

Abstract
Background
Although advanced heart failure (HF) is a clinically documented risk factor for vascular cognitive impairment, the occurrence and pathomechanisms of vascular cognitive impairment in early stages of HF are equivocal. Here, we characterize vascular cognitive impairment in the early stages of HF development and assess whether cerebral hypoperfusion or prothrombotic conditions are involved.
Methods and Results
Tgαq*44 mice with slowly developing isolated HF triggered by cardiomyocyte‐specific overexpression of G‐αq*44 protein were studied before the end‐stage HF, at the ages of 3, 6, and 10 months: before left ventricle dysfunction; at the stage of early left ventricle diastolic dysfunction (with preserved ejection fraction); and left ventricle diastolic/systolic dysfunction, respectively. In 6‐ to 10‐month‐old but not in 3‐month‐old Tgαq*44 mice, behavioral and cognitive impairment was identified with compromised blood‐brain barrier permeability, most significantly in brain cortex, that was associated with myelin sheet loss and changes in astrocytes and microglia. Brain endothelial cells displayed increased E‐selectin immunoreactivity, which was accompanied by increased amyloid‐β1‐42 accumulation in piriform cortex and increased cortical oxidative stress (8‐OHdG immunoreactivity). Resting cerebral blood flow measured by magnetic resonance imaging in vivo was preserved, but ex vivo NO‐dependent cortical arteriole flow regulation was impaired. Platelet hyperreactivity was present in 3‐ to 10‐month‐old Tgαq*44 mice, but it was not associated with increased platelet‐dependent thrombogenicity.
Conclusions
We report for the first time that vascular cognitive impairment is already present in the early stage of HF development, even before left ventricle systolic dysfunction. The underlying pathomechanism, independent of brain hypoperfusion, involves preceding platelet hyperreactivity and brain endothelium inflammatory activation.
Keywords: blood‐brain barrier, cognitive impairment, endothelium, heart failure, platelet
Subject Categories: Cognitive Impairment, Blood-Brain Barrier, Animal Models of Human Disease, Endothelium/Vascular Type/Nitric Oxide, Heart Failure
Clinical Perspective
What Is New?
Vascular cognitive impairment (VCI) occurs in the early stage of heart pathology, before the development of systolic impairment of left ventricle function.
Underlying pathology of VCI at the early stage of heart failure development is independent of pathomechanisms of VCI identified in advanced heart failure such as brain hypoperfusion or prothrombotic state.
Underlying pathomechanisms of VCI in early heart pathology involve preceding platelet hyperreactivity and subsequent brain endothelium inflammatory activation that result in blood‐brain barrier leakage, cortical oxidative stress, and β‐amyloid cortical accumulation as well as impairment of endothelial NO‐dependent regulation of vascular tone in cortical arterioles.
What Are the Clinical Implications?
VCI development at the preclinical stage of heart failure is important in the clinical setting because it potentially enables identification and early treatment of a large population of patients at risk of VCI.
The platelet hyperreactivity that precedes VCI symptoms suggests a potential role of platelets in VCI treatment.
Impairment of major brain endothelium functions (permeability, blood flow regulation, inflammatory activation) in heart failure–induced VCI appears to be critical for patients with cognitive impairment of vascular origin.
Disability related to cognitive impairment and dementia is recognized as one of the greatest social and economic challenges of the 21st century worldwide. It is estimated that in 2010, over 35 million people lived with dementia, and due to aging of the population, this number is projected to grow to over 115 million in 2050.1 Alzheimer disease is the most common form of dementia, the second (>20% of cases) being represented by vascular dementia—the most severe form of vascular cognitive impairment (VCI). In the majority of older patients, VCI coexists with and accelerates the onset of Alzheimer disease, and this mixed vascular and neurodegenerative form of dementia was recently identified as the primary cause of age‐related cognitive impairment.2, 3
Heart failure (HF) is one of the leading causes of morbidity and mortality in developed countries.4 It is a progressive and multifactorial condition leading to functional impairment of ventricular diastolic and/or systolic function, which is differentiated based on left ventricle (LV) ejection fraction (EF). In the advanced stage around half of patients suffer from diastolic failure—HF with preserved EF (HFpEF)—and the remaining half suffers from systolic failure—HF with reduced EF (HFrEF). Based on population studies, HF was recognized as contributing to the development of cognitive impairment and dementia.5, 6, 7, 8
Clinical results show correlation between features of HF and functional or structural brain pathology.5, 9, 10 The majority of these findings relate to HFrEF patients with an advanced stage of HF with insufficient systolic heart function. Based on indirect evidence from clinical trials in which cognitive function was improved after pharmacological compensation of HF,11 improved adherence to treatment12 and successful cardiac resynchronization therapy13 or left ventricular assist device implantation,14 2 major pathomechanisms of cognitive impairment associated with HF were identified. These mechanisms are cerebral hypoperfusion and/or microemboli originating from the left heart.15 Although there are reports on development of cognitive impairment in preclinical stages of HF8 as well as in HFpEF patients,6, 16 underlying pathologies are yet to be identified. Furthermore, pathomechanisms of VCI at preclinical stages of HF and HFpEF seem to differ from pathomechanisms of VCI at HFrEF. Indeed, the treatment successful for HFrEF patients failed to demonstrate improvement either in cognition or in HF progression in HFpEF patients.17, 18
Genetically modified animal models are optimal for studying the role of isolated pathomechanisms in the development of clinically heterogeneous diseases. In VCI studies, genetic models of HF have not yet been implemented.19 So far, experimental studies aimed at mimicking pathology present in HFrEF patients (advanced systolic failure) have employed mouse models in which HF or HF‐pathology was induced surgically or pharmacologically.19, 20 Mice in these models develop VCI pathology due to global brain hypoperfusion and global reduction in cerebral blood flow (CBF). In turn, in humans, before the development of advanced HF as well as in HFpEF, global CBF remains unaltered in slowly progressing heart deterioration.21, 22
In this study we assessed (1) whether slowly progressing heart pathology—before the development of LV systolic dysfunction and end‐stage HF—leads to VCI, and (2) what roles the 2 previously proposed major cardiac pathomechanisms—global cerebral hypoperfusion and prothrombotic condition—play in VCI development. We employed Tgαq*44 mice in which HF develops due to slowly progressing cardiomyopathy, which mimics human HF on a molecular, morphological, phenotypic, and functional level.23, 24, 25, 26, 27, 28, 29, 30, 31 Their pathology is triggered by postnatal activation of a cardiomyocyte‐specific transgene, causing agonist‐independent, constitutive activation of the Gαq pathway, present only in cardiomyocytes.23, 32 This genetic modification mimics constant neurohormonal overstimulation of cardiomyocytes by renin–angiotensin‐aldosterone, sympathetic, and endothelin‐1–dependent systems, mediated by angiotensin AT1, adrenergic α1, and endothelin ET‐A receptor stimulation, respectively.23, 24, 25
We ascertained that VCI occurs in the early stage of heart pathology and before the development of systolic impairment of LV function; hence, before the end‐stage HF. Our results suggest that, in Tgαq*44 mice at early stages of HF development, a pathomechanism leading to brain endothelial dysfunction and dysregulation of neurovascular coupling occurs before the development of LV systolic dysfunction and is not due to brain hypoperfusion or a prothrombotic state but instead to brain endothelium pathology preceded by platelet hyperreactivity. The brain endothelial dysfunction was evidenced by inflammatory activation, blood‐brain barrier (BBB) permeability, oxidative stress, and β‐amyloid cortical accumulation as well as impairment of endothelial NO‐dependent regulation of vascular tone in cortical arterioles.
Methods
The data, analytic methods, and study materials will be made available on request to other researchers for purposes of reproducing the results or replicating the procedure.
Animals
The animal protocol was reviewed and approved by the Local Ethical Committee for Jagiellonian University; hence, all experimental procedures were in accordance with institutional guidelines. Tgαq*44 mice, generated and genotyped as described previously,24 as well as wild‐type control mice (FVB) were bred at the Animal Laboratory of the Institute of Experimental and Clinical Medicine of the Polish Academy of Sciences in Warsaw. Mice were housed in pathogen‐free conditions, fed a standard laboratory diet, and given water ad libitum. Due to a higher level of aggression in aging males then females, which could influence behavior and cognitive function, all experiments were performed on female Tgαq*44 and age‐matched female FVB mice at the ages of 3, 6, and 10 months. Each experimental age group was studied on a separate group of mice: 3‐month FVB n=24, 6‐month FVB n=28, 10‐month FVB n=50, 3‐month Tgαq*44 n=25, 6‐month Tgαq*44 n=31, and 10‐month Tgαq*44 n=51. In Tgαq*44 mice, systolic and diastolic heart function deteriorates over time and becomes significantly impaired at the age of 14 to 16 months, which represents the end stage of HF in this model.24, 25, 26, 27, 28, 29 It is then characterized not only by severe impairment of systolic and diastolic cardiac function but also by impaired dobutamine reserve.30 Therefore, Tgαq*44 mice at the ages of 3, 6, and 10 months represent relatively early stages of HF development. At the age of 4 months, Tgαq*44 mice have preserved global cardiac function (EF=63%, cardiac output [CO]=15 mL/min, preserved high‐dose dobutamine response) and show no functional difference from age‐matched control FVB mice; hence, younger‐than‐3‐month Tgαq*44 mice represent a functionally normal heart.28, 29, 31 At the age of 6 months, Tgαq*44 mouse heart function parameters are lower (EF=59%, CO=14.4 mL/min, preserved high‐dose dobutamine response), but global cardiac function is still fully maintained. Yet, at this stage we detect changes characteristic for atrial pathology—lower atrial late flow velocity and subtle diastolic dysfunction of LV—detectable only as decreased myocardial radial strain but not as hemodynamic parameter changes (EF/CO).29, 31 At 10 months, global cardiac performance is decreased (ejection rate, fraction area change).26, 27, 31 Therefore, Tgαq*44 mice at the ages of 3, 6, and 10 months represent sequential stages of heart failure development: normal heart, early diastolic, and systolic/diastolic cardiac dysfunction, respectively. Furthermore, because there is significant deterioration of heart function parameters from 6 to 14 months, but transition to overt heart failure occurs in 8‐ to 12‐month‐old Tgαq*44 mice (EF=54%, CO=13 mL/min, to EF=46%, CO=10 mL/min, respectively) with the critical period between the ages of 10 and 12 months,33 we claim that Tgαq*44 mice at the age of 10 months still represent a relatively early stage of HF.31
Behavioral Tests
Behavioral tests were conducted between 9 am and 2 pm in constant white light. Before each test, all elements were cleaned with alcohol to remove olfactory clues. Tests were video recorded for subsequent analysis.
The open field test (OFT), adapted from Schiavone et al,34 was carried out in a circular arena (60 cm diameter) divided into 6 equal fields. The test was conducted 1 day after habituation (6 minutes in the arena). To perform the test, animals were placed in the arena for 6 minutes, and a trained and blinded observer counted the number of fields crossed, which led to the test result.
The novel object recognition (NOR) test was adapted from Giovannini et al.35 Animals were tested in a plastic box (28×40×22 cm). On the first day (adaptation), animals were allowed to explore the open field for 10 minutes. The next day, animals were placed in the box (training) to familiarize them with 2 identical objects for 10 minutes. After 60 minutes, animals were placed in the box again (choice trial). In the choice trial, 1 of the familiar objects was replaced with a novel object and animals were left to explore the box for 10 minutes. A trained, blinded observer measured the time spent on exploration of each object (ie, touching, sniffing, sitting in close proximity with nose directed to the object). The NOR index was calculated using the following formula: (N−F)/(N+F) (where N=time spent on exploration of the novel object during the choice trial; F=time spent on exploration of the familiar object in the choice trial).35
Immunohistochemistry
Mice were anesthetized (100 mg/kg ketamine+10 mg/kg xylazine IP) and transcardially perfused with 0.01 mol/L phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde in PBS. The brains were removed, postfixed in paraformaldehyde overnight, transferred to 30% sucrose in 0.01 PBS, and kept in a refrigerator for 3 to 5 days. The right hemisphere of each brain was then frozen in −80°C, and sections were cut at 40 μm in the coronal plane with a freezing microtome. The left hemisphere of each brain was paraffin embedded, and sections were cut at 7 μm in the coronal plane with a microtome for processing on a light/fluorescence microscope.
Sections from frozen brain were blocked with NDS (normal donkey serum). After the wash, sections were incubated in 0.1% Triton X‐100 in PBS (PBST) for 15 minutes. Then, after preincubation in NDS‐buffer (5% NDS in PBST) for 1 hour, the following antibodies (Ab) were added (all from Abcam, Cambridge, UK): rabbit anti‐BMP (1:1000), goat anti‐Iba‐1 (1:500), chicken anti‐GFAP (1:800) for overnight incubation at 4°C. After washing cycles in PBST, sections were incubated for 2 hours at room temperature with secondary antibodies (Jackson ImmunoResearch, West Grove, PA): Cy3 to detect BMP, 488 Dylight to detect Iba‐1, and 647 Alexa Fluor to detect GFAP, all at 1:400. After being washed in PBST, sections were incubated in Hoechst blue/PBS (Invitrogen, Carlsbad, CA) at a concentration of 20 μg/mL for 15 minutes and rinsed in PBST. BMP/Iba‐1/GFAP/Hoechst stain was acquired with a scanning confocal microscope (Leica TCS SP 8, Wetzlar, Germany). For each mouse, 1 z‐stack from 1 slide stained with 4 dyes was collected for each of the studied brain regions: prefrontal cortex (2.10 mm anterior to bregma), anterior commissure (1.10 mm anterior to bregma), lateral ventricle and striatum (0.02 mm anterior to bregma), corpus callosum and cingulum (0.94 mm posterior to bregma), DG, CA3, and CA1 (1.94 mm posterior to bregma). The z‐stacks were composed of 1‐μm‐thick optical sections for all regions and were collected using a ×20 oil objective on a Leica TCS SP 8 confocal microscope using a 470‐ to 670‐nm white light laser, 488‐nm argon laser, and 405‐nm diode laser. The pinhole, photomultiplier, and hybrid detectors’ gain and contrast settings were kept constant for all image stacks acquired from all animals.
Paraffinized 7‐μm‐thick sections from the region of primary and secondary motor cortex as well as piriform cortex (0.96 mm anterior to bregma) were automatically deparaffinized through graded alcohols, subjected to heat‐induced epitope retrieval for 15 minutes in 1:100 citrate buffer (10 nmol/L, pH 6.0), and incubated for 1 hour at 4°C in PBST buffer containing single antibodies raised against the following: rabbit anti β‐amyloid1‐42 (BA 1‐42; 1:50; Millipore, Billerica, MA), rat anti E‐selectin (CD62‐E; 1:50, BD, Franklin Lakes, NJ), and mouse anti 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG, 1:10, JaICA, Shizuoka, Japan). After washing cycles, sections were incubated for 30 minutes at room temperature with secondary and/or tertiary Ab: Cy3 to detect BA 1‐42 (1:800, Jackson ImmunoResearch), first with streptavidin (1:600) and then with Cy3 to detect CD62‐E (1:800, Jackson ImmunoResearch). 8‐OHdG staining was visualized employing DAB (3,3′‐diaminobenzidinetetrahydrochloride hydrate) according to the manufacturer's protocol, which involved prestain blockage with 0.1% peroxide. BA 1‐42 and CD62‐E stains were acquired using a ×40 objective on an AxioObserver.D1 inverted fluorescent microscope (Zeiss, Oberkochen, Germany). 8‐OHdG stain was acquired with an automated Olympus BX51 light microscope with a ×40 objective. The photomultiplier and contrast settings were kept constant for all images acquired from all animals. ImageJ (National Institutes of Health, Bethesda, MD) was used to normalize and semiquantitatively analyze all images. The GFAP, Iba‐1, and MBP immunoreactivities in the selected brain regions were analyzed as the percentage of area occupied by the signal. Additionally, MBP immunoreactivity in anterior commissure was analyzed as a texture pattern.
Detailed characteristics of antibodies can be found in Table S1, region of interest (ROI) selection in Figures S1 and S2 and analyses in Data S1.
In Vivo BBB Permeability
Anesthetized mice (100 mg/kg ketamine+10 mg/kg xylazine, IP) were injected (femoral vein) with 4 mL/kg of a solution comprising Evans Blue (EB, 70 kDa, Sigma Aldrich, St. Louis, MO) and sodium fluorescein (NaF 0.45 kDa, Sigma Aldrich) and saline (2% EB and 2% NaF in 0.9% saline). The dye mixture was allowed to circulate for 10 minutes, before simultaneous perfusion of left (systemic circulation) and right ventricles (pulmonary circulation) with PBS for 15 minutes. Lungs and brain were isolated. Cortex, hippocampus, cerebellum, and pons were further isolated from each brain. Isolated brain structures and lungs were dry‐weighed (for result normalization) and homogenized in 200 μL of 50% TCA (dissolved in distillated water). Homogenate was frozen and kept at −20°C for subsequent dye concentration measurements. Thawed homogenates were centrifuged (at 10625 g for 12 minutes at 4°C), collected supernatant was diluted with 1:3 volumes of 95% ethanol before photospectrometric (Synergy 4, Bio‐Tek, Winooski, VT) determination of EB concentration (fluorescence: excitation at 590 nm, emission at 645 nm; absorbance at 620 nm) and NaF concentration (fluorescence: excitation at 460 nm, emission at 515 nm).
In Vivo CBF Imaging
CBF was measured using magnetic resonance imaging on a 9.4 T/21 cm (BioSpec 94/20 USR, Bruker, Ettlingen, Germany) horizontal scanner equipped with a 39 mm diameter 1H quadrature transmit/receive radio‐frequency volume coil.
Animals were anesthetized with 2% isoflurane in a mixture of air and oxygen delivered via a nose cone and injected IP with 400 μL of saline to avoid dehydration. Throughout the experiment lasting 1.5 hours, animals were in a prone position, and their temperature was maintained at 37°C. Electrocardiography, respiration, and temperature were monitored with the Monitoring and Gating System (SA Inc, Stony Brook, NY).
Anatomical imaging was conducted with RARE (Rapid Acquisition with Relaxation Enhancement) and short echo time sequence before arterial spin labeling. The perfusion map was placed in the same plane as the anatomical image for each animal. RARE short echo time sequence parameters were echo time (TE)=5 ms, repetition time (TR)=3508 ms, number of averages (NA)=4, field of view (FOV)=20×20 mm, matrix size=128×128 pixels, rare factor=16, slice thickness=2 mm.
Arterial spin labeling imaging was conducted using the Flow‐Sensitive Alternative Inversion Recovery RARE pulse sequence with the following parameters: TE=4.642 ms, TR=18 000 ms, NA=6, FOV=20×20 mm, matrix size=128×128, rare factor=72, slice thickness=2 mm, selective inversion slice thickness=4 mm, and 15 inversion recovery times from 30 to 8000 ms. Fat suppression was used with a frequency‐selective 90‐degree pulse. A 20×20 mm field of view and 128×128 pixel image matrix resulted in 0.15625×0.15625 mm/pixel spatial resolution of perfusion images. The total arterial spin labeling measurement time was ≈50 minutes.
Perfusion maps were created using ASL Perfusion Macro software (ParaVision 5.1, Bruker, Ettlingen, Germany) and further processed with ImageJ software (NIH, Bethesda, MD). The threshold was applied to the perfusion maps to reject false values (higher than 300 mL/min per 100 g or negative). As ROI, cortex, hippocampus, and thalamus were chosen based on anatomical images (Figure S3) acquired using RARE short echo time sequence. CBF (mL/min per 100 g) for each ROI was expressed as median value of analyzed ROI perfusion.
Endothelial Function in Isolated Intracerebral Arterioles
Mice were euthanized by cervical dislocation. Subsequently, isolation of intracerebral arterioles branching from the middle cerebral artery was performed under a stereomicroscope using Dumant forceps. The specimens were transferred in a pipette to a thermostated chamber (Warner Instruments, Hamden, CT) and mounted in a Luigs and Neumann perfusion system (Ratigen, Germany). Vessels were aspirated into a holding pipette (40‐45 μm) and cannulated with a perfusion pipette (20‐22 μm). The driving pressure was increased until the vessel opened and perfusion was established. The perfusate consisted of PSS36 with 1% BSA in 5% CO2 in atmospheric air and was driven from a reservoir pressurized to 20 to 40 mm Hg. All experimental protocols started with a 30‐minute period of equilibration after the perfusion had been established (37°C), and during the equilibration vessels did not develop myogenic tone. Viability was tested by administration of a high KCl to the organ chamber (55 mmol/L), containing 10−5 mol/L phentolamine. Cumulative concentration response curves to U46619 (from 10−9 to 10−6 mol/L) were performed on each vessel. The concentration of U46619 at which the second highest contraction was observed was chosen as the submaximal concentration. This resulted in a submaximal concentration of U46619 between 10−7 and 10−6 mol/L. The concentration of U46619 with the submaximum effect was used to measure arteriole constriction with and without L‐NAME (Nω‐nitro‐l‐arginine methyl ester) (10−4 mol/L). Furthermore, effects of a single dose of sodium nitroprusside (SNP) (10−5 mol/L) were tested on top of constriction to U46619 with and without L‐NAME. Last, contraction to high potassium was tested in the presence of L‐NAME.
Systemic Platelet Reactivity
Platelet reactivity was assessed according to a previously published protocol with modification.37 Briefly, subsequent to anesthesia, blood was collected on heparin from the right heart ventricle. Whole blood was diluted and either directly processed according to a “wash blood” protocol38 or incubated at 37°C for 60 minutes, with constant stirring in a Xyzyk apparatus (Xyzyk Company, Krakow, Poland) and then processed according to the “wash blood” protocol. Washed blood, both stirred and unstirred, was incubated (30 minutes, 37°C) with ADP and with 2 Ab panels (Table S1): first, FITC‐conjugated anti‐CD41/61 (platelets linage marker GpIIbIIIa) and PE‐conjugated anti‐CD41/61 active form (platelet activation marker GpIIbIIIa active form) Ab and second, with FITC‐conjugated anti‐CD41/61 and PE‐conjugated anti‐CD62P (platelet activation marker P‐selectin). Flow cytometry data (2 000 000 events per sample) were collected on an LSRII cytometer. The gating strategy, pictured in Figure S4, started with platelet identification based on GpIIbIIIa expression and forward scatter event size; then singlet platelets, selected from the forward‐scatter‐H versus the forward‐scatter‐W plots, were studied for GpIIbIIIa active form and P‐selectin surface expression. Results were expressed as the percentage of platelets positive for activation marker and as median fluorescent intensity of activation marker. Raw data were compensated and analyzed using FlowJo software version 9.2 for Mac (Tree Star, San Carlos, CA). Abs were titrated to achieve optimal resolution, and gates for GpIIbIIIa active form and P‐selectin were set using the “fluorescence minus one” method.39
Ex Vivo Platelet‐Dependent Thrombus Formation
Platelet thrombus formation under arterial flow conditions was measured quantitatively ex vivo with a microchip‐based flow‐chamber system T‐TAS (Total Thrombus‐formation Analysis System; Fujimori Kogyo Co Ltd, Tokyo, Japan). Subsequent to anesthesia (100 mg/kg ketamine+10 mg/kg xylazine, IP), whole blood was sampled via cardiac puncture, immediately anticoagulated with hirudin (525 ATU hirudin/mL blood), and left to rest for 1 hour. Platelet count was measured in duplicate by a hematology analyzer (ABC Vet, Horiba, Germany). Before the measurement, blood was gently mixed, 300 μL was applied to T‐TAS and perfused over a PL microchip coated with collagen at a flow rate of 12 μL/min. Development of thrombus was measured with 3 parameters: time to onset of thrombus formation (time required to reach the pressure of 10 kPa in the microcapillary; seconds), occlusion time (time required to reach the pressure 60 kPa in the microcapillary; seconds), and area under the flow‐pressure curve for 10 minutes after the start of the assay; this parameter characterizes thrombus stability and hence defines total thrombogenicity (PL12‐AUC10, less than 60 kPa).40
Statistical Analysis
The data were analyzed using R version 3.3.1. The Shapiro test was used to assess for the normality of the data. Two groups with normally distributed data were compared using a 2‐tailed Student t‐test; data from relative CBF and from arteriole perfusion experiments were analyzed as paired (Paired Student t‐test). Two groups with nonnormally distributed data were analyzed with a Mann‐Whitney test. ANOVA and Tukey test were used to perform multiple group comparisons. Box plots were used to show data distribution (median, Q1, Q3, interquartile range, and outliers). Probability values (P) of less than 0.05 were considered statistically significant.
Results
Behavioral and Cognitive Impairment in the Early Stage of HF Development
To address the possible role of early‐stage HF in the development of cognitive impairment, we employed OFT and NOR tests to compare the cognitive function of Tgαq*44 mice versus age‐matched FVB control mice at 3, 6, and 10 months (n=8 per each group). OFT measured locomotor and behavioral activity, whereas NOR assessed complex cognition independent of motor activity. In OFT, activity of Tgαq*44 mice at 6 and 10 months was reduced by 48.2% (P=0.002) and 60.2% (P=0.002), respectively, compared to age‐matched controls (Figure 1A). The NOR index at 6 months was lower by 62.2% (P=0.003) and at 10 months by 65.9% (P=0.04) in Tgαq*44 mice compared to age‐matched controls (Figure 1B). Importantly, there was no difference between performance of 3‐month‐old Tgαq*44 mice and control mice at all ages (FVB at 3, 6, and 10 months) in OFT (P=0.996) and NOR (P=0.93) tests.
Figure 1.
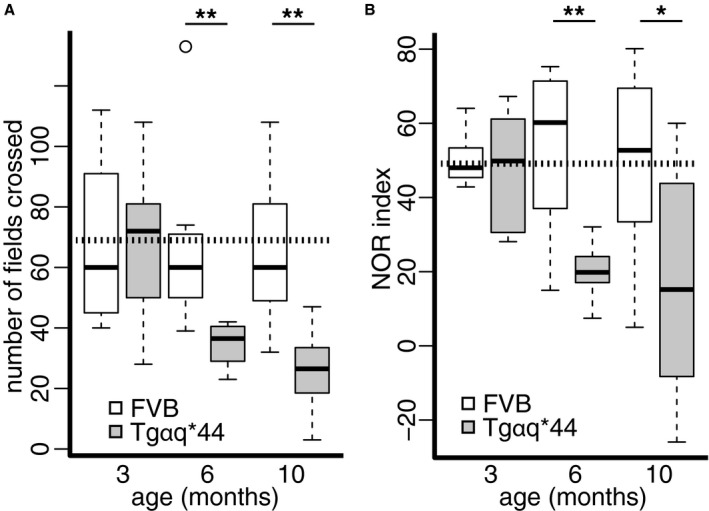
Development of behavioral and cognitive impairment in early stages of heart failure in Tgαq*44 mice. Tgαq*44 and FVB (wild‐type control) mice at 3, 6, and 10 months of age (n=8) were studied with (A) open field test and (B) novel object recognition (NOR) test. Data distribution is presented as box plots (median, Q1, Q3, interquartile range, and outliers); *P<0.05; **P<0.005; horizontal dashed line represents mean value for all FVB. Q1, Q3 indicate 25th and 75th percentiles, respectively.
Changes in Brain Nervous Tissue Associated With Cognitive Impairment in the Early Stage of HF Development
Because brain nervous tissue plays a crucial role in cognitive processes, we assessed whether structural changes in its components, myelin and glia, correspond with identified cognitive deficits.41 Brains of Tgαq*44 mice at the age of 6 and 10 months (n=6) and brains of age‐matched FVB mice (n=4) were multiply stained for DAPI (nucleus) and for IBA‐1, GFAP, and MBP proteins specific for microglia, astroglia, and axonal myelin sheath, respectively. Analysis of gray matter revealed that brains from 6‐month‐old Tgαq*44 mice had a reduced area occupied by IBA‐1–immunopositive signal (microglia) by 70% in the prefrontal cortex (P=0.047) as compared to FVB brains. In both groups of Tgαq*44 and FVB mice, microglia had a rather similar ramified appearance. Comparison of white matter structures between Tgαq*44 and FVB mice at 6 and 10 months of age showed decreased area occupied by fluorescence signal from microglia in both the anterior commissure (by 44%, P=0.0006 and by 52%, P=0.008, respectively, for age group) and the corpus callosum (by 27%, P=0.047 and by 52%, P=0.007, respectively, for age group). At the age of 6 months, Tgαq*44 versus FVB mice had a 56% reduced area of GFAP immunoreactivity (astrocytes, P=0.006) in the anterior commissure and a 32% reduced area occupied by MBP immunoreactivity (myelin sheath, P=0.009) in the cingulum. Furthermore, the texture of MBP‐immunopositive fibers in the anterior commissure was significantly different between Tgαq*44 and FVB brains at the ages of 6 (P=0.001) and 10 months (P=0.02). The area occupied by GFAP immunoreactivity of astrocytes adjoining the lateral ventricle walls was reduced by 38% in 6‐month‐old Tgαq*44 mice as compared to FVB mice, whereas in 10‐month‐old Tgαq*44 mice the percentage of GFAP immunoreactivity was increased 2‐fold compared to 6‐month‐old Tgαq*44 mice (P=0.008) and showed a tendency to be stronger compared to age‐matched FVB mice (P=0.09). Results are shown in Figure 2, Table, and Table S2.
Figure 2.
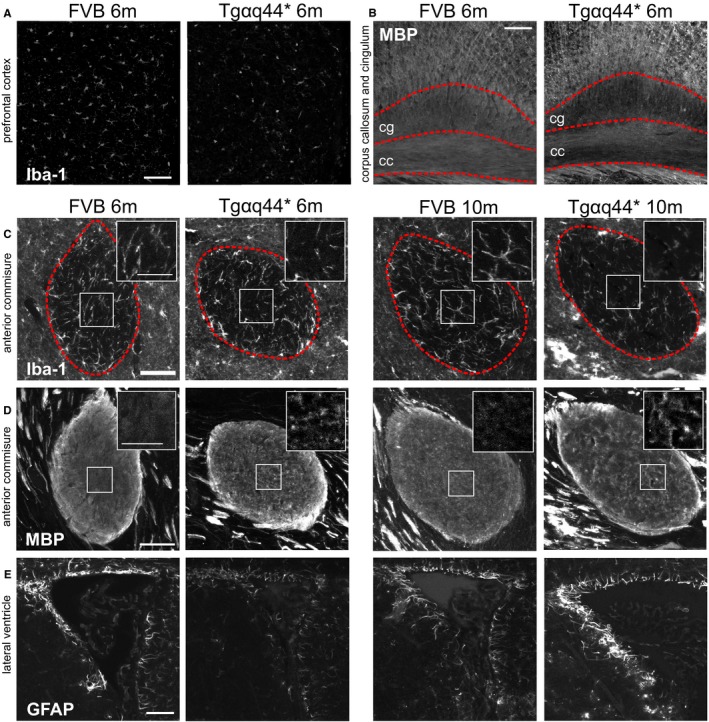
Representative microphotographs of brain demonstrating changes in neuroglia and white matter in early stages of heart failure in Tgαq*44 (n=6) as compared to FVB (n=4) mice. A, Decreased IBA‐1 (microglia) immunoreactivity in prefrontal cortex in Tgαq*44 compared to FVB (wild‐type control) mice; (B) decreased MBP (myelin) immunoreactivity in cingulum (cg‐dotted area) in Tgαq*44 compared to FVB mice; (C) decreased IBA‐1 immunoreactivity in anterior commissure (within dashed area) in Tgαq*44 compared to FVB mice and inserted magnification of square area in upper right; (D) changes in myelin fiber texture in anterior commissure in Tgαq*44 compared to FVB mice and inserted magnification of square area in upper right; (E) a decrease and an increase of GFAP (astrocyte) immunoreactivity in periventricular area in 6‐ and 10‐month‐old Tgαq*44 mice, respectively, compared to FVB mice; scale bar for large microphotographs indicates 50 μm, for inserted magnifications 25 μm; m, animal age in months; cg, cingulum; cc, corpus callosum.
Table 1.
Microglia‐, Astroglia‐, and Myelin‐Specific Protein Immunoreactivity in Selected Brain Regions in Tgαq*44 (n=6) and FVB (n=6) Mice at 6 and 10 Months of Age
| Prefrontal Cortex | Dentate Gyrus | Anterior Commissure | Corpus Callosum | Cingulum | Lateral Ventricle | |
|---|---|---|---|---|---|---|
| IBA‐1 immunoreactivity for microglia, mean (SD)/median (IQR)* | ||||||
| FVB 6 m | 4.7 (2.15) | 1.27 (0.22) | 5.76 (0.18) | 2.09 (0.31)* | 1.23 (0.33) | N/A |
| Tgαq*44 6 m | 1.41 (1.03)† | 1.15 (0.49) | 3.81 (0.7)† | 1.67 (0.32)† | 1.41 (0.26) | N/A |
| FVB 10 m | 3.49 (1.09) | 1.19 (0.8) | 5.62 (1.16) | 2.62 (0.55) | 1.83 (0.68) | N/A |
| Tgαq*44 10 m | 2.43 (1.23) | 0.86 (0.36) | 2.69 (0.71)‡ | 1.26 (0.54)† | 1.44 (0.32) | N/A |
| GFAP immunoreactivity for astroglia, mean (SD) | ||||||
| FVB 6 m | N/A | 9.16 (2.81) | 7.61 (1.62) | 6.2 (2.09) | 4.34 (1.3) | 25.58 (9.09) |
| Tgαq*44 6 m | N/A | 7.49 (2.24) | 3.35 (1.19)† | 3.32 (2.03) | 2.73 (1.35) | 15.89 (4.21)† |
| FVB 10 m | N/A | 6.07 (3.24) | 3.76 (1.9) | 3.58 (0.57) | 3.07 (1.09) | 20.94 (7.46) |
| Tgαq*44 10 m | N/A | 8.1 (1.22) | 4.18 (1.74) | 3.7 (1.55) | 3.31 (1.11) | 31.39 (9.52) |
| MBP immunoreactivity for myelin, mean (SD)/median (IQR)* | ||||||
| FVB 6 m | N/A | N/A | 0.19 (0.01)§ | 45.83 (9.2) | 56.7 (14.29)* | N/A |
| Tgαq*44 6 m | N/A | N/A | 0.12 (0.01)‡, § | 42.33 (5.8) | 40.43 (6.43)† | N/A |
| FVB 10 m | N/A | N/A | 0.1 (0.01)§ | 41.76 (10.12) | 40.67 (9.27) | N/A |
| Tgαq*44 10 m | N/A | N/A | 0.12 (0.01)†, § | 50.64 (9.42) | 32.81 (4.81) | N/A |
FVB indicates wild‐type control mice; IQR, interquartile range; m, months; N/A, measurement not applicable/assessed.
*Refers to nonnormally distributed data and represents median and interquartile range (IQR).
P‐value †<0.05, ‡<0.005.
Results represent percentage of signal‐positive area, except for §texture analysis (IDM parameter).
Impairment of BBB in Early Stage of HF Development
Selective permeability of the BBB is a critical function of the neurovascular unit, which structurally depends on intracellular tight junctions between brain microvascular endothelial cells.42 Dysfunction of the BBB is associated with development of cognitive impairment and structural changes in neuroglia.2 In vivo permeabilities of BBB and lung vessels were compared between Tgαq*44 (n=7 per group) and FVB mice (n=6 per group) at 3, 6, and 10 months of age. Tgαq*44 mice at 6 and 10 months of age had global impairment in BBB integrity as evidenced by increased BBB permeability in Tgαq*44 mice compared to age‐matched FVB controls in all studied brain structures (cortex, hippocampus, pons, and cerebellum), whereas lung vessel permeability was largely preserved (Figure 3, Figure S5). Similar to behavioral test results, BBB permeability in Tgαq*44 mice at the age of 3 months was fully preserved and comparable to the value in 3‐month‐old FVB control mice. A pathological increase in BBB permeability in Tgαq*44 mice was most significant in the cortex for both small (NaF, 0.45 kDa) and large (EB, 70 kDa) permeability markers, and it increased from 141% for NaF (P=0.006) and 150% for EB (P=0.02) to 156% for NaF (P=0.002) and 171% for EB (P=0.002) in 6‐ and 10‐month‐old Tgαq*44 mice, respectively, as compared to age‐matched FVB.
Figure 3.
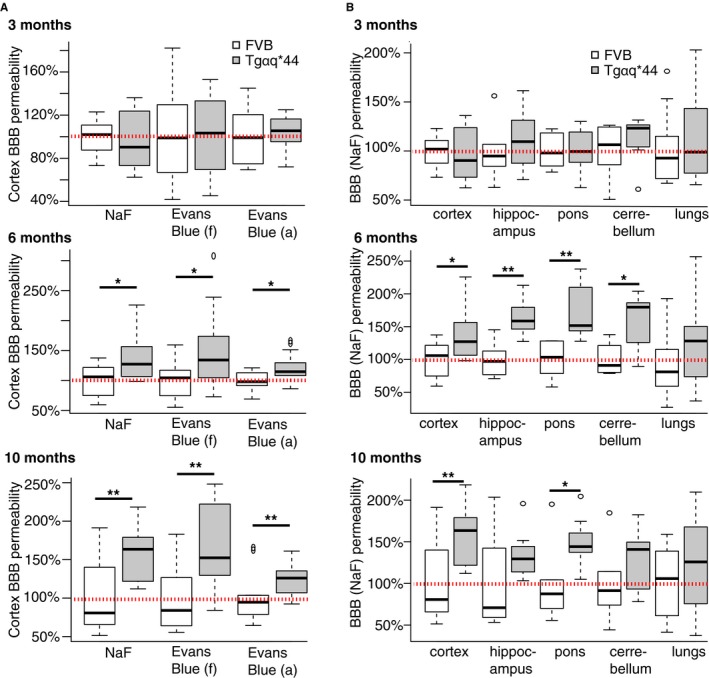
Development of changes in BBB permeability in early stages of heart failure in Tgαq*44 mice. Increase in relative (to age‐matched FVB mice) BBB permeability to (A) NaF and Evans Blue dyes in cortex of Tgαq*44 mice (n=7) at 6 and 10 months of age; and to (B) NaF in cortex, hippocampus, pons, and cerebellum but not in lungs in Tgαq*44 mice (n=7) at 6 and 10 months of age. Data distribution is presented as box plots (median, Q1, Q3, interquartile range, and outliers). Red dotted line indicates average permeability in FVB control mice (n=6); *P<0.05, **P<0.005; BBB indicates blood‐brain barrier; (a), based on absorbance; (f), based on fluorescence; Q1, Q3, 25th and 75th percentiles.
Progressive Cortical Brain Endothelial Cell Inflammation in Early Stage of HF Development
Brain endothelial cells comprise the vascular element of BBB, the dysfunction of which is caused by inflammation. Therefore, we measured cortical vessel immunoreactivity to E‐selectin, which as a protein is expressed almost exclusively on inflammatory activated endothelium.43 Frontal cortex sections of brains from Tgαq*44 mice at the ages of 6 and 10 months (n=4) and corresponding sections of age‐matched FVB mice (n=4) were stained for E‐selectin (CD62‐E). Analysis (pictured in detail in Figure S2) was based on 14 cortical vessels identified in slices from 10‐month‐old FVB mice and 9 cortical vessels identified in each of the remaining groups (each section represented 1 animal; an entire hemisphere cortex was scanned for vessels with lumen diameters >20 μm). Tgαq*44 mice at the age of 6 months had over 2‐fold higher (P=0.02) and at the age of 10 months almost 6‐fold higher (P<0.0001) E‐selectin immunoreactivity of the small cortical vessels compared to age‐matched FVB mice. Thus, there was a progressive ≈2‐fold increase in E‐selectin expression from 6‐ to 10‐month‐old Tgαq*44 mice (P=0.02) (Figure 4A).
Figure 4.
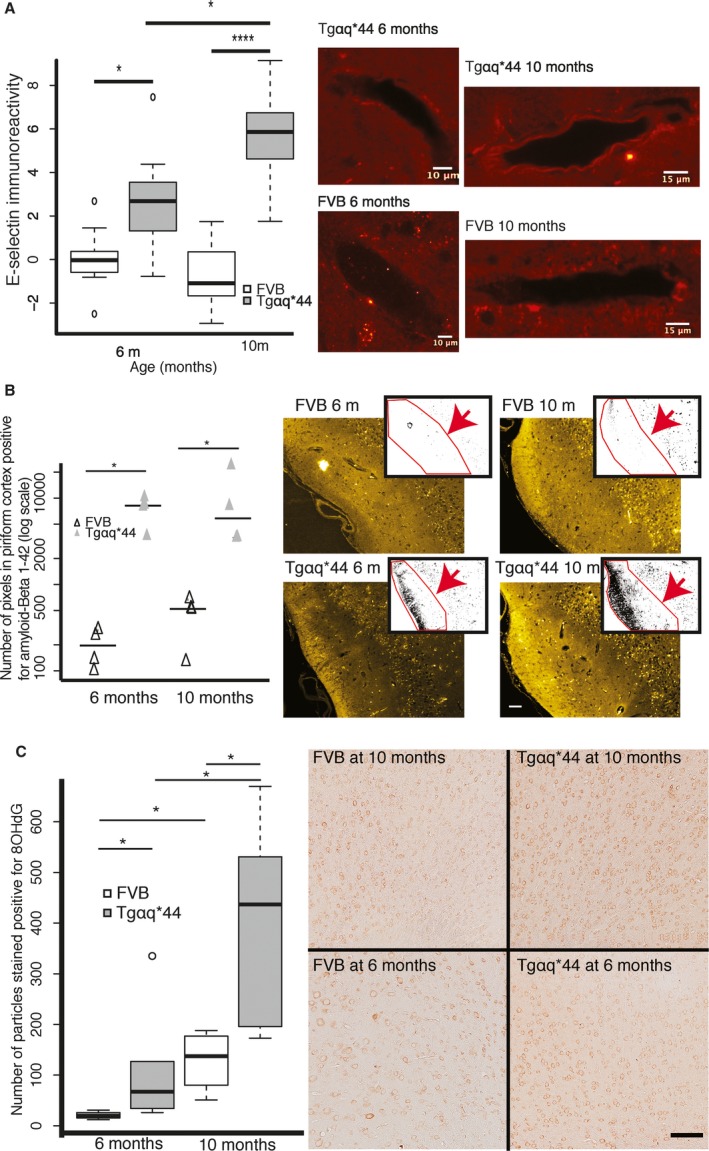
Endothelial inflammation of cortical vessel wall and related cortical pathology in early stage heart failure in Tgαq*44 mice. A, Progressive increase in E‐selectin immunoreactivity in cortical vessel endothelial cells in 6‐ and 10‐month‐old Tgαq*44 (n=4) vs FVB (n=4) mice; representative images illustrate E‐selectin immunoreactivity in cortical vessel walls; (B) increased accumulation of β‐amyloid1‐42 in 6‐ and 10‐month‐old Tgαq*44 (n=4) vs FVB (n=4) mice in piriform cortex; representative images (yellow) illustrate raw immunoreactivity of β‐amyloid1‐42 in piriform cortex, whereas overlapping images represent quantitative measure (black dots in ROI, pointed by red arrow) of β‐amyloid1‐42 immunoreactivity; scale bar indicates 50 μm; (C) progressive increase in cortical oxidative stress in 6‐ and 10‐month‐old Tgαq*44 (n=6) vs FVB (n=4) mice at 6 and 10 months; representative raw images illustrate 8‐OHdG signal intensity; scale bar indicates 100 μm; data distributions in (A) and (C) are presented as box plots (median, Q1, Q3, interquartile range, and outliers); in (B) each result and group median is shown. Q1 and Q3 indicate 25th and 75th percentiles, respectively; ROI, region of interest; *P<0.05, **P<0.005.
Increased Accumulation of β‐Amyloid1‐42 in Piriform Cortex and Increased Cortical Oxidant Stress in Early Stage of HF Development
To further verify whether brain vascular pathology is accompanied by brain parenchymal degeneration, we assessed 2 common cortical markers of neurodegeneration, oxidative stress and β‐amyloid1‐42 deposition,2 in Tgαq*44 mice at the ages of 6 and 10 months. Frontal cortex sections of Tgαq*44 mice and age‐matched FVB mice were stained either for β‐amyloid1‐42 (n=4 per group) or 8‐OHdG (FVB n=4 and Tgαq*44 n=6). Figure 4B illustrates the accumulation of pathologic β‐amyloid1‐42 in the piriform cortex of Tgαq*44 mice, which was 37‐fold higher in 6‐month‐old Tgαq*44 mice (P=0.01) and 21‐fold higher in 10‐month‐old Tgαq*44 mice (P=0.03) compared to age‐matched FVB mice (measured as the number of pixels with signal above background within each ROI). Figure 4C illustrates an over 2‐fold increase in oxidative stress in Tgαq*44 mice compared to FVB mice at 6 (P=0.02) and at 10 months of age (P=0.01) as well as a general age‐related increase in β‐amyloid1‐42 deposition from 6 to 10 months of age in both Tgαq*44 (P=0.01) and in FVB mice (P=0.04).
Lack of Impairment of Resting CBF in Early Stage of HF Development
Brain hypoperfusion is 1 of 2 major mechanisms underlying development of cognitive impairment.44 Therefore, we compared resting CBF in the cortex, hippocampus, and thalamus in 10‐month‐old Tgαq*44 (n=6) and FVB mice (n=7) (Figure 5A) using in vivo magnetic resonance imaging. CBF was not different between Tgαq*44 and FVB mice in the cortex (mean flow 101.3 versus 105.4 mL/min per 100 g; 95% confidence interval [CI] −27.1 to 35.4, respectively), hippocampus (mean flow 124.7 versus 110.4 mL/min per 100 g; 95% CI −58 to 29.4, respectively), and thalamus (mean flow 136.2 versus 133.8 mL/min per 100 g; 95% CI −44.7 to 39.9, respectively) (Figure 5B). After pooling CBF results from Tgαq*44 and FVB mice, the difference between absolute CBF in the thalamus (mean flow 134.9 mL/min per 100 g) and cortex (mean flow 103.5 mL/min per 100 g) was borderline significant (P=0.07; 95% CI −2.88 to 65.73). To further assess whether blood was distributed equally among the studied structures, the cortex and hippocampus CBFs were expressed as relative to CBF in the thalamus (ie, relative cortex CBF equals thalamus CBF minus cortex CBF) in each animal. Relative CBF in the cortex and in the hippocampus did not differ between Tgαq*44 and FVB mice (Figure 5C). A comparison of relative CBF in Tgαq*44 mice revealed significantly (P=0.02; 95% CI −42.9 to −3.9) lower CBF in the cortex (relative mean flow −34.9 mL/min per 100 g) compared to CBF in the hippocampus (relative mean flow −11.5 mL/min per 100 g).
Figure 5.
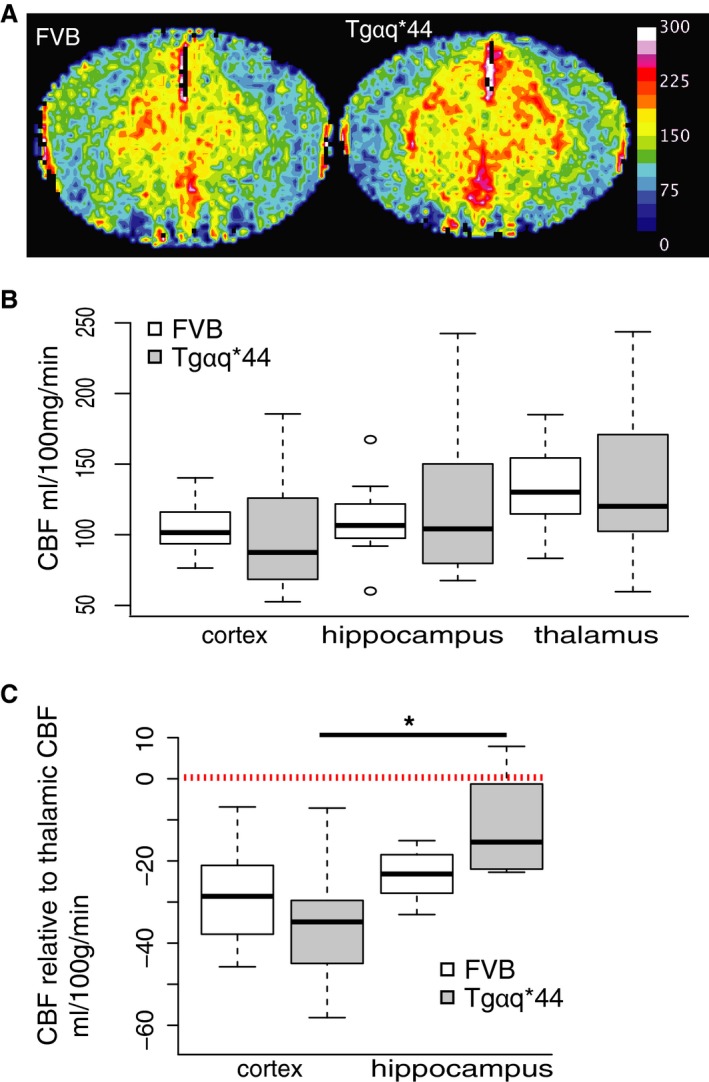
Preserved CBF in early stages of HF in Tgαq*44 mice. A, Representative cerebral blood perfusion maps of brains of 10‐month‐old Tgαq*44 and FVB mice; color bar represents blood perfusion in mL/100 mg per minute. B, CBF in cortex, hippocampus, and thalamus does not differ between Tgαq*44 (n=6) and FVB (n=7) mice; (C) CBF in cortex and hippocampus expressed as relative to thalamic CBF does not differ between Tgαq*44 (n=6) and FVB (n=7) mice; relative cortical CBF is lower than relative hippocampal CBF in Tgαq*44 mice; data distribution is presented as box plots (median, Q1, Q3, interquartile range, and outliers); dashed line indicates average thalamic CBF. CBF indicates cerebral blood flow; HF, heart failure; Q1 and Q3 indicate 25th and 75th percentiles, respectively; *P<0.05.
Impairment of NO‐Dependent Regulation of Vascular Tone in Isolated and Perfused Cortical Arterioles in the Early Stage of HF Development
To assess whether HF pathology leads to local impairment of brain arteriole autoregulation, which is often present in advanced VCI,19, 45 we assessed NO‐dependent endothelial function in isolated, perfused cerebral arterioles. Contraction of isolated brain arterioles (40‐50 μm in diameter) from Tgαq*44 (n=6) and FVB (n=7) mice at the age of 10 months was measured ex vivo in the isolated cannulated arterioles. There was no significant difference between cumulative vasoconstrictor response curves to thromboxane A2 receptor agonist U46619 (Figure 6A). Arteriole contraction induced by U46619 was significantly increased in the presence of L‐NAME (a nonspecific NOS inhibitor) in FVB mice only (mean difference 8.68%; P=0.03; 95% CI 1.3‐16.05), whereas Tgαq*44 mice were unresponsive to L‐NAME (mean difference −1.4%; P=0.75; 95% CI −12.1 to 9.3) (Figure 6B). To assess whether NO‐dependent pathology in Tgαq*44 mice is caused by endothelial and/or vascular muscle dysfunction, we tested vascular muscle response to SNP (NO donor). After precontraction of arterioles with U46619, a significant vascular muscle‐dependent relaxation response to SNP in the presence of L‐NAME was recorded in both Tgαq*44 (mean difference 15.3%; P=0.02; 95% CI 3.6‐27.1) and FVB mice (mean difference 15.5%; P=0.03; 95% CI 1.7‐29.2). Furthermore, arteriole dilation was significantly greater in the presence than in the absence of L‐NAME for both Tgαq*44 (mean difference 9.9%; P=0.004; 95% CI 4.7‐15.1) and FVB mice (mean difference 16.7%; P=0.01; 95% CI 5.2‐28.1) (Figure 6C). An impaired effect of L‐NAME on U46619‐induced vasoconstriction in Tgαq*44 mice was observed, but a comparable response to SNP in Tgαq*44 and FVB mice suggests an endothelial NO deficiency in Tgαq*44 mice. On the other hand, there was no difference in arteriole contraction and secondary dilation induced by high potassium before and after addition of L‐NAME (Figure S6).
Figure 6.
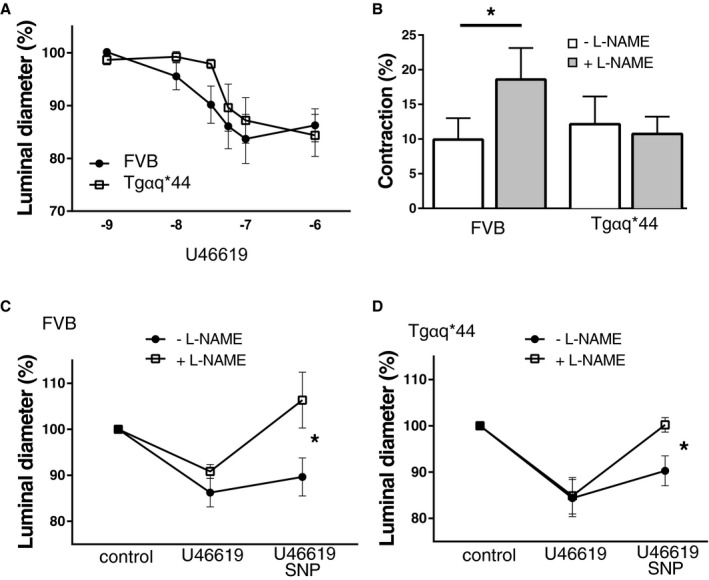
Impairment of NO‐dependent regulation of vascular tone in isolated perfused cerebral arterioles in early stages of heart failure in 10‐month‐old Tgαq*44 as compared to FVB mice. A, Contraction response curve to increasing U46619 concentration (from 10−9 to 10−5 mol/L); (B) effect of suboptimal U46619 dose on arteriole contraction with and without L‐NAME; (C and D) the effect of SNP (10−5 mol/L) on cortical arterioles before and after L‐NAME in FVB (C) and Tgαq*44 mice (D); FVB mice n=7, Tgαq*44 mice n=6; data distribution is presented as mean and SD. L‐NAME indicates Nω‐nitro‐l‐arginine methyl ester; SNP, sodium nitroprusside. *P<0.05.
Platelet Hyperreactivity in Early Stage of HF Development
A prothrombotic state is 1 of 2 major mechanisms responsible for VCI development in HF.44 Furthermore, platelet activation, independent of preserved or reduced EF, is known to be present in cardiomyopathy, and HF and was shown to be linked to cardiac inflammation.46, 47, 48 Therefore, we measured platelet responsiveness to stimulation in Tgαq*44 versus FVB mice at the ages of 3, 6, and 10 months (n=10 per group) by measuring surface expression of P‐selectin and GpIIbIIIa after 2 types of stimulations (ADP and stirring). The first stimulation with ADP (Figure 7A) induced an increase in frequency of platelets expressing P‐selectin in Tgαq*44 mice at 10 months (P=0.001) and increased median fluorescence intensity, representing level of platelet surface expression, of P‐selectin in 6‐ and 10‐month‐old Tgαq*44 mice (P=0.05, 0.0009, respectively). The second stimulation, with stirring and ADP (Figure 7B), further increased P‐selectin expression in Tgαq*44 mice. At 6 and 10 months, the increase in both frequency of platelets expressing P‐selectin (P=0.02, 0.002, respectively) and platelets median fluorescence intensity of P‐selectin (P=0.009, 0.001, respectively) was identified in Tgαq*44 mice compared to FVB mice. GpIIbIIIa platelet expression after the first stimulation (Figure 7C) was increased in Tgαq*44 mice in terms of frequency of platelets expressing GpIIbIIIa at 3, 6, and 10 months (P=0.0002, 0.02, and 0.0004, respectively) as well as in terms of platelet median fluorescence intensity of GpIIbIIIa at 3 and 10 months (P=0.01, 0.0004, respectively). The second stimulation (stirring and ADP) (Figure 7D) did not induce significant changes in GpIIbIIIa platelet expression in Tgαq*44 or FVB mice.
Figure 7.
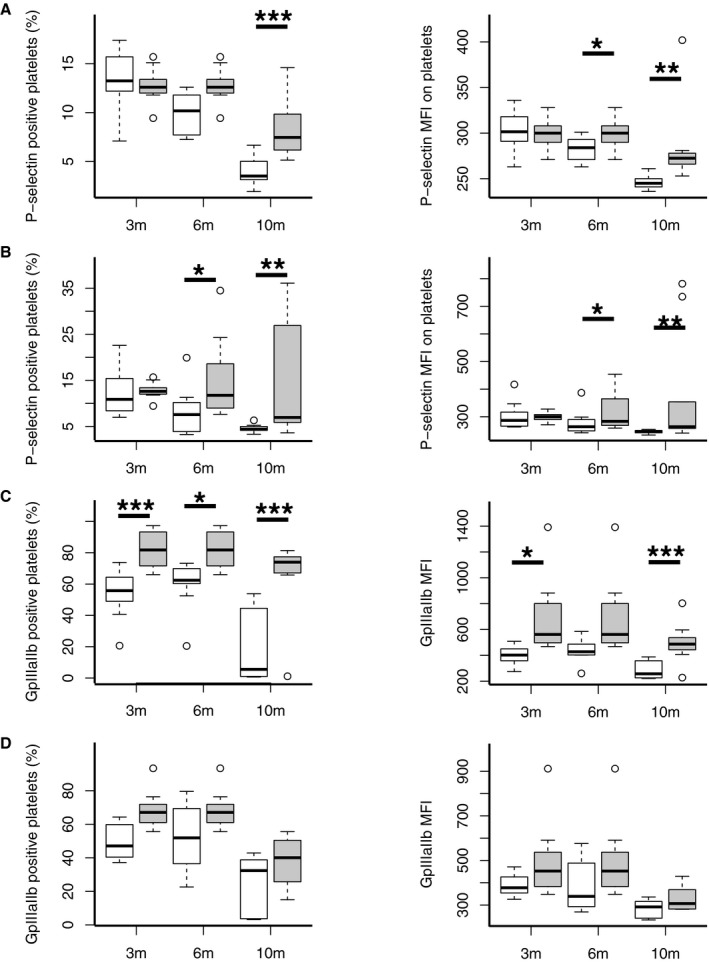
Systemic inflammatory activation of platelets in early stages of heart failure in Tgαq*44 mice. Platelets response to 2‐step activation first via ADP and second via ADP and 60 minutes of stirring in FVB (n=10 per group) and Tgαq*44 (n=10 per group) mice at 3, 6, and 10 months of age. Level of platelet activation is based on surface expression of either P‐selectin (CD62p) or GpIIbIIIa, which are measured as percentage of platelets expressing activation markers (marker‐positive platelets) and level of activation markers expression (marker MFI). A, Increased frequency of P‐selectin–positive platelets and increased P‐selectin MFI expression after ADP stimulation in Tgαq*44 at 6 and 10 months. B, Increased frequency of P‐selectin–positive platelets and increased P‐selectin MFI expression after 60 minutes mechanical and ADP stimulation in Tgαq*44 at 6 and 10 months. C, Increased frequency of GpIIbIIIa‐positive platelets and increased GpIIbIIIa MFI expression after ADP stimulation in 3‐, 6‐, and 10‐month‐old Tgαq*44. D, GpIIbIIIa expression after ADP and 60 minutes of mechanical stimulation; data distribution is presented as box plots (median, Q1, Q3, interquartile range, and outliers). *P<0.05, **P<0.005, ***P<0.0005. 3m, 6m, and 10m indicate 3, 6, and 10 months, respectively; MFI, median fluorescence intensity; Q1 and Q3 indicate 25th and 75th percentiles, respectively.
Lack of Increased Platelet‐Dependent Thrombogenicity in the Early Stage of HF Development
To further assess whether identified platelet hyperreactivity induces a prothrombotic state, we compared platelet thrombus formation between 10‐month‐old Tgαq*44 (n=8) and FVB mice (n=8) in the microchip‐based flow‐chamber system T‐TAS. There was no significant difference (P=0.76; 95% CI −185 to 138) between platelet count in Tgαq*44 and control FVB mice (Figure 8A). There was also no significant difference in platelet thrombus formation between Tgαq*44 and FVB mice based on all studied parameters: time to onset of thrombi formation (P=0.8; 95% CI −29 to 37), occlusion time (P=0.79; 95% CI −46 to 59), and area under the flow pressure curve (P=0.43; 95% CI −45 to 20) (Figure 8B through 8D).
Figure 8.
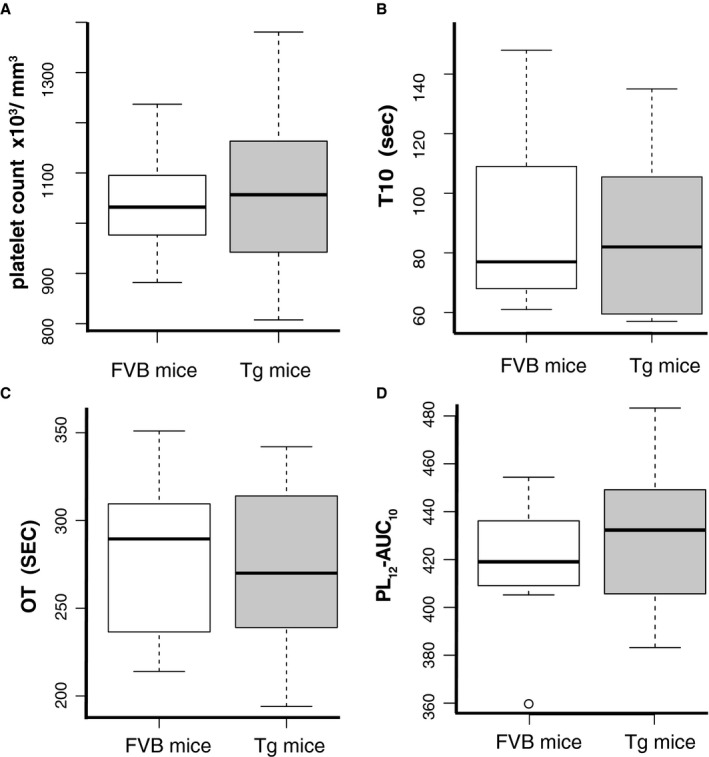
Lack of ex vivo platelet‐dependent prothrombotic state in early stage of heart failure development in 10‐month‐old Tgαq*44 as compared to FVB mice. Comparison of platelet count and platelet thrombus formation process in whole blood between FVB (n=8) and Tgαq*44 (n=8) mice at 10 months of age employing PL‐microchips coated with collagen and T‐TAS. A, Platelet count (×103/mm3); (B) T10 (seconds); (C) OT (seconds); and (D) PL 12‐AUC 10. Data distribution is presented as boxplots (median, Q1, Q3, interquartile range and outliers). OT indicates occlusion time; PL 12‐AUC 10, total thrombogenicity; Tg, Tgαq*44; T10, time to onset of thrombus formation; T‐TAS, Total Thrombus‐formation Analysis System (Fujimori Kogyo Co Ltd, Tokyo, Japan).
Discussion
This study, based on an experimental model, presents for the first time the finding that VCI with vascular and brain pathology is already present during the early stage of HF development in normotensive Tgαq*44 mice, before LV systolic dysfunction. Identification of hyperreactive platelets before VCI development without detectable impairment of resting CBF or severe prothrombotic state suggests a possible inflammatory pathomechanism of VCI pathology in Tgαq*44 mice. Development of cognitive impairment appears to be driven by dysfunction of the BBB due to progressive brain endothelium inflammatory activation and is associated with impairment of NO‐dependent regulation of brain arteriolar microcirculation.
HF—a common consequence of most heart diseases, manifesting as structural or functional impairment of ventricular filing and ejection of blood—is a recognized independent risk factor for cognitive impairment and dementia.49 Ventricular dysfunction in HF patients develops due to various vascular risk factors: hypertension, diabetes mellitus, coronary heart disease, obesity, or valvular heart disease.4 These HF risk factors are also independent risk factors for cognitive impairment.45 Concomitant presence of vascular diseases made it difficult to identify and validate HF‐specific pathology underlying cognitive impairment other than that related to advanced functional hemodynamic heart impairment. As indicated earlier, compensation of advanced HF (HFrEF) does improve cognition. However, in the case of the preclinical stage of HF, or HFpEF, in which heart function is preserved, knowledge of mechanisms responsible for cognitive impairment is significantly limited, and no treatment is yet available.
Tgαq*44 mice are a unique model because cardiomyocyte pathology leading to end‐stage HF develops over a long period of time (over 12 months), without the need for surgical or therapeutic intervention. These interventions, in other HF models, first cause heart decompensation (decompensated HF) and subsequent heart remodeling that secondly lead to heart compensation (compensated HF)—a baseline condition for experimental VCI studies.19 Therefore, slowly progressing pathology in Tgαq*44 mice uniquely enables the study of VCI development before development of advanced, end‐stage HF in the absence of preexisting vascular risk factors. Pathological cardiac hypertrophy is known to be caused by humoral activation, mechanical stress, transcription switch to fetal gene program and is linked to cardiac fibrosis.50, 51 These changes are also induced in Tgαq*44 mice via constitutive cardiomyocyte‐specific, postnatal activation of the Gαq pathway, which mimics the effects of neurohormonal activation in cardiomyocytes.23 Subsequently, at different time points, but before development of end‐stage HF, other signs of pathological cardiac hypertrophy occur and progress. As early as the fourth month, mRNA for the β‐myosin heavy chain is expressed in cardiomyocytes, indicating activation of fetal genes24, 26; cardiac fibrosis is already 4‐fold higher compared to control mice—indicating pathological tissue reorganization25 and increased compensatory expression of cardiomyocyte desmin,25 subsequently resulting in extracellular matrix remodeling and sarcomere pathological reorganization as in mechanical stress.50 Altogether, cardiomyocyte pathology in Tgαq*44 mice, while maintaining normotension,24 leads to slowly progressing heart deterioration and HF that was previously characterized,23, 25, 26, 27, 28, 29, 30, 31 but it was not known whether HF in this model results in VCI.
Evidence presented here suggests that VCI develops in Tgαq*44 mice at the early stages of HF development and is not due to brain hypoperfusion or the overt prothrombotic state identified so far as the major pathomechanisms of HF‐induced VCI, but to brain endothelium pathology preceded by platelet hyperreactivity. The brain endothelial dysfunction was evidenced by inflammatory activation, BBB permeability, oxidative stress, and β‐amyloid cortical accumulation as well as impairment of endothelial NO‐dependent regulation of vascular tone in cortical arterioles. Clinically, cardiomyopathies are known to cause platelet activation, a phenomenon independent of left ventricular size, EF, or heart function.46, 47, 48 Underlying mechanisms of this activation are not yet fully understood, but platelet activation was shown to correlate with myocardial inflammation.46 On the other hand, thromboembolism is 1 of the 2 recognized mechanisms of cognitive decline in advanced HF patients and occurs commonly in patients with arrhythmia, causing micro and macro brain infarcts, and both adherence to pharmacological treatment and surgical cardiac resynchronization improve patients’ cognition.4, 12, 13 In Tgαq*44 mice, development of cardiomyopathy is associated with co‐occurrence of numerous cardiomyocyte pathologies (fibrosis, fetal genes expression, sarcomere pathology) associated with heart inflammation51 and could be responsible for the chronic activation of platelets that mechanistically contribute to pathological cardiac remodeling as shown in other models.52 Interestingly, we found that platelets were hyperresponsive (increased GpIIbIIIa expression), already at the age of 3 months before development of detectable BBB pathology and cognitive deficits. A lack of significant differences in platelet‐dependent thrombogenicity in Tgαq*44 mice as compared with FVB at 10 months of age, assessed ex vivo in the microchip‐based flow‐chamber system T‐TAS, suggests that it is not the prothrombotic but rather the proinflammatory role of platelets that may contribute to VCI. However, T‐TAS may not have the sensitivity to detect subtle alterations in the prothrombotic phenotype of platelets; therefore prothrombotic mechanisms cannot definitely be excluded. Furthermore, as evidenced in atherosclerosis or diabetes mellitus type 2, platelet‐related proinflammatory mechanisms frequently coexist with or lead to prothrombotic pathomechanisms.53, 54
Altogether we suggest that platelet‐dependent mechanisms contribute to the activation of endothelial inflammation (E‐selectin brain vessel expression), to the impairment of brain endothelial integrity and function (increased BBB permeability), and to the impairment of NO regulation. Similarly, in type 2 diabetes mellitus, platelets are hypersensitive to agonists and respond to subthreshold stimuli before development of detectable vascular wall damage.53 Furthermore, GpIIbIIIa receptor inhibition with tirofiban in diabetic patients undergoing percutaneous cardiac interventions resulted in decreased inflammatory response.55 Experimental and clinical results show that activated platelets interact via surface molecules (ie, selectins) with circulating leukocytes and endothelium, causing inflammation and vascular diseases and contributing to disruption of endothelial barrier integrity.56 In our model, BBB permeability was compromised in all studied brain areas with the most significant permeability increase in the brain cortex. Moreover, brain endothelial cells—the vascular element of BBB—expressed an inflammatory phenotype (E‐selectins). Dysfunction of BBB and brain endothelium is commonly found in postmortem brain tissues of people suffering from cognitive impairment.57 In our study cognitive impairment and increased BBB permeability were both present by the age of 6 months, at which time Tgαq*44 mice display fully preserved systolic and global cardiac function with early signs of diastolic dysfunction. Furthermore, cognitive impairment at 6 and 10 months was accompanied by reduced myelin signal and disarrangement of myelinated fibers in major white‐matter tracts (corpus callosum, cingulum, anterior commissure). In parallel, we observed changes in neuroglial protein immunoreactivity (astrocytes, microglia) in cortex and above‐mentioned white‐matter structures compared to age‐matched controls, which we believe reflects the state of chronic pathological activation. Given that integrity of white matter and neuron‐glia interaction are essential for learning and memory processes,41 changes identified in Tgαq*44 mice may reflect or be responsible for identified cognitive impairment. Our further studies of cortical implications of BBB dysfunction and brain endothelium inflammation revealed increased β‐amyloid1‐42 accumulation in piriform cortex, increased cortical oxidative stress, and impairment in NO‐dependent cortical arteriole blood flow regulation. It is known that endothelial dysfunction causes impairment in neurovascular coupling, partly mediated by oxidative stress and endothelial NO production impairment.2 Furthermore, decreased NO production was linked to increased expression of amyloid precursor protein and β‐site amyloid precursor protein–cleaving enzyme 1, which further increases secretion and brain deposition of β‐amyloid.58 In our model, β‐amyloid1‐42 was found to be increased in piriform cortex, the region of the highest amyloid plaque concentration in the Alzheimer disease model.59
One of the study limitations is that it is based on an artificial model in which cognitive impairment arises from a single pathology driven by the cardiomyocyte‐specific activation of the Gαq pathway, whereas clinically, cognitive impairment is a multifactorial disease. We believe that this limitation is also a strength of our work because it gave a unique opportunity to characterize the role of a single risk factor such as HF in complex pathomechanisms of VCI development, exclusively at the relatively early stages of HF development in this model.23, 25, 26, 27, 28, 29, 30, 31 Another study limitation is the sensitivity of the CBF assay, on the basis of which we excluded brain hypoperfusion as a VCI pathomechanism. Magnetic resonance–based CBF assessment did not detect a difference between Tgαq*44 and FVB mice, but we cannot exclude the presence of minor regional and functional CBF impairment due to subtle alterations in the NO‐dependent function evidenced in the isolated cerebral arterioles in ex vivo studies. A significant reduction in CBF is present in the majority of vascular and neurodegenerative diseases linked to cognitive impairment; in advanced HF, CBF was reduced by 30%, and in Alzheimer disease by 20%,60, 61 whereas in experimental models mimicking hypoperfusion‐based VCI (ie, bilateral common carotid artery stenosis, transverse carotid constriction), CBF reduction reaches 70%.19, 20 Therefore, the identified 4% reduction in cortical CBF (based on mean CBF value) in Tgαq*44 compared to FVB mice, if it proved significant in larger experimental groups, would still play a marginal role as a contributor to VCI development in this model. Yet another study limitation is the possible involvement of other mechanisms of VCI, not studied here, as well as the descriptive nature of our findings, suggestive of a role of platelets in disease pathomechanism. Nevertheless, our conclusions are supported by the time sequence in which VCI pathology occurs. It begins with platelet hyperresponsiveness in Tgαq*44 mice at the age of 3 months, at which time the cardiac function is fully preserved but cardiac inflammation and fibrosis ensue; this is followed by BBB impairment, brain pathology, and impaired cognition in Tgαq*44 mice at the age of 6 months, at which age early‐stage diastolic but not systolic cardiac dysfunction is present; and finally, VCI changes develop further at the age of 10 months, at which stage both diastolic and systolic cardiac dysfunctions are present, and brain endothelium inflammation is further potentiated, but still this stage does not represent end‐stage HF as it occurs later.29, 30, 33 Moreover, Tgaq*44 mice at 3 months first show no changes in BBB permeability and, second, have cognitive function comparable to wild‐type mice at 3, 6, and 10 months. In contrast, at later time points (6 and 10 months), Tgaq*44 mice show significant and mostly progressive functional and structural deterioration in comparison to wild‐type mice.
Conclusions
In conclusion, we report, to our knowledge for the first time, that VCI develops in the early stage of experimental HF in Tgaq*44 mice independently of deterioration of systolic cardiac function and brain hypoperfusion. Development of cognitive impairment is preceded by platelet hyperactivity, which we believe could contribute to dysfunction of BBB, that is associated with brain endothelium inflammatory activation, and impairment of NO‐dependent regulation of arteriolar brain microcirculation, oxidative stress, and β‐amyloid cortical accumulation.
Sources of Funding
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska‐Curie grant agreement No. 660946 (Adamski) and from Polish National Science Centre grant No. DEC‐2015/16/W/NZ4/00070 (Chlopicki).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. Characteristics of Antibodies Used for Immunohistochemistry and Flow Cytometry
Table S2. Statistical Difference (P‐Values) in Microglia‐, Astroglia‐, and Myelin‐Specific Protein Immunoreactivity in Selected Brain Regions Between Tgαq*44 and FVB Mice at 6 and 10 Months of Age
Figure S1. ROI selection based on DAPI stain.
Figure S2. E‐selectin analysis protocol.
Figure S3. ROI for cortex, hippocampus, and thalamus for in vivo CBF imaging based on representative sample.
Figure S4. Platelet‐gating strategy based on representative sample.
Figure S5. Development of BBB permeability impairment in early stages of heart failure.
Figure S6. High‐potassium‐induced contraction and secondary dilation before and after L‐NAME in FVB and Tgαq*44 mice.
(J Am Heart Assoc. 2018;7:e007694 DOI: 10.1161/JAHA.117.007694.)
Contributor Information
Mateusz G. Adamski, Email: adamskimateusz@yahoo.com.
Stefan Chlopicki, Email: stefan.chlopicki@jcet.eu.
References
- 1. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75.e2. [DOI] [PubMed] [Google Scholar]
- 2. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barone FC, Gustafson DD, Crystal HA, Moreno H, Adamski MG, Arai K, Baird AE, Balucani C, Brickman AM, Cechetto D, Gorelick PP, Biessels GJ, Kiliaan A, Launer LJ, Schneider JA, Sorond FA, Whitmer R, Wright CB, Zhang ZG, Zhang G. First translational “Think Tank” on cerebrovascular disease, cognitive impairment and dementia. J Transl Med. 2016;14:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Writing Group Members , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J‐P, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee, Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 5. Sabayan B, van Buchem MA, Sigurdsson S, Zhang Q, Harris TB, Gudnason V, Arai AE, Launer LJ. Cardiac hemodynamics are linked with structural and functional features of brain aging: the age, gene/environment susceptibility (AGES)‐Reykjavik Study. J Am Heart Assoc. 2015;4:e001294 DOI: 10.1161/JAHA.114.001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cermakova P, Lund LH, Fereshtehnejad S‐M, Johnell K, Winblad B, Dahlström U, Eriksdotter M, Religa D. Heart failure and dementia: survival in relation to types of heart failure and different dementia disorders. Eur J Heart Fail. 2015;17:612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levin SN, Hajduk AM, McManus DD, Darling CE, Gurwitz JH, Spencer FA, Goldberg RJ, Saczynski JS. Cognitive status in patients hospitalized with acute decompensated heart failure. Am Heart J. 2014;168:917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alwerdt J, Edwards JD, Athilingam P, O'Connor ML, Valdés EG. Longitudinal differences in cognitive functioning among older adults with and without heart failure. J Aging Health. 2013;25:1358–1377. [DOI] [PubMed] [Google Scholar]
- 9. Arangalage D, Ederhy S, Dufour L, Joffre J, Van der Vynckt C, Lang S, Tzourio C, Cohen A. Relationship between cognitive impairment and echocardiographic parameters: a review. J Am Soc Echocardiogr. 2015;28:264–274. [DOI] [PubMed] [Google Scholar]
- 10. Ogren JA, Fonarow GC, Woo MA. Cerebral impairment in heart failure. Curr Heart Fail Rep. 2014;11:321–329. [DOI] [PubMed] [Google Scholar]
- 11. Kindermann I, Fischer D, Karbach J, Link A, Walenta K, Barth C, Ukena C, Mahfoud F, Köllner V, Kindermann M, Böhm M. Cognitive function in patients with decompensated heart failure: the Cognitive Impairment in Heart Failure (CogImpair‐HF) study. Eur J Heart Fail. 2012;14:404–413. [DOI] [PubMed] [Google Scholar]
- 12. Alosco ML, Spitznagel MB, Cohen R, Sweet LH, Josephson R, Hughes J, Rosneck J, Gunstad J. Better adherence to treatment recommendations in heart failure predicts improved cognitive function at a one‐year follow‐up. J Clin Exp Neuropsychol. 2014;36:956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoth KF, Poppas A, Ellison KE, Paul RH, Sokobin A, Cho Y, Cohen RA. Link between change in cognition and left ventricular function following cardiac resynchronization therapy. J Cardiopulm Rehabil Prev. 2010;30:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhat G, Yost G, Mahoney E. Cognitive function and left ventricular assist device implantation. J Heart Lung Transplant. 2015;34:1398–1405. [DOI] [PubMed] [Google Scholar]
- 15. Ampadu J, Morley JE. Heart failure and cognitive dysfunction. Int J Cardiol. 2015;178:12–23. [DOI] [PubMed] [Google Scholar]
- 16. Almeida OP, Garrido GJ, Beer C, Lautenschlager NT, Arnolda L, Flicker L. Cognitive and brain changes associated with ischaemic heart disease and heart failure. Eur Heart J. 2012;33:1769–1776. [DOI] [PubMed] [Google Scholar]
- 17. Cermakova P, Eriksdotter M, Lund LH, Winblad B, Religa P, Religa D. Heart failure and Alzheimer's disease. J Intern Med. 2015;277:406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2014;11:507–515. [DOI] [PubMed] [Google Scholar]
- 19. Bink DI, Ritz K, Aronica E, van der Weerd L, Daemen MJAP. Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition. J Cereb Blood Flow Metab. 2013;33:1666–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madigan JB, Wilcock DM, Hainsworth AH. Vascular contributions to cognitive impairment and dementia: topical review of animal models. Stroke. 2016;47:1953–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Erkelens CD, van der Wal HH, de Jong BM, Elting J‐W, Renken R, Gerritsen M, van Laar PJ, van Deursen VM, van der Meer P, van Veldhuisen DJ, Voors AA, Luijckx G‐J. Dynamics of cerebral blood flow in patients with mild non‐ischaemic heart failure. Eur J Heart Fail. 2017;19:261–268. [DOI] [PubMed] [Google Scholar]
- 22. Georgiadis D, Sievert M, Cencetti S, Uhlmann F, Krivokuca M, Zierz S, Werdan K. Cerebrovascular reactivity is impaired in patients with cardiac failure. Eur Heart J. 2000;21:407–413. [DOI] [PubMed] [Google Scholar]
- 23. Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin‐dependent and independent pathways. Proc Natl Acad Sci USA. 1998;95:13893–13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mende U, Semsarian C, Martins DC, Kagen A, Duffy C, Schoen FJ, Neer EJ. Dilated cardiomyopathy in two transgenic mouse lines expressing activated G protein αq: lack of correlation between phospholipase C activation and the phenotype. J Mol Cell Cardiol. 2001;33:1477–1491. [DOI] [PubMed] [Google Scholar]
- 25. Mackiewicz U, Czarnowska E, Brudek M, Pająk B, Duda M, Emanuel K, Csanyi G, Fedorowicz A, Grochal E, Tyrankiewicz U, Skórka T, Mende U, Lewartowski B, Chłopicki S. Preserved cardiomyocyte function and altered desmin pattern in transgenic mouse model of dilated cardiomyopathy. J Mol Cell Cardiol. 2012;52:978–987. [DOI] [PubMed] [Google Scholar]
- 26. Drelicharz Ł, Woźniak M, Skórka T, Tyrankiewicz U, Heinze‐Paluchowska S, Jabłońska M, Gebska A, Chłopicki S. Application of magnetic resonance imaging in vivo for the assessment of the progression of systolic and diastolic dysfunction in a mouse model of dilated cardiomyopathy. Kardiol Pol. 2009;67:386–395. [PubMed] [Google Scholar]
- 27. Elas M, Bielanska J, Pustelny K, Plonka PM, Drelicharz L, Skorka T, Tyrankiewicz U, Wozniak M, Heinze‐Paluchowska S, Walski M, Wojnar L, Fortin D, Ventura‐Clapier R, Chlopicki S. Detection of mitochondrial dysfunction by EPR technique in mouse model of dilated cardiomyopathy. Free Radic Biol Med. 2008;45:321–328. [DOI] [PubMed] [Google Scholar]
- 28. Drelicharz L, Kozlovski V, Skorka T, Heinze‐Paluchowska S, Jasinski A, Gebska A, Guzik T, Olszanecki R, Wojnar L, Mende U, Csanyi G, Chlopicki S. NO and PGI2 in coronary endothelial dysfunction in transgenic mice with dilated cardiomyopathy. Basic Res Cardiol. 2008;103:417–430. [DOI] [PubMed] [Google Scholar]
- 29. Czarnowska E, Bierła JB, Toczek M, Tyrankiewicz U, Pająk B, Domal‐Kwiatkowska D, Ratajska A, Smoleński RT, Mende U, Chłopicki S. Narrow time window of metabolic changes associated with transition to overt heart failure in Tgaq*44 mice. Pharmacol Rep. 2016;68:707–714. [DOI] [PubMed] [Google Scholar]
- 30. Tyrankiewicz U, Skorka T, Jablonska M, Petkow‐Dimitrow P, Chlopicki S. Characterization of the cardiac response to a low and high dose of dobutamine in the mouse model of dilated cardiomyopathy by MRI in vivo. J Magn Reson Imaging. 2013;37:669–677. [DOI] [PubMed] [Google Scholar]
- 31. Tyrankiewicz U, Olkowicz M, Skorka T, Jablonska M, Orzyłowska A, Bar A, Gonet M, Berkowicz P, Jasinski K, Zoladz JA, Smolenski RT, Chlopicki S. Activation pattern of ACE2/Ang‐(1‐7) and ACE/Ang II pathway in course of heart failure assessed by multiparametric MRI in vivo in Tgαq*44 mice. J Appl Physiol. 2018;124:52–65. [DOI] [PubMed] [Google Scholar]
- 32. Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. Myocardial expression of a constitutively active alpha 1B‐adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc Natl Acad Sci USA. 1994;91:10109–10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grassi B, Majerczak J, Bardi E, Buso A, Comelli M, Chlopicki S, Guzik M, Mavelli I, Nieckarz Z, Salvadego D, Tyrankiewicz U, Skorka T, Bottinelli R, Zoladz JA, Pellegrino MA. Exercise training in Tgαq*44 mice during the progression of chronic heart failure: cardiac vs. peripheral (soleus muscle) impairments to oxidative metabolism. J Appl Physiol. 2017;123:326–336. [DOI] [PubMed] [Google Scholar]
- 34. Schiavone S, Sorce S, Dubois‐Dauphin M, Jaquet V, Colaianna M, Zotti M, Cuomo V, Trabace L, Krause K‐H. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry. 2009;66:384–392. [DOI] [PubMed] [Google Scholar]
- 35. Giovannini MG, Bartolini L, Kopf SR, Pepeu G. Acetylcholine release from the frontal cortex during exploratory activity. Brain Res. 1998;784:218–227. [DOI] [PubMed] [Google Scholar]
- 36. Thuesen AD, Lyngsø KS, Rasmussen L, Stubbe J, Skøtt O, Poulsen FR, Pedersen CB, Rasmussen LM, Hansen PBL. P/Q‐type and T‐type voltage‐gated calcium channels are involved in the contraction of mammary and brain blood vessels from hypertensive patients. Acta Physiol (Oxf). 2017;219:640–651. [DOI] [PubMed] [Google Scholar]
- 37. Przyborowski K, Kassassir H, Wojewoda M, Kmiecik K, Sitek B, Siewiera K, Zakrzewska A, Rudolf AM, Kostogrys R, Watala C, Zoladz JA, Chlopicki S. Effects of a single bout of strenuous exercise on platelet activation in female ApoE/LDLR–/– mice. Platelets. 2017;28:657–667. [DOI] [PubMed] [Google Scholar]
- 38. Rozalski M, Kassassir H, Siewiera K, Klepacka A, Sychowski R, Watala C. Platelet activation patterns are different in mouse models of diabetes and chronic inhibition of nitric oxide synthesis. Thromb Res. 2014;133:1097–1104. [DOI] [PubMed] [Google Scholar]
- 39. Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7:681–685. [DOI] [PubMed] [Google Scholar]
- 40. Hosokawa K, Ohnishi T, Fukasawa M, Kondo T, Sameshima H, Koide T, Tanaka KA, Maruyama I. A microchip flow‐chamber system for quantitative assessment of the platelet thrombus formation process. Microvasc Res. 2012;83:154–161. [DOI] [PubMed] [Google Scholar]
- 41. Fields RD, Araque A, Johansen‐Berg H, Lim S‐S, Lynch G, Nave K‐A, Nedergaard M, Perez R, Sejnowski T, Wake H. Glial biology in learning and cognition. Neuroscientist. 2014;20:426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood‐brain barrier. Neurobiol Dis. 2010;37:13–25. [DOI] [PubMed] [Google Scholar]
- 43. Petrovic‐Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res. 2016;119:142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ritz K, van Buchem MA, Daemen MJ. The heart‐brain connection: mechanistic insights and models. Neth Heart J. 2013;21:55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S; American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia . Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bobbert P, Weikert U, Schmidt‐Lucke C, Skurk C, Meyer A, Steffens D, Schultheiss HP, Rauch U. Platelet activation and thrombus formation relates to the presence of myocardial inflammation in patients with cardiomyopathy. J Cardiol. 2014;63:379–384. [DOI] [PubMed] [Google Scholar]
- 47. Weikert U, Kuhl U, Schultheiss HP, Rauch U. Platelet activation is increased in patients with cardiomyopathy: myocardial inflammation and platelet reactivity. Platelets. 2002;13:487–491. [DOI] [PubMed] [Google Scholar]
- 48. Gibbs CR, Blann AD, Watson RD, Lip GY. Abnormalities of hemorheological, endothelial, and platelet function in patients with chronic heart failure in sinus rhythm: effects of angiotensin‐converting enzyme inhibitor and beta‐blocker therapy. Circulation. 2001;103:1746–1751. [DOI] [PubMed] [Google Scholar]
- 49. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128:1810–1852. [DOI] [PubMed] [Google Scholar]
- 50. Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, Popov A‐F, Dohmen PM, Choi Y‐H, Wahlers T, Weymann A. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res. 2016;22:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu C, Zhao W, Meng W, Zhao T, Chen Y, Ahokas RA, Liu H, Sun Y. Platelet‐derived growth factor blockade on cardiac remodeling following infarction. Mol Cell Biochem. 2014;397:295–304. [DOI] [PubMed] [Google Scholar]
- 53. Natarajan A, Zaman AG, Marshall SM. Platelet hyperactivity in type 2 diabetes: role of antiplatelet agents. Diab Vasc Dis Res. 2008;5:138–144. [DOI] [PubMed] [Google Scholar]
- 54. Von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. [DOI] [PubMed] [Google Scholar]
- 55. Azar RR, Badaoui G, Sarkis A, Kassab R, Salamé E, Aboujaoudé S, Hamdan R, Barakett V, Germanos M. Effect of high bolus dose tirofiban on the inflammatory response following percutaneous coronary intervention. Clin Cardiol. 2010;33:E14–E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. 2013;112:1506–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, Schneider JA, Wellington C, Wilcock DM, Zipfel GJ, Zlokovic B, Bain LJ, Bosetti F, Galis ZS, Koroshetz W, Carrillo MC. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement. 2015;11:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2013;35:888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J‐M, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid‐β deposition. Nat Neurosci. 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gruhn N, Larsen FS, Boesgaard S, Knudsen GM, Mortensen SA, Thomsen G, Aldershvile J. Cerebral blood flow in patients with chronic heart failure before and after heart transplantation. Stroke. 2001;32:2530–2533. [DOI] [PubMed] [Google Scholar]
- 61. Roher AE, Debbins JP, Malek‐Ahmadi M, Chen K, Pipe JG, Maze S, Belden C, Maarouf CL, Thiyyagura P, Mo H, Hunter JM, Kokjohn TA, Walker DG, Kruchowsky JC, Belohlavek M, Sabbagh MN, Beach TG. Cerebral blood flow in Alzheimer's disease. Vasc Health Risk Manag. 2012;8:596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Characteristics of Antibodies Used for Immunohistochemistry and Flow Cytometry
Table S2. Statistical Difference (P‐Values) in Microglia‐, Astroglia‐, and Myelin‐Specific Protein Immunoreactivity in Selected Brain Regions Between Tgαq*44 and FVB Mice at 6 and 10 Months of Age
Figure S1. ROI selection based on DAPI stain.
Figure S2. E‐selectin analysis protocol.
Figure S3. ROI for cortex, hippocampus, and thalamus for in vivo CBF imaging based on representative sample.
Figure S4. Platelet‐gating strategy based on representative sample.
Figure S5. Development of BBB permeability impairment in early stages of heart failure.
Figure S6. High‐potassium‐induced contraction and secondary dilation before and after L‐NAME in FVB and Tgαq*44 mice.