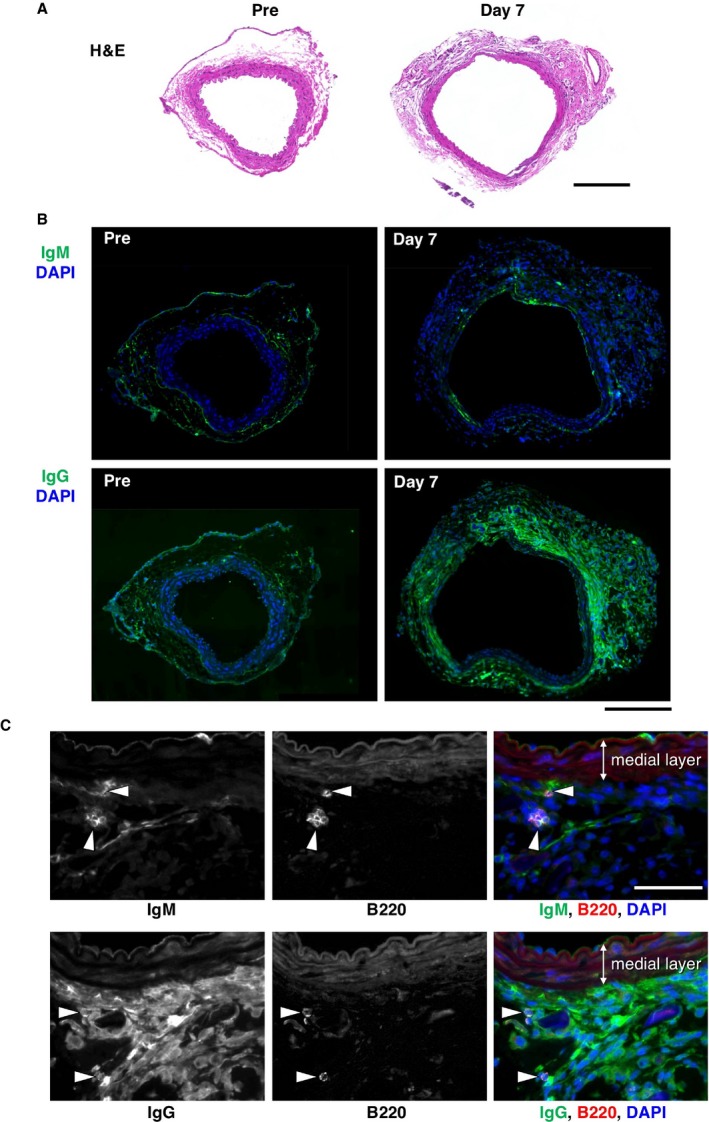
Deposition of immunoglobulins in mouse AAA. A, Tissue sections are shown for mouse aorta without (Pre) and with (Day 7) CaCl2 challenge by hematoxylin and eosin staining (H&E). Bar 200 μm. B, Deposition of IgM or IgG was detected by immunofluorescence staining (green color) and shown with nuclear staining (DAPI, blue color). Bar 200 μm. C, Deposition of IgM or IgG is visualized by immunofluorescence staining (green color in the rightmost panels) with B‐cell marker B220 staining (arrowheads, red color in the rightmost panels) in aortic tissue 7 days after CaCl2 challenge. Bar 50 μm. AAA indicates abdominal aortic aneurysm; DAPI, 4′,6‐Diamidino‐2‐Phenylindole, Dihydrochloride.
Because it is well established that Syk, a hematopoietic lineage‐specific tyrosine kinase, is involved in the inflammatory response including B‐cell activation and effector function of immunoglobulins downstream of Fc receptors,18 we examined the activation status of Syk in the aorta by immunofluorescence staining of activated (phosphorylated) Syk (pSyk) before and after CaCl2 challenge (Figure 3). Before CaCl2 challenge, pSyk was barely detectable but was readily detectable mainly in adventitia 1 week after CaCl2 challenge (Figure 3A). To localize pSyk in specific cell types, we stained the AAA tissue for B220, a B‐cell marker (Figure 3B), and Iba‐1, a macrophage marker (Figure 3C). Although B220‐positive B cells were positive for pSyk, most of the pSyk‐positive cells were non‐B cells. Iba‐1‐positive macrophages were abundantly detected in the adventitia of AAA tissue, many of which were positive for pSyk. Collectively, the deposition of IgG on most of the adventitial cells and Syk activation in the infiltrating cells suggested that they may participate in AAA pathogenesis.
Figure 3.
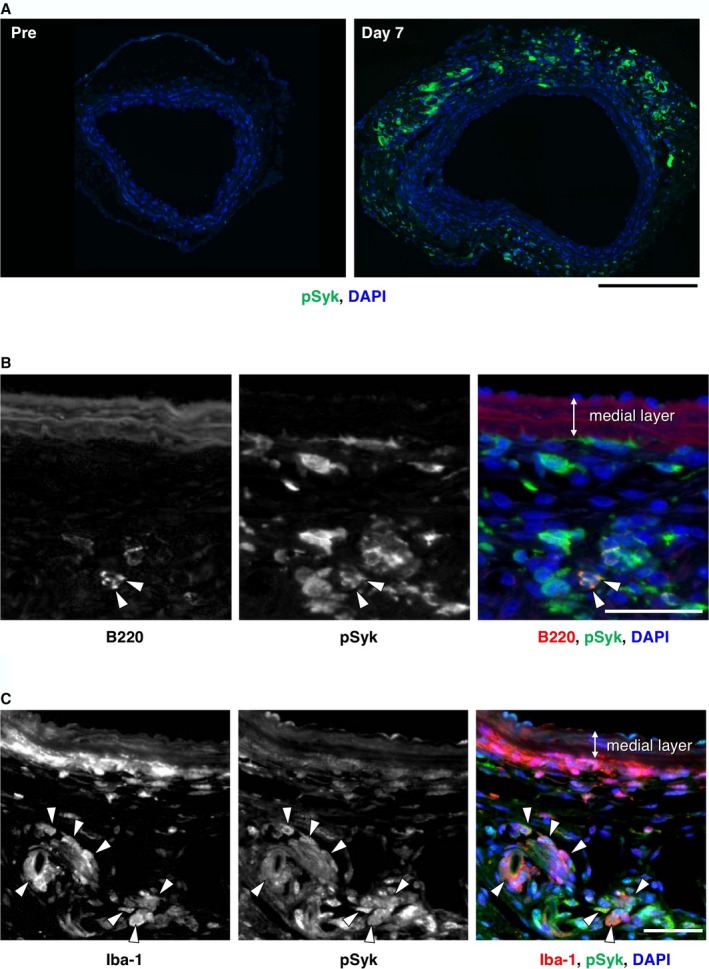
Activation of Syk in mouse AAA. A, Representative images are shown for immunofluorescence staining for pSyk (green color) without (Pre) and with (Day 7) CaCl2 challenge. Bar 200 μm. B, Localization of B cells (B220, arrowheads, red color in the rightmost panel) is shown with pSyk (green color in the rightmost panel) in aortic tissue 7 days after CaCl2 challenge. Bar 50 μm. C, Localization of macrophages (Iba‐1, arrowheads, red color in the rightmost panel) is shown with pSyk (green color in the rightmost panel) in aortic tissue 7 days after CaCl2 challenge. Bar 50 μm. AAA indicates abdominal aortic aneurysm; pSyk, phosphorylated Syk.
Involvement of B Cells in AAA Pathogenesis
We examined the involvement of B cells in AAA pathogenesis using B cell–deficient muMT mice, which had undergone gene targeting to delete the heavy chain of IgM.25 IgM serves as a B‐cell receptor required for early B‐cell development. Thus, in muMT mice, B‐cell development is arrested, and the mice are deficient in mature B cells. When we treated muMT mice with CaCl2 to induce AAA, the AAA lesions were significantly smaller than those observed in WT (Figure 4A). This result indicated that B cells were required for the full development of AAA.
Figure 4.
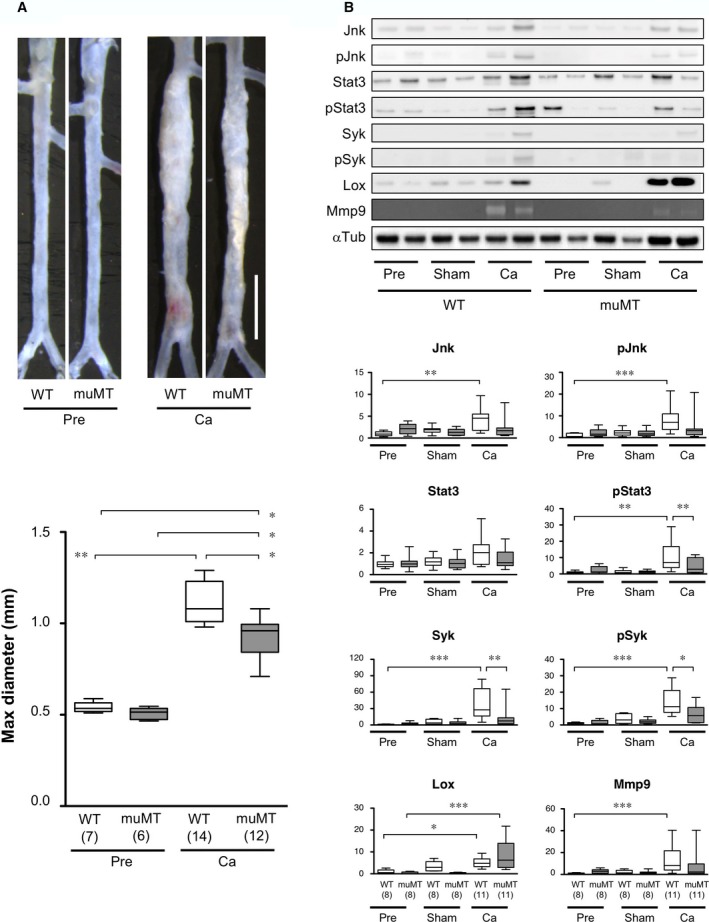
AAA model in B cell‐deficient muMT mice. AAA mouse models were induced with a periaortic CaCl2 treatment. A Upper, Representative images of abdominal aortas before (Pre) and 6 weeks after CaCl2 treatment (Ca) in wild‐type (WT) and B cell–deficient (muMT) mice. White scale bar 2 mm. Lower, Quantitative analysis of the maximal diameters of abdominal aortas. Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plot. B, Upper, Protein expression levels were examined on immunoblots for the indicated proteins and with gelatin zymography for MMP9. Representative image shows aortic samples acquired before (Pre) and 7 days after CaCl2 treatment (Ca) from WT and muMT mice. Sham samples were treated with vehicle only. Lower, White and gray columns indicate WT and muMT mice, respectively. Observation numbers in each experimental group are shown in parentheses at the bottom of the box‐and‐whisker plots. Values indicate fold expression levels relative to expression in WT Pre samples. *P<0.05, **P<0.01, ***P<0.001. AAA indicates abdominal aortic aneurysm; MMP9, matrix metalloproteinase; WT, wild type.
Next, we examined the expression levels of key molecules in AAA pathogenesis (Figure 4B). The small AAA lesions in muMT mice were associated with less significant activations of Jnk, Stat3, and Syk than those observed in WT AAA. Consistent with that finding, compared to WT AAA tissues, muMT AAA tissues showed less significant expression of MMP9, an ECM protease that is regulated by Jnk in AAA,26 and a more significant increase in the expression of lysyl oxidase (Lox), an ECM‐synthesizing enzyme that can ameliorate AAA development.26 The observed changes in the protein expressions were specific to CaCl2 challenge because they were not observed in sham‐operated mice, confirming the results of our previous article that saline treatment did not induce strong inflammation or AAA formation.23 These results indicated that a B‐cell deficiency resulted in the suppression of inflammatory signaling pathways including Syk and impeded development of AAA, possibly by suppressing ECM destruction and enhancing ECM synthesis.
Role of B Cells in Gene Expression During AAA Development
We performed a transcriptome analysis in mouse tissues extracted before and 7 days after CaCl2 challenge to gain insight into B‐cell involvement in AAA pathogenesis. Among the 55 681 probes on the DNA microarray, at 7 days after CaCl2 challenge, 3750 probes showed a significant increase (P<0.05, fold change ≥2), and 3457 probes showed a significant decrease (P<0.05, fold change ≤0.5) in expression in WT aortas (Table 1). At 7 days after CaCl2 challenge, muMT aortas showed higher expression in 299 probes and lower expression in 1244 probes compared to WT aortas at the corresponding time points. Among the probes with elevated expression in muMT aortas after CaCl2 challenge, 180 (60.2%) showed suppressed expression in WT aortas after CaCl2 challenge. Among the 1244 probes with suppressed expression in muMT aortas after CaCl2 challenge, 839 probes (67.4%) showed increased expression in WT aortas after CaCl2 challenge. These results indicated that a major effect of B‐cell deletion was to suppress the effects of CaCl2 challenge. Among the CaCl2 challenge‐induced and ‐suppressed probes in WT AAA aortas, 22.4% and 5.2%, respectively, were ameliorated by B‐cell deletion.
Table 1.
Changes in Gene Expression in a Mouse Model of AAA
| Number of Genes | muMT Day 7/WT Day 7 | ||||
|---|---|---|---|---|---|
| Up | Down | No Change | |||
| 299 | 1244 | ||||
| WT day7/WT pre | Up | 3750 | 1 | 839 | 2910 |
| Down | 3457 | 180 | 12 | 3265 | |
| No change | 118 | 393 | |||
The number of genes that showed significant expression changes are shown for WT mice before (pre) and after CaCl2 treatment (day 7). The number of genes that showed significant expression changes after CaCl2 treatment are shown for muMT mice relative to WT mice. AAA indicates abdominal aortic aneurysm; WT, wild type.
We analyzed the functional annotations of the genes that were induced by CaCl2 challenge in WT tissues and suppressed by CaCl2 challenge in muMT tissues. Functional annotations were retrieved from DAVID (the Database for Annotation, Visualization, and Integrated Discovery).27 The gene ontology terms in DAVID revealed functional annotations that clustered with “immune response” and “defense response” (Table 2). We also performed a DAVID analysis of genes that were suppressed by CaCl2 challenge in WT tissues and restored by CaCl2 challenge in muMT tissues. The gene ontology terms revealed functional annotations that clustered with “myofibril,” “muscle system process,” and “muscle structure development” (Table 3). These results suggested that, in the mouse AAA model, immune and inflammatory responses were induced, and muscle components, probably representing aortic wall smooth muscle cells, were suppressed. These findings underscored the importance of B cells in AAA pathogenesis.
Table 2.
Functional Annotation Clusters of CaCl2‐Induced Genes With an Influence of B‐Cell Deletion
| Annotation Cluster 1 | Enrichment Score: 33.77128000272969 |
|---|---|
| Category | Term |
| GOTERM_BP_FAT | GO:0006955—immune response |
| GOTERM_BP_FAT | GO:0045321—leukocyte activation |
| GOTERM_BP_FAT | GO:0006952—defense response |
| GOTERM_BP_FAT | GO:0002682—regulation of immune system process |
| GOTERM_BP_FAT | GO:0002684—positive regulation of immune system process |
| GOTERM_BP_FAT | GO:0050776—regulation of immune response |
| GOTERM_BP_FAT | GO:0050778—positive regulation of immune response |
| GOTERM_BP_FAT | GO:0002252—immune effector process |
| GOTERM_BP_FAT | GO:0048584—positive regulation of response to stimulus |
| Annotation Cluster 2 | Enrichment Score: 20.427032466893277 |
|---|---|
| Category | Term |
| GOTERM_CC_FAT | GO:0009897—external side of plasma membrane |
| GOTERM_CC_FAT | GO:0098552—side of membrane |
| GOTERM_CC_FAT | GO:0009986—cell surface |
| Annotation Cluster 3 | Enrichment Score: 15.990954240580606 |
|---|---|
| Category | Term |
| GOTERM_BP_FAT | GO:0006952—defense response |
| GOTERM_BP_FAT | GO:0045087—innate immune response |
| GOTERM_BP_FAT | GO:0050776—regulation of immune response |
| GOTERM_BP_FAT | GO:0050778—positive regulation of immune response |
| GOTERM_BP_FAT | GO:0006954—inflammatory response |
| GOTERM_BP_FAT | GO:0031349—positive regulation of defense response |
| GOTERM_BP_FAT | GO:0031347—regulation of defense response |
| GOTERM_BP_FAT | GO:0045089—positive regulation of innate immune response |
| GOTERM_BP_FAT | GO:0045088—regulation of innate immune response |
| GOTERM_BP_FAT | GO:0002757—immune response‐activating signal transduction |
Functional annotation clusters are shown for genes with induction by CaCl2 treatment in wild‐type mice that were suppressed in muMT mice. GO term: gene ontology term provided in DAVID (the Database for Annotation, Visualization, and Integrated Discovery) analysis. Only the first 10 terms are shown for simplicity.
Table 3.
Functional Annotation Clusters of CaCl2‐Suppressed Genes With an Influence of B‐Cell Deletion
| Annotation Cluster 1 | Enrichment Score: 8.508519489022873 |
|---|---|
| Category | Term |
| GOTERM_CC_FAT | GO:0030016—myofibril |
| GOTERM_CC_FAT | GO:0043292—contractile fiber |
| GOTERM_CC_FAT | GO:0030017—sarcomere |
| GOTERM_CC_FAT | GO:0044449—contractile fiber part |
| GOTERM_CC_FAT | GO:0031674—I band |
| GOTERM_CC_FAT | GO:0030018—Z disc |
| Annotation Cluster 2 | Enrichment Score: 3.5140614375419363 |
|---|---|
| Category | Term |
| GOTERM_BP_FAT | GO:0003012—muscle system process |
| GOTERM_BP_FAT | GO:0006936—muscle contraction |
| GOTERM_BP_FAT | GO:0006941—striated muscle contraction |
| GOTERM_BP_FAT | GO:0044057—regulation of system process |
| GOTERM_BP_FAT | GO:0090257—regulation of muscle system process |
| GOTERM_BP_FAT | GO:0006937—regulation of muscle contraction |
| GOTERM_BP_FAT | GO:0008016—regulation of heart contraction |
| GOTERM_BP_FAT | GO:0008015—blood circulation |
| GOTERM_BP_FAT | GO:0003013—circulatory system process |
| GOTERM_BP_FAT | GO:0060047—heart contraction |
| Annotation Cluster 3 | Enrichment Score: 2.981185764056471 |
|---|---|
| Category | Term |
| GOTERM_BP_FAT | GO:0061061—muscle structure development |
| GOTERM_BP_FAT | GO:0055002—striated muscle cell development |
| GOTERM_BP_FAT | GO:0051146—striated muscle cell differentiation |
| GOTERM_BP_FAT | GO:0055001—muscle cell development |
| GOTERM_BP_FAT | GO:0060537—muscle tissue development |
| GOTERM_BP_FAT | GO:0014706—striated muscle tissue development |
| GOTERM_BP_FAT | GO:0042692—muscle cell differentiation |
| GOTERM_BP_FAT | GO:0030239—myofibril assembly |
| GOTERM_BP_FAT | GO:0031032—actomyosin structure organization |
| GOTERM_BP_FAT | GO:0007507—heart development |
Functional annotation clusters for genes with suppression by CaCl2 treatment in wild‐type mice that were restored in muMT mice. GO term: gene ontology term provided in DAVID (the Database for Annotation, Visualization, and Integrated Discovery) analysis. Only the first 10 terms are shown for simplicity.
Involvement of Immunoglobulins in AAA Pathogenesis
Based on our finding that immunoglobulins, the major effector molecules of B cells, were deposited in the aortic walls of Ca‐treated mice, we reasoned that immunoglobulins might be involved in AAA pathogenesis. To test this hypothesis, we administered mouse polyclonal IgG to muMT mice with the CaCl2 challenge. IgG with Ca administration caused muMT mice to develop AAA comparable to that observed in WT mice (Figure 5A). This result indicated that IgG was sufficient for the full development of AAA in muMT mice. We examined the expression and activities of inflammatory and ECM metabolic molecules in muMT AAA tissues (Figure 5B). CaCl2 challenge caused an increase in the expression of Stat3, Lox, and MMP9 and activation of Jnk and Syk. Administration of exogenous IgG caused more significant expression of Jnk, Stat3, Syk, and MMP9; more significant activation of Syk; and less significant expression of Lox.
Figure 5.
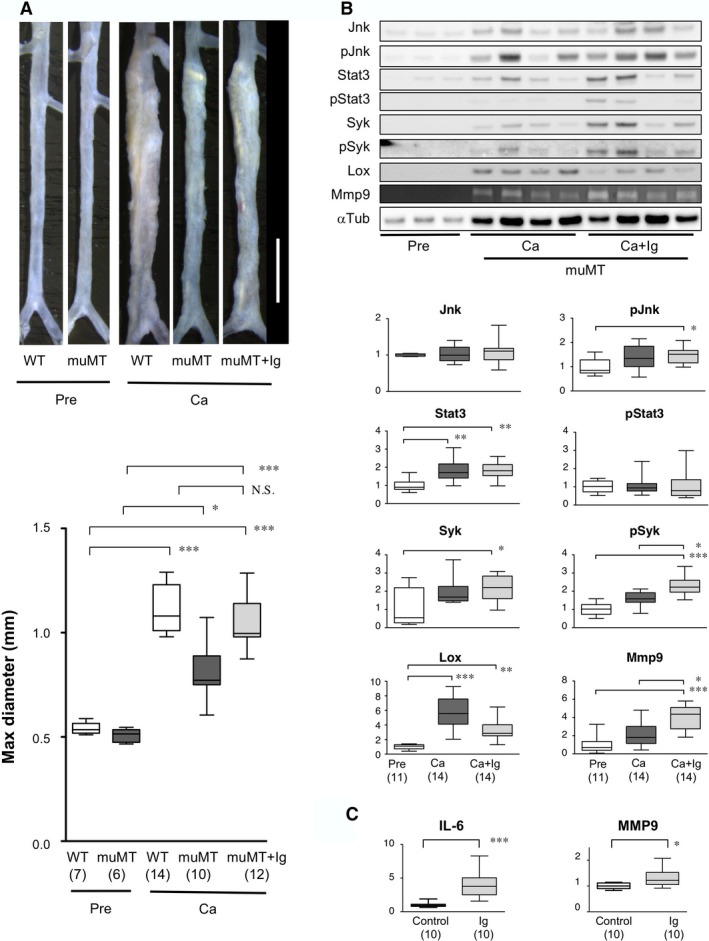
Effect of exogenous immunoglobulins on AAA in muMT mice and human AAA tissue in culture. A, Upper, Representative images of abdominal aortas before (Pre) and 6 weeks after CaCl2 treatment (Ca) in wild‐type (WT) and mutant B cell‐deficient (muMT) mice, treated with or without exogenous immunoglobulins (Ig). White scale bar 2 mm. Lower, Quantitative analysis of maximal diameters of abdominal aortas. Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plot. B, Upper, Representative images of immunoblots and zymography show protein expression levels in samples acquired before and 7 days after CaCl2 treatment, alone (Ca) or with exogenous immunoglobulins (Ca+Ig) in muMT mice. Lower, Quantitative analyses of the indicated molecules. Columns indicate protein expression levels before treatment (Pre, white), after CaCl2 treatment (Ca, dark gray), and after CaCl2 with immunoglobulin treatment (Ca+Ig, light gray). Observation numbers are shown in parentheses at the bottom of the box and whisker plots. Values represent fold expression relative to expression in Pre samples. C, Human AAA aortic wall tissue samples were minced and cultured in serum‐free conditions for 48 hours. Basal IL‐6 and MMP9 secretions were measured in culture supernatants. Cultures were then treated without (Control) or with 0.5 mg/mL exogenous human immunoglobulins (Ig) for another 48 hours. Culture supernatants were measured to determine stimulated secretion levels. The ratios of stimulated/basal secretions are shown; Control samples were set to 1. Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plots. Human immunoglobulins of different lots showed essentially identical results. *P<0.05, **P<0.01, ***P<0.001. AAA indicates abdominal aortic aneurysm; MMP9, matrix metalloproteinase‐9; N.S., not significant.
To test whether our findings in mouse AAA models were relevant to human AAA, we examined the effect of immunoglobulins on human AAA tissue in culture. We found that the addition of exogenous immunoglobulin significantly increased the secretions of IL‐6 and MMP9 (Figure 5C). This result indicated that exogenous immunoglobulins had a proinflammatory effect on human AAA tissues, consistent with our findings in the mouse AAA model. These results suggested that the effects of B cells seemed to be mediated by IgG.
Involvement of Syk in AAA Pathogenesis
To explore a potential therapeutic target for treating AAA, we focused on the role of Syk in AAA pathogenesis because Syk plays a pivotal role in B‐cell activation and in the effector function of immunoglobulins.19, 28 Syk is known to modulate various biological processes including apoptosis, cell cycle, inflammatory response, and expression of matrix metalloproteinases.18, 29 We examined the effect of B‐cell deletion on these biological processes by examining the expression pattern of genes with these annotations before and 7 days after CaCl2 challenge in WT and muMT aortas (Figure 6, Tables S1 through S4). Hierarchical clustering analysis of the gene expression pattern showed that a significant portion of these genes were either induced or suppressed by CaCl2 challenge in WT mice, and these changes were ameliorated in muMT mice, indicating that these reportedly Syk‐dependent biological processes were modulated by deletion of B cells.
Figure 6.
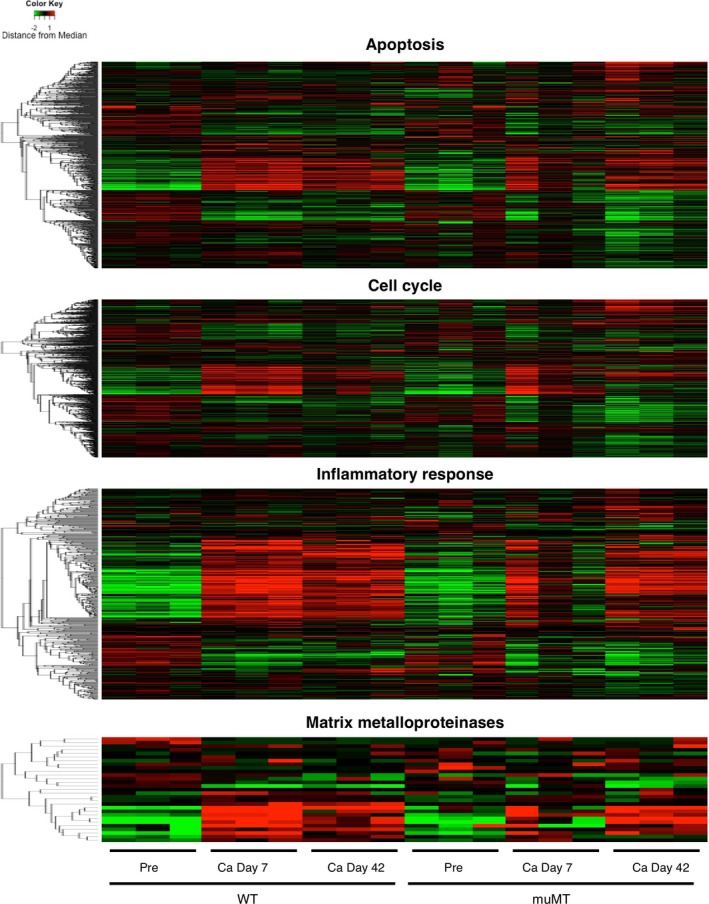
Gene expression profile in mouse aorta. Results of the hierarchical clustering analysis of the genes with the indicated annotations are shown by heat map. After log conversion of the signal value, the distance from the median is shown in green, black, and red when lower, intermediate, and higher than those of other samples, respectively, within a given gene.
Next, we treated mice with R788 (fostamatinib), an oral Syk inhibitor,24 and examined its effect on AAA development after CaCl2 challenge. The administration of R788 in drinking water resulted in a significant reduction in the diameter of CaCl2‐induced AAA lesions (Figure 7A). In contrast, CaCl2 challenge with vehicle administration induced AAA development comparable to CaCl2 alone. These results indicated that Syk was involved in AAA pathogenesis. We examined the inflammatory response 7 days after CaCl2 challenge with and without the administration of R788 (Figure 7B). CaCl2 challenge caused a marked increase in IgG deposition. Administration of R788 caused a significant decrease in tissue IgG deposition, whereas it showed no significant effect on the serum level of IgG; this result suggested that IgG deposition was regulated locally in aortic tissue. We also examined Syk, Jnk, Stat3, and Lox with an immunoblot analysis and the MMP9 with gelatin zymography. Mice treated with R788 showed less significant activations of Jnk, Stat3, and Syk, less significant induction of MMP9, and more significant induction of Lox in response to CaCl2 challenge. These findings suggested that Syk promoted AAA development by activating the inflammatory response and ECM degradation.
Figure 7.
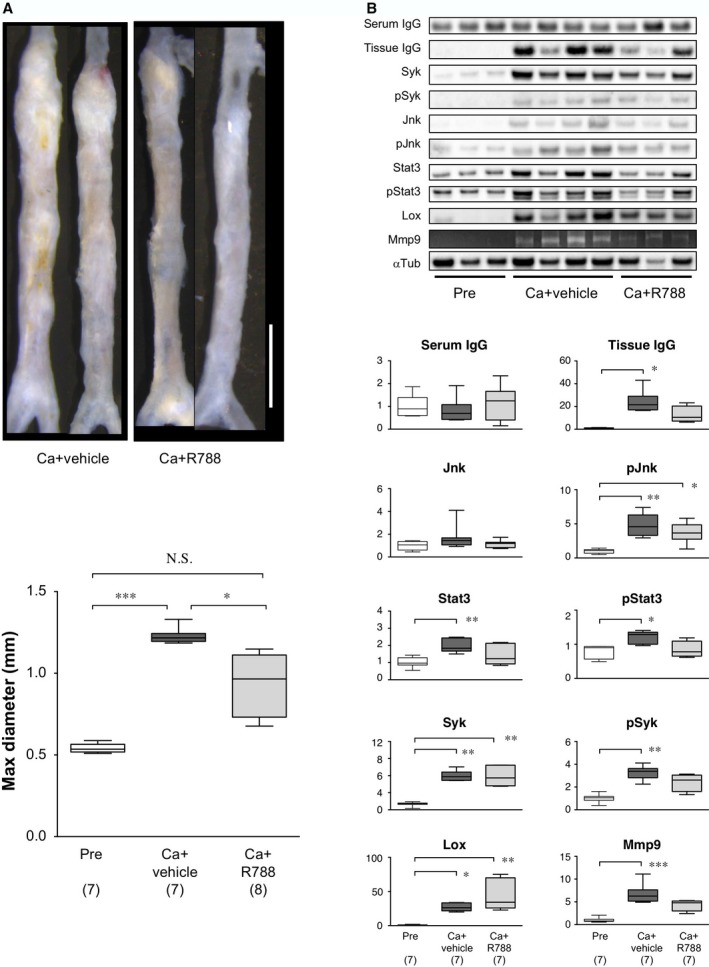
Effect of Syk inhibitor on murine AAA. A, Upper, Representative images of abdominal aortas, 6 weeks after CaCl2 treatment, without (Ca+vehicle) or with R788 administration (Ca+R788). White bar 2 mm. Lower, Quantitative analysis of maximal diameters of abdominal aortas. Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plot. B, Upper, Representative images of immunoblots and zymography show protein levels and MMP9 activity, respectively, in samples acquired before (Pre) and 7 days after CaCl2 treatment, without (Ca+vehicle) or with R788 administration (Ca+R788). Lower, Quantitative analyses of the indicated molecules on immunoblots and gelatin zymography (for MMP9). Columns indicate protein expression levels measured before (pre, white columns) or after periaortic application of CaCl2, without (dark gray columns) or with R788 treatment (light gray columns). Observation numbers are shown in parentheses at the bottom of the box‐and‐whisker plots. Values indicate fold expression levels relative to expression measured in Pre samples. *P<0.05, **P<0.01, ***P<0.001. AAA indicates abdominal aortic aneurysm; MMP9, matrix metalloproteinase‐9; N.S., not significant.
Discussion
In this study we demonstrated that, in a mouse AAA model induced with a periaortic application of CaCl2, B cells promoted the development of AAA through the proinflammatory effect of immunoglobulins. B cell–deficient muMT mice showed reduced AAA expansion, which was associated with blunted inflammatory and tissue destructive activities, compared to WT mice. Administration of exogenous immunoglobulins restored the molecular and morphological phenotypes in muMT to the levels observed in WT AAA mice; moreover, exogenous immunoglobulins stimulated IL‐6 and MMP9 secretions in human AAA tissue cultures. Conversely, inhibition of Syk, a tyrosine kinase essential in B cell–mediated inflammation,18, 19, 20, 21 ameliorated the inflammatory response and the expansion of AAA in WT mice. These findings indicated that B cells promoted inflammation and an imbalance in ECM metabolism to enhance tissue destruction, and these activities appeared to involve immunoglobulins and Syk.
Several animal models of AAA have been described previously. In WT mice AAA can be induced by exposing the aorta to elastase or CaCl2. In Apoe‐deficient or Ldlr‐deficient hyperlipidemic mice, AAA can be induced with a continuous infusion of angiotensin II. Our finding that B cells were required for the full development of AAA after CaCl2 challenge was consistent with previous reports that showed that muMT mice were protected from elastase‐induced AAA14 and that B cell depletion by anti‐CD20 antibody abrogated AAA development induced by angiotensin II in Apoe‐deficient mice.15 On the other hand, a state of total lymphocyte deficiency attenuated atherosclerosis in Apoe‐deficient mice but not angiotensin II–induced AAA30; furthermore, muMT mice were not protected from elastase‐induced AAA.16 A potential explanation for these controversial results could be that T cells participated in AAA pathogenesis by modulating the effect of B cells.15, 16 For example, the deletion of all T cells has been reported to abrogate the effect of regulatory T cells in suppressing AAA formation in an angiotensin II–induced model.31, 32, 33 Because no animal model can recapitulate all the aspects of human AAA, further research is required to elucidate the role of B cells in human AAA. In this study we found that administration of exogenous polyclonal immunoglobulins was sufficient to promote IL‐6 and MMP9 secretions in human AAA tissue. Therefore, circulating immunoglobulins may contribute to the maintenance of chronic inflammation in human patients with AAA. B cells residing in AAA tissues may also be important for antigen recognition and continuous production of pathogenic immunoglobulins, including those against fibrinogen, as previously reported, and other endogenous antigens in AAA tissue.13, 14 Our finding that immunoglobulins were deposited on AAA tissue and localized to most of adventitial cells suggests that they may activate diverse cell types in AAA tissue.
Most patients with AAA exhibit no symptoms. Therefore, therapeutic interventions should aim to prevent or slow AAA expansion, which may require years of treatment. Our findings and previous reports have indicated that B cells and immunoglobulins have AAA‐promoting effects. However, it would be deleterious to remove whole B cells or immunoglobulins from patients with AAA because of the resulting immunosuppression. Therefore, it is imperative to elucidate how B cells and immunoglobulins exert AAA‐promoting effects. This information will facilitate identification of a therapeutic target that can be modulated, preferably by small therapeutic compounds, to achieve flexibility and adjustability for individual treatments. Of note, in this study we found that Syk was activated in a manner dependent on the presence of B cells and immunoglobulins. Activated Syk was localized in B cells and macrophages, suggesting its involvement in the inflammatory response. Furthermore, R788, a small‐molecule inhibitor of Syk, suppressed the inflammatory response and expansion of AAA. Due to the diverse functions of Syk,29, 34, 35, 36 the mechanism underlying R788 suppression of AAA expansion may not be limited to its function in B cells. Indeed, we found that Syk is activated not only in B cells but also in other cell types including macrophages. Because the macrophage is an important driver of AAA37 and Syk is involved in macrophage‐mediated inflammation,29 activation of macrophage Syk, possibly by the effect of immunoglobulins, may be involved in AAA pathogenesis. Elucidation of such a Syk‐dependent disease mechanism may reveal a new avenue of AAA research. It would be important to examine the effect of a Syk inhibitor on normal aorta and other organs to validate its safety. Although depletion of B cells by anti‐CD20 antibody is another potential option, its effect lasts for months, which may raise a concern for unwanted side effects. The feasibility of Syk inhibition or B‐cell suppression needs to be further tested in a context that more closely mimics a clinical setting.
Other issues that must be addressed, when one is developing pharmacotherapies for AAA, are the relevant time window and duration for applying therapeutic interventions. A potential therapeutic window could be the acute period after introducing a stent‐graft in an EVAR. It is well known that the EVAR procedure causes an acute inflammatory reaction, demonstrated by an increase in circulating C‐reactive protein and MMP9 levels.38, 39 That inflammatory response might cause acute degradation in the stent‐graft landing zone, which could compromise long‐term stability.39 Acute and short‐term suppression of the AAA‐promoting functions of B cells or immunoglobulins, possibly with a Syk inhibitor, may be a feasible intervention to promote long‐term stability after an EVAR. Another potential scenario might be a biomarker‐directed pharmacotherapy, where therapeutic reagents are administered only when the biomarker indicates high disease activity.40, 41
In conclusion, we demonstrated that B cells promoted AAA expansion by enhancing the proinflammatory function of immunoglobulins and that Syk represents a potential therapeutic target in AAA. Further studies are required to understand how B cells are activated and how immunoglobulins and Syk exert proinflammatory functions in the context of AAA. Understanding the B cell–mediated promotion of AAA could lead to the identification of molecular targets for future diagnostic and therapeutic approaches.
Sources of Funding
This work was funded, in part, by grants from the Japan Society for the Promotion of Science (25861237, 15K19928, and 17K10770) to Furusho; grants from the Japan Society for the Promotion of Science (21390367, 24390334, 24659640, 26670621, and 16H05428) to Aoki; grants from the Daiichi Sankyo Foundation of Life Science, the Uehara Memorial Foundation to Aoki, the Vehicle Racing Commemorative Foundation to Aoki, and Bristol‐Myers Squibb to Aoki; and a TaNeDS grant from Daiichi Sankyo to Aoki.
Disclosures
None.
Supporting information
Table S1. Expression Analysis of Apoptosis‐Related Genes Expression of apoptosis‐related genes as determined by DNA microarray are shown, which correspond to the top panel of Figure 6
Table S2. Expression Analysis of Cell Cycle–Related Genes Expression of cell cycle–related genes as determined by DNA microarray are shown, which correspond to the second panel of Figure 6
Table S3. Expression Analysis of Inflammatory Response–Related Genes. Expression of inflammatory response–related genes as determined by DNA microarray are shown, which correspond to the third panel of Figure 6
Table S4. Expression Analysis of Matrix Metalloproteinase Genes. Expression of genes for matrix metalloproteinases as determined by DNA microarray are shown, which correspond to the bottom panel of Figure 6
Acknowledgments
We would like to thank Kiyohiro, Nishigata, Nakao, Shiramizu, Nakayama, and Yamamoto for technical assistance.
(J Am Heart Assoc. 2018;7:e007750 DOI: 10.1161/JAHA.117.007750.)29545260
References
- 1. Cronenwett JL, Johnston KW. Vascular Surgery. Philadelphia: Elsevier Health Sciences; 2014. [Google Scholar]
- 2. Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, Hiratzka LF, Murphy WR, Olin JW, Puschett JB, Rosenfield KA, Sacks D, Stanley JC, Taylor LM Jr, White CJ, White J, White RA, Antman EM, Smith SC Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Gibbons RJ, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter‐Society Consensus; and Vascular Disease Foundation. Circulation. 2006;113:e463–e654. [DOI] [PubMed] [Google Scholar]
- 3. EVAR Trial Participants . Endovascular aneurysm repair versus open repair in patients with abdominal aortic aneurysm (EVAR trial 1): randomised controlled trial. Lancet. 2005;365:2179–2186. [DOI] [PubMed] [Google Scholar]
- 4. EVAR Trial Participants . Endovascular aneurysm repair and outcome in patients unfit for open repair of abdominal aortic aneurysm (EVAR trial 2): randomised controlled trial. Lancet. 2005;365:2187–2192. [DOI] [PubMed] [Google Scholar]
- 5. Bergoeing MP, Thompson RW, Curci JA. Pharmacological targets in the treatment of abdominal aortic aneurysms. Expert Opin Ther Targets. 2006;10:547–559. [DOI] [PubMed] [Google Scholar]
- 6. Senemaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2017;37:401–410. [DOI] [PubMed] [Google Scholar]
- 7. Emeto TI, Seto SW, Golledge J. Targets for medical therapy to limit abdominal aortic aneurysm progression. Curr Drug Targets. 2014;15:860–873. [DOI] [PubMed] [Google Scholar]
- 8. Aoki H, Yoshimura K, Matsuzaki M. Turning back the clock: regression of abdominal aortic aneurysms via pharmacotherapy. J Mol Med. 2007;85:1077–1088. [DOI] [PubMed] [Google Scholar]
- 9. Guessous I, Periard D, Lorenzetti D, Cornuz J, Ghali WA. The efficacy of pharmacotherapy for decreasing the expansion rate of abdominal aortic aneurysms: a systematic review and meta‐analysis. PLoS One. 2008;3:e1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ocana E, Bohorquez JC, Perez‐Requena J, Brieva JA, Rodriguez C. Characterisation of T and B lymphocytes infiltrating abdominal aortic aneurysms. Atherosclerosis. 2003;170:39–48. [DOI] [PubMed] [Google Scholar]
- 11. Zhang L, Wang Y. B lymphocytes in abdominal aortic aneurysms. Atherosclerosis. 2015;242:311–317. [DOI] [PubMed] [Google Scholar]
- 12. Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. [DOI] [PubMed] [Google Scholar]
- 13. Zhou HF, Yan H, Bertram P, Hu Y, Springer LE, Thompson RW, Curci JA, Hourcade DE, Pham CT. Fibrinogen‐specific antibody induces abdominal aortic aneurysm in mice through complement lectin pathway activation. Proc Natl Acad Sci USA. 2013;110:E4335–E4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou HF, Yan H, Stover CM, Fernandez TM, Rodriguez de Cordoba S, Song WC, Wu X, Thompson RW, Schwaeble WJ, Atkinson JP, Hourcade DE, Pham CT. Antibody directs properdin‐dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm. Proc Natl Acad Sci USA. 2012;109:E415–E422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schaheen B, Downs EA, Serbulea V, Almenara CC, Spinosa M, Su G, Zhao Y, Srikakulapu P, Butts C, McNamara CA, Leitinger N, Upchurch GR Jr, Meher AK, Ailawadi G. B‐cell depletion promotes aortic infiltration of immunosuppressive cells and is protective of experimental aortic aneurysm. Arterioscler Thromb Vasc Biol. 2016;36:2191–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meher AK, Johnston WF, Lu G, Pope NH, Bhamidipati CM, Harmon DB, Su G, Zhao Y, McNamara CA, Upchurch GR Jr, Ailawadi G. B2 cells suppress experimental abdominal aortic aneurysms. Am J Pathol. 2014;184:3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Krishna S, Golledge J. The calcium chloride‐induced rodent model of abdominal aortic aneurysm. Atherosclerosis. 2013;226:29–39. [DOI] [PubMed] [Google Scholar]
- 18. Deng GM, Kyttaris VC, Tsokos GC. Targeting Syk in autoimmune rheumatic diseases. Front Immunol. 2016;7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009;228:132–148. [DOI] [PubMed] [Google Scholar]
- 20. Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. [DOI] [PubMed] [Google Scholar]
- 21. Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci. 2014;35:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Furusho A, Aoki H, Ohno‐Urabe S, Nishihara M, Hirakata S, Nishida N, Ito S, Hayashi M, Imaizumi T, Hiromatsu S, Akashi H, Tanaka N, Fukumoto Y. Involvement of B cells, immunoglobulins, and Syk in the pathogenesis of abdominal aortic aneurysm. 2018. Gene Expression Omnibus, NCBI. GSE109640. [DOI] [PMC free article] [PubMed]
- 23. Nishihara M, Aoki H, Ohno S, Furusho A, Hirakata S, Nishida N, Ito S, Hayashi M, Imaizumi T, Fukumoto Y. The role of IL‐6 in pathogenesis of abdominal aortic aneurysm in mice. PLoS One. 2017;12:e0185923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Colonna L, Catalano G, Chew C, D'Agati V, Thomas JW, Wong FS, Schmitz J, Masuda ES, Reizis B, Tarakhovsky A, Clynes R. Therapeutic targeting of Syk in autoimmune diabetes. J Immunol. 2010;185:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell‐deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature. 1991;350:423–426. [DOI] [PubMed] [Google Scholar]
- 26. Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c‐Jun N‐terminal kinase. Nat Med. 2005;11:1330–1338. [DOI] [PubMed] [Google Scholar]
- 27. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 28. Tsubata T, Wienands J. B cell signaling. Introduction. Int Rev Immunol. 2001;20:675–678. [DOI] [PubMed] [Google Scholar]
- 29. Yi YS, Son YJ, Ryou C, Sung GH, Kim JH, Cho JY. Functional roles of Syk in macrophage‐mediated inflammatory responses. Mediators Inflamm. 2014;2014:270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, Daugherty A. Total lymphocyte deficiency attenuates AngII‐induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis. 2010;211:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ait‐Oufella H, Wang Y, Herbin O, Bourcier S, Potteaux S, Joffre J, Loyer X, Ponnuswamy P, Esposito B, Dalloz M, Laurans L, Tedgui A, Mallat Z. Natural regulatory T cells limit angiotensin II‐induced aneurysm formation and rupture in mice. Arterioscler Thromb Vasc Biol. 2013;33:2374–2379. [DOI] [PubMed] [Google Scholar]
- 32. Meng X, Yang J, Zhang K, An G, Kong J, Jiang F, Zhang Y, Zhang C. Regulatory T cells prevent angiotensin II‐induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension. 2014;64:875–882. [DOI] [PubMed] [Google Scholar]
- 33. Yodoi K, Yamashita T, Sasaki N, Kasahara K, Emoto T, Matsumoto T, Kita T, Sasaki Y, Mizoguchi T, Sparwasser T, Hirata K. Foxp3+ regulatory T cells play a protective role in angiotensin II‐induced aortic aneurysm formation in mice. Hypertension. 2015;65:889–895. [DOI] [PubMed] [Google Scholar]
- 34. Denyer J, Patel V. Syk kinase inhibitors in allergic diseases. Drug News Perspect. 2009;22:146–150. [DOI] [PubMed] [Google Scholar]
- 35. Lucas MC, Tan SL. Small‐molecule inhibitors of spleen tyrosine kinase as therapeutic agents for immune disorders: will promise meet expectations? Future Med Chem. 2014;6:1811–1827. [DOI] [PubMed] [Google Scholar]
- 36. Navara CS. The spleen tyrosine kinase (Syk) in human disease, implications for design of tyrosine kinase inhibitor based therapy. Curr Pharm Des. 2004;10:1739–1744. [DOI] [PubMed] [Google Scholar]
- 37. Raffort J, Lareyre F, Clement M, Hassen‐Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. [DOI] [PubMed] [Google Scholar]
- 38. Arnaoutoglou E, Kouvelos G, Milionis H, Mavridis A, Kolaitis N, Papa N, Papadopoulos G, Matsagkas M. Post‐implantation syndrome following endovascular abdominal aortic aneurysm repair: preliminary data. Interact Cardiovasc Thorac Surg. 2011;12:609–614. [DOI] [PubMed] [Google Scholar]
- 39. Ng E, Morris DR, Golledge J. The association between plasma matrix metalloproteinase‐9 concentration and endoleak after endovascular aortic aneurysm repair: a meta‐analysis. Atherosclerosis. 2015;242:535–542. [DOI] [PubMed] [Google Scholar]
- 40. Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nat Rev Cardiol. 2009;6:543–552. [DOI] [PubMed] [Google Scholar]
- 41. Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of AAA progression. Part 1: extracellular matrix degeneration. Nat Rev Cardiol. 2009;6:464–474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Expression Analysis of Apoptosis‐Related Genes Expression of apoptosis‐related genes as determined by DNA microarray are shown, which correspond to the top panel of Figure 6
Table S2. Expression Analysis of Cell Cycle–Related Genes Expression of cell cycle–related genes as determined by DNA microarray are shown, which correspond to the second panel of Figure 6
Table S3. Expression Analysis of Inflammatory Response–Related Genes. Expression of inflammatory response–related genes as determined by DNA microarray are shown, which correspond to the third panel of Figure 6
Table S4. Expression Analysis of Matrix Metalloproteinase Genes. Expression of genes for matrix metalloproteinases as determined by DNA microarray are shown, which correspond to the bottom panel of Figure 6