

Abstract
Background
Short QT syndrome (SQTS), a disorder associated with characteristic ECG QT‐segment abbreviation, predisposes affected patients to sudden cardiac death. Despite some progress in assessing the organ‐level pathophysiology and genetic changes of the disorder, the understanding of the human cellular phenotype and discovering of an optimal therapy has lagged because of a lack of appropriate human cellular models of the disorder. The objective of this study was to establish a cellular model of SQTS using human‐induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs).
Methods and Results
This study recruited 1 patient with short QT syndrome type 1 carrying a mutation (N588K) in KCNH2 as well as 2 healthy control subjects. We generated hiPSCs from their skin fibroblasts, and differentiated hiPSCs into cardiomyocytes (hiPSC‐CMs) for physiological and pharmacological studies. The hiPSC‐CMs from the patient showed increased rapidly activating delayed rectifier potassium channel current (IK r) density and shortened action potential duration compared with healthy control hiPSC‐CMs. Furthermore, they demonstrated abnormal calcium transients and rhythmic activities. Carbachol increased the arrhythmic events in SQTS but not in control cells. Gene and protein expression profiling showed increased KCNH2 expression in SQTS cells. Quinidine but not sotalol or metoprolol prolonged the action potential duration and abolished arrhythmic activity induced by carbachol.
Conclusions
Patient‐specific hiPSC‐CMs are able to recapitulate single‐cell phenotype features of SQTS and provide novel opportunities to further elucidate the cellular disease mechanism and test drug effects.
Keywords: arrhythmia (heart rhythm disorders), arrhythmia (mechanisms), ion channel, short QT syndrome
Subject Categories: Arrhythmias, Sudden Cardiac Death, Electrophysiology, Cellular Reprogramming, Translational Studies
Clinical Perspective
What Is New?
This is the first cellular model of short QT‐syndrome type 1 using human induced pluripotent stem cell–derived cardiomyocytes.
The site mutation (p.N588K) in KCNH2 increases the expression of HERG channels and thus enhances outward potassium current, shortens action potential duration, and promotes proarrhythmic activity in short QT‐syndrome–cardiomyocytes.
What Are the Clinical Implications?
Patient‐specific pluripotent stem cell–derived cardiomyocytes may provide opportunities to gain new insights into the cellular mechanisms of short QT‐syndrome and discover new effective drugs for treating the disease.
It has been reported that an alteration of QTc interval over 24 hours, either shortening or prolongation, is associated with sudden cardiac death (SCD).1 Short QT syndrome (SQTS) is a rare, inheritable cardiac electrical disease characterized by a shortened corrected QT interval (QTc) and associated with a risk of SCD. The prevalence of SQTS is 0.02% to 0.1% with a male predominance.2 The majority of cardiac events (>80%) in SQTS patients occur under resting conditions or during sleep. Even survivors of SCD are at high risk for recurrent ventricular tachyarrhythmia events. So SCD remains a major public health burden despite revolutionary progress in the past 3 decades in the treatment of ventricular tachyarrhythmia with the use of implantable cardioverter defibrillator (ICD) therapy.
The QT interval of the human ECG presents the duration of the cellular action potential. It depends on the heart rate and therefore a correction should be made before its interpretation. The latest European Society of Cardiology guideline produced in 2015 suggests upper and lower limits of 480 and 360 ms, respectively, for both males and females.3The presence of QTc of ≤340 ms in ECG is a risk marker of SQTS. SQTS should also be considered when a patient has QTc≤360 ms and 1 or more of the following criteria: (1) confirmed gene mutation, (2) a family history of SQTS, (3) a family history of SCD at the age of 40 years, and (4) survival of SCD. Additionally, patients who have SQTS may be asymptomatic or report palpitations and dizziness caused by atrial fibrillation.4, 5 It has been recommended to perform an ECG screening in schools to detect the disease. Furthermore, when a patient is diagnosed, clinical assessment is recommended in all family members.6, 7
SQTS can have a congenital cause or acquired causes. The first family with the entity of SQTS was described in 2000.8 After that, more patients with SQTS have been reported.4, 9, 10 Until now, 6 subtypes of SQTS are known. SQTS1, SQTS2, and SQTS3 are associated with gain of function of potassium channels, whereas SQTS4‐6 can be caused by loss of function of calcium channels. The first genetic alteration in SQTS was identified in the KCNH2 gene encoding a pore‐forming subunit of voltage‐activated potassium channel belonging to the ether‐a‐go‐go family, which mediates the rapidly delayed rectifying potassium current (IKr) in the heart. Currently, 3 genes encoding potassium channels, KCNH2 (for SQTS1), KCNQ1 (for SQTS2), KCNJ2 (for SQTS3), and 3 more genes encoding calcium channels, CACNA1C (for SQTS4), CACNB2 (for SQTS5), and CACNA2D1 (for SQTS6) are identified and linked to SQTS. The genetic analysis, however, does not identify any genetic cause in up to 40% of SQTS cases.11 Additionally, overlap syndrome, SQTS with Brugada syndrome, has been reported.12, 13 Causes of acquired SQTS are electrolyte imbalance including hyperkalemia or hypercalcemia, myocardial ischemia, acidosis, or carnitine deficiency. Hyperthermia can also cause a shortened QT interval, as can drugs such as digitalis, acetylcholine, catecholamines, or ATP‐sensitive K+ channel activators. Short QT intervals have also been associated with epilepsy, particularly during the ictal and postictal states.14, 15, 16
In spite of rapid advances in understanding the mechanisms and genetic bases of SQTS, much less is known about the appropriate treatments for SQTS patients. Because of the high risk of SCD, it has been recommended to implant ICD in patients with a strong family history of SCD and evidence of short QTc interval. However, ICD is not always feasible or adequate for every patient. Therefore, effective drug therapies are needed for some patients. Until now, there has been very limited clinic research to test drug effects in SQTS patients. A few antiarrhythmic drugs such as disopyramide, nifekalant, quinidine, flecainide, sotalol, and ibutilide have been tested in small patient series with SQTS1, but only quinidine has been shown to be effective in the treatment.10, 17, 18, 19
For studies on cardiac functions, especially the cardiac ion channel functions, human induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) have important advantages over heterologous expression systems such as Xenopus oocytes, human embryonic kidney cells, and Chinese Hamster Ovary cells lacking important constituents of cardiac ion channel macromolecular complexes that might be necessary for normal electrophysiological characteristics. Transgenic animals possess cardiac electrophysiological properties crucially different from that in humans. Thus, taking into account the hurdle for obtaining human ventricular cardiomyocytes, hiPSC‐CMs could be a good alternative for SQTS studies, either mechanistic or therapeutic. Indeed, hiPSC‐CMs have been successfully used to recapitulate the phenotype of some genetic heart diseases such as long QT syndromes, Brugada syndrome, arrhythmogenic right ventricular cardiomyopathy, catecholaminergic polymorphic ventricular tachycardia, and hypertrophic and dilated cardiomyopathy.20, 21, 22, 23, 24 Therefore, we aimed in the present study to establish a cellular model of SQTS, which is still lacking, by using hiPSC‐CMs from a short QT patient with a mutation (N588K) in the KCNH2 gene.
Materials and Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure; they will be provided on request to the corresponding author.
Ethics Statement
The skin biopsies from 2 healthy donors and 1 SQTS patient were obtained with written informed consent. The study was approved by the Ethics Committee of the Medical Faculty Mannheim, University of Heidelberg (approval number: 2009‐350N‐MA) and by the Ethics Committee of University Medical Center Göttingen (approval number: 10/9/15). The study was carried out in accordance with the approved guidelines and conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983.
Clinical Data
Skin biopsy was performed in a 29‐year‐old male patient with familial SQTS1 caused by identified missense mutation (C to G substitution at nucleotide 1764) in the KCNH2 gene. The SQTS was diagnosed many years ago. Two patients from a family were referred for syncope, palpitations, and a strong family history of SCD, which was present in 3 generations. The family pedigree is demonstrated in Figure 1A. Three of the family members including our patient presented a short QT interval (270–300 ms corrected with Bazett's formula (QTc) at ECG (Figure 1B). Extensive clinical and instrumental evaluation, including physical examination, serial ECGs, 24‐hour Holter monitoring, echocardiogram, exercise testing, and cardiac magnetic resonance imaging confirmed familial SQTS. The electrophysiology study of this patient showed a ventricular effective refractory period of 140 ms at any pacing site and at different pacing cycle lengths. Programmed stimulation did not induce malignant arrhythmia. Atrial effective refractory period was 160 ms at a basal cycle length of 600 ms. In consideration of the familial history and presence of SQT, the patient prophylactically received an ICD. Nineteen months after implantation the patient had an appropriate ICD shock because of a documented ventricular fibrillation.
Figure 1.
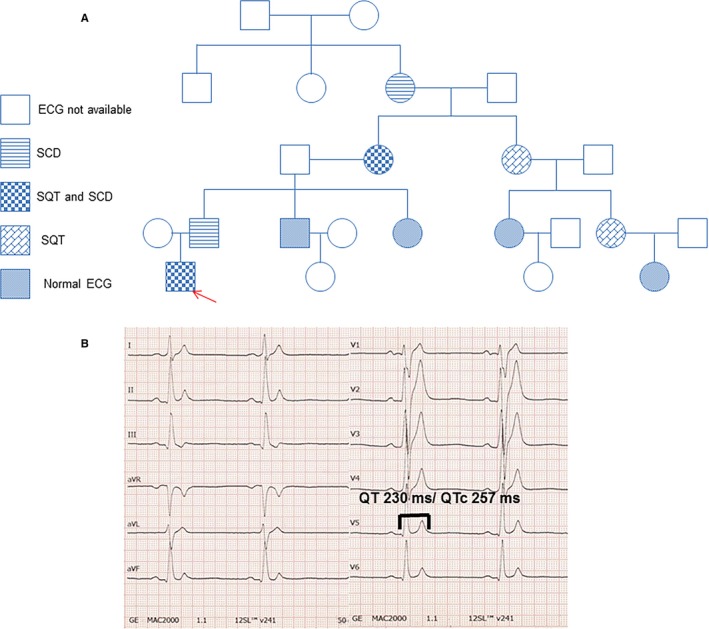
A, The pedigree of the family with SQTS. The patient recruited for this study is marked by the arrow. B, The ECG from the SQTS patient shows a classic SQTS‐ECG pattern. SCD indicates sudden cardiac death; SQT, short QT syndrome (SQTS).
Generation of Human iPS Cells
Human iPS cells (hiPSCs) were generated from primary human fibroblasts derived from skin biopsies. HiPSC line D1 (here abbreviated as D1) was generated using lentiviral particles carrying the transactivator rtTA and an inducible polycistronic cassette containing the reprogramming factors OCT4, SOX2, KLF4, and c‐MYC as described previously.25, 26 HiPSC lines ipWT1.1 (GOEi014‐B.1), ipWT1.3 (GOEi014‐B.3, here abbreviated as D2), and ipWT1.6 (GOEi014‐B.6) were generated in feeder‐free culture conditions using the integration‐free episomal 4‐in‐1 CoMiP reprogramming plasmid (Addgene, #63726) with the reprogramming factors OCT4, KLF4, SOX2, c‐MYC, and short hairpin RNA against p53, as described previously with modifications.27 HiPSC lines isSTQSa1.7 (GOEi091‐A.7, here abbreviated as SQTS), isSQTSa1.8 (GOEi091‐A.8), and isSQTSa1.15 (GOEi091‐A.15) were generated in feeder‐free culture conditions using the integration‐free CytoTune‐iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, #A16517) with the reprogramming factors OCT4, KLF4, SOX2, and c‐MYC according to manufacturer's instructions with modifications. The generated hiPSCs were characterized for their pluripotency and their in vitro differentiation potential (Figures S1 and S2). See Data S1 for further details.
Generation of hiPSC‐CMs
Frozen aliquots of hiPSCs were thawed and cultured without feeder cells and differentiated into hiPSC‐CMs as described with some modifications.28, 29 At 40 to 50 days of culture with basic culture medium, cardiomyocytes were dissociated from 24‐well plates and plated on Matrigel‐coated 3.5‐cm petri dishes for patch‐clamp measurements.
Polymerase‐Chain‐Reaction Assays
To quantify the steady‐state mRNA expression of the hiPSC‐CMs, RNA was reverse transcribed and quantitative polymerase chain reaction was performed as described.29 Gene symbols, RefSeq No., and Cat. No. of the primers used for quantitative polymerase chain reaction analyses in hiPSC‐CMs characterization are listed in Table S1. For evaluation of the characteristics of the used hiPSC lines, reverse transcription‐polymerase chain reaction was performed as follows: Total RNA was isolated using the SV Total RNA Isolation System (Promega, #Z3105) according to manufacturer's instructions. One hundred nanograms RNA was used for the first‐strand cDNA synthesis by using MULV Reverse Transcriptase (Thermo Fisher Scientific, #N8080018) and Oligo d(T)16 (Thermo Fisher Scientific, #N8080128). One tenth of cDNA was used as polymerase chain reaction template and amplified using the GoTaq G2 DNA polymerase (Promega, #M7845) according to manufacturer's instructions. Primer sequences, annealing temperatures, and cycles used for reverse transcription polymerase chain reaction analyses of the hiPSC lines are listed in Table S2.
Immunofluorescence Staining
Immunofluorescence staining was performed using appropriate primary antibodies and AlexaFluor conjugated secondary antibodies (ThermoFisher) according to the manufacturer's instructions. The primary antibodies used in hiPSC‐CMs were α‐actinin (Sigma Aldrich), TNNT (Sigma Aldrich), and KCNH2 (Sigma Aldrich). All the other antibodies used for characterization of hiPSC lines are listed in Table S3.
Patch‐Clamp
Standard patch‐clamp recording techniques were used to measure the rapidly delayed rectifier potassium (IKr), slowly delayed rectifier potassium (IKs), transient outward potassium (Ito), inward rectifier (IK1), late sodium (INa), and L‐type calcium (ICaL) channel currents as well as action potential (AP) in the whole‐cell configuration at room temperature.
Measurement of Intracellular Calcium Concentration
To measure the intracellular Ca2+ concentration ([Ca2+]i), cells were loaded with the fluorescent Ca2+‐indicator Fluo‐3 AM. The fluorescence of the cells was measured by using Cairn Optoscan calcium imaging system (Cairn Research, UK). Fluorescence is excited by 488 nm and emitted at 520 nm. Changes in [Ca2+]i were described by
where kd=dissociation constant of Fluo‐3 (400‐nmol/L), F=Fluo‐3 fluorescence, and Fmax=Ca2+‐saturated fluorescence obtained at the end of each experiment.30
Statistical Analysis
If not otherwise indicated, data are shown as mean±SEM and were analyzed using InStat© (GraphPad, San Diego, CA) and SigmaPlot 11.0 (Systat GmbH, Germany). By analyzing the data with the Kolmogorov Smirnov test, it was decided whether parametric or nonparametric tests were used for analysis. Student t test and the Mann–Whitney U test were used to compare continuous variables with normal and non‐normal distributions, respectively. The χ2 test for independence was used to compare categorical variables. For parametric data, 1‐way ANOVA with Bonferroni post‐test for multiple comparisons was performed. For nonparametric data, the Kruskal–Wallis test with Dunn's multiple comparisons post‐test was used. Paired t test was used for comparisons of data before and after application of a drug. P<0.05 (2‐tailed) was considered significant.
Results
Characterization of Patient‐Specific hiPSCs and hiPSC‐CMs
To confirm the successful generation of hiPSCs reprogrammed from skin fibroblasts of a patient with SQTS and a healthy donor, 3 iPSC lines of each subject were first characterized for pluripotency. Generated iPSCs displayed characteristic human embryonic stem cell morphology and showed expression of pluripotency markers at mRNA and protein level (Figure 2A through 2D, Figure S1A through S1C and S2A through S2C). Spontaneous differentiation of generated iPSC lines via embryoid bodies confirmed expression of germ layer–specific genes (Figure 2E, Figures S1D through S1E and S2D through S2E). A previously generated iPSC line D1 from a healthy donor was additionally used as a second control.
Figure 2.
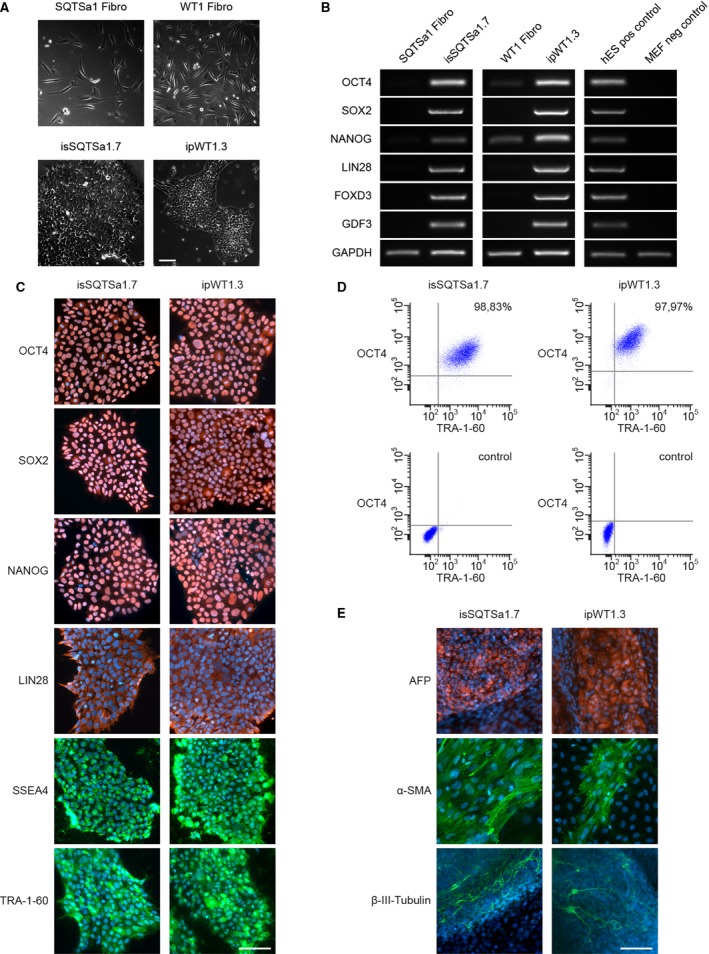
Pluripotent characteristics of human induced pluripotent stem cell (hiPSC) lines. A, The hiPSC lines of short QT syndrome (SQTS) (SQTSa1.7) and donor 2 (D2) (ipWT1.3) generated from skin fibroblasts (upper panel) display a typical morphology for human pluripotent stem cells (lower panel). B, In comparison to donor's fibroblasts, generated iPSC lines show expression of endogenous pluripotency markers SOX2 (sex determining region Y‐ box 2), OCT4 (octamer‐binding transcription factor 4), NANOG (pron. nanOg, homeobox protein), LIN28 (lin‐28 homolog A), FOXD3 (Forkhead Box D3), and GDF3 (growth differentiation factor‐3) at mRNA level proven by reverse transcription‐polymerase chain reaction. Human embryonic stem cells (hESCs) were used as positive control, and mouse embryonic fibroblasts (MEFs) were used as negative control. C, Generated hiPSC lines express pluripotency markers OCT4, SOX2, NANOG, LIN28, SSEA4 (stage‐specific embryonic antigen 4), and TRA‐1‐60 as shown by immunofluorescence staining. Nuclei are co‐stained with DAPI (4′, 6‐diamidino‐2‐phenylindole). D, Flow cytometry analysis of pluripotency markers OCT4 and TRA‐1‐60 reveals a homogeneous population of pluripotent cells in generated iPSC lines. E, Spontaneous differentiation potential of generated hiPSC lines was analyzed by embryoid body formation and germ‐layer specific marker expression. Immunocytochemical staining of spontaneously differentiated hiPSC lines shows expression of endodermal marker AFP (α‐fetoprotein), mesodermal‐specific α‐SMA (α‐smooth muscle actin), and ectodermal βIII‐tubulin. Nuclei are co‐stained with DAPI. Scale bars: 100 μm.
SQTS and control iPSCs (D1 and D2) were directly differentiated into functional iPSC‐CMs in feeder‐free culture conditions for subsequent functional analyses. Beating cardiomyocytes were observed 8 to 12 days after starting the differentiation. No differences in differentiation efficiency were observed between control and SQTS iPSCs. Typical cardiac markers such as α‐actinin and human cardiac troponin T were presented at day 40 of differentiation (Figure 3A and 3B). The beating cardiomyocytes 40 to 50 days after differentiation were used for patch‐clamp and calcium transient imaging.
Figure 3.
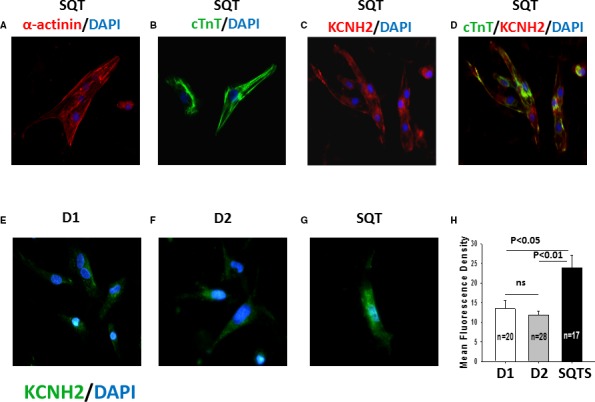
KCNH2 channels are upregulated in SQTS (short QT syndrome)‐cardiomyocytes. Shown are the immunostaining for detecting cardiomyocyte markers (SQTS; A and B) and KCNH2 (rapidly activating delayed rectifier potassium channel) proteins located in the whole cell (SQTS; C and D) and the cell membrane of hiPSC‐CMs (human induced pluripotent stem cell–derived cardiomyocytes) from the healthy donors (D1, D2; E and F) and the patient (SQTS; G). Nuclear staining was induced with DAPI (4′, 6‐diamidino‐2‐phenylindole) (blue). A, Red: Fluorescein isothiocyanate (FITC)‐conjugated α‐actinin antibody at day 40 after differentiation (α‐actinin). B, Green: FITC‐conjugated cTnT2 (human cardiac troponin T) antibody at day 40 after differentiation (cTnT). C, Red: Human KCNH2 antibody at day 40 after differentiation. D, Green: FITC ‐conjugated cTnT2 antibody, red: human KCNH2 antibody at day 40 after differentiation. Nuclear staining was induced with DAPI (blue). E through G, Green: Human KCNH2 antibody located in the cell membrane at day 40 after differentiation. H, The mean fluorescence density of hiPSC‐CMs from the donors (D1, D2) and the patient (SQTS). ns indicates not significant difference.
Action Potential Duration Was Shortened in hiPSC‐CMs From the Patient
AP characterizations are summarized in Table S4. Action potential amplitude, resting potential, and maximum depolarization velocity (Vmax) were similar in the measured hiPSC‐CMs from the patient and healthy donors (Figure 4B through 4D). Action potential durations (APD 50 and 90), however, were significantly shorter in SQTS cells compared with that in the cells from the 2 healthy donors (Figure 4E and 4F). Of note, cells from the 2 donors displayed very similar AP parameters.
Figure 4.
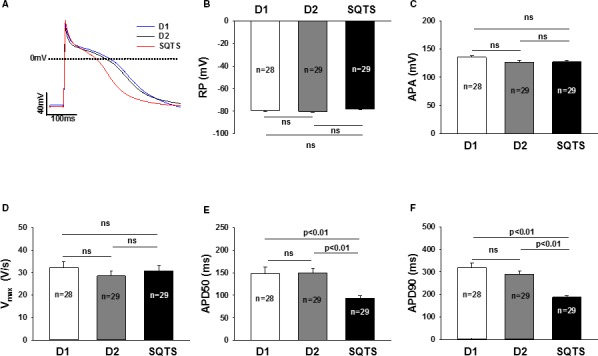
Shortened action potential duration (APD) in SQTS (short QT syndrome)‐cardiomyocytes. A, Representative traces of action potentials (AP) in control‐ (D1 and D2) and SQTS‐cardiomyocytes (SQTS). B, Mean values of resting potentials (RP). C, Mean values of action potential amplitude (APA). D, Mean values of maximal upstroke velocity of AP (Vmax). E, Mean values of APD at 50% repolarization (APD50). F, Mean values of APD at 90% repolarization (APD90). Values given are mean±SEM. n, number of cells. ns indicates not significant difference.
IKr was Enhanced in hiPSC‐CMs From the Patients
To investigate the underlying currents for APD shortening, we assessed different ion channel currents that can influence APD. First, we used patch‐clamp technique and specific ion channel blockers to measure and compare the K+ currents in SQTS cells and donor cells. The transient outward current (Ito), the rapidly activating delayed rectifier potassium current (Ikr) and slowly activating delayed rectifier potassium current (Iks) as well as inward rectifier potassium channel current (IK1) were assessed by 4‐AP‐, E‐4031‐, chromanol 293B‐, and Ba2+‐sensitive currents, respectively. As expected, IKr was enhanced in SQTS cells, but Ito and IKs as well as IK1 were similar among all the cells (Figures 5 and 6, Figures S3 and S4). Notably, the steady‐state currents of IKr were enhanced but the tail currents were reduced in SQTS cells (Figure 5C), indicating an impairment of inactivation of KCNH2 channels. In addition, we also checked L‐type calcium channel current (ICaL) and late sodium channel current (INa), which can also influence APD. Both currents were not significantly changed in SQTS cells when compared with donor cells (Figure 6D and 6E, Figure S5).
Figure 5.
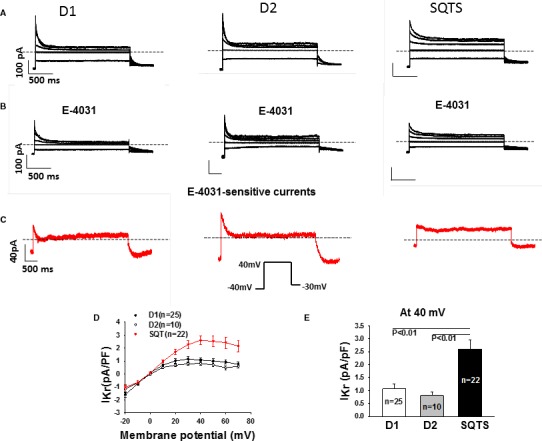
Enhanced IKr in SQTS (short QT syndrome)‐cardiomyocytes. Ikr (rapidly activating delayed rectifier potassium channel) was recorded D1, D2, and SQTS‐cells depolarized from −20 to +70 mV with a holding potential of −40 mV to measure the steady‐state currents. The pulses were repolarized to −30 mV to check the tail currents. Symmetrical Cs+ concentrations (140/140 mmol/L) were used for the current recordings. E‐4031 (3 μmol/L) was applied to isolate IKr. A and B, Representative families of IKr traces (at −20, 0, 20, 40, and 60 mV) in a D1, D2, and SQTS‐cell in absence (A) and presence (B) of E‐4031. C, Representative traces of E‐4031 sensitive currents (IKr). D, Comparison of I–V (current–voltage relationship) curves of Ikr from D1, D2, and SQTS‐cells. E, Comparison of IKr density at 40 mV between donor and the patient. Values given are mean±SEM. n, number of cells.
Figure 6.
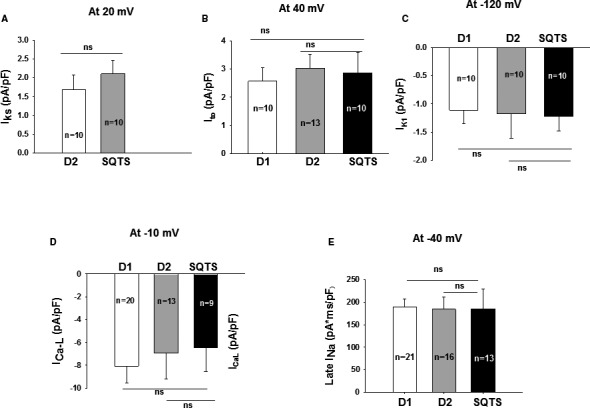
Changes in ion channel currents in SQTS (short QT syndrome)‐cardiomyocytes. Ion channel currents were recorded in the control‐ (D1, D2) and SQTS‐cells (SQTS). A, Mean values of IK s (slowly activating delayed rectifier potassium channel). B, Mean values of peak Ito (transient outward potassium channel current). C, Mean values of IK 1 (inward rectifier potassium channel current). D, Mean values of peak IC aL (L‐type calcium channel current). E, Mean values of the area under curve of TTX (tetrodotoxin) sensitive late IN a (sodium channel current). Values given are mean±SEM; n, number of cells; ns (not significant), P>0.05.
Considering that steady‐state inward currents were always observed in all the cell lines and all the depolarizing pulses induced instantaneous peak currents when the holding potential was set at −40 mV, which are different from that observed in a previous study,31 some IKr recordings were performed at a holding potential of −80 mV. When the holding potential was changed to −80 mV, the basal inward currents disappeared and the instantaneous currents were replaced by currents showing time‐dependent activation (Figure S6).
Next, by using quantitative polymerase chain reaction analysis, we evaluated the gene expression of the assessed ion channels. The mRNA levels of KCND3/KCHIP2 (for Ito), CACNA1C (for α‐subunit of ICaL), and KCNH2 (for IKr) were significantly increased in SQTS‐hiPSC‐CMs, whereas the gene expression of SCN5A (for INa), KCNJ2 (for IK1), and of KCNQ1 (for IKs) were similar compared with donor hiPSC‐CMs (Figure 7). The quantitative polymerase chain reaction data except for CACNA1C and KCND3/KCHIP2 were in agreement with patch‐clamp data. More strikingly, besides enhanced current and mRNA level, the increased protein level of KCNH2 channels in the cell membrane was also detected by the immunostaining of KCNH2 (Figure 3E through 3H).
Figure 7.
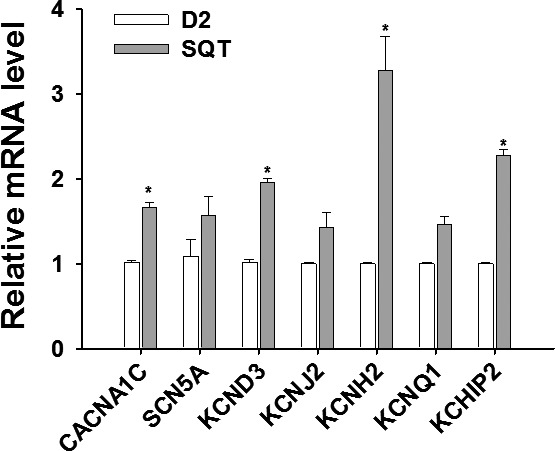
Changes in mRNA expression of ion channels in SQTS (short QT syndrome)‐cardiomyocytes. The relative mRNA levels of each ion channel were analyzed by qPCR (quantitative PCR) in hiPSC‐CMs (human induced pluripotent stem cell–derived cardiomyocytes) of the patient (STQS) and compared with control cells (D2). Values given are mean±SEM (from 3 biological replicates). *P<0.05 vs control (D2).
Calcium Handling Was Changed and Arrhythmia Events Were Increased in hiPSC‐CMs From the Patients
To check whether the calcium handling is normal in SQTS‐hiPSC‐CMs, we examined the intracellular calcium concentration. An increasing of systolic and diastolic concentration has been observed (Figure 8A through 8E). These results reveal altered Ca2+ homeostasis in human SQTS cardiomyocytes with mutation in the HERG‐gene.
Figure 8.
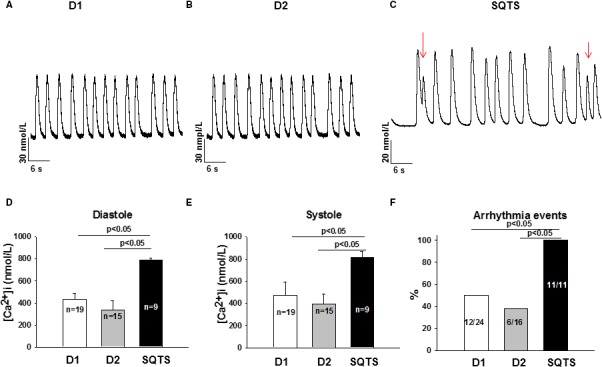
Increased intracellular Ca2+ level and arrhythmic events in SQTS (short QT syndrome)‐cardiomyocytes. A and B, Representative Ca2+ transients in donor (D1, D2) cells. C, Representative Ca2+ transients in a SQTS‐human induced pluripotent stem cell–derived cardiomyocytes with early afterdepolarization‐ and delayed afterdepolarization‐like triggered activities (marked by red arrows). D, Mean values of diastolic Ca2+ concentration. E, Mean values of systolic Ca2+ concentration. F, Percentages of cells showing arrhythmic (triggered) events. Values given are mean±SEM; n, number of cells; ns (not significant), P>0.05.
Interestingly, during measurements of spontaneous calcium transient, all SQTS cardiomyocytes presented irregular, delayed afterdepolarization–like and early afterdepolarization–like arrhythmic events, whereas many fewer donor cells showed those phenomena. The percentage of cells showing arrhythmic events in SQTS, D1, and D2 cells are 100%, 50%, and 37.5%, respectively (P<0.05, Figure 8F). Given that arrhythmias in SQTS patients happen frequently at rest or in sleep, we checked whether cholinergic stimulation can be a trigger for arrhythmias. Indeed, 10 μmol/L carbachol (CCh), a muscarinic receptor agonist, provoked arrhythmic events in cardiomyocytes from the SQTS patient (percentage of cells showing CCh‐induced arrhythmic events: SQT, 100%; D1, 0%; D2, 14%; P<0.05; Figure 9C).
Figure 9.
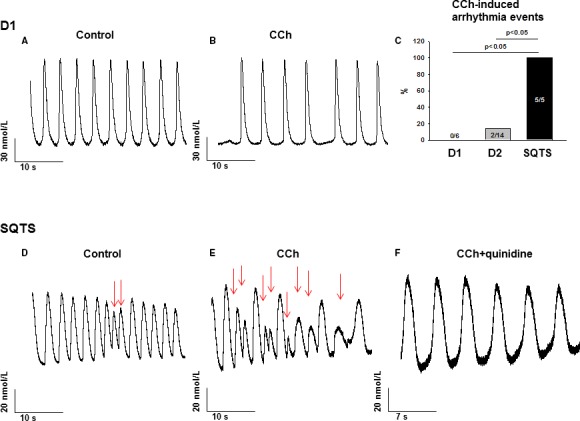
Carbachol increased and quinidine abolished arrhythmic events in SQTS (short QT syndrome)‐cardiomyocytes. A and B, Representative Ca2+ transients in a donor (D1) cell in absence (A) and presence (B) of 10 μmol/L carbachol (CCh). C, Percentages of cells showing CCh‐induced arrhythmia events. D and E, Representative Ca2+ transients in a SQTS‐cell in absence (D) and presence (E) of 10 μmol/L CCh. The red arrows indicate the arrhythmia events. F, Representative Ca2+ transients in a SQTS‐cell in presence of 10 μmol/L CCh plus 10 μmol/L quinidine.
Quinidine Attenuated Arrhythmic Events in hiPSC‐CMs From the Patients
To test the responses of the SQTS‐hiPSC‐CMs to antiarrhythmic drugs, we chose 3 drugs, quinidine, sotalol, and metoprolol, which have been assessed in patients or cells transfected with the same mutated IKr channels in previous studies. All 3 drugs did not change resting potential and action potential amplitude (Figure S7). Ten micromoles per liter quinidine reduced Vmax (Figure S7A), prolonged APD (Figure S8A through S8C), and also abolished CCh‐induced arrhythmic events in SQTS cardiomyocytes (Figure 9D through 9F), while sotalol and metoprolol were not effective in prolonging APD (Figure S8D through S8F, Table S4), consistent with previous in vitro and in vivo studies. Taken together, our data indicate that our hiPSC‐CMs from the SQTS patient mimicked the phenotype of SQTS and recapitulated the drug effects in SQTS patents.
Discussion
We have for the first time generated patient‐specific induced pluripotent stem cell–derived cardiomyocytes from a SQTS patient with “aborted sudden cardiac death” carrying a mutation in the KCNH2 gene and compared their cellular physiological and pharmacological properties with that of healthy cells. We have found the following in SQTS‐hiPSC‐CMs: (1) a shortening of APD and enhancement of IKr; (2) elevated expression level of KCNH2 channels; (3) a high vulnerability of SQTS cardiomyocytes with arrhythmia events; (4) carbachol‐provoked arrhythmia events in SQTS cardiomyocytes; and (5) the antiarrhythmic drug quinidine but not metoprolol or sotalol prolonged APD and attenuated arrhythmia events.
SQTS is a newly recognized rare cardiac disorder. This disease has not been well investigated yet. Many data, either clinical or experimental, which are important for understanding or treating the disease, are not available. One obstacle for studies on SQTS is the low prevalence. It is difficult to have enough patients or tissues from patients for mechanistic or drug‐testing studies. Therefore, an optimal experimental model for SQTS will be useful for studying this rare disease. Recently, an animal model of drug‐induced SQTS has been established to test candidate antiarrhythmic agents that may be effective for treatment of SQTS patients.32, 33, 34 Because of the differences between animals and humans, animal models are not ideal for drug testing. In the current study, we used patient‐specific hiPSC‐CMs to model SQTS and test drug effects.
The patient chosen for this study is from a family with familial SQTS1 carrying a missense mutation (C to G substitution at nucleotide 1764) in KCNH2 (HERG/IKr) gene, which results in an amino acid change in the S5‐P loop region (p.N588K) in HERG/IKr channels. The clinical characteristics of the family including syndromes, family history, gene investigation, ECG properties, and treatment have been described in detail in previous publications.5, 19, 35 Importantly, all the family members showed SQTS or sudden cardiac arrest but none of the unaffected members in the family carry the mutation.35 So it is already clear that this mutation is associated with SQTS in this family. The influence of this mutation on the channel current (IKr) has been experimentally investigated.35, 36, 37, 38 Effects of some antiarrhythmic drugs have also been tested in this family.38 All the published data can be useful for determining the success of the planned modeling of the SQTS phenotype by using hiPSC‐CMs. The first aim of this study was to check whether the hiPSC‐CMs from a SQTS patient of this family can model the disease. Indeed, the hiPSC‐CMs from the patient of the family displayed enhanced IKr, abbreviation of APD, and increased arrhythmia events, indicating successful recapitulation of the main phenotype of SQTS in the patient.
Regarding the mechanism underlying the shortening of QT‐interval in the family, the HERG channel was mutated at the same site as in the patients (p.N588K‐HERG) and was expressed in modified human embryonic kidney cells (TSA201) to check the effects of the mutation on channel current.35 Interestingly, it was revealed that the p.N588K‐mutation enhanced the channel currents (IKr), suggesting that gain of function mutation of HERG channel, which is an important repolarizing current and determinant for APD, may be the reason for the abbreviation of QT‐interval. Also, it was proved that the p.N558K‐mutation led to impairment of channel inactivation and thus enhanced the steady‐state currents.18, 35, 36 This is until now the explanation for IKr enhancement. In the present study, we also detected the impairment of inactivation and enhanced IKr in the SQTS‐hiPSC‐CMs, in good agreement with previous studies. In addition, we found that the expression of HERG channel in SQTS‐hiPSC‐CMs was increased at both mRNA and protein levels, indicative of a second mechanism for IKr enhancement. These novel data may expand our knowledge for further mechanistic and therapeutic target–searching studies. We also examined IKs, IK1, and ICaL, which have been linked to SQTS, as well as late INa that may influence APD. As expected, none of them were significantly different in SQTS cells and donor cells. In addition, we surprisingly observed in SQTS cells an elevated systolic and diastolic intracellular Ca2+ level. The reason for and pathological meaning of the changed Ca2+ handling remain to be addressed.
To reduce the possible influences of other ion channels on the recorded IKr to minimal, we used Cs+ as charge carrier for IKr to block some K+ currents and set the holding potential at −40 mV to inactivate some voltage‐dependent ion channels. With the holding potential of −40 mV, some HERG channels are activated and stay in open state. Therefore, steady‐state inward currents at baseline were observed in all the cell lines and every depolarizing pulse induced an instantaneous current through the opened channels (Figure 5). When the membrane potential was held at −80 mV, the channels were in resting state and there were no basal currents. Depolarizing pulses induced time‐dependent but not instantaneous peak currents, representing the process of the channel activation from resting state to open state (Figure S6). Of note, the enhanced outward steady‐state currents and reduced tail currents in SQTS cells could be observed when the holding potential was set at either −40 mV or −80 mV.
Tachyarrhythmias in SQTS patients occur frequently at rest or in sleep, suggesting involvement of increased vagal tone. A previous study showed that acetylcholine applied to coronary‐perfused canine left‐ventricular wedge preparations, mimicking increased vagal tone, precipitated drug‐ (an Ito activator plus a ICaL blocker) induced polymorphic ventricular tachycardia/ventricular fibrillation.32 Until now, this effect has not been tested in a SQTS model. We observed in SQTS‐hiPSC‐CMs that a muscarinic receptor agonist, CCh, increased arrhythmia events compared with cells from 2 healthy donors. This finding supports the role played by vagal tone in occurrence of tachyarrhythmias in SQTS patients. Given that CCh alone did not show any effect on INa, ICaL, Ito, IKr, and IKs, the mechanisms underlying the CCh‐induced arrhythmia events need to be clarified in future studies.
For the treatment of SQTS, there is still no optimal strategy. Because of SCD risk, ICD has been recommended and is also effective in some patients. Because of the limitations of ICD and to avoid inappropriate ICD shock delivery that is also associated with increased mortality, drug therapy is also needed. Therefore, different antiarrhythmic drugs were tested in different SQTS families including our patient. Surprisingly, the well‐known IKr blocker, sotalol, did not effectively prolong QT‐interval, whereas quinidine, a multiple‐channel blocker, corrected QT‐abbreviation and prevented arrhythmias.19, 35 The reason for ineffectiveness of sotalol has been proved in a heterologous expression system (TSA201 cells) to be the largely reduced sensitivity of the mutated channel to sotalol.18, 35 The second aim of the present study was to test whether the hiPSC‐CMs from the SQTS patient exhibit the same or similar responses to the examined drugs. Actually, we observed in SQTS‐hiPSC‐CMs that sotalol failed but quinidine succeeded in prolonging APD and reversed CCh‐induced arrhythmia events, again in good agreement with previous data of either clinical or experimental studies. This finding highlighted the ability of iPSC‐CMs to recapitulate not only the phenotype but also the drug responses of the disorder. Furthermore, another study tested the effects of 2 widely used β‐blockers, carvedilol and metoprolol, on p.N588K‐HERG and p.V307L‐KCNQ1 channels expressed in mammalian cells.39 It was found that metoprolol is more potent than carvedilol in inhibiting both mutant K+ channel currents. This suggests that metoprolol may be a new candidate drug for treating SQTS. However, in the current study, metoprolol failed to prolong APD in SQTS‐hiPSC‐CMs. This suggests effects of metoprolol on other ion channels that counteracted the IKr and IKs inhibition. Thus, our data argue against the application of metoprolol for SQTS treatment.
Conclusion
Our data demonstrate that the main phenotypic features of SQTS were successfully recapitulated in the hiPSC‐CMs from a SQTS1 patient, including increased IKr, shortened APD, and increased arrhythmia events as well as changed responses to antiarrhythmic drugs. In addition, a new mechanism for IKr enhancement in SQTS1 with p.N588K mutation was identified. This study supports an application of hiPSC‐CMs in future studies on SQTS including examination of disease mechanisms, identification of new therapeutic targets, and test of drug effects.
Study Limitations
HiPSC‐CMs from 2 healthy donors and 1 SQTS patient were used for this study. Differences among individuals cannot be ruled out. The results from the 3 individuals of this study should, from a statistical point of view, not be interpreted as that from the whole population of SQT patients and healthy people. Statistical analyses did not take into account possible correlation between measurements taken from the same individual. hiPSC‐CMs possess similarities but also distinct differences in their physiological properties when compared with adult human cardiomyocytes. In particular, the immature characteristics of iPSC‐CMs are a well‐known limitation. In addition, in the single‐cell studies, many factors such as cell‐to‐cell interaction, nerve and hormone regulations are not involved. The differences between a single‐cell and whole organ system should be considered in interpretation of the experimental data from hiPSC‐CMs.
It is known that both Cs+ and the mutation (N588K) reduced the sensitivity of the channel to E‐4031. Therefore, the analysis of E‐4031‐sensitive currents may lead to underestimation of Ikr, especially in SQT‐hiPSC‐CMs. Furthermore, how Cs+ influences the sensitivity of the mutant HERG channel to E‐4031 and how the mutation influences the effects of Cs+ on the sensitivity of the channel to E‐4031 still need to be investigated.
Sources of Funding
This study was supported by the DZHK (German Center for Cardiovascular Research), the BMBF (German Ministry of Education and Research) (grants to Borggrefe, Utikal, Wieland, and Zimmermann).
Disclosures
None.
Supporting information
Data S1. Material and Methods.
Table S1. Gene Symbols, RefSeq No., and Cat. No. of the Primers Used for qPCR Analyses in hiPSC‐CMs Characterization
Table S2. Primer Sequences, Annealing Temperatures, and Cycles Used for RT‐PCR Analyses in hiPSC Pluripotency Characterization
Table S3. Antibodies and Dilutions Used for Immunocytochemistry of iPSC Pluripotency Characterization
Table S4. Action Potential Parameters
Figure S1. The generated hiPSC lines from the patient with short QT syndrome (SQTS) display pluripotent characteristics. A, The hiPSC (human induced pluripotent stem cell) lines isSQTSa1.7, isSQTSa1.8, and isSQTSa1.15 generated from skin fibroblasts of the SQTS patient display a typical morphology for human pluripotent stem cells (upper panel) and are positive for alkaline phosphatase (lower panel). B, In comparison to fibroblasts, generated hiPSC lines show expression of endogenous pluripotency markers SOX2 (sex determining region Y‐ box 2), OCT4 (octamer‐binding transcription factor 4), NANOG (pron. nanOg, homeobox protein), LIN28 (lin‐28 homolog A), FOXD3 (Forkhead Box D3), and GDF3 (growth differentiation factor‐3) at mRNA level proven by RT‐PCR (reverse transcription‐polymerase chain reaction). Human embryonic stem cells (hESCs) were used as positive control, mouse embryonic fibroblasts (MEFs) were used as negative control. C, Generated iPSC lines express pluripotency markers OCT4, SOX2, NANOG, LIN28, SSEA4 (stage‐specific embryonic antigen 4) and TRA‐1‐60 as shown by immunofluorescence staining. Nuclei are co‐stained with DAPI (4’,6‐diamidino‐2‐phenylindole). D, Spontaneous differentiation potential of generated iPSC lines was analyzed by embryoid body (EB) formation. Germ layer–specific genes such as α‐fetoprotein (AFP) and albumin (ALB) (endoderm), cardiac troponin T and α‐MHC (mesoderm), and tyrosine hydroxylase (TH) and MAP2 (ectoderm) are expressed in a developmentally controlled manner during differentiation of EBs (days 0, 8, or 8+25), whereas endogenous OCT4 expression is decreased during spontaneous differentiation. MEFs were used as negative control. E, Immunocytochemical staining of spontaneously differentiated hiPSC lines shows expression of endodermal marker AFP, mesodermal‐specific α‐SMA (α‐smooth muscle actin), and ectodermal βIII‐tubulin. Nuclei are co‐stained with DAPI. Scale bars: 100 μm.
Figure S2. The generated hiPSC lines from a healthy donor display pluripotent characteristics. A, The hiPSC (human induced pluripotent stem cell) lines ipWT1.1, ipWT1.3 and ipWT1.6 generated from skin fibroblasts of a healthy donor (D2) display a typical morphology for human pluripotent stem cells (upper panel) and are positive for alkaline phosphatase (lower panel). B, In comparison to fibroblasts, generated hiPSC lines show expression of endogenous pluripotency markers SOX2 (sex determining region Y‐ box 2), OCT4 OCT4 (octamer‐binding transcription factor 4), NANOG (pron. nanOg, homeobox protein), LIN28 (lin‐28 homolog A), FOXD3 (Forkhead Box D3) and GDF3 (growth differentiation factor‐3) at mRNA level proven by RT‐PCR (reverse transcription‐polymerase chain reaction). Human embryonic stem cells (hESCs) were used as positive control, mouse embryonic fibroblasts (MEFs) were used as negative control. C, Generated hiPSC lines express pluripotency markers OCT4, SOX2, NANOG, LIN28, SSEA4 (stage‐specific embryonic antigen 4) and TRA‐1‐60 as shown by immunofluorescence staining. Nuclei are co‐stained with DAPI (4’,6‐diamidino‐2‐phenylindole). D, Spontaneous differentiation potential of generated hiPSC lines was analyzed by embryoid body (EB) formation. Germ layer–specific genes such as α‐fetoprotein (AFP) and albumin (ALB) (endoderm), cTNT (cardiac troponin T) and α‐MHC (mesoderm), and tyrosine hydroxylase (TH) and MAP2 (ectoderm) are expressed in a developmentally controlled manner during differentiation of EBs (days 0, 8, or 8+25), whereas endogenous OCT4 expression is decreased during spontaneous differentiation. MEFs were used as negative control. E, Immunocytochemical staining of spontaneously differentiated hiPSC lines shows expression of endodermal marker AFP, mesodermal‐specific α‐SMA (α‐smooth muscle actin), and ectodermal βIII‐tubulin. Nuclei are co‐stained with DAPI. Scale bars: 100 μm.
Figure S3. Slowly activating delayed rectifier currents (IKs) in donor‐ and SQTS‐ (short QT syndrome) cells. IKs (slowly activating delayed rectifier potassium channel) were evoked by the indicated protocol (B) in absence (control) and presence of a channel blocker. Chromanol 293B (10 μmol/L) was used to isolate IKs from other currents. A, Representative IKs at +40 mV recorded in a cell from a donor (D2) and the patient (SQTS) with and without (Ctr) 293B. B, Currents that are 293B sensitive. (C) I–V curves of IKs from the donor‐ and SQTS‐cells. n, number of cells. *P<0.05 vs donor cells.
Figure S4. The transient outward currents (Ito) and inward rectifier currents (IK1) in donor‐and SQTS (short QT syndrome)‐cells. Ito and IK1 were evoked by the indicated protocol (A and C) in absence (Ctr) and presence of a channel blocker. 4‐Aminopyridine (4‐AP, 5 mmol/L) and BaCl2 (0.1 mmol/L) was used to isolate Ito and IK1 from other currents. A, Representative Ito at +40 mV recorded in a cell from the donors (D1 and D2) and the patient (SQTS). (B) I–V curves of Ito from the donor‐ and SQTS‐cells. C, Representative IK1 at −120 mV recorded in a cell from the donors (D1 and D2) and the patient (SQTS). D, I–V curves of IK1 from the donor‐ and SQTS‐cells. n, number of cells.
Figure S5. The L‐type calcium channel currents (ICaL) in donor‐ and SQTS (short QT syndrome)‐cells. ICaL was evoked by the indicated protocol (A). A, Representative ICaL at different potentials recorded in a cell from the donors (D1 and D2) and the patient (SQTS). B, I–V (current–voltage relationship) curves of ICaL from the donor‐ and SQTS‐cells.
Figure S6. Ikr recorded in human induced pluripotent stem cell–derived (short QT syndrome)‐cardiomyocytes with a holding potential of −80 mV. Ikr (rapidly activating delayed rectifier potassium channel) was evoked by 2‐s depolarizing pulses from −70 to +70 mV with 10‐mV increments and repolarized to the holding potential of −80 mV. A, Representative traces of IKr in a donor (D2) cell. B, Representative traces of IKr in a SQTS (SQT) cell.
Figure S7. Effects of quinidine, sotalol, and metoprolol on AP parameters. APs (action potentials) were recorded in a cell before (control) and after application of a drug. A–C, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of quinidine. D–F, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of sotalol. G–I, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of metoprolol. n, number of cells. ns indicates .
Figure S8. Effects of antiarrhythmic drugs on action potential duration (APD) in SQTS (short QT syndrome)‐cardiomyocytes. A, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 10 μmol/L quinidine. B, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 10 μmol/L quinidine. C, Representative traces of action potentials in absence (control) and presence of 10 μmol/L quinidine. D, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 100 μmol/L sotalol. E, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 100 μmol/L sotalol. F, Representative traces of action potentials in absence (control) and presence of 100 μmol/L sotalol. G, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 10 μmol/L metoprolol. H, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 10 μmol/L metoprolol. I, Representative traces of action potentials in absence (control) and presence of 10 μmol/L metoprolol. Values given are mean±SEM. n, number of cells. ns, P>0.05.
Acknowledgments
We gratefully thank C. Liebetrau, M. Grohe, L. Rogge, and Y. Wiegräfe for excellent technical assistance. We acknowledge the financial support of the Deutsche Forschungsgemeinschaft and Ruprecht‐Karls‐Universität Heidelberg within the funding program Open Access Publishing. We thank the China Scholarship Council (CSC) for the financial support for Zhihan Zhao and Xin Li.
(J Am Heart Assoc.2018;7:e007394 DOI: 10.1161/JAHA.117.007394.)
References
- 1. Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. Qt interval variables from 24 hour electrocardiography and the two year risk of sudden death. Br Heart J. 1993;70:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guerrier K, Kwiatkowski D, Czosek RJ, Spar DS, Anderson JB, Knilans TK. Short QT interval prevalence and clinical outcomes in a pediatric population. Circ Arrhythm Electrophysiol. 2015;8:1460–1464. [DOI] [PubMed] [Google Scholar]
- 3. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 4. Giustetto C, Di Monte F, Wolpert C, Borggrefe M, Schimpf R, Sbragia P, Leone G, Maury P, Anttonen O, Haissaguerre M, Gaita F. Short QT syndrome: clinical findings and diagnostic‐therapeutic implications. Eur Heart J. 2006;27:2440–2447. [DOI] [PubMed] [Google Scholar]
- 5. Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. [DOI] [PubMed] [Google Scholar]
- 6. Brugada J, Gussak I, Brugada P. Short QT syndrome: a predictable story. Cardiology. 2014;128:231–233. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki H, Hoshina S, Ozawa J, Sato A, Minamino T, Aizawa Y, Saitoh A. Short QT syndrome in a boy diagnosed on screening for heart disease. Pediatr Int. 2014;56:774–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102. [DOI] [PubMed] [Google Scholar]
- 9. Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle‐aged Finnish population. Circulation. 2007;116:714–720. [DOI] [PubMed] [Google Scholar]
- 10. Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, Probst V, Blanc JJ, Sbragia P, Dalmasso P, Borggrefe M, Gaita F. Long‐term follow‐up of patients with short QT syndrome. J Am Coll Cardiol. 2011;58:587–595. [DOI] [PubMed] [Google Scholar]
- 11. Perez Riera AR, Paixao‐Almeida A, Barbosa‐Barros R, Yanowitz FG, Baranchuk A, Dubner S, Palandri Chagas AC. Congenital short QT syndrome: landmarks of the newest arrhythmogenic cardiac channelopathy. Cardiol J. 2013;20:464–471. [DOI] [PubMed] [Google Scholar]
- 12. Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V, Miceli C, O'Rourke S, Borio G, Zienciuk‐Krajka A, Curcio A, Surducan AE, Colombo M, Napolitano C, Priori SG. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss‐of‐function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST‐segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Workman AJ, MacKenzie I, Northover BJ. A K(ATP) channel opener inhibited myocardial reperfusion action potential shortening and arrhythmias. Eur J Pharmacol. 2001;419:73–83. [DOI] [PubMed] [Google Scholar]
- 15. Roussel J, Labarthe F, Thireau J, Ferro F, Farah C, Roy J, Horiuchi M, Tardieu M, Lefort B, Francois Benoist J, Lacampagne A, Richard S, Fauconnier J, Babuty D, Le Guennec JY. Carnitine deficiency induces a short QT syndrome. Heart Rhythm. 2016;13:165–174. [DOI] [PubMed] [Google Scholar]
- 16. Surges R, Taggart P, Sander JW, Walker MC. Too long or too short? New insights into abnormal cardiac repolarization in people with chronic epilepsy and its potential role in sudden unexpected death. Epilepsia. 2010;51:738–744. [DOI] [PubMed] [Google Scholar]
- 17. Mizobuchi M, Enjoji Y, Yamamoto R, Ono T, Funatsu A, Kambayashi D, Kobayashi T, Nakamura S. Nifekalant and disopyramide in a patient with short QT syndrome: evaluation of pharmacological effects and electrophysiological properties. Pacing Clin Electrophysiol. 2008;31:1229–1232. [DOI] [PubMed] [Google Scholar]
- 18. Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro J, Dumaine R, Brugada R, Hong K, Bauersfeld U, Gaita F, Borggrefe M. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, Brugada R, Antzelevitch C, Borggrefe M, Wolpert C. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494–1499. [DOI] [PubMed] [Google Scholar]
- 20. Itzhaki I, Maizels L, Huber I, Zwi‐Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. [DOI] [PubMed] [Google Scholar]
- 21. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott‐Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient‐specific induced pluripotent stem‐cell models for long‐QT syndrome. N Engl J Med. 2010;363:1397–1409. [DOI] [PubMed] [Google Scholar]
- 22. Novak A, Barad L, Zeevi‐Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz‐Eldor J, Binah O. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta‐adrenergic stimulation. J Cell Mol Med. 2012;16:468–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Savla JJ, Nelson BC, Perry CN, Adler ED. Induced pluripotent stem cells for the study of cardiovascular disease. J Am Coll Cardiol. 2014;64:512–519. [DOI] [PubMed] [Google Scholar]
- 24. Liang P, Sallam K, Wu H, Li Y, Itzhaki I, Garg P, Zhang Y, Vermglinchan V, Lan F, Gu M, Gong T, Zhuge Y, He C, Ebert AD, Sanchez‐Freire V, Churko J, Hu S, Sharma A, Lam CK, Scheinman MM, Bers DM, Wu JC. Patient‐specific and genome‐edited induced pluripotent stem cell‐derived cardiomyocytes elucidate single‐cell phenotype of Brugada syndrome. J Am Coll Cardiol. 2016;68:2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larribere L, Wu H, Novak D, Galach M, Bernhardt M, Orouji E, Weina K, Knappe N, Sachpekidis C, Umansky L, Beckhove P, Umansky V, De Schepper S, Kaufmann D, Ballotti R, Bertolotto C, Utikal J. NF1 loss induces senescence during human melanocyte differentiation in an iPSC‐based model. Pigment Cell Melanoma Res. 2015;28:407–416. [DOI] [PubMed] [Google Scholar]
- 26. Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K. A high‐efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diecke S, Lu J, Lee J, Termglinchan V, Kooreman NG, Burridge PW, Ebert AD, Churko JM, Sharma A, Kay MA, Wu JC. Novel codon‐optimized mini‐intronic plasmid for efficient, inexpensive, and xeno‐free induction of pluripotency. Sci Rep. 2015;5:8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tiburcy M, Zimmermann WH. Modeling myocardial growth and hypertrophy in engineered heart muscle. Trends Cardiovasc Med. 2014;24:7–13. [DOI] [PubMed] [Google Scholar]
- 29. El‐Battrawy I, Lang S, Zhao Z, Akin I, Yucel G, Meister S, Patocskai B, Behnes M, Rudic B, Tulumen E, Liebe V, Tiburcy M, Dworacek J, Zimmermann WH, Utikal J, Wieland T, Borggrefe M, Zhou XB. Hyperthermia influences the effects of sodium channel blocking drugs in human‐induced pluripotent stem cell‐derived cardiomyocytes. PLoS One. 2016;11:e0166143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trafford AW, Diaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time‐course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 1999;437:501–503. [DOI] [PubMed] [Google Scholar]
- 31. Zhang S. Isolation and characterization of i(kr) in cardiac myocytes by Cs+ permeation. Am J Physiol Heart Circ Physiol. 2006;290:H1038–H1049. [DOI] [PubMed] [Google Scholar]
- 32. Koncz I, Gurabi Z, Patocskai B, Panama BK, Szel T, Hu D, Barajas‐Martinez H, Antzelevitch C. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol. 2014;68:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Milberg P, Tegelkamp R, Osada N, Schimpf R, Wolpert C, Breithardt G, Borggrefe M, Eckardt L. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol. 2007;18:658–664. [DOI] [PubMed] [Google Scholar]
- 34. Frommeyer G, Ellermann C, Dechering DG, Kochhauser S, Bogeholz N, Guner F, Leitz P, Pott C, Eckardt L. Ranolazine and vernakalant prevent ventricular arrhythmias in an experimental whole‐heart model of short QT syndrome. J Cardiovasc Electrophysiol. 2016;27:1214–1219. [DOI] [PubMed] [Google Scholar]
- 35. Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short‐QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. [DOI] [PubMed] [Google Scholar]
- 36. McPate MJ, Duncan RS, Hancox JC, Witchel HJ. Pharmacology of the short QT syndrome N588k‐hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br J Pharmacol. 2008;155:957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McPate MJ, Duncan RS, Witchel HJ, Hancox JC. Disopyramide is an effective inhibitor of mutant HERG K+ channels involved in variant 1 short QT syndrome. J Mol Cell Cardiol. 2006;41:563–566. [DOI] [PubMed] [Google Scholar]
- 38. Abriel H, Rougier JS. Beta‐blockers in congenital short‐QT syndrome as ion channel blockers. J Cardiovasc Electrophysiol. 2013;24:1172–1174. [DOI] [PubMed] [Google Scholar]
- 39. Bodi I, Franke G, Pantulu ND, Wu K, Perez‐Feliz S, Bode C, Zehender M, zur Hausen A, Brunner M, Odening KE. Differential effects of the beta‐adrenoceptor blockers carvedilol and metoprolol on SQT1‐ and SQT2‐mutant channels. J Cardiovasc Electrophysiol. 2013;24:1163–1171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Material and Methods.
Table S1. Gene Symbols, RefSeq No., and Cat. No. of the Primers Used for qPCR Analyses in hiPSC‐CMs Characterization
Table S2. Primer Sequences, Annealing Temperatures, and Cycles Used for RT‐PCR Analyses in hiPSC Pluripotency Characterization
Table S3. Antibodies and Dilutions Used for Immunocytochemistry of iPSC Pluripotency Characterization
Table S4. Action Potential Parameters
Figure S1. The generated hiPSC lines from the patient with short QT syndrome (SQTS) display pluripotent characteristics. A, The hiPSC (human induced pluripotent stem cell) lines isSQTSa1.7, isSQTSa1.8, and isSQTSa1.15 generated from skin fibroblasts of the SQTS patient display a typical morphology for human pluripotent stem cells (upper panel) and are positive for alkaline phosphatase (lower panel). B, In comparison to fibroblasts, generated hiPSC lines show expression of endogenous pluripotency markers SOX2 (sex determining region Y‐ box 2), OCT4 (octamer‐binding transcription factor 4), NANOG (pron. nanOg, homeobox protein), LIN28 (lin‐28 homolog A), FOXD3 (Forkhead Box D3), and GDF3 (growth differentiation factor‐3) at mRNA level proven by RT‐PCR (reverse transcription‐polymerase chain reaction). Human embryonic stem cells (hESCs) were used as positive control, mouse embryonic fibroblasts (MEFs) were used as negative control. C, Generated iPSC lines express pluripotency markers OCT4, SOX2, NANOG, LIN28, SSEA4 (stage‐specific embryonic antigen 4) and TRA‐1‐60 as shown by immunofluorescence staining. Nuclei are co‐stained with DAPI (4’,6‐diamidino‐2‐phenylindole). D, Spontaneous differentiation potential of generated iPSC lines was analyzed by embryoid body (EB) formation. Germ layer–specific genes such as α‐fetoprotein (AFP) and albumin (ALB) (endoderm), cardiac troponin T and α‐MHC (mesoderm), and tyrosine hydroxylase (TH) and MAP2 (ectoderm) are expressed in a developmentally controlled manner during differentiation of EBs (days 0, 8, or 8+25), whereas endogenous OCT4 expression is decreased during spontaneous differentiation. MEFs were used as negative control. E, Immunocytochemical staining of spontaneously differentiated hiPSC lines shows expression of endodermal marker AFP, mesodermal‐specific α‐SMA (α‐smooth muscle actin), and ectodermal βIII‐tubulin. Nuclei are co‐stained with DAPI. Scale bars: 100 μm.
Figure S2. The generated hiPSC lines from a healthy donor display pluripotent characteristics. A, The hiPSC (human induced pluripotent stem cell) lines ipWT1.1, ipWT1.3 and ipWT1.6 generated from skin fibroblasts of a healthy donor (D2) display a typical morphology for human pluripotent stem cells (upper panel) and are positive for alkaline phosphatase (lower panel). B, In comparison to fibroblasts, generated hiPSC lines show expression of endogenous pluripotency markers SOX2 (sex determining region Y‐ box 2), OCT4 OCT4 (octamer‐binding transcription factor 4), NANOG (pron. nanOg, homeobox protein), LIN28 (lin‐28 homolog A), FOXD3 (Forkhead Box D3) and GDF3 (growth differentiation factor‐3) at mRNA level proven by RT‐PCR (reverse transcription‐polymerase chain reaction). Human embryonic stem cells (hESCs) were used as positive control, mouse embryonic fibroblasts (MEFs) were used as negative control. C, Generated hiPSC lines express pluripotency markers OCT4, SOX2, NANOG, LIN28, SSEA4 (stage‐specific embryonic antigen 4) and TRA‐1‐60 as shown by immunofluorescence staining. Nuclei are co‐stained with DAPI (4’,6‐diamidino‐2‐phenylindole). D, Spontaneous differentiation potential of generated hiPSC lines was analyzed by embryoid body (EB) formation. Germ layer–specific genes such as α‐fetoprotein (AFP) and albumin (ALB) (endoderm), cTNT (cardiac troponin T) and α‐MHC (mesoderm), and tyrosine hydroxylase (TH) and MAP2 (ectoderm) are expressed in a developmentally controlled manner during differentiation of EBs (days 0, 8, or 8+25), whereas endogenous OCT4 expression is decreased during spontaneous differentiation. MEFs were used as negative control. E, Immunocytochemical staining of spontaneously differentiated hiPSC lines shows expression of endodermal marker AFP, mesodermal‐specific α‐SMA (α‐smooth muscle actin), and ectodermal βIII‐tubulin. Nuclei are co‐stained with DAPI. Scale bars: 100 μm.
Figure S3. Slowly activating delayed rectifier currents (IKs) in donor‐ and SQTS‐ (short QT syndrome) cells. IKs (slowly activating delayed rectifier potassium channel) were evoked by the indicated protocol (B) in absence (control) and presence of a channel blocker. Chromanol 293B (10 μmol/L) was used to isolate IKs from other currents. A, Representative IKs at +40 mV recorded in a cell from a donor (D2) and the patient (SQTS) with and without (Ctr) 293B. B, Currents that are 293B sensitive. (C) I–V curves of IKs from the donor‐ and SQTS‐cells. n, number of cells. *P<0.05 vs donor cells.
Figure S4. The transient outward currents (Ito) and inward rectifier currents (IK1) in donor‐and SQTS (short QT syndrome)‐cells. Ito and IK1 were evoked by the indicated protocol (A and C) in absence (Ctr) and presence of a channel blocker. 4‐Aminopyridine (4‐AP, 5 mmol/L) and BaCl2 (0.1 mmol/L) was used to isolate Ito and IK1 from other currents. A, Representative Ito at +40 mV recorded in a cell from the donors (D1 and D2) and the patient (SQTS). (B) I–V curves of Ito from the donor‐ and SQTS‐cells. C, Representative IK1 at −120 mV recorded in a cell from the donors (D1 and D2) and the patient (SQTS). D, I–V curves of IK1 from the donor‐ and SQTS‐cells. n, number of cells.
Figure S5. The L‐type calcium channel currents (ICaL) in donor‐ and SQTS (short QT syndrome)‐cells. ICaL was evoked by the indicated protocol (A). A, Representative ICaL at different potentials recorded in a cell from the donors (D1 and D2) and the patient (SQTS). B, I–V (current–voltage relationship) curves of ICaL from the donor‐ and SQTS‐cells.
Figure S6. Ikr recorded in human induced pluripotent stem cell–derived (short QT syndrome)‐cardiomyocytes with a holding potential of −80 mV. Ikr (rapidly activating delayed rectifier potassium channel) was evoked by 2‐s depolarizing pulses from −70 to +70 mV with 10‐mV increments and repolarized to the holding potential of −80 mV. A, Representative traces of IKr in a donor (D2) cell. B, Representative traces of IKr in a SQTS (SQT) cell.
Figure S7. Effects of quinidine, sotalol, and metoprolol on AP parameters. APs (action potentials) were recorded in a cell before (control) and after application of a drug. A–C, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of quinidine. D–F, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of sotalol. G–I, Mean values of maximal depolarization speed (Vmax), resting potential (RP), and amplitude of AP (APA) in absence and presence of metoprolol. n, number of cells. ns indicates .
Figure S8. Effects of antiarrhythmic drugs on action potential duration (APD) in SQTS (short QT syndrome)‐cardiomyocytes. A, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 10 μmol/L quinidine. B, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 10 μmol/L quinidine. C, Representative traces of action potentials in absence (control) and presence of 10 μmol/L quinidine. D, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 100 μmol/L sotalol. E, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 100 μmol/L sotalol. F, Representative traces of action potentials in absence (control) and presence of 100 μmol/L sotalol. G, Mean values of APD at 50% repolarization (APD50) in absence (control) and presence of 10 μmol/L metoprolol. H, Mean values of APD at 90% repolarization (APD90) in absence (control) and presence of 10 μmol/L metoprolol. I, Representative traces of action potentials in absence (control) and presence of 10 μmol/L metoprolol. Values given are mean±SEM. n, number of cells. ns, P>0.05.