

Abstract
Background
A clear understanding of the molecular mechanisms underlying hemodynamic stress‐initiated cardiac hypertrophy is important for preventing heart failure. Interferon‐γ (IFN‐γ) has been suggested to play crucial roles in various diseases other than immunological disorders by modulating the expression of myriad genes. However, the involvement of IFN‐γ in the pathogenesis of cardiac hypertrophy still remains unclear.
Methods and Results
In order to elucidate the roles of IFN‐γ in pressure overload–induced cardiac pathology, we subjected Balb/c wild‐type (WT) or IFN‐γ‐deficient (Ifng −/−) mice to transverse aortic constriction (TAC). Three weeks after TAC, Ifng −/− mice developed more severe cardiac hypertrophy, fibrosis, and dysfunction than WT mice. Bone marrow–derived immune cells including macrophages were a source of IFN‐γ in hearts after TAC. The activation of PI3K/Akt signaling, a key signaling pathway in compensatory hypertrophy, was detected 3 days after TAC in the left ventricles of WT mice and was markedly attenuated in Ifng −/− mice. The administration of a neutralizing anti‐IFN‐γ antibody abrogated PI3K/Akt signal activation in WT mice during compensatory hypertrophy, while that of IFN‐γ activated PI3K/Akt signaling in Ifng −/− mice. TAC also induced the phosphorylation of Stat5, but not Stat1 in the left ventricles of WT mice 3 days after TAC. Furthermore, IFN‐γ induced Stat5 and Akt phosphorylation in rat cardiomyocytes cultured under stretch conditions. A Stat5 inhibitor significantly suppressed PI3K/Akt signaling activation in the left ventricles of WT mice, and aggravated pressure overload–induced cardiac hypertrophy.
Conclusions
The IFN‐γ/Stat5 axis may be protective against persistent pressure overload–induced cardiac hypertrophy by activating the PI3K/Akt pathway.
Keywords: cell signaling, cytokine, hypertrophy, interferon‐γ, PI3K/Akt, protein kinase B, signal transducer and activator of transcription 5
Subject Categories: Basic Science Research, Cell Signalling/Signal Transduction, Growth Factors/Cytokines, Myocardial Biology
Clinical Perspective
What Is New?
Our study demonstrated a novel function of interferon‐γ in the pathogenesis of pressure overload–induced heart failure.
Three weeks after transverse aortic constriction, Ifng −/− mice developed more severe cardiac hypertrophy, fibrosis, and dysfunction than WT mice. Interferon‐γ produced by bone marrow–derived cells including CD68+ macrophages were involved in PI3K/Akt signaling activation through Stat5 phosphorylation in compensatory cardiac hypertrophy.
These observations would indicate that interferon‐γ/Stat5 signal pathway can be protective against transverse aortic constriction–induced cardiac hypertrophy.
What Are the Clinical Implications?
The molecules involved in the interferon‐γ/Stat5/PI3K/Akt pathway may be targets for the prevention and/or treatment of pressure overload–induced cardiac hypertrophy and eventual cardiac failure.
Thus, we believe that these findings will interest cardiologists and other clinical physicians.
Introduction
The mammalian heart responds to environmental demands, and displays different growth patterns in response to various stimuli. Cardiac hypertrophy is induced by an increased workload caused by a pathological or physiological stimulation, and is initiated as an adaptive response to preserve cardiac function by reducing myocardial wall stress and energy expenditure.1 In contrast, impairments in adaptive mechanisms may ultimately result in heart failure.2, 3 Thus, it is important to clearly understand the molecular mechanisms underlying hemodynamic stress–initiated cardiac hypertrophy in order to prevent heart failure.
Evidence is accumulating to imply that chronic inflammation is an underlying cause of various lifestyle‐related diseases and cancer. In patients with heart failure, the levels of a number of inflammatory cytokines, such as tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6,4, 5 were shown to be significantly elevated in plasma and circulating leukocytes, as well as in the failing myocardium itself.6 Moreover, besides these proinflammatory cytokines, additional inflammatory mediators have been proposed to play important roles in the pathogenesis of chronic heart failure arising from maladaptive cardiac remodeling based on the findings obtained from experimental studies7, 8 and clinical trials.9
Interferon (IFN)‐γ is a proinflammatory cytokine and is known as a key player in the immune system because it exerts pleiotropic effects on T cells, NK cells, and macrophages, including enhancements in their antiviral and bactericidal activities and the upregulation of major histocompatibility complex class II expression on macrophages. Moreover, IFN‐γ may be crucially involved in various types of diseases other than immunological disorders10, 11, 12 by modulating the expression of myriad genes. We also previously revealed the protective roles of IFN‐γ against sodium arsenite–induced and cisplatin‐induced renal injury through its enhancement of ABC transporter expression and activation of autophagic flux, respectively.13, 14
Similar to other pro‐inflammatory cytokines,15, 16, 17 IFN‐γ appears to be involved in the pathogenesis of chronic heart failure. However, inconsistent observations in chronic heart failure models18, 19, 20 prompted us to investigate the roles of endogenous IFN‐γ in cardiac hypertrophy, particularly persistent hemodynamic stress–induced cardiac hypertrophy. Hence, we herein examined the effects of sustained pressure overload on the hearts of IFN‐γ‐deficient (Ifng −/−) and wild‐type (WT) mice. The results obtained demonstrated that endogenous IFN‐γ has protective roles in the compensatory responses of cardiomyocytes to prolonged hemodynamic stress.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure because some materials are used for other unpublished projects.
Reagents and Antibodies
Recombinant mouse and rat IFN‐γ were purchased from PeproTech, Inc (Rocky Hill, NJ), while a Stat5 inhibitor (N′‐((4‐Oxo‐4H‐chromen‐3‐yl) methylene) nicotinohydrazide) was obtained from Calbiochem (San Diego, CA). A wheat germ agglutinin‐Alexa Fluor 488 conjugate was purchased from Invitrogen (Carlsbad, CA). The following monoclonal antibodies (mAbs) and polyclonal Abs (pAbs) were used for neutralization, immunofluorescence staining, and Western blotting: rat anti‐mouse IFN‐γ mAb (R&D Systems, Minneapolis, MN), rabbit anti‐human IFN‐γ pAb (Novus Biologicals, Littleton, CO), rat anti‐mouse CD68 mAb (Bio‐Rad Laboratories, Hercules, CA), rabbit anti‐PDK1 mAb, rabbit anti‐phospho(p)‐PDK1 (Ser241) mAb, rabbit anti‐Akt mAb, rabbit anti‐p‐Akt (Ser473) mAb, rabbit anti‐Gsk‐3β mAb, rabbit anti‐p‐Gsk‐3β (Ser9) mAb, rabbit anti‐Gab2 mAb, rabbit anti‐p‐Gab2 (Tyr452) mAb, rabbit anti‐FoxO1 mAb, rabbit‐anti‐p‐FoxO1(Thr24)/3a(Thr32) pAb, rabbit anti‐Stat5 mAb, rabbit anti‐p‐Stat5 (Tyr694) mAb, rabbit anti‐GAPDH mAb (Cell Signaling Technology, Danvers, MA), rabbit anti‐GATA4 pAbs (Abcam Japan, Tokyo, Japan), and horseradish peroxidase–conjugated goat anti‐rabbit pAb and biotin‐conjugated rabbit anti‐rat IgG pAb (Dako, Glostrup, Denmark).
Animals
Pathogen‐free 8‐ to 10‐week‐old male BALB/c mice were obtained from Japan SLC, Inc (Hamamatsu, Japan) and designated as WT mice in the present study. Age‐ and sex‐matched Ifng −/− mice, backcrossed to BALB/c mice for at least 8 generations,10 were used in experiments. All mice were housed individually in cages under specific pathogen‐free conditions during experiments. All animal experimental procedures were approved by the Committee on Animal Care and Use in Wakayama Medical University.
Transverse Aortic Constriction
Transverse aortic constriction (TAC) was performed in order to induce heart failure as described previously.21 Briefly, mice were anesthetized by an intraperitoneal injection of avertin (2, 2, 2‐tribromoethanol) at a dose of 240 mg/kg. Mice were then placed in a supine position, received endotracheal intubation, and were ventilated using a Model 687 Mouse Ventilator (Harvard Apparatus, Holliston, MA) with a tidal volume of 0.4 mL oxygen containing 1.5% isoflurane and a respiratory rate of 110 breaths/min. The thoracic cavity was exposed by the incision of the proximal portion of the sternum. The aortic arch was constricted between the innominate and left common carotid arteries with a 7‐0 silk suture tied firmly twice against a 27‐gauge blunted needle. Sham‐operated mice underwent a similar surgical procedure without the constriction of the aorta. In some experiments, Ifng −/− mice received a continuous infusion of recombinant IFN‐γ (15 U/d per mouse) using a subcutaneously implanted Alzet micro‐osmotic pump (Model 1007D; Muromachi Kikai Co, Ltd, Tokyo, Japan) from 2 days before TAC. In another series of experiments, WT mice received an intraperitoneal injection of rat anti‐mouse IFN‐γ mAb (50 μg/mouse) once every 2 days from 1 day before TAC until the end of the experiments, or the intraperitoneal administration of the Stat5 inhibitor (250 μg) or vehicle (4% dimethyl sulfoxide in PBS) once a day from 1 day before TAC through to the end of the experiments.
Histopathological Analyses
Heart tissues were obtained at the indicated time intervals after TAC and were fixed in 4% formaldehyde buffered with PBS (pH 7.2) in order to prepare 4‐μm‐thick paraffin‐embedded sections. Thereafter, sections were subjected to hematoxylin and eosin or Masson's trichrome staining.
Immunohistochemical Staining and Quantification of Intracardiac Inflammatory Cells After TAC
Deparaffinized heart sections were incubated with rabbit anti‐human CD3 pAbs (DAKO), rabbit anti‐mouse CD4 mAb (Abcam), rabbit anti‐mouse CD68 pAb (Abcam, Tokyo, Japan), and rabbit anti‐myeloperoxidase pAbs (NeoMarkers, Fremont, CA) at 4°C overnight. After incubation with peroxidase‐conjugated goat anti‐rabbit pAb (DAKO), positive signal was developed by incubation with 3, 3‐diaminobenzidine. Intracardiac inflammatory cells were evaluated semiquantitatively. Briefly, the positive cells were enumerated in 8 high‐magnification fields (×400) in each specimen. All measurements were performed by 2 examiners without prior knowledge of the experimental procedures.
Double‐Color Immunofluorescence Analysis
Paraffin‐embedded heart sections were incubated with rabbit anti‐human IFN‐γ pAb and rat anti‐mouse CD68 mAb at 4°C overnight. The anti‐human IFN‐γ pAb on the sections was detected with horseradish peroxidase–conjugated goat anti‐rabbit IgG pAb and cyanine 3‐tyramide signal amplification system (PerkinElmer Japan, Yokohama, Japan). After quenching horseradish peroxidase activity on the slides, the anti‐mouse CD68 mAb on the sections was detected with biotin‐conjugated rabbit anti‐rat IgG pAb, streptavidin–horseradish peroxidase, and fluorescein‐tyramide signal amplification system (PerkinElmer). Images were obtained using a Fluorescence Microscope BZ‐X700 (KEYENCE, Osaka, Japan).
Wheat Germ Agglutinin Staining
In order to delineate the membrane boundary of each cardiomyocyte, paraffin‐embedded heart sections were incubated in 0.1 mg/mL Alexa Fluor 488‐conjugated wheat germ agglutinin for 2 hours in the dark. After washing with PBS, images were obtained as described in the immunohistochemical staining procedure.
Echocardiography
Three weeks after TAC, mice were subjected to transthoracic echocardiography to examine cardiac function and structure. In order to measure left ventricular (LV) systolic function and chamber dimensions, echocardiography was performed with TOSHIBA AplioTM MX (SSA‐780A) or AplioTM 500 (TUS‐A500) using a 12‐MHz probe (Toshiba Medical Systems, Otawara, Japan). The diastolic intraventricular septum, LV end diastolic diameter, diastolic posterior wall thickness, LV internal dimension in systole, and percent LV fractional shortening were assessed from M‐mode images.
Measurement of Hydroxyproline Contents
Heart tissues were removed 21 days after TAC in order to measure the contents of hydroxyproline, a major component of collagen, as previously described.22 Data were expressed as the amount (μg) per dry weight of the heart sample (mg).
Primary Culture of Neonatal Rat Ventricular Myocytes
Left ventricular cardiomyocytes were obtained from 1‐day‐old SD rats (Kiwa Laboratory Animals Co, Ltd, Kiminocho, Japan) as described by Matsui et al.23 A cardiomyocyte‐enriched cell fraction was prepared by centrifugation through a discontinuous Percoll gradient23 and the purity of the resultant cells was ≈95%. Cardiomyocytes were plated on collagen I‐coated 60‐mm culture dishes and collagen I‐coated 10‐cm2 silicon chambers (STREX, Inc, Osaka, Japan) at 2.5×106 and 2×105 cells, respectively. Cells were subjected to a 20% cyclic stretch in a uniaxial strain at 60 cycles/min for 20 hours in a 37°C incubator under a 5% CO2 atmosphere in the presence or absence of rat IFN‐γ (10 U/mL). In another series of experiments, cardiomyocytes were cultured with or without the Stat5 inhibitor (100 μmol/L) under the stretch condition. Control samples were cultured in the collagen I‐coated silicon chamber without mechanical stretch.
Ifngr1 Gene Expression Analysis on Murine and Rat Cardiomyocytes
Murine cardiomyocytes from 8‐week‐old Balb/c mice (n=4) were isolated as described previously.24 Both murine and rat cardiomyocytes were immediately subjected to total RNA isolation. Cardiomyocyte total RNA was subjected to real‐time polymerase chain reaction (PCR) analysis of Ifngr1 expression with or without reverse transcription. Myh7 gene expression in the samples was analyzed in the same way as a reference gene expression in cardiomyocytes.
Quantitative Reverse Transcription‐PCR
Total RNA was extracted from mouse hearts or rat cardiomyocytes using ISOGENE (Nippon Gene, Toyama, Japan). One microgram of total RNA was reverse‐transcribed at 37°C for 15 minutes in 20 μL of the reaction mixture with the PrimeScript® RT Reagent Kit (TAKARA BIO, Otsu, Japan) with random primers (hexadeoxyribonucleotide mixture). Quantitative PCR was performed on Thermal Cycler Dice® Real Time System II (TAKARA BIO) with SYBR® Premix Ex Taq™ II (TAKARA BIO) using specific sets of primers (Table S1). The expression levels of the examined transcripts were compared with that of GAPDH and normalized to the mean value of the controls.
Western Blotting Analysis
At the indicated time intervals after TAC, heart tissues were homogenized with lysis buffer (10 mmol/L PBS, pH 7.4 containing 0.01% Triton X‐100, 0.5% sodium deoxycholate, and 0.1% SDS) containing Complete Protease Inhibitor Mixture®, and Phosphatase Inhibitor Cocktails for serine/threonine protein phosphatases and tyrosine protein phosphatases (P2850 and P5726; Sigma‐Aldrich) and then centrifuged to obtain lysates. Equal amounts of extracted proteins were separated using 12% SDS‐polyacrylamide gel electrophoresis and transferred to an Immobilon‐P transfer membrane (Millipore, Billerica, MA). The membranes were treated with primary Abs at 4°C overnight and incubated with a horseradish peroxidase–conjugated anti‐rabbit secondary antibody (Dako, Japan) at a dilution of 1:2000 at room temperature for 1 hour. Bound antibody complexes were detected using an enhanced chemiluminescence reagent (Millipore) according to the manufacturer's instructions.
Generation of Bone Marrow Chimeric Mice
The following bone marrow (BM) chimeric mice were prepared: Ifng −/− BM to WT mice, WT BM to Ifng −/− mice, WT BM to WT mice, and Ifng −/− BM to Ifng −/− mice. BM cells were collected from the femurs of donor mice by aspiration and flushing. Recipient mice were irradiated to 15 Gy using an RX‐650 irradiator (Faxitron X‐ray, Wheeling, IL), and then intravenously received 5×106 BM cells from donor mice in a volume of 200 μL of sterile PBS under anesthesia. Mice were then housed in sterilized microisolator cages and were fed normal chow and autoclaved hyperchlorinated water for 60 days. The successful engraftment and reconstruction of BM in transplanted mice were confirmed by PCR analyses for the WT or mutant IFN‐γ gene in the peripheral blood of each chimeric mouse 30 days after BM transplantation. After durable BM engraftment was confirmed, mice were subjected to TAC.
Statistical Analyses
Means and SEMs were calculated for all parameters assessed in the present study. The significance of differences was evaluated using ANOVA or the Mann–Whitney U test. P<0.05 was accepted as significant.
Results
Intracardiac IFN‐γ Expression After TAC
We initially examined whether TAC enhances intracardiac Ifng gene expression. Intracardiac Ifng gene expression was weakly detected in WT mice without any surgical treatment. TAC enhanced Ifng gene expression, starting 1 day after TAC and persisting until 1 week (Figure 1A). We also performed double‐color immunofluorescence analyses using the heart samples of WT mice 1 week after TAC in order to identify IFN‐γ‐expressing cells. Subsequently, IFN‐γ was expressed by CD68+ macrophages in the heart (Figure S1). These results implied that IFN‐γ‐produced immune cells including macrophages would be involved in pressure overload–induced cardiac hypertrophy.
Figure 1.
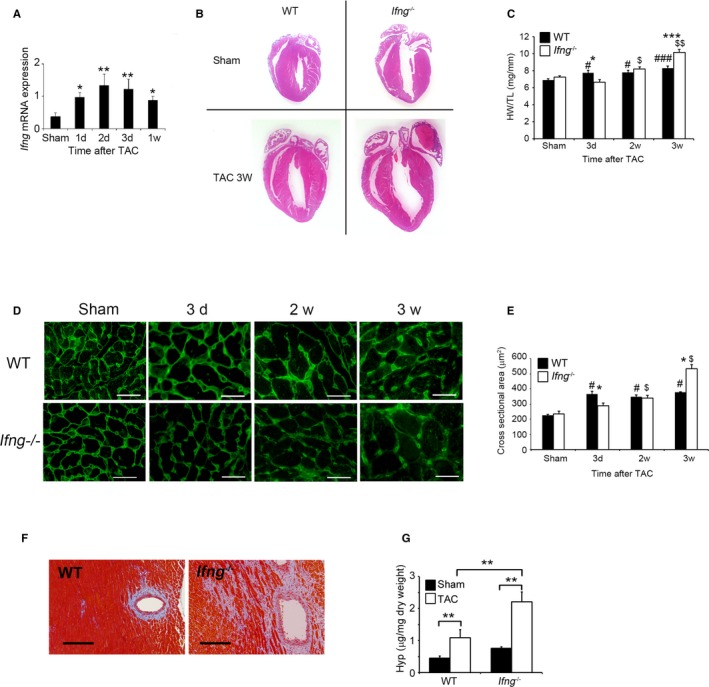
Evaluation of TAC‐induced cardiac hypertrophy in WT and Ifng −/− mice. A, Intracardiac Ifng gene expression was analyzed by quantitative RT‐PCR. Total RNA was extracted from the LV of WT mice at the indicated time points after TAC and was subjected to quantitative RT‐PCR. All values represent means±SEM (n=6–10 at each time point). *P<0.05, **P<0.01. TAC mice vs sham‐operated mice. B, Macroscopic sagittal views of H&E‐stained hearts of WT and Ifng −/− mice 3 weeks after TAC. Representative results from at least 6 animals are shown here. C, Changes in the heart weight/tibial length ratio (HW/TL ratio) of WT and Ifng −/− mice after TAC. All values represent means±SEM (n=8–10). *P<0.05, ***P<0.0005 WT vs Ifng −/−; # P<0.05, ### P<0.005 sham‐ vs TAC‐operated WT mice; $ P<0.05, $$ P<0.005, sham vs TAC operated Ifng −/− mice. D, Wheat germ agglutinin‐Alexa Fluor 488 conjugate staining of transverse sections of the left ventricular epicardial free walls, illustrating the membrane boundary of individual myocytes. Original magnification, ×400 (scale bar: 25 μm). E, Quantification of the myocyte cross‐section area of the LV wall as shown in (C). All values represent means±SEM (n=6). *P<0.05 WT vs Ifng −/−; # P<0.05 sham vs TAC operated WT mice; $ P<0.05 sham vs TAC‐operated Ifng −/− mice. F, Masson's trichrome staining images of the LV of WT and Ifng −/− mice 3 weeks after TAC. Representative results from at least 6 animals are shown here. Original magnification, ×400 (scale bar: 200 μm). G, Hydroxyproline contents in the LV of WT and Ifng −/− mice 3 weeks after TAC. All values represent means±SEM (n=4). **P<0.01. LV indicates left ventricle; RT‐PCR, reverse transcription polymerase chain reaction; TAC, transverse aortic constriction; WT, wild type.
Cardiac Maladaptation to Pressure Overload in the Absence of IFN‐γ
In order to clarify the pathophysiological roles of endogenous IFN‐γ in pressure overload–induced cardiac hypertrophy, Ifng −/− mice were subjected to TAC. No significant differences were observed in cardiac size or the heart weight/tibial length ratio between sham‐operated WT and Ifng −/− mice (Figure 1B and 1C). WT mice adaptively developed compensatory cardiac hypertrophy within 3 days in response to TAC. In contrast, Ifng −/− mice exhibited gradual increases in the heart weight/tibial length ratio until 3 weeks after TAC, ultimately resulting in severe cardiac hypertrophy with higher heart weight/tibial length ratios and larger left ventricle expansion 3 weeks after TAC (Figure 1B and 1C). Consistent with these macroscopic changes, wheat germ agglutinin staining showed enlarged cardiomyocytes in WT mice 3 days after TAC, whereas TAC‐induced enlargement was significantly attenuated in Ifng −/− mice (Figure 1D and 1E). However, 3 weeks after TAC, cardiomyocytes were larger in Ifng −/− mice than in WT mice (Figure 1D and 1E). Moreover, intracardiac fibrotic changes were exaggerated in Ifng −/− mice as evidenced by accentuated blue coloration on Masson's trichrome staining (Figure 1F) and enhanced hydroxyproline contents (Figure 1G), whereas no significant differences were observed in the intracardiac recruitment of leukocyte subpopulations such as neutrophils, macrophages, and T cells between WT and Ifng −/− mice after TAC, as revealed by immunohistochemical analyses and quantitative reverse transcription PCR to determine leukocyte type‐specific gene expression (Figure S2). Furthermore, echocardiographic analyses revealed that WT mice developed concentric cardiac hypertrophy as evidenced by increased intraventricular septum and diastolic posterior wall thickness, but without significant impairments in contractile functions; LV end diastolic diameter, LV internal dimension in systole, and LV fractional shortening remained unchanged (Figure 2A through 2G). In contrast, Ifng −/− mice developed marked LV dilatation and contractile dysfunction, but not concentric cardiac hypertrophy (Figure 2A through 2F). Consistently, the intracardiac mRNA level of natriuretic peptide B (Nppb), a marker of heart failure, was significantly increased in Ifng −/− mice, but not in WT mice (Figure 2G). When WT mice were administered neutralizing anti‐IFN‐γ Ab, TAC‐induced cardiac hypertrophy was significantly exaggerated, with enhanced fibrosis and worsened cardiac function, over that in WT mice treated with control IgG (Figure 3A through 3E). In contrast, exogenous IFN‐γ administration rescued cardiac pressure overload–induced maladaptation in Ifng −/− mice (Figure 3A through 3E). These results implied that the lack of IFN‐γ causes maladaptation to pressure overload and exaggerates pathological cardiac remodeling without any effect on intracardiac leukocyte recruitment, ultimately resulting in severe heart failure. Thus, TAC‐induced intracardiac IFN‐γ expression may protect cardiac hypertrophy induced by sustained pressure overload.
Figure 2.
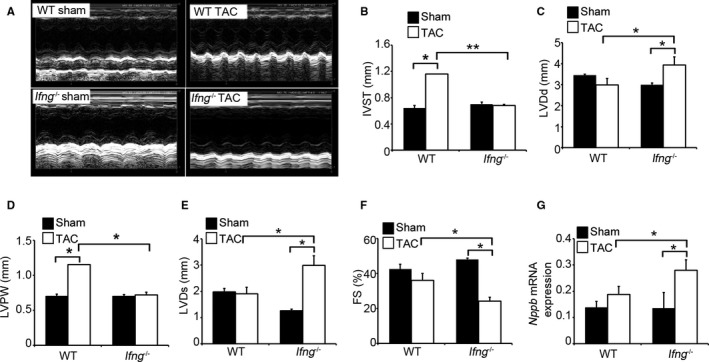
Effects of endogenous IFN‐γ on cardiac function after TAC. A, M‐mode echocardiographic images in WT and Ifng −/− mice 3 weeks after TAC. Representative images from at least 5 animals are shown here. B through G, The following parameters were analyzed and calculated: (B) IVST, interventricular septum; (C) LVDd, end‐diastolic left ventricular internal dimension; (D) LVPW, left ventricular posterior wall; (E) LVDs, end‐systolic left ventricular internal dimension, and (F) LVFS, left ventricular fractional shortening. All values represent means±SEM (n=5–11 in each group). *P<0.05; **P<0.01. G, Intracardiac Nppb gene expression in WT and Ifng −/− mice. Nppb mRNA expression was analyzed by quantitative RT‐PCR as described in Materials and Methods. All values represent means±SEM (n=6). *P<0.05. FS indicates fractional shortening; Nppb, natriuretic peptide B; IFN‐γ, interferon‐γ; RT‐PCR, reverse transcription polymerase chain reaction; TAC, transverse aortic constriction; WT, wild type.
Figure 3.
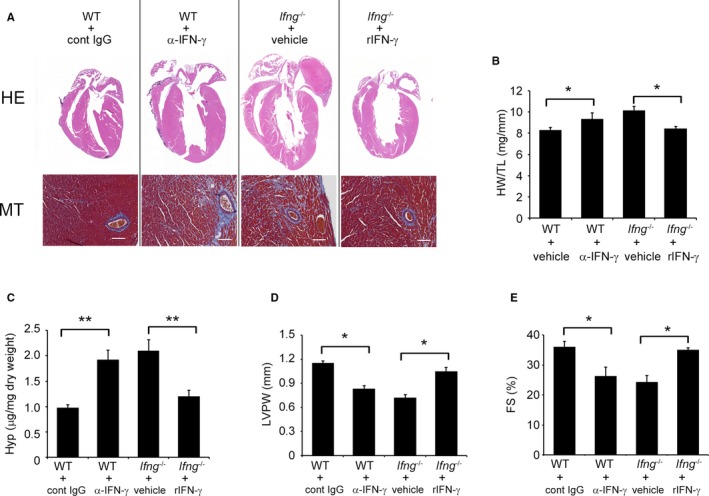
Effects of anti‐IFN‐γ mAb on WT mice and recombinant IFN‐γ on Ifng −/− mice in the process of TAC‐induced cardiac hypertrophy. A, Macroscopic sagittal views of the H&E‐stained hearts of WT and Ifng −/− mice 3 weeks after TAC (upper panels). MT staining images of the LV of WT and Ifng −/− mice 3 weeks after TAC (lower panels). Representative results from at least 6 animals are shown here. Original magnification: upper panels, ×2; lower panels, ×400 (scale bar: 50 μm). B, The HW/TL ratio was calculated in WT and Ifng −/− mice 3 weeks after TAC. All values represent means±SEM (n=5–6). *P<0.05. C, Hydroxyproline contents in the LV of WT and Ifng −/− mice 3 weeks after TAC. All values represent means±SEM (n=5–6). **P<0.01. D and E, Echocardiographic analysis of cardiac function in mice 3 weeks after TAC. LVPW (D) and FS (E) were analyzed and calculated. All values represent means±SEM (n=5–6). *P<0.05. FS indicates percent LV fractional shortening; Hyp, hydroxyproline; HW/TL ratio, heart weight/tibial length ratio; LV, left ventricle; LVPW, diastolic posterior wall thickness; mAb, monoclonal antibody; MT staining, Masson's trichrome staining; TAC, transverse aortic constriction.
Contribution of BM Cell–Derived IFN‐γ to Compensatory Cardiac Hypertrophy
We conducted TAC on BM chimeric mice generated between WT and Ifng −/− mice. When WT mice were transplanted with Ifng −/−‐BM cells, TAC‐induced cardiac hypertrophy was significantly aggravated, with enhanced cardiac fibrosis and dysfunction, over that in those transplanted with WT‐BM cells (Figure 4A through 4E). In contrast, maladapted cardiac hypertrophy and subsequent cardiac dysfunction appeared to have improved more in Ifng −/− mice transplanted with WT‐BM cells than in those bearing Ifng −/−‐BM cells (Figure 4A through 4E). Based on the expression of IFN‐γ by intracardiac macrophages, these results implied that IFN‐γ produced by BM‐derived immune cells including macrophages is indispensable for compensatory cardiac hypertrophy induced by TAC.
Figure 4.
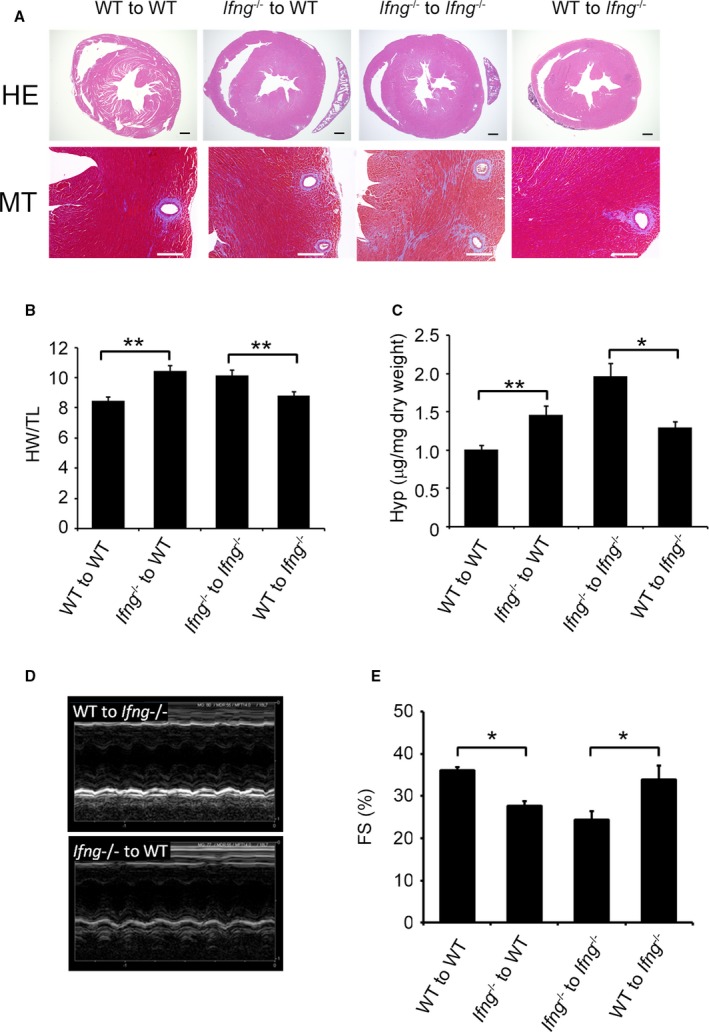
Effects of BM transplantation on compensatory cardiac hypertrophy. A, The upper panels show representative cross‐sectional views of hearts from BM chimeric mice 3 weeks after TAC. Original magnification ×2 (scale bar: 500 μm). The lower panels show Masson's trichrome staining images of the LV of BM chimeric mice 3 weeks after TAC. Original magnification, ×400 (scale bar: 200 μm). B, The HW/TL was calculated in BM chimeric mice 3 weeks after TAC. All values represent means±SEM (n=6). **P<0.01. C, Hydroxyproline contents in the LV of BM chimeric mice 3 weeks after TAC. All values represent means±SEM (n=6). *P<0.05; **P<0.01. D and E, M‐mode echocardiographic analysis of hearts from BM chimeric mice 3 weeks after TAC. Representative images from at least 6 animals are shown in (D). E, FS, left ventricular fractional shortening was analyzed and calculated. All values represent means±SEM (n=6–11 mice in each group). *P<0.05. BM indicates bone marrow; FS, percent LV fractional shortening; HW/TL, heart weight/tibial length ratio; Hyp, hydroxyproline; LV, left ventricle; TAC, transverse aortic constriction.
Involvement of IFN‐γ in PI3K/Akt Signaling Activation in Compensatory Cardiac Hypertrophy
Several lines of evidence indicate that the TAC‐induced activation of PI3K/Akt signaling pathways contributes to compensatory cardiac hypertrophy, which may prevent ensuing cardiac dysfunction.25, 26 Hence, we investigated PI3K/Akt pathways in this TAC model. Three days after TAC, the LV in WT mice exhibited the enhanced phosphorylation of PDK1, Akt, Gsk3‐β, and FoxO1 (Figure 5A through 5E), which are downstream of PI3K. The attenuated phosphorylation of these molecules was significantly greater in Ifng −/− mice than in WT mice (Figure 5A through 5E). Moreover, the expression levels of intracardiac GATA4, a transcription factor that is essentially involved in compensatory cardiac hypertrophy and is downstream of Akt,27, 28 were higher in WT mice than in Ifng −/− mice (Figure 5A and 5F). The IFN‐γ infusion further restored the TAC‐induced phosphorylation of Akt and GSK‐3β in Ifng −/− mice (Figure 5G through 5I). Reciprocally, the immunoneutralization of IFN‐γ significantly reduced the TAC‐induced phosphorylation of Akt and GSK‐3β in WT mice (Figure 5G through 5I). Furthermore, WT mice transplanted with Ifng −/− BM cells showed the weaker activation of Akt and GSK‐3β after TAC (Figure 5J through 5L). Collectively, these results implied that IFN‐γ is indispensable for the activation of the PI3K/Akt signaling pathway and subsequent compensatory hypertrophy.
Figure 5.
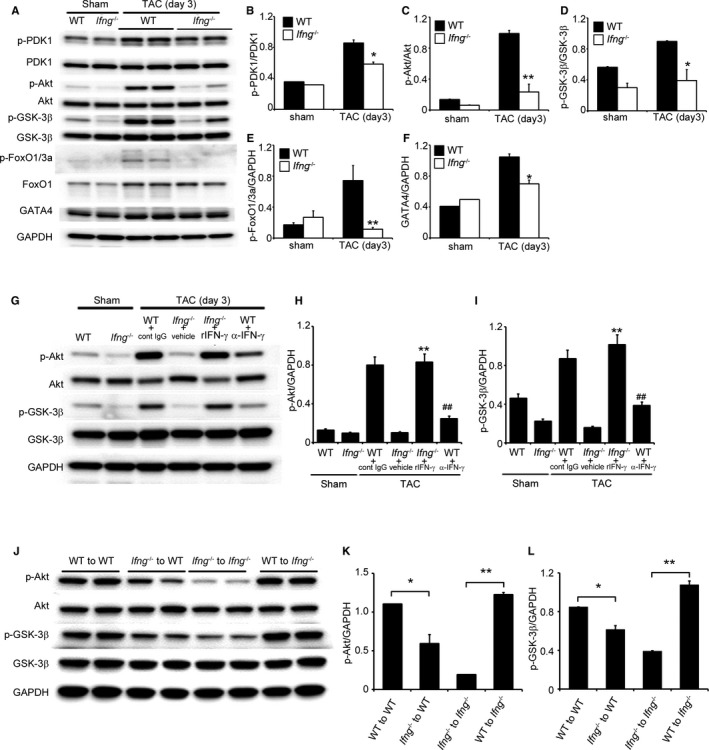
PI3K/Akt signaling in the LV after TAC. A, Western blotting analysis of PI3K/Akt signaling‐related molecules in the LV of WT and Ifng −/− mice 3 days after TAC. Representative results from 6 independent experiments are shown here. Relative amounts of p‐PDK1 (B), p‐Akt (C), p‐GSK‐3β (D), p‐FoxO1/3a (E), and GATA4 (F) to GAPDH were calculated by densitometry and shown here. All values represent means±SEM (n=6). *P<0.05; **P<0.01. G, Effects of anti‐IFN‐γ mAb and recombinant IFN‐γ on TAC‐induced PI3K/Akt signaling in the LV of WT and Ifng −/− mice, respectively. Western blotting analysis of Akt and GSK‐3β in the LV of mice 3 days after TAC. Representative results from 6 independent experiments are shown here. Relative amounts of p‐Akt (H) and p‐GSK‐3β (I) to GAPDH were analyzed by densitometry and calculated. All values represent means±SEM (n=6). **P<0.01, vs Ifng −/− mice treated with vehicle; ## P<0.01, vs WT treated with control IgG. J, Effects of BM transplantation on TAC‐induced PI3K/Akt signaling in the LV. Western blotting analysis of the phosphorylation of Akt and GSK‐3β in the LV of BM chimeric mice 3 days after TAC. Representative results from 6 independent experiments are shown here. Relative amounts of p‐Akt (K) and p‐GSK‐3β (L) to GAPDH were analyzed by densitometry and calculated. All values represent means±SEM (n=6). *P<0.05; **P<0.01. Akt indicates protein kinase B; BM, bone marrow; FoxO1/3a, forkhead box protein 1/3a; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GATA4, transcription factor GATA4; GSK‐3β, glycogen synthase kinase 3β; IFN‐γ, interferon‐γ; IgG, immunoglobulin G; LV, left ventricle; mAb, monoclonal antibody; PDK1, phosphoinositide‐dependent kinase‐1; PI3K, phosphoinositide 3‐kinase; TAC, transverse aortic constriction.
Essential Involvement of the IFN‐γ/Stat5 Signal Pathway in the Activation of PI3K/Akt Signaling After TAC
We failed to detect the enhanced phosphorylation of Stat1 (data not shown), a main signal transducer for IFN‐γ, in the LV of WT and Ifng −/− mice after TAC. Since IFN‐γ may induce the phosphorylation of Stat5, ultimately resulting in the activation of PI3K/Akt signaling through the phosphorylation of GRB2‐associated‐binding protein 2 (Gab2),29, 30, 31 we investigated the phosphorylation of Stat5 and Gab2. TAC induced Stat5 and Gab2 phosphorylation in WT mice after 3 days; however, this phosphorylation was diminished in Ifng −/− mice (Figure 6A through 6C).
Figure 6.
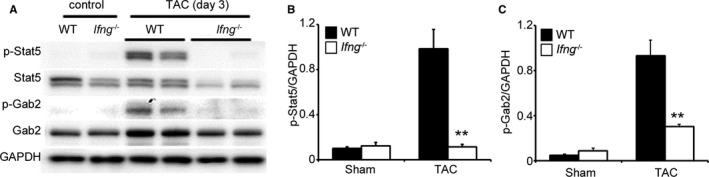
Stat5 and Gab2 in the LV of mice 3 days after TAC. A through C, Western blotting analysis of Stat5 and Gab2 in the LV of WT and Ifng −/− mice 3 days after TAC. Representative results from 6 independent experiments are shown in (A). Relative amounts of p‐Stat5 (B) and p‐Gab2 (C) to GAPDH were analyzed by densitometry and calculated. All values represent means±SEM (n=6). **P<0.01 WT vs Ifng −/− mice. Gab2 indicates GRB2‐associated‐binding protein 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; LV, left ventricle; Stat5, signal transducer and activator of transcription 5; TAC, transverse aortic constriction; WT, wild type.
Stretch‐Induced IFN‐γ/Stat5‐Dependent PI3K/Akt Activation in Cardiomyocytes
In addition to the results obtained from immunofluorescence analyses (Figure 1B), the analysis of BM chimeric mice revealed that an IFN‐γ deficiency restricted to radiosensitive BM cells recapitulated the TAC‐induced phenotypes observed in IFN‐γ−/− mice (Figures 4 and 5), implicating BM cells, but not cardiomyocytes as a major source of IFN‐γ in this TAC model. Consistently, rat primary cardiomyocytes failed to express Ifng mRNA even under stretch conditions (data not shown). However, we found that cardiomyocytes constitutively expressed the IFN‐γ receptor, suggesting that IFN‐γ directly acts on cardiomyocytes (Figure S3). Thus, we examined the direct effects of exogenous IFN‐γ on the Stat5 and Akt signaling pathways in cardiomyocytes under static and stretch conditions. The addition of IFN‐γ enhanced the phosphorylation of Stat5 in cardiomyocytes under static and stretch conditions, whereas IFN‐γ enhanced Akt phosphorylation under stretch, but not static conditions (Figure 7A through 7C). Moreover, under stretch conditions, the specific Stat5 inhibitor abrogated IFN‐γ‐induced Akt phosphorylation in cardiomyocytes (Figure 7D through 7F). Thus, the IFN‐γ/Stat5 signal pathway appears to be essential for the activation of PI3K/Akt signaling in cultured cardiomyocytes under stretch conditions.
Figure 7.
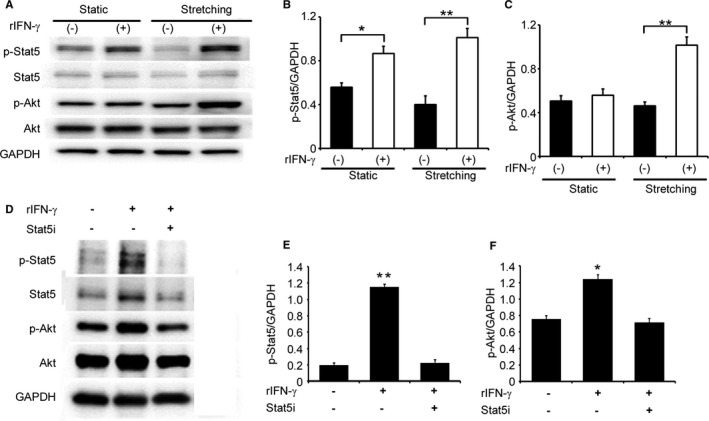
Effects of IFN‐γ on the activation of Stat5 and Akt in in vitro cultured neonatal rat cardiomyocytes. A, Western blotting analyses were conducted to detect Stat5 and Akt in neonatal rat cardiomyocytes with or without mechanical stretch. Representative results from 5 independent experiments are shown here. Relative amounts of p‐Stat5 (B) and p‐Akt (C) to GAPDH were analyzed by densitometry and calculated. All values represent means±SEM (n=5). *P<0.05; **P<0.01. D through F, Effects of the Stat5 inhibitor on the IFN‐γ‐induced phosphorylation of Stat5 and Akt in rat cardiomyocytes, which were cultured under the stretch condition. Relative amounts of p‐Stat5 (E) and p‐Akt (F) to GAPDH were analyzed by densitometry and calculated. All values represent means±SEM (n=5). *P<0.05; **P<0.01. Akt indicates protein kinase B; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IFN‐γ, interferon‐γ; Stat5, signal transducer and activator of transcription 5.
Effects of the Stat5 Inhibitor on TAC‐Induced Cardiac Hypertrophy and Heart Failure
We examined the in vivo effects of the Stat5 inhibitor on TAC‐induced cardiac hypertrophy and heart failure. The administration of the Stat5 inhibitor reduced the phosphorylation of Stat5, Akt, and Gab2 in the LV of WT mice to levels similar to those in vehicle‐treated Ifng −/− mice 3 days after TAC (Figure 8A and 8B). Consistently, the Stat5 inhibitor suppressed the rapid enlargement of cardiomyocytes in cross‐sectional areas in the LV of WT within 3 days of TAC, to a level similar to that in vehicle‐treated Ifng −/− mice (Figure 8C and 8D). Moreover, the Stat5 inhibitor accentuated pathological cardiac hypertrophy (Figure 8E) with augmented cardiac dysfunction in WT mice to a similar extent as that observed in vehicle‐treated Ifng −/− mice (Figure 8F and 8G). In contrast, the Stat5 inhibitor did not exert any effects on the phosphorylation of Stat5, Gab2, or Akt in the LV of Ifng −/− mice 3 days after TAC (Figure S4A and S4B). Moreover, the Stat 5 inhibitor failed to affect TAC‐induced cardiac hypertrophy (Figure S4C) or cardiac function 3 weeks after TAC (Figure S4D and S4E). Thus, the activation of IFN‐γ‐dependent Stat5 phosphorylation appears to be indispensable for pressure overload–induced Akt activation in cardiomyocytes.
Figure 8.
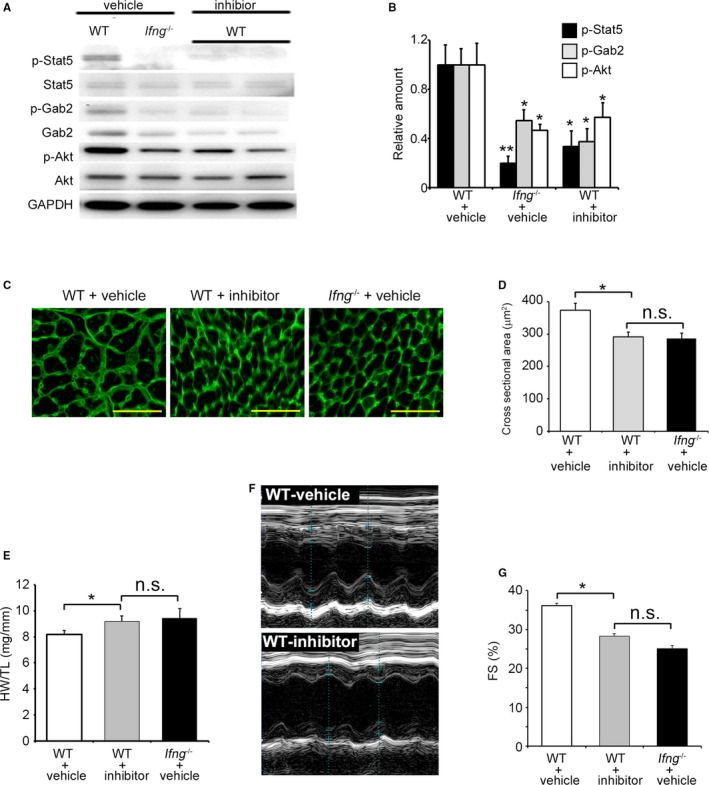
Effects of the Stat5 inhibitor on TAC‐induced cardiac hypertrophy and heart failure in WT mice. A and B, Phosphorylation of Akt and Gab2 in LV in mice treated with the Stat5 inhibitor or vehicle. A Western blotting analysis was conducted to detect Stat5, Gab2, and Akt in the LV of mice 3 days after TAC. Representative results from 6 independent experiments are shown in (A). Relative amounts of p‐Stat5, p‐Gab2, and p‐Akt to GAPDH were analyzed by densitometry and calculated, and are shown in (B). All values represent means±SEM (n=6). *P<0.05, **P<0.01, vs WT treated with vehicle in each column. C, Wheat germ agglutinin‐Alexa Fluor 488 conjugate staining of transverse sections of the LV epicardial free walls of mice 3 days after TAC. Original magnification, ×400 (scale bar: 50 μm). D, Quantification of the myocyte cross‐section area of the LV wall shown in (C). All values represent means±SEM (n=6). *P<0.05. E, The HW/TL ratio in WT mice treated with the Stat5 inhibitor or vehicle and Ifng −/− mice treated with vehicle 3 weeks after TAC was calculated. All values represent means±SEM (n=6). *P<0.05. F, M‐mode echocardiographic images of WT mice treated with the Stat5 inhibitor or vehicle 3 weeks after TAC. Representative images from at least 5 animals are shown here. G, LVFS were calculated for WT mice treated with vehicle or the Stat5 inhibitor, or Ifng −/− mice treated with vehicle 3 weeks after TAC. All values represent means±SEM (n=6). *P<0.05. Akt indicates protein kinase B; FS, fractional shortening; Gab2, GRB2‐associated‐binding protein 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HW/TL ratio, heart weight/tibial length ratio; LV, left ventricle; LVFS, percent LV fractional shortening; n.s., not significant; Stat5, signal transducer and activator of transcription 5; TAC, transverse aortic constriction; WT, wild type.
Discussion
IFN‐γ is a pleiotropic cytokine produced by NK cells, T cells, and macrophages,32 and is essentially involved in several types of inflammatory diseases as well as tissue repair by several organs.10, 33, 34 Hemodynamic stress may cause compensatory cardiac hypertrophy, the associated impairments of which may subsequently induce pathological cardiac hypertrophy, resulting in severe heart failure.26, 35, 36 Several lines of evidence suggest the involvement of IFN‐γ in hemodynamic stress‐induced compensatory cardiac hypertrophy.18, 19, 20, 37, 38 However, the mechanisms by which IFN‐γ contributes to ensuing pathological cardiac dysfunction have not yet been elucidated. Hence, we investigated the pathophysiological roles of IFN‐γ in sustained pressure overload–induced cardiac pathology using mice lacking IFN‐γ. We revealed that persistent pressure overload induced more severe cardiac hypertrophy and fibrosis in Ifng −/− mice than in WT mice. These results implied that endogenous IFN‐γ plays a beneficial role in compensatory adaptive responses to hemodynamic stress. Moreover, the administration of exogenous IFN‐γ abrogated pathological heart failure in Ifng −/− mice. Thus, IFN‐γ and/or its related molecules may be used to prevent heart failure arising from sustained pressure overload.
In line with the present results, several independent groups reported protective roles for IFN‐γ in the pathogenesis of pressure overload–induced cardiac hypertrophy.18, 39 In another cardiac hypertrophy model, namely, that induced by aldosterone, the protective roles of IFN‐γ were demonstrated using Ifng −/− mice with a genetic background of BALB/c.19 In contrast, Markó and colleagues demonstrated that the genetic disruption of receptors for IFN‐γ in 129S6 mice alleviated angiotensin II‐induced cardiac damage, in spite of the lack of a difference in Nppb mRNA expression.20 However, the observation period in the latter study was shorter than that in other studies, including ours. Thus, these discrepancies may be attributed to differences in the genetic backgrounds of the mice used and/or the observation period.
Liu and colleagues similarly demonstrated that BCG and TLR4 agonists prevented abdominal aortic constriction–induced pathological cardiac hypertrophy and fibrosis and further revealed that their protective actions were mediated by IFN‐γ expressed by cardiomyocytes in the LV.39 In contrast, evidence is accumulating to implicate inflammatory cells as a major source of IFN‐γ in heart failure.38 T cells have been identified as a major cell source of IFN‐γ in angiotensin II–induced cardiac remodeling.40 In the present study, we demonstrated that BM‐derived CD68+ macrophages were a source of IFN‐γ (Figure S1, Figures 4 and 5). Thus, infiltrating immune cells including macrophages appear to have been responsible for intracardiac INF‐γ expression in our present model.
The pathophysiological roles of IFN‐γ in fibrotic changes in tissues and organs remain unclear. Endogenous IFN‐γ promoted pulmonary fibrosis and venous thrombotic fibrosis.34 In contrast, several groups reported that IFN‐γ inhibited collagen synthesis by fibroblasts.41 In line with these findings, we observed that the lack of endogenous IFN‐γ significantly accelerated the wound healing process as evidenced by enhanced granulation tissue formation,33 similarly in this model. Thus, although endogenous IFN‐γ may be anti‐ or profibrotic in a context‐dependent manner, it may dampen cardiac fibrosis in this model.
IFN‐γ may transduce its signals mainly by phosphorylating Stat1.42 However, Stat1‐deficient monocytes display a similar level of IFN‐γ‐stimulated adhesion to WT monocytes,43 indicating the presence of a Stat1‐independent IFN‐γ‐mediated signaling pathway. IFN‐γ promoted the differentiation of the human myelomonocytic cell line, U937, through the activation of Stat5.44 Moreover, IFN‐γ induced increases in intestinal epithelial permeability by activating Stat5.31 In line with these findings, TAC induced the robust phosphorylation of Stat5, but not Stat1 in WT mouse hearts, and its phosphorylation was abrogated in Ifng −/− mice. Moreover, rat cardiomyocytes exhibited Stat5, but not Stat1 phosphorylation when they were cultured in vitro in the presence of IFN‐γ only under stretch conditions. Furthermore, the Stat5 inhibitor may have attenuated TAC‐induced cardiac hypertrophy to a similar level as that observed in Ifng −/− mice. These findings indicate the crucial involvement of the IFN‐γ/Stat5 pathway in this TAC‐induced cardiac pathology.
When hemodynamic stress‐induced compensatory cardiac hypertrophy is impaired, pathological cardiac hypertrophy develops together with heart failure.26, 35, 36 Pressure overload may induce maladaptive remodeling of the heart when temporal PI3K/Akt signaling activation in the early period (≈4 days after TAC surgery) is impaired.35 Similarly, in the presence of pressure overload, the depletion of IL‐18, a potent IFN‐γ‐inducing factor, attenuated compensatory hypertrophy by suppressing Akt activation.28 These findings indicate that PI3K/Akt signaling activation is indispensable for compensatory, but not pathological cardiac hypertrophy.26, 28, 45, 46 Moreover, IFN‐γ may activate Akt/PI3K in human monocytes,43 and Stat5 activation has been shown to activate the PI3K/Akt pathway.29, 47 Hence, we examined PI3K/Akt signaling 3 days after TAC when enhanced Ifng expression was observed in hearts. TAC induced PDK and Akt phosphorylation in WT mouse hearts and their phosphorylation was depressed in Ifng −/− mice. Moreover, the expression of GATA4, a transcription factor that is downstream of Akt signal pathways and contributes to compensatory cardiac hypertrophic remodeling in response to pressure overload,28 was weaker in Ifng −/− mice than in WT mice. Thus, IFN‐γ may activate the PI3K/Akt signal pathway, which is crucially involved in compensatory cardiac adaptation to pressure overload and the prevention of ensuing cardiac hypertrophy arising from persistent pressure overload.
Evidence is accumulating to indicate the essential roles of the interaction between Stat5 and p85, a regulatory subunit of PI3K, in Stat5‐mediated PI3K/Akt signal pathway activation.29 Subsequent studies revealed that this interaction may be mediated by Gab2, a scaffolding adaptor, and requires the phosphorylation of Stat5 and Gab2.30, 31 We also observed the IFN‐γ‐dependent phosphorylation of Stat5, Gab2, and Akt in WT mice 3 days after TAC, and the administration of the Stat5 inhibitor significantly reduced the phosphorylation of Akt and Gab2, resulting in augmented cardiac hypertrophy with impaired cardiac functions. These observations would indicate that the IFN‐γ/Stat5 signal pathway can be protective against TAC‐induced cardiac hypertrophy, by inducing adaptation of the heart to hemodynamic stress through activating PI3K/Akt signal pathways in Gab2 phosphorylation‐dependent manner. Thus, the molecules involved in the IFN‐γ/Stat5/PI3K/Akt pathway may be targets for the prevention and/or treatment of pressure overload–induced cardiac hypertrophy with cardiac failures.
Sources of Funding
This work was supported in part by Grants‐in‐Aids for Scientific Research (A) (grant 20249040, to Kondo) and for Challenging Exploratory Research (grant 26670358, to Kimura) from the Ministry of Education, Culture, Science, and Technology of Japan and by Research Grant on Priority Areas (to Kondo) from Wakayama Medical University.
Disclosures
None.
Supporting information
Table S1. Sequences of the Primers Used for RT‐PCR
Figure S1. A double‐color immunofluorescence analysis was performed on the heart 1 week after TAC. The analysis was performed using anti‐CD68 (FITC) and anti‐IFN‐γ (Cy3), followed by observations under a fluorescence microscope, and signals were merged digitally. Representative results from 6 individual animals are shown here. Original magnification, ×400 (scale bar: 100 μm). Cy3 indicates Cyanine3; FITC, fluorescein isothiocyanate; IFN‐γ, interferon‐γ; TAC, transverse aortic constriction.
Figure S2. Quantification of intracardiac inflammatory cells. Representative microscopic images and quantification of CD3‐positive T cell (A and B), CD4‐positive T cell (C and D), CD68‐positive macrophages (E and F), and MPO‐positive neutrophil (G and H) in the heart paraffin sections of sham‐operated and TAC‐operated (day 3) mice. Original magnification, ×400 (scale bar: 100 μm). All values represent means±SEM (n=6). Relative gene expression of Cd3 (I), Cd4 (J), Cd68 (K), and Mpo (L) in the LV of sham‐operated and TAC‐operated mice (day 3) was analyzed by real‐time RT‐PCR. All values represent means±SEM (n=5). LV indicates left ventricle; MPO, myeloperoxidase; RT‐PCR, reverse transcription polymerase chain reaction; Sham, sham‐operated mice; TAC, transverse aortic constriction–operated mice.
Figure S3. Gene expressions of Myh7 (A and C) and Ifngr1 (B and D) in isolated mouse (A and B) and rat (C and D) cardiomyocytes were analyzed by real‐time PCR with or without reverse transcription. All values represent means±SEM (n=4). Akt indicates protein kinase B; FS, fractional shortening; Gab2, GRB2‐associated‐binding protein 2; HW/TL ratio, heart weight/tibial length ratio; Ifngr1 indicates interferon gamma receptor 1; Myh7, myosin heavy chain 7; n.s., not significant; PCR, polymerase chain reaction; Stat5, signal transducer and activator of transcription 5; WT, wild type. * , ** ,
Figure S4. Effects of a Stat5 inhibitor on TAC‐induced cardiac hypertrophy and heart failure in Ifng −/− mice. A and B, Phosphorylation of Akt and Gab2 in LV in mice treated with a Stat5 inhibitor or vehicle. Western blotting analysis was conducted to detect Stat5, Gab2, and Akt in LV of mice at 3 days after TAC operation. Representative results from 6 independent experiments are shown in (A). Relative amounts of p‐Stat5, p‐Gab2, and p‐Akt to GAPDH were analyzed by densitometry and calculated, and are shown in (B). All values represent means±SEM (n=6). *P<0.05, **P<0.01, vs WT treated with vehicle in each column. C, The HW/TL ratio in WT mice treated with vehicle and Ifng −/− mice treated with the Stat5 inhibitor or vehicle 3 weeks after TAC was calculated. All values represent means±SEM (n=6). *P<0.05. WT treated with vehicle. D, M‐mode echocardiographic images of Ifng −/− mice treated with a Stat5 inhibitor or vehicle, 3 weeks after TAC operation. Representative images from at least 5 animals are shown here. E, LVFS were calculated on WT mice treated with vehicle or Ifng −/− mice treated with a Stat5 inhibitor or vehicle at 3 weeks after TAC surgery. All values represent means±SEM (n=6). *P<0.05. WT treated with vehicle. Akt indicates protein kinase B; Gab2, GRB2‐associated‐binding protein 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HW/TL ratio, heart weight/tibial length ratio; Ifng, interferon gamma; LV, left ventricle; LVFS, percent LV fractional shortening; n.s., not significant; Stat5, signal transducer and activator of transcription 5; TAC, transverse aortic constriction; WT, wild type.
(J Am Heart Assoc. 2018;7:e008145 DOI: 10.1161/JAHA.117.008145.)29555642
References
- 1. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. [DOI] [PubMed] [Google Scholar]
- 2. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. [DOI] [PubMed] [Google Scholar]
- 4. Mann DL. Targeted anticytokine therapy and the failing heart. Am J Cardiol. 2005;95:9C–16C; discussion 38C–40C. [DOI] [PubMed] [Google Scholar]
- 5. Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, LeJemtel TH. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–971. [DOI] [PubMed] [Google Scholar]
- 6. Vanderheyden M, Paulus WJ, Voss M, Knuefermann P, Sivasubramanian N, Mann D, Baumgarten G. Myocardial cytokine gene expression is higher in aortic stenosis than in idiopathic dilated cardiomyopathy. Heart. 2005;91:926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, Fukai T, Matsuzaki M. Angiotensin activates MCP‐1 and induces cardiac hypertrophy and dysfunction via Toll‐like receptor 4. J Atheroscler Thromb. 2015;22:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, Bin J, Kitakaze M. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. 2011;92:385–393. [DOI] [PubMed] [Google Scholar]
- 9. Gullestad L, Aukrust P. Review of trials in chronic heart failure showing broad‐spectrum anti‐inflammatory approaches. Am J Cardiol. 2005;95:17C–23C; discussion 38C–40C. [DOI] [PubMed] [Google Scholar]
- 10. Hayashi T, Ishida Y, Kimura A, Iwakura Y, Mukaida N, Kondo T. IFN‐gamma protects cerulein‐induced acute pancreatitis by repressing NF‐kappa B activation. J Immunol. 2007;178:7385–7394. [DOI] [PubMed] [Google Scholar]
- 11. Tsuji H, Mukaida N, Harada A, Kaneko S, Matsushita E, Nakanuma Y, Tsutsui H, Okamura H, Nakanishi K, Tagawa Y, Iwakura Y, Kobayashi K, Matsushima K. Alleviation of lipopolysaccharide‐induced acute liver injury in Propionibacterium acnes‐primed IFN‐gamma‐deficient mice by a concomitant reduction of TNF‐alpha, IL‐12, and IL‐18 production. J Immunol. 1999;162:1049–1055. [PubMed] [Google Scholar]
- 12. Murphy SP, Tayade C, Ashkar AA, Hatta K, Zhang J, Croy BA. Interferon gamma in successful pregnancies. Biol Reprod. 2009;80:848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kimura A, Ishida Y, Hayashi T, Wada T, Yokoyama H, Sugaya T, Mukaida N, Kondo T. Interferon‐gamma plays protective roles in sodium arsenite‐induced renal injury by up‐regulating intrarenal multidrug resistance‐associated protein 1 expression. Am J Pathol. 2006;169:1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura A, Ishida Y, Inagaki M, Nakamura Y, Sanke T, Mukaida N, Kondo T. Interferon‐gamma is protective in cisplatin‐ induced renal injury by enhancing autophagic flux. Kidney Int. 2012;82:1093–1104. [DOI] [PubMed] [Google Scholar]
- 15. Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K, Isoda K, Riches DW, Matoba S, Okigaki M, Matsubara H. Pressure‐mediated hypertrophy and mechanical stretch induces IL‐1 release and subsequent IGF‐1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res. 2009;105:1149–1158. [DOI] [PubMed] [Google Scholar]
- 16. Jobe LJ, Melendez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF‐alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol. 2009;297:H1462–H1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janssen SP, Gayan‐Ramirez G, Van A, Herijgers P, Maes K, Verbeken E, Decramer M. Interleukin‐6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111:996–1005. [DOI] [PubMed] [Google Scholar]
- 18. Jin H, Li W, Yang R, Ogasawara A, Lu H, Paoni NF. Inhibitory effects of interferon‐gamma on myocardial hypertrophy. Cytokine. 2005;31:405–414. [DOI] [PubMed] [Google Scholar]
- 19. Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F. Interferon‐γ ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol. 2012;303:H587–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN. Interferon‐γ signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension. 2012;60:1430–1436. [DOI] [PubMed] [Google Scholar]
- 21. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:e7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishida Y, Kimura A, Kondo T, Hayashi T, Ueno M, Takakura N, Matsushima K, Mukaida N. Essential roles of the CC chemokine ligand 3‐CC chemokine receptor 5 axis in bleomycin‐induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am J Pathol. 2007;170:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsui H, Yokoyama T, Tanaka C, Sunaga H, Koitabashi N, Takizawa T, Arai M, Kurabayashi M. Pressure mediated hypertrophy and mechanical stretch up‐regulate expression of the long form of leptin receptor (ob‐Rb) in rat cardiac myocytes. BMC Cell Biol. 2012;13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ackers‐Johnson M, Li PY, Holmes AP, O'Brien SM, Pavlovic D, Foo RS. A simplified, Langendorff‐free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ Res. 2016;119:909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. [DOI] [PubMed] [Google Scholar]
- 27. Pikkarainen S, Tokola H, Majalahti‐Palviainen T, Kerkela R, Hautala N, Bhalla SS, Charron F, Nemer M, Vuolteenaho O, Ruskoaho H. GATA‐4 is a nuclear mediator of mechanical stretch‐activated hypertrophic program. J Biol Chem. 2003;278:23807–23816. [DOI] [PubMed] [Google Scholar]
- 28. Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, Trevino RJ, Freeman GL, Chandrasekar B. Interleukin‐18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun. 2007;354:552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santos SC, Lacronique V, Bouchaert I, Monni R, Bernard O, Gisselbrecht S, Gouilleux F. Constitutively active STAT5 variants induce growth and survival of hematopoietic cells through a PI 3‐kinase/Akt dependent pathway. Oncogene. 2001;20:2080–2090. [DOI] [PubMed] [Google Scholar]
- 30. Nyga R, Pecquet C, Harir N, Gu H, Dhennin‐Duthille I, Regnier A, Gouilleux‐Gruart V, Lassoued K, Gouilleux F. Activated STAT5 proteins induce activation of the PI 3‐kinase/Akt and Ras/MAPK pathways via the Gab2 scaffolding adapter. Biochem J. 2005;390:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smyth D, Phan V, Wang A, McKay DM. Interferon‐gamma‐induced increases in intestinal epithelial macromolecular permeability requires the Src kinase Fyn. Lab Invest. 2011;91:764–777. [DOI] [PubMed] [Google Scholar]
- 32. Farrar MA, Schreiber RD. The molecular cell biology of interferon‐gamma and its receptor. Annu Rev Immunol. 1993;11:571–611. [DOI] [PubMed] [Google Scholar]
- 33. Ishida Y, Kondo T, Takayasu T, Iwakura Y, Mukaida N. The essential involvement of cross‐talk between IFN‐gamma and TGF‐beta in the skin wound‐healing process. J Immunol. 2004;172:1848–1855. [DOI] [PubMed] [Google Scholar]
- 34. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Inui M, Mukaida N, Kondo T. Absence of IFN‐gamma accelerates thrombus resolution through enhanced MMP‐9 and VEGF expression in mice. J Clin Invest. 2011;121:2911–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sbroggio M, Carnevale D, Bertero A, Cifelli G, De BE, Mascio G, Hirsch E, Bahou WF, Turco E, Silengo L, Brancaccio M, Lembo G, Tarone G. IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc Res. 2011;91:456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perez LI, Cariolato L, Maric D, Gillet L, Abriel H, Diviani D. A‐kinase anchoring protein Lbc coordinates a p38 activating signaling complex controlling compensatory cardiac hypertrophy. Mol Cell Biol. 2013;33:2903–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun. 2013;45:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levick SP, Goldspink PH. Could interferon‐gamma be a therapeutic target for treating heart failure? Heart Fail Rev. 2014;19:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu YY, Cai WF, Yang HZ, Cui B, Chen ZR, Liu HZ, Yan J, Jin W, Yan HM, Xin BM, Yuan B, Hua F, Hu ZW. Bacillus Calmette‐Guerin and TLR4 agonist prevent cardiovascular hypertrophy and fibrosis by regulating immune microenvironment. J Immunol. 2008;180:7349–7357. [DOI] [PubMed] [Google Scholar]
- 40. Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, Zhang HJ, Du J. Reciprocal interaction between macrophages and T cells stimulates IFN‐gamma and MCP‐1 production in Ang II‐induced cardiac inflammation and fibrosis. PLoS One. 2012;7:e35506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amento EP, Bhan AK, McCullagh KG, Krane SM. Influences of gamma interferon on synovial fibroblast‐like cells. Ia induction and inhibition of collagen synthesis. J Clin Invest. 1985;76:837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling‐does it mean JAK‐STAT? Cytokine Growth Factor Rev. 2008;19:383–394. [DOI] [PubMed] [Google Scholar]
- 43. Navarro A, Anand‐Apte B, Tanabe Y, Feldman G, Larner AC. A PI‐3 kinase‐dependent, Stat1‐independent signaling pathway regulates interferon‐stimulated monocyte adhesion. J Leukoc Biol. 2003;73:540–545. [DOI] [PubMed] [Google Scholar]
- 44. Meinke A, Barahmand‐Pour F, Wohrl S, Stoiber D, Decker T. Activation of different Stat5 isoforms contributes to cell‐type‐restricted signaling in response to interferons. Mol Cell Biol. 1996;16:6937–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. [DOI] [PubMed] [Google Scholar]
- 46. McMullen JR, Amirahmadi F, Woodcock EA, Schinke‐Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL. Protective effects of exercise and phosphoinositide 3‐kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, Gastier‐Foster JM, Willman CL, Hunger SP, Mullighan CG, Loh ML. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2‐rearranged B‐precursor acute lymphoblastic leukemia. Blood. 2012;120:833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of the Primers Used for RT‐PCR
Figure S1. A double‐color immunofluorescence analysis was performed on the heart 1 week after TAC. The analysis was performed using anti‐CD68 (FITC) and anti‐IFN‐γ (Cy3), followed by observations under a fluorescence microscope, and signals were merged digitally. Representative results from 6 individual animals are shown here. Original magnification, ×400 (scale bar: 100 μm). Cy3 indicates Cyanine3; FITC, fluorescein isothiocyanate; IFN‐γ, interferon‐γ; TAC, transverse aortic constriction.
Figure S2. Quantification of intracardiac inflammatory cells. Representative microscopic images and quantification of CD3‐positive T cell (A and B), CD4‐positive T cell (C and D), CD68‐positive macrophages (E and F), and MPO‐positive neutrophil (G and H) in the heart paraffin sections of sham‐operated and TAC‐operated (day 3) mice. Original magnification, ×400 (scale bar: 100 μm). All values represent means±SEM (n=6). Relative gene expression of Cd3 (I), Cd4 (J), Cd68 (K), and Mpo (L) in the LV of sham‐operated and TAC‐operated mice (day 3) was analyzed by real‐time RT‐PCR. All values represent means±SEM (n=5). LV indicates left ventricle; MPO, myeloperoxidase; RT‐PCR, reverse transcription polymerase chain reaction; Sham, sham‐operated mice; TAC, transverse aortic constriction–operated mice.
Figure S3. Gene expressions of Myh7 (A and C) and Ifngr1 (B and D) in isolated mouse (A and B) and rat (C and D) cardiomyocytes were analyzed by real‐time PCR with or without reverse transcription. All values represent means±SEM (n=4). Akt indicates protein kinase B; FS, fractional shortening; Gab2, GRB2‐associated‐binding protein 2; HW/TL ratio, heart weight/tibial length ratio; Ifngr1 indicates interferon gamma receptor 1; Myh7, myosin heavy chain 7; n.s., not significant; PCR, polymerase chain reaction; Stat5, signal transducer and activator of transcription 5; WT, wild type. * , ** ,
Figure S4. Effects of a Stat5 inhibitor on TAC‐induced cardiac hypertrophy and heart failure in Ifng −/− mice. A and B, Phosphorylation of Akt and Gab2 in LV in mice treated with a Stat5 inhibitor or vehicle. Western blotting analysis was conducted to detect Stat5, Gab2, and Akt in LV of mice at 3 days after TAC operation. Representative results from 6 independent experiments are shown in (A). Relative amounts of p‐Stat5, p‐Gab2, and p‐Akt to GAPDH were analyzed by densitometry and calculated, and are shown in (B). All values represent means±SEM (n=6). *P<0.05, **P<0.01, vs WT treated with vehicle in each column. C, The HW/TL ratio in WT mice treated with vehicle and Ifng −/− mice treated with the Stat5 inhibitor or vehicle 3 weeks after TAC was calculated. All values represent means±SEM (n=6). *P<0.05. WT treated with vehicle. D, M‐mode echocardiographic images of Ifng −/− mice treated with a Stat5 inhibitor or vehicle, 3 weeks after TAC operation. Representative images from at least 5 animals are shown here. E, LVFS were calculated on WT mice treated with vehicle or Ifng −/− mice treated with a Stat5 inhibitor or vehicle at 3 weeks after TAC surgery. All values represent means±SEM (n=6). *P<0.05. WT treated with vehicle. Akt indicates protein kinase B; Gab2, GRB2‐associated‐binding protein 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HW/TL ratio, heart weight/tibial length ratio; Ifng, interferon gamma; LV, left ventricle; LVFS, percent LV fractional shortening; n.s., not significant; Stat5, signal transducer and activator of transcription 5; TAC, transverse aortic constriction; WT, wild type.