

Abstract
Background
Development of right ventricular (RV) hypertension eventually contributes to RV and left ventricular (LV) myocardial fibrosis and dysfunction. The molecular mechanisms are not fully elucidated.
Methods and Results
Pulmonary artery banding was used to induce RV hypertension in rats in vivo. Then, we evaluated cardiac function and regional remodeling 6 weeks after pulmonary artery banding. To further elucidate mechanisms responsible for regional cardiac remodeling, we also mimicked RV hypertensive stress by cyclic mechanical stretching applied to confluent cultures of cardiac fibroblasts, isolated from the RV free wall, septal hinge points, and LV free wall. Echocardiography and catheter evaluation demonstrated that rats in the pulmonary artery banding group developed RV hypertension with leftward septal displacement, LV compression, and increased LV end‐diastolic pressures. Picrosirius red staining indicated that pulmonary artery banding induced marked RV fibrosis and dysfunction, with prominent fibrosis and elastin deposition at the septal hinge points but less LV fibrosis. These changes were associated with proportionally increased expressions of integrin‐β1 and profibrotic signaling proteins, including phosphorylated Smad2/3 and transforming growth factor‐β1. Moreover, mechanically stretched fibroblasts also expressed significantly increased levels of α‐smooth muscle actin, integrin‐β1, transforming growth factor‐β1, collagen I deposition, and wrinkle formation on gel assays, consistent with myofibroblast transformation. These changes were not observed in parallel cultures of mechanically stretched fibroblasts, preincubated with the integrin inhibitor (BTT‐3033).
Conclusions
Experimentally induced RV hypertension triggers regional RV, hinge‐point, and LV integrin β1‐dependent mechanotransduction signaling pathways that eventually trigger myocardial fibrosis via transforming growth factor‐β1 signaling. Reduced LV fibrosis and preserved global function, despite geometrical and pressure aberrations, suggest a possible elastin‐mediated protective mechanism at the septal hinge points.
Keywords: fibrosis, integrin, pressure overload, regional stress
Subject Categories: Pulmonary Hypertension, Cell Signalling/Signal Transduction, Fibrosis, Hypertrophy, Remodeling
Clinical Perspective
What Is New?
Isolated right ventricular (RV) hypertension leads to geometrical changes that promote regional RV and septal hinge‐point integrin‐β1 and profibrotic signaling.
However, despite left ventricular geometrical distortion and increased end‐diastolic pressures, mechanical‐molecular remodeling is attenuated in the left ventricle, suggesting a possible mechanotransduction adaptive mechanism mediated by integrin‐β1, possibly through upregulation of elastin.
Thus, β1‐integrins may mediate coexistent adaptive and maladaptive mechanotransduction mechanisms, and the balance between these may determine biventricular function in RV pressure loading.
What Are the Clinical Implications?
In RV pressure loading, treatments modulating integrin‐β1 expression and function or those increasing elastin production and decreasing RV and left ventricular fibrosis may be beneficial.
Furthermore, there may a therapeutic window before more extensive development of left ventricular fibrosis and dysfunction.
The exact prevalence of pediatric heart failure is largely unknown, but it is increasing. Development of progressive right ventricular (RV) hypertension initially triggers RV dysfunction, and, if persistent, it eventually transduces to the left ventricular (LV) myocardium, causing its dysfunction and increasing mortality.1, 2, 3, 4, 5, 6 However, the molecular mechanisms of this pathologic phenomenon are not yet fully elucidated. Observations in human cases of pulmonary arterial hypertension have shown the development of peculiar fibrosis at the septal hinge points (HPs), which is associated with RV and pulmonary hemodynamics and clinical outcomes.7, 8 We recently demonstrated, in an experimental model of pulmonary artery banding (PAB)–induced RV hypertension, marked upregulation of profibrotic signaling and severe collagenous fibrosis at the septal HPs; we hypothesized that this mechanically stressed region, where the RV and LV adjoin, plays a pivotal role in mediating, or possibly mitigating, LV injury secondary to RV hypertension.9
The integrin transmembrane glycoprotein receptors, and their molecular cross‐talk with transforming growth factor (TGF)‐β1 pathways, have previously been recognized in the LV as translating pressure‐stress into biochemical signaling (mechanotransduction) in various clinical and experimental models.10, 11, 12 Integrins are also major regulators of intracellular profibrotic signaling, including connective tissue growth factor (CTGF) and TGF‐β1. More important, we have recently demonstrated, in an experimental PAB model, that triggering of TGF‐β1 signaling pathways preceded generation of RV and LV fibrosis and their ultimate dysfunction in response to excessive RV pressure load; this suggested that β1‐integrins and other mechanotransduction pathways may play a pivotal role in triggering RV and LV fibrosis in response to RV hypertension.13, 14, 15, 16, 17
However, our experiments showed that, although LV geometry and pressure are substantially affected by RV hypertension, the LV free wall was substantially less affected by collagenous fibrosis compared with the RV and septal hinge regions.9 This suggests the presence of a possible adaptive mechanotransduction mechanism to reduce LV myocardial remodeling and maintain LV function.
Consequently, the aim of this study was to investigate regional β1‐integrin mechanotransduction in RV hypertension and its relation to the development of RV and LV fibrosis. Herein, we hypothesized that regional integrin‐β1A+D expression mediates RV and LV fibrosis secondary to RV hypertension.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure because the tissue samples are limited.
In Vivo PAB Model
Animal experiments were approved by the institutional animal ethics committee. Thirty‐six male Sprague‐Dawley rats (200 g body weight) were randomized to either sham (n=10) surgery (thoracotomy) or to PAB (n=26). Anesthesia was induced by IP pentobarbital sodium (60 mg/kg). After intubation, animals were mechanically ventilated using a volume‐controlled respirator and oxygen‐enriched room air. Positive end‐expiratory pressure was maintained at 4 cm H2O. Through a left thoracotomy, the pulmonary artery (PA) was separated from the aorta. Then, an 18‐gauge needle was placed under the PA, and a silk suture was tied around the needle and vessel. Subsequent removal of the needle produced a fixed and standardized external PA constriction, proportional to the needle diameter. Both groups, sham and PAB rats, were then maintained for the following 6 weeks when the PAB animals developed permanent PA constriction, leading to RV pressure overload.
Assessment of Cardiac Function
Echocardiography
Rats from both experimental groups were monitored at the time of PAB placement and at the terminal experiment using a Vivid E9 ultrasound system with a 12‐MHz phased‐array transducer (GE Healthcare, Wauwatosa, WI), achieving 2‐dimensional frame rates of 275 frames per second. After 6 weeks, at the terminal experiment, before euthanasia, 2‐dimensional, M‐mode, color Doppler, and conventional pulsed Doppler images were obtained with simultaneous ECGs. Digitally recorded images were stored for offline analysis with the EchoPac, version 8.0 (GE Healthcare) system.
Interventricular septum and LV free‐wall thickness and shortening fraction were measured by M‐mode from a short‐axis view at the papillary muscle level. The LV eccentricity index, defined as the ratio between the LV anteroposterior and septolateral dimensions,18 was measured at end systole and end diastole as an index of RV hypertension severity and geometric distortion of the LV caused by leftward septal shift (Figure 1). RV functional parameters included fractional area change, tricuspid annular plane systolic excursion, and RV myocardial performance index.
Figure 1.
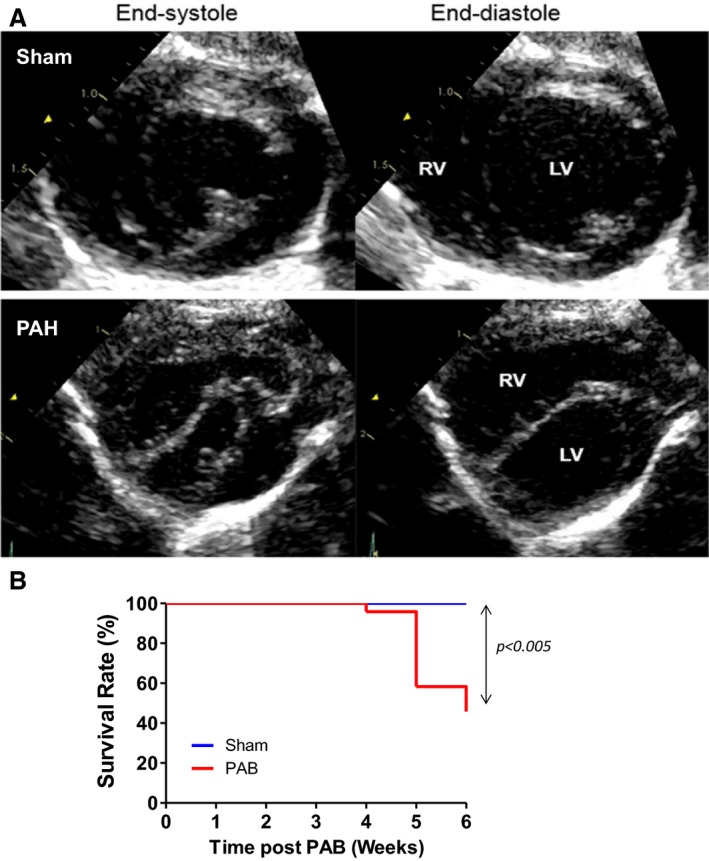
A, Representative echocardiography indicating that increasing the right ventricular (RV) pressure load induces RV dilatation, septal flattening, and left ventricular (LV) compression. Septal flattening changes geometry at the septal hinge‐point regions and LV. Short‐axis view obtained at the papillary muscle level at end systole and end diastole in a sham (A) and Pulmonary Arterial Hypertension (PAH) (B) rat. The sham rat shows a circular LV with normal round interventricular septal curvature and position throughout the cardiac cycle; in PAH, the RV is markedly enlarged and the LV is flattened and “D shaped” throughout the cardiac cycle, with the interventricular septum displaced leftward in systole and flattened into end diastole. B, Kaplan‐Meier survival curves in sham (n=8) and pulmonary artery banding (PAB) (n=26) at 6 weeks in rats. The median survival in PAB is 6. P<0.005 vs sham. Echocardiographic parameters are summarized in table 1.
Hemodynamic Measurements
After 6 weeks, RV and LV hemodynamics were assessed immediately after echocardiography. A 2F high‐fidelity pressure‐tipped catheter (Millar Instruments, Inc, Houston, TX) was inserted into the RV and LV through the apex. The maximal rate of ventricular pressure increase was recorded as an index of systolic function. Ventricular relaxation was reflected by τ, the time constant of monoexponential decay of ventricular pressure during isovolumic relaxation.
Histochemical Detection of Collagen I and Hydroxyproline as Markers of Fibrosis
Transverse sections of the hearts (5‐mm thick) from both experimental groups (euthanized after 6 weeks) were fixed in 10% formalin for 24 hours, dehydrated, and embedded in paraffin. Then, 4‐μm microtome sections were prepared (Leica Microsystems A/S, Herlev, Denmark) and subjected to histochemical staining with Picrosirius red to visualize interstitial collagen, as previously described.19 The proportional comparison of areas occupied by Picrosirius red–positive collagen versus the entire visual field was quantified morphometrically.
We also quantified hydroxyproline content in the heart tissues, using a commercial kit, according to the manufacturer's protocol (Sigma Aldrich, St Louis, MO). Briefly, the RV, LV, and septal HP regions of sham and PAB hearts were dissected separately, weighed, homogenized in sterile water, and hydrolyzed in 12 N HCl at 120°C for 3 hours. Then, hydrolyzed samples were incubated with 4‐(dimethylamino) benzaldehyde for 90 minutes at 60°C, and the absorbance of oxidized hydroxyproline was determined photometrically at 560 nm.
Detection of Elastin Deposition by Movat's Pentachrome Staining
Cryostat sections (5‐μm thick) were fixed in Bouin's solution for 30 minutes, followed by staining with Alcian Blue solution for 30 minutes. Slides were then washed in running water for 3 minutes and incubated with ammonium hydroxide in 95% alcohol (pH >8) for 2 hours. After washing with running water and rinsing in 70% alcohol, the slides were incubated with Weigert‐Hart Resorcin solution for 3 hours at 58°C and then stained with Weigert's hematoxylin for 15 minutes. Next, they were rinsed with running water and stained with Woodstain Scarlet Fuchsin solution for 5 minutes, followed by rinsing in 0.5% aqueous glacial acetic acid. Slides were then rinsed thoroughly in 3 changes of absolute alcohol and incubated with alcoholic saffron for 15 minutes. This was followed by washing with 4 changes of absolute alcohol. Finally, the slides were washed with several changes of xylene and then mounted in Permount.20
Western Blot Analysis
Protein was extracted from heart tissue in in vivo experiments and from cultured fibroblasts with lysis buffer and diluted 1:1 with 2× SDS sample buffer (Invitrogen Canada Inc). Equal amounts of protein (20 μg) were loaded in each lane of Novex 4% to 12% Tris‐Glycinegel (Invitrogen Canada Inc). Proteins were separated by electrophoresis and transferred from the gel to a nitrocellulose membrane with an electroblotting apparatus. Membranes were incubated with 5% nonfat dry milk for 1 hour to decrease nonspecific sites. Then, they were incubated overnight at 4°C with the following: primary antibodies to integrin‐β1A and integrin‐β1D, which were generated in our laboratory and used in previously published studies21; α‐integrins 1, 2, 10, and 11 (Santa Cruz, Inc); focal adhesion kinase (FAK), phosphorylated FAK, integrin‐linked kinase, extracellular signal–regulated kinase (ERK), and phosphorylated ERK (Cell Signaling Technology); TGF‐β1 and CTGF (Abcam, Inc); Smad2/3 and phosphorylated Smad 2 and 3 (Cell Signaling Technology). Samples were then washed and incubated with peroxidase‐conjugated secondary antibody and detected by an electrochemiluminescence detection kit (Bio‐Rad, Canada).
Immunohistochemistry and Immunofluorescence
Cryostat sections (5‐μm thick), fixed in 4% paraformaldehyde/PBS, were incubated with 0.3% hydrogen peroxide and 10% BSA for 15 minutes. Then, they were incubated with primary antibodies recognizing natriuretic peptides A preproprotein at 4°C overnight. The sections were then incubated with a matching biotinylated secondary antibody (Vector, Burlingame, CA) for 45 minutes at room temperature. Negative controls were performed for all immunological staining by omission of the primary antibody. Similar procedures were followed for fibroblast proliferation using antibodies to vimentin and Ki‐67. Wheat germ agglutinin was additionally used to visualize cardiomyocyte cell membranes. Sections were then incubated with appropriate secondary or fluorescein‐conjugated secondary antibodies for 60 minutes at room temperature. Negative controls were performed for all immunological staining by omission of the primary antibody. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole, dilactate.
Primary Cardiac Fibroblast Cultures
To evaluate the effect of mechanical stress in fibroblasts isolated from RV hypertensive animals, we isolated cardiac fibroblasts from a Sugen plus hypoxia rat model. For these experiments, pulmonary hypertension (PH) was induced in male Sprague‐Dawley rats (200 g body weight) with SC Sugen5416 (20 mg/kg), followed by long‐term hypoxia (10% O2) for 3 weeks. Rats were then kept at 3 weeks of normoxia before euthanasia. Control animals were placed in room air for the same duration.
Region‐specific primary cardiac fibroblasts were isolated by collagenase digestion from dissected fragments of the RV free wall, LV free wall, and septal HPs derived from 200‐ to 250‐g adult healthy and PH (Sugen‐hypoxia) male Sprague‐Dawley rats. Isolated cells were resuspended in fresh medium consisting of DMEM with 10% fetal bovine serum and plated on 100‐mm2 culture dishes at 37°C with 5% CO2 for 2 hours. Nonadherent cells were washed out, and the adherent cells (95% fibroblasts) were incubated in medium containing 10% fetal bovine serum and 1% antibiotics. The cells were passaged 3 times (passage 1–passage 3) for all experiments and used for quantitative assessment of protein expression and immunofluorescence analyses.
Mechanical Stretch of Cardiac Region‐Specific Fibroblasts
To explore the possibility that integrin‐β1 plays a crucial role in RV‐LV mechanotransduction, we mimicked the mechanical stretch induced by RV hypertension in cardiac fibroblasts plated on membranes of the stretch apparatus (Flexcell 4000; Hillsborough, NC). Cells were plated at a density of 0.5×106 cells/well in DMEM and maintained at 37°C in humid air with 5% CO2. When cells were ≈80% confluent, the culture medium was changed to 2% serum DMEM, and separate cultures were maintained in the presence and absence of integrin‐β1 inhibitor (BTT‐3033; 10 μmol/L; Tocris Bioscience). Stretched cell cultures were exposed to cyclic 20% equiaxial stretch at 1.7 Hz for 24 hours in a Flexcell FX‐4000 strain unit equipped with loading posts. Nonstretched fibroblasts were cultured on the same stretch chambers in the same incubator.
After treatment, cells on stretch membranes were washed twice with PBS, fixed in 4% paraformaldehyde in PBS or cold methanol for 15 minutes, and blocked in 10% normal BSA in PBS for 15 minutes. Then, they were incubated with primary antibodies recognizing collagen type I, integrin‐β1A, TGF‐β1, α‐smooth muscle actin, and Ki‐67 at 4°C overnight. This step was followed by a 45‐minute‐long incubation with the appropriate secondary antibodies conjugated with green fluorescein isothiocyanate or red tetrarhodamine isothiocyanate (Sigma). Nuclei were counterstained with a blue 4′,6‐diamidino‐2‐phenylindole. Secondary antibodies alone were used as an additional control. The cells were washed twice with PBS, and the wells were separated with a scalpel and mounted onto slides with cover slips. All cultures were examined with a Nikon Eclipse E1000 microscope attached to a cooled charge‐coupled device camera (Retiga EX; QImaging, Surrey, BC, Canada).
Isolated Cardiac Fibroblast Contraction Assays
To evaluate the functional properties of active (myo)fibroblasts, cells were obtained from 3 different regions in the heart (RV, LV, and septal HPs) and cultured on stretchable substrates. Cells were subjected to 24‐hour stretching or no‐stretching conditions in the presence or absence of the integrin inhibitor BTT‐3033. Cells were then trypsinized and reseeded onto deformable silicone substrates coated with gelatin (2 μg/cm2).22 Cell force transmission to these substrates results in surface distortions (“wrinkles”) that are visible as white lines in phase‐contrast micrographs.23 After attachment overnight and development of wrinkles because of cell contraction, 20 images were taken per condition (12 conditions in total) and wrinkle area was quantified per cell after thresholding images for bright lines. After elimination of technically inadequate points, 18 to 19 data points were analyzed per condition.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism 5.0. Survival between groups was analyzed using Kaplan‐Meier survival analysis. For nonnormally distributed data, the Mann‐Whitney or Kruskal‐Wallis test was applied to test for significance. Values are expressed as medians and interquartile range. Statistical significance is recognized at P<0.05.
Results
Echocardiography and Hemodynamics
Sham rats showed a circular LV with round interventricular septal curvature and position throughout the cardiac cycle. In PAB rats, the RV was significantly enlarged, and the LV was flattened and geometrically distorted, with a “D‐shaped” configuration throughout the cardiac cycle, where the interventricular septum was displaced leftward in systole and flattened through to end diastole (Figure 1A). Echocardiographic and hemodynamic parameters are summarized in Tables 1 and 2. They show that RV peak systolic and end‐diastolic pressures were increased in the PAB group versus controls, with increased LV systolic and diastolic eccentricity index and smaller LV end‐diastolic diameter. The eccentricity index was increased in systole, but did not reach statistical significance (because of a large SD). Of 26 PAB rats that entered the protocol, the survival rate was 100% after 3 weeks, 88% after 4 weeks, 53% after 5 weeks, and 42% after 6 weeks after PAB versus 100% in sham controls (Figure 1B). Of these rats, 5 PAB rats with complete imaging, hemodynamic, histologic, and molecular data were analyzed.
Table 1.
Comparison of Echocardiographic Measurements in PAB Rats Versus Shams
| Variables | Sham Group (n=8) | PAB Group (n=5) |
|---|---|---|
| HR, bpm | ||
| Median | 341.1 | 264* |
| Interquartile range | 320.4–373.4 | 244.5–346.5 |
| RV parameter | ||
| RVEDD, cm | ||
| Median | 0.25 | 0.54† |
| Interquartile range | 0.24–0.2625 | 0.515–0.62 |
| RVESD, cm | ||
| Median | 0.155 | 0.49† |
| Interquartile range | 0.12–0.19 | 0.37–0.515 |
| RV FAC, % | ||
| Median | 44 | 21† |
| Interquartile range | 37.5–47 | 18–22 |
| TAPSE, cm | ||
| Median | 0.28 | 0.09† |
| Interquartile range | 0.2425–0.3075 | 0.065–0.155 |
| RV‐MPI | ||
| Median | 0.13 | 0.59† |
| Interquartile range | 0.02–0.31 | 0.475–0.645 |
| LV parameter | ||
| LV end‐systolic ECC | ||
| Median | 1.095 | 2.22 |
| Interquartile range | 1.06–1.163 | 0.73–2.545 |
| LV end‐diastolic ECC | ||
| Median | 1.065 | 1.57 |
| Interquartile range | 1.045–1.113 | 1.075–1.585 |
| LVPW, cm | ||
| Median | 0.15 | 0.11 |
| Interquartile range | 0.13–0.17 | 0.09–0.145 |
| IVS, cm | ||
| Median | 0.135 | 0.13 |
| Interquartile range | 0.1275–0.1525 | 0.085–0.155 |
| LVEDD, cm | ||
| Median | 0.885 | 0.71 |
| Interquartile range | 0.7175–0.6225 | 0.645–0.92 |
| LVESD, cm | ||
| Median | 0.55 | 0.61 |
| Interquartile range | 0.485–0.66 | 0.385–0.645 |
| LV SF, % | ||
| Median | 33.5 | 28 |
| Interquartile range | 27–39 | 20–42 |
Quantification of the echocardiograms and presented as the LV eccentricity index. These geometrical LV changes are expected to induce increased mechanical stress at the RV and septal hinge‐point regions and mimic findings in human RV hypertension. Two‐dimensional measurements: ECC and FAC. M‐mode measurements: PW, SF, and TAPSE. Pulsed‐Doppler measurement: MPI. Bpm indicates beats per minute; ECC, eccentricity index; FAC, fractional area change; HR, heart rate; IVS, interventricular septum; LV, left ventricle; LVEDD, LV end‐diastolic diameter; LVESD, LV end‐systolic diameter; MPI, myocardial performance index; PAB, pulmonary artery band; PW, posterior wall; RV, right ventricle; RVEDD, RV end‐diastolic diameter; RVESD, RV end‐systolic diameter; SF, shortening fraction; TAPSE, tricuspid systolic annular excursion.
*P<0.05 vs sham.
† P<0.01 vs shams.
Table 2.
Comparison of Invasive Hemodynamic Parameters in PAB Rats Versus Shams
| Variable | Sham Group (n=8) | PAB Group (n=5) |
|---|---|---|
| HR, bpm | ||
| Median | 343.6 | 270† |
| Interquartile range | 325.1–391.5 | 262–315 |
| RV parameters | ||
| RV peak systolic pressure, mm Hg | ||
| Median | 19 | 55† |
| Interquartile range | 18.25–22.5 | 52.5–79 |
| RV end‐diastolic pressure, mm Hg | ||
| Median | 1.3 | 6† |
| Interquartile range | 1.1–3.475 | 4–6.5 |
| RV dP/dTmax, mm Hg/s | ||
| Median | 995 | 2895* |
| Interquartile range | 889.3–1455 | 1413–3293 |
| RV τ, ms | ||
| Median | 10.5 | 19* |
| Interquartile range | 5.25–15.75 | 14.5–21.5 |
| LV parameters | ||
| LV peak systolic pressure, mm Hg | ||
| Median | 77 | 110 |
| Interquartile range | 64.5–92 | 66–117 |
| LV end‐diastolic pressure, mm Hg | ||
| Median | 4.35 | 7* |
| Interquartile range | 3.073–4.448 | 5–13 |
| LV dP/dTmax, mm Hg/s | ||
| Median | 3495 | 6454 |
| Interquartile range | 2456–3425 | 3170–6211 |
| LV τ, ms | ||
| Median | 8 | 20* |
| Interquartile range | 8–10.5 | 11–23.5 |
Comparison of hemodynamic parameters in PAB vs sham rats. RV peak systolic and end‐diastolic pressures were increased in the PAB vs sham group, with increased systolic and diastolic eccentricity index and smaller LV end‐diastolic diameter. In PAB rats, eccentricity index increased in systole, but did not reach statistical significance (because of a large SD). Bpm indicates beats per minute; dP/dTmax, maximum intraventricular rate of pressure rise; HR, heart rate; LV, left ventricle; PAB, pulmonary artery band; and RV, right ventricle.
*P<0.05, † P<0.01 vs sham rats.
Regional Cardiac Hypertrophy and Fibrosis
Given markedly altered heart geometry after PAB, with septal flattening and displacement at the septal HP regions, we also evaluated regional heart morphological features and changes in expression of integrin‐β1 pathway and profibrotic signaling components. The histologic assessment and morphometry of serial hematoxylin and eosin–stained transverse sections indicated that PAB induced a significantly increased RV free‐wall thickness versus shams, whereas LV free‐wall thickness was similar between groups (Figure 2).
Figure 2.
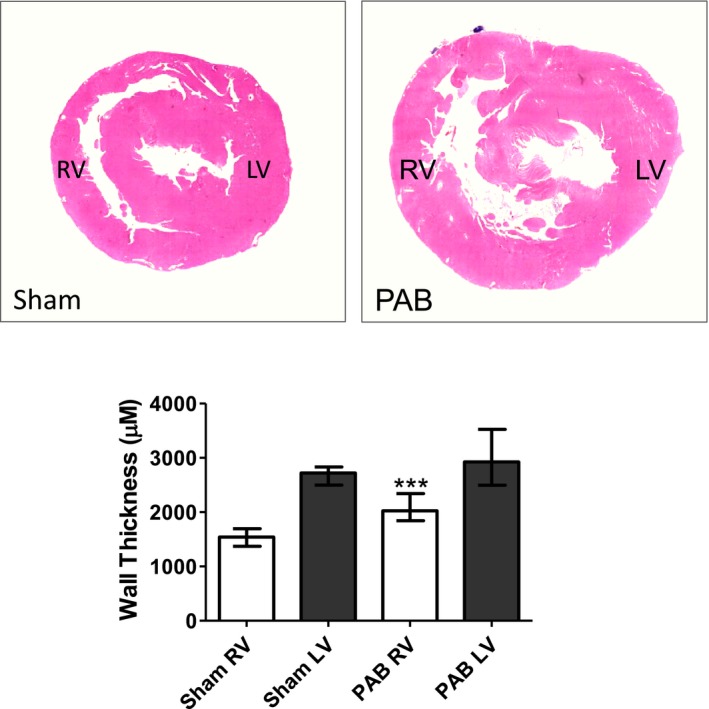
Representative hematoxylin and eosin (H&E)–stained histologic morphometry of rat hearts 6 weeks after sham and pulmonary artery banding (PAB) procedures. H&E‐stained heart cross‐sections derived from sham and PAB animals (top). Morphometric quantification indicated that PAB induced a significantly increased right ventricular (RV) free‐wall thickness vs shams, whereas left ventricular (LV) free‐wall thickness was similar between groups. Values are expressed as medians and interquartile range (n=5). ***P<0.0001 vs sham.
We then established that the RV free wall and HP regions of PAB rats demonstrated heightened deposition of Picrosirius red–positive collagen I, compared with sham controls. This finding was additionally confirmed by the fact that tissue fragments dissected from these regions demonstrated higher concentrations of hydroxyproline. Histochemical analysis also showed that the LV of PAB rats demonstrated increased collagen I fibrosis; however, this was significantly less compared with the RV and septal HP regions and was mostly limited to perivascular areas (Figure 3).
Figure 3.
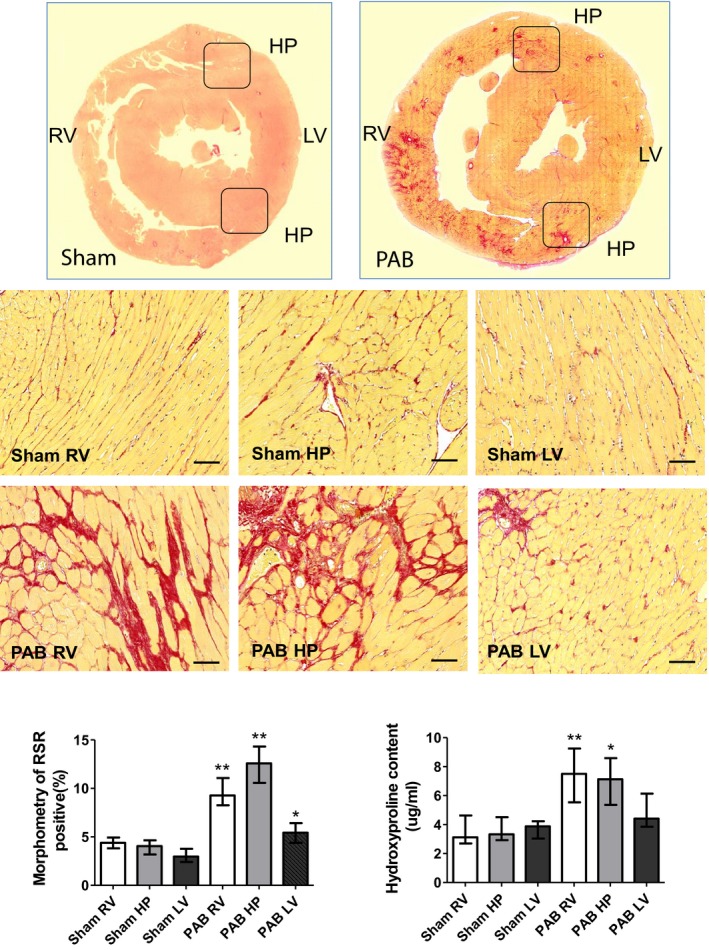
Representative Picrosirius red (PSR) staining of rat hearts 6 weeks after sham and pulmonary artery banding (PAB) procedures. PSR staining demonstrates that PAB‐induced right ventricular (RV) pressure load is associated with a remarkable accumulation of PSR‐positive collagen and myocyte hypertrophy in the RV and septal hinge‐point (HP) regions. In contrast, the left ventricle (LV) displays only disseminated foci of PSR‐positive material (top panel). Low magnification of heart histological cross‐sections derived from sham and PAB animals and stained with PSR (middle panels). Higher magnification of representative sections derived from the RV, hinge‐point (HP), and LV heart regions (bottom panels). Bars=50 μm. The bar graphs depict morphometric quantification of the areas occupied by PSR‐positive collagen, as well as values of hydroxyproline content. Values are expressed as medians and interquartile range (n=5). *P<0.05, **P<0.005 vs sham.
To further evaluate the effect of PAB on extracellular matrix (ECM) composition, we also evaluated regional changes in myocardial elastin content. In comparison to sham controls, Movat's staining in PAB rats showed increased elastin content in the RV and most prominently at the septal HPs, but much less in the LV (Figure 4).
Figure 4.
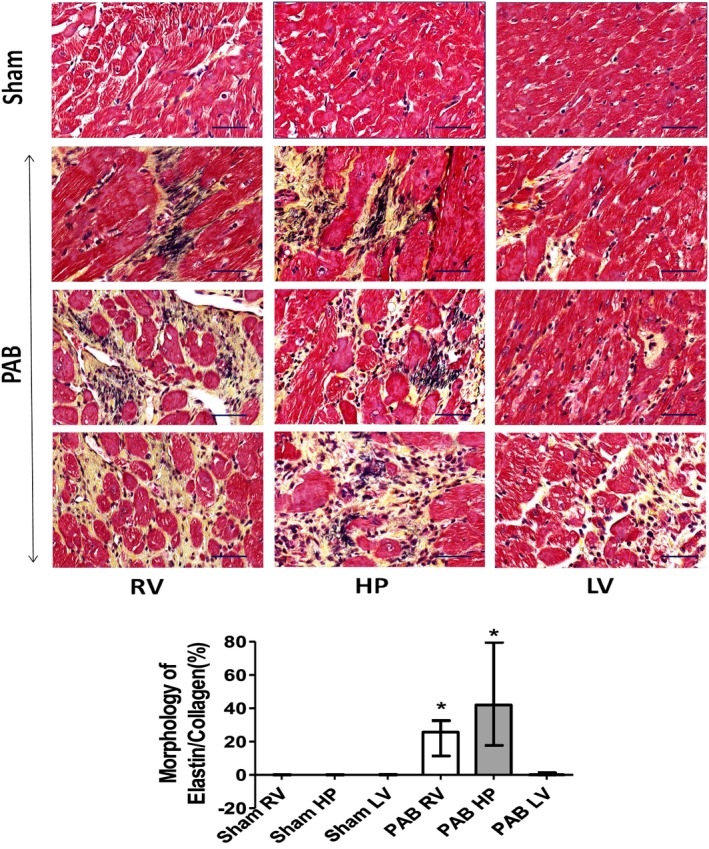
Representative Movat's staining of rat hearts 6 weeks after sham and pulmonary artery banding (PAB) procedures. Movat's staining depicting collagen I (yellow) and elastin (black) marking differences in regional cardiac fibrosis and extracellular matrix composition in fragments dissected from the right ventricle (RV), hinge‐point (HP), and left ventricle (LV) heart regions of sham and PAB rats (top). There is increased elastin deposition most prominently at the septal HP. Bars=50 μm. The bar graphs depict morphometric quantification of areas occupied by Movat's‐positive elastin and collagen (bottom). Values are expressed as medians and interquartile range (n=5). *P<0.05 vs sham.
Further immunohistochemical studies showed that all cardiac regions in PAB rats had increased numbers of interstitial cardiac fibroblasts, marked by the presence of immune‐detected vimentin and an increased number colocalized with the presence of Ki‐67 proliferative antigen (Figure 5A). Interestingly, detection of fluorescent wheat germ agglutinin, which marks the cell membranes of cardiomyocytes (Figure 5B) and hematoxylin and eosin staining (Figure 5C), in parallel sections, additionally demonstrated that, in PAB rats, individual RV and HP cardiomyocytes showed significant hypertrophy; LV cardiomyocytes hypertrophied, but less than RV and HP cells (Figure 5D). Furthermore, Natural antisense transcript of natriuretic peptide precursor A (NPPA), a sensitive marker for local stress, displayed granular cytoplasmic expression in cardiac muscle of PAB rats more at the HP and RV regions, whereas the LV showed only a mild increase in immune‐positive NPPA staining (Figure 5E).
Figure 5.
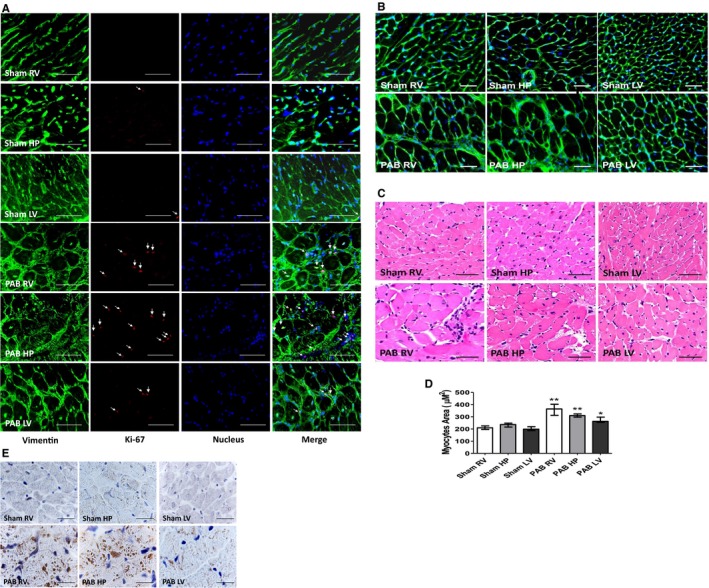
Representative micrographs of transverse sections of rat hearts sham and 6 weeks after pulmonary artery banding (PAB) procedures. A, Immunofluorescent detection of vimentin‐positive fibroblasts (green) and those displaying the presence of Ki‐67 proliferative antigen (red). Cell nuclei were stained blue, with 4′,6‐diamidino‐2‐phenylindole (DAPI). Bars=15 μm. B, Wheat germ agglutinin (WGA) interacting with cardiac myocyte cell membranes, detected with green fluorescein isothiocyanate (FITC; fluorescein) staining. Bar=20 μm. C, Hematoxylin and eosin staining for cardiomyocyte cross‐sectional area. Bar=20 μm. D, The bar graphs depict morphometric assessment of cardiac myocyte areas (n=5, with 200 cells per section). Values are expressed as medians and interquartile range. E, Immunohistochemical analysis of natriuretic peptide (NPPA) in cardiac muscle of sham and PAB rats. Bar=50 μm. HP indicates hinge point; LV, left ventricle; NPPA, Natural antisense transcript of natriuretic peptide precursor A; and RV, right ventricle. *P<0.05, **P<0.001 vs sham.
Regional Expression of Diverse Integrins
Given the varying regional hypertrophy and fibrosis after PAB, we evaluated whether varying levels of regional mechanotransduction molecules would diversely modulate downstream signaling pathways, likely engaging activation of different integrins, including the collagen binding α1 and 11 integrins. Indeed, although we observed rather similar levels of PAB‐induced increases in β1A‐ and α1‐integrin protein expression (detected by Western blots) in all regions, levels of β1D‐ and α11‐integrin were even higher in the septal HP regions (Figure 6). Moreover, integrin‐β1A (detected by immunofluorescence) could be localized to interstitial fibroblasts, myofibroblasts, and smooth muscle cells in those regions, but not to the cardiomyocytes (data not shown). In contrast, integrin‐β1D was predominantly expressed at the cardiomyocyte cell membrane.
Figure 6.
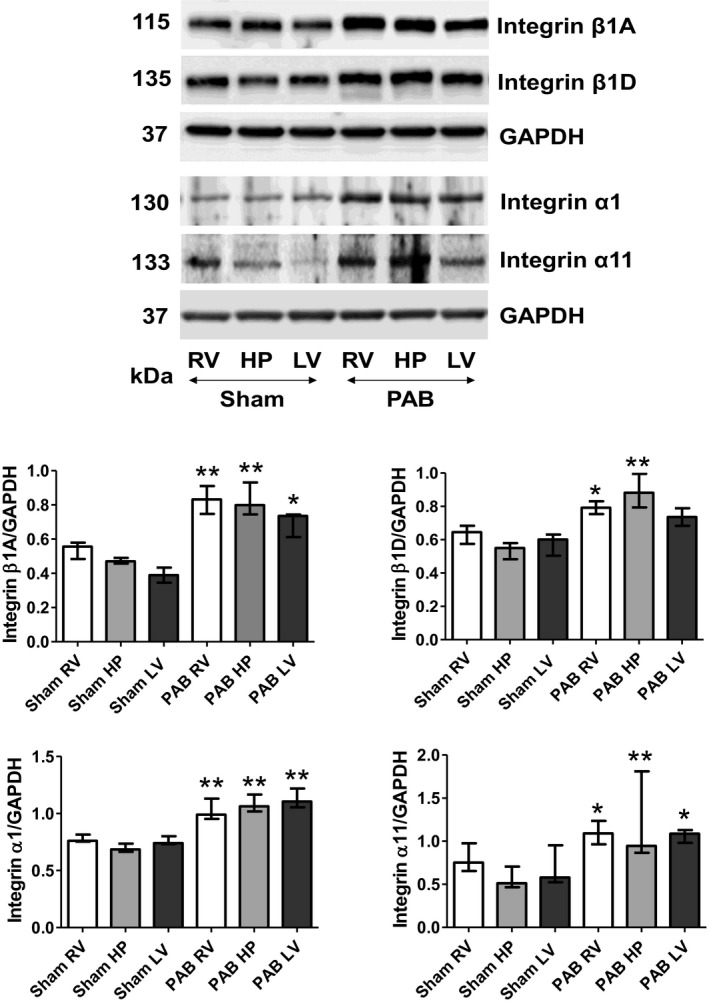
Right ventricular (RV) pressure load diversely upregulates levels of indicated integrins in heart regions 6 weeks after sham and pulmonary artery banding (PAB) procedures. Representative Western blots detecting indicated integrins and their quantitative assessments in the RV, hinge‐point (HP), and left ventricle (LV) regions of sham and PAB rats. Values are expressed as medians and interquartile range (n=5). *P<0.05, **P<0.001 vs sham.
Assessment of Integrin‐Induced Downstream Signaling and Profibrotic Signaling
Given the biventricular increase in integrin‐β1 signaling after PAB, we also assessed 3 major components of downstream integrin‐mediated signaling. We established that expression of phosphorylated FAK, integrin‐linked kinase, and phosphorylated ERK were significantly increased in the RV free‐wall and septal HP regions, compared with their counterparts in sham controls. Meaningfully, these proteins were only slightly unregulated in the LV of PAB rats (Figure 7A). Moreover, we also found that the RV and septal HP of PAB hearts demonstrated heightened protein expression of the profibrotic signaling pathway components TGF‐β1, Smad‐2, Smad‐3, phosphorylated Smad‐2/3, and CTGF. These results are consistent with activation of TGF‐β1. However, expression of these proteins was only mildly elevated in the LV (Figure 7B).
Figure 7.
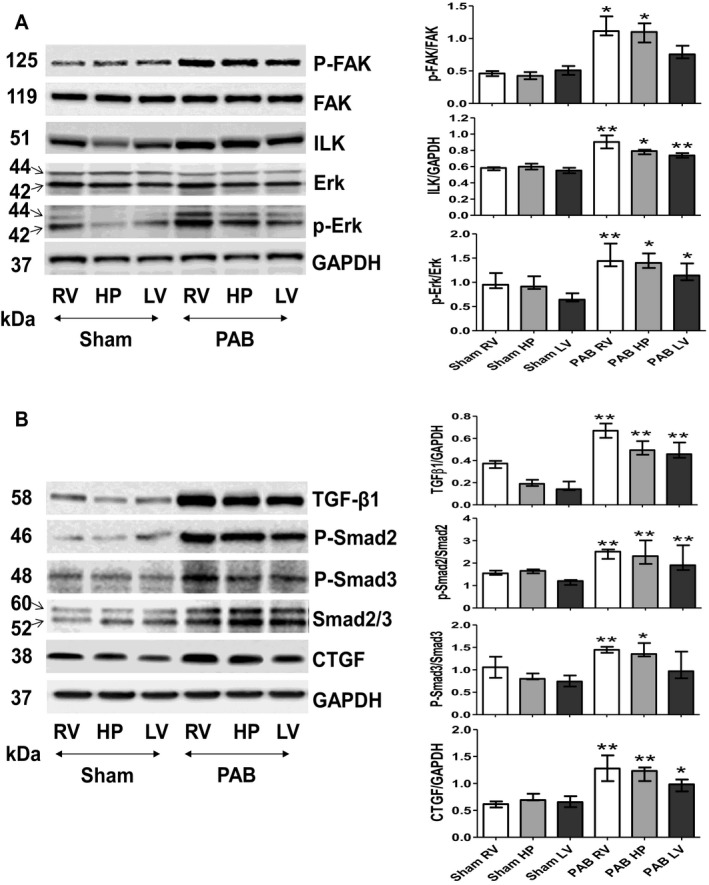
Application of pulmonary artery banding (PAB) induces a diverse upregulation in activity of the integrin‐dependent signals that consequently trigger profibrotic pathway 6 weeks after sham and PAB procedures. A, Representative Western blots detecting basic regional expression of protein levels and phosphorylation rates of the integrin‐induced downstream signaling pathway components (focal adhesion kinase [FAK], integrin‐linked kinase [ILK], and extracellular signal–regulated kinase [ERK]). Values are expressed as medians and interquartile range (n=5). * P<0.05, ** P<0.001 vs sham. B, Representative Western blots detecting basic regional expression of protein levels and phosphorylation rates of profibrotic components transforming growth factor (TGF)‐β1, Smad 2, Smad 3, and connective tissue growth factor (CTGF) in sham and PAB‐exposed hearts. Quantification analyses (n=5). Values are expressed as medians and interquartile range. *P<0.05, **P<0.001 vs sham; . HP indicates hinge point; LV, left ventricle; and RV, right ventricle.
Integrins Upregulate TGF‐β1 Expression and Collagen Type I Deposition in RV, LV, and HP Region Fibroblasts in Response to in Vitro Mechanical Stress
Given our previously mentioned data acquired from in vivo experiments, we next studied the possible effects of mechanical stress on confluent cultures of normal cardiac fibroblasts, derived from control (healthy rats) and in cardiac fibroblasts isolated from the RV, HP, and LV from Sugen‐hypoxia induced PH rats. Fibroblasts that were plated on membranes of the stretch apparatus chamber (Flexcell FX‐4000) were kept for 24 hours in the presence or absence of cyclic 20% equiaxial stretch at 1.7 Hz, as previously described. All cultures were also kept in the presence or absence of the integrin inhibitor BTT‐3033. We first found that primary cultures of cardiac fibroblasts isolated from 3 myocardial regions of normal rats showed no morphologic differences (data not shown). However, we observed that 24‐hour culture of fibroblasts isolated from all 3 myocardial regions significantly upregulated their production of collagen type I in response to mechanical stress, compared with nonstretched controls (Figure 8A and 8C). Nonstretched and stretched cells from the RV and HP regions from PH rats showed increased collagen production, with no further increase in response to stretch. Collagen production in cell cultures isolated from the LV in PH animals showed significant upregulation of collagen type I production in response to mechanical stress (Figure 8B and 8C). Addition of the integrin inhibitor (BTT‐3033) completely suppressed collagen production in response to mechanical stretch in both control and PH rats (Figure 8A through 8C). Because BTT‐3033 also inhibited collagen I production in nonstretched cultures, this suggests that even basal production of collagen in the heart could be modulated by integrin signaling.
Figure 8.
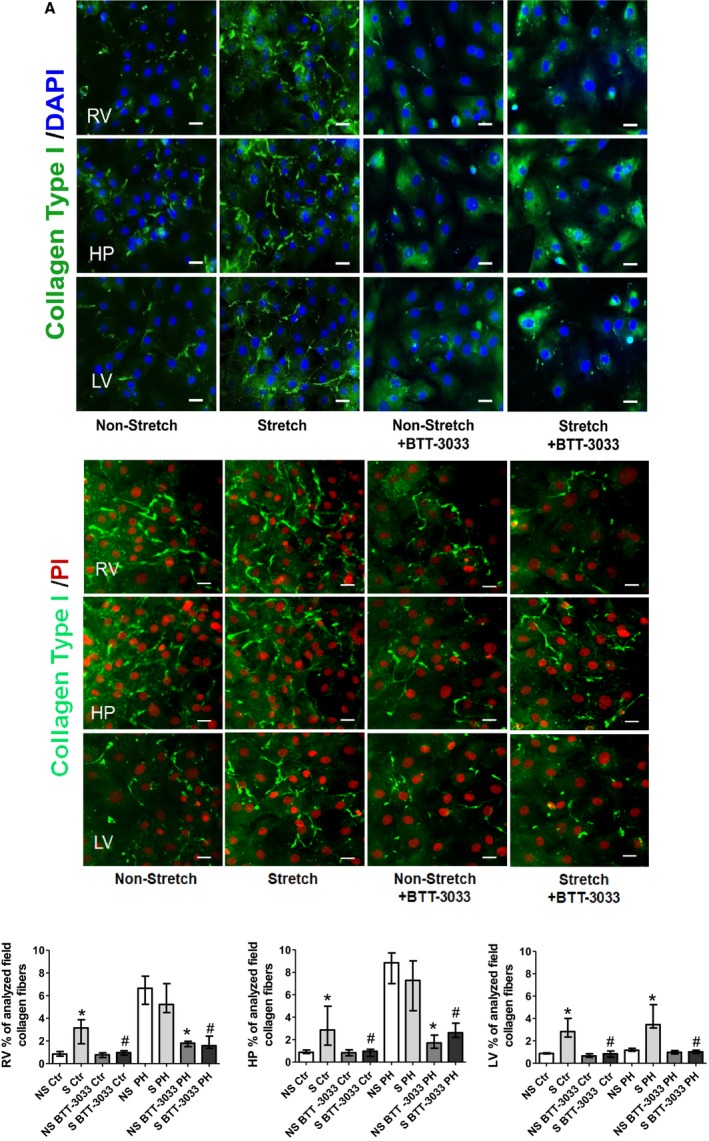
Cyclic mechanical stretch (24 hours long) of cultured cardiac fibroblasts isolated from the indicated heart regions. A, From control rats, induced a strong upregulation of immune‐detected collagen I deposition, compared with the nonstretched counterparts. This phenomenon has been suppressed in parallel cultures exposed to integrin inhibitor (BTT‐3033) (top panels). B, From pulmonary hypertension rats, indicated strong immune‐detected collagen I deposition in non‐stretched and stretched cells of right ventricular (RV) and hinge‐point (HP) regions, but not in left ventricle (LV). C, Representative fields of cardiac fibroblasts immune stained with collagen type I (green) and nuclear 4′,6‐diamidino‐2‐phenylindole (DAPI) staining (blue)/propidium iodide (PI) (red). Bar=50 μm. The bar graph represents the quantification data of the percentage of analyzed collagen fibers per field. Values are expressed as medians and interquartile range (n=4). Ctr indicates control; NS, nonstretch; PH, pulmonary hypertension; and S, stretch. * P<0.05 vs NS group; # P<0.05 vs S group.
Results of the next series of experiments, in which we tested RV‐derived fibroblasts, provided more details on RV integrin β1‐dependent profibrotic signaling. First, they demonstrated that cultures of RV‐derived fibroblasts, stretched for 24 hours, contained more pronounced immune staining of antibodies to integrin‐β1. They also contained more α‐smooth muscle actin–positive cells that could be defined as myofibroblasts and displayed more pronounced expression of TGF‐β1 as well as increased wrinkles in gel assays, suggesting myofibroblast activity (Figures 9 and 10). Moreover, stretched cultures of cardiac fibroblasts also demonstrated higher expression of immune‐detected CTGF than nonstretched controls. We also found that 24‐hour‐long treatment with the integrin inhibitor BTT‐3033 markedly decreased their expression of integrin‐β1, α‐smooth muscle actin, and TGF‐β1 in both nonstretched and stretched cultures and contractile function in gel wrinkle assays. Meaningfully, treatment with BTT‐3033 did not diminish the stretch‐induced upregulation in CTGF expression. This latest observation indicates that mechanical forces might also induce parallel profibrotic pathways, containing CTGF, but not dependent on the prior activation of integrin‐β1. The additional quantitative assessments of Western blots, detecting components of profibrotic pathways, confirmed immunohistochemistry results (Figure 10).
Figure 9.
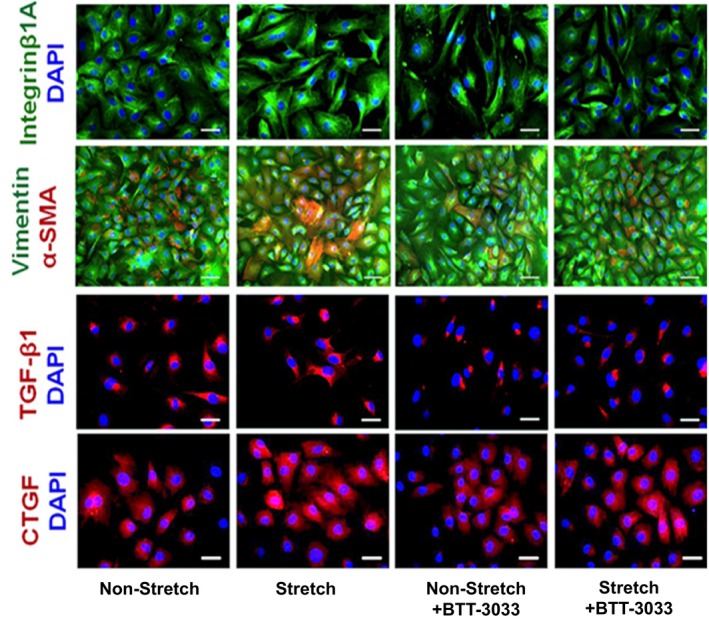
Representative micrographs depicting cultures of cardiac fibroblasts isolated from right ventricle (RV) that were either kept still or subjected to 24‐hour‐long mechanical stretching in the presence or absence of integrin inhibitor (BTT‐3033). The parallel cultures were immune stained with specific antibodies recognizing integrin‐β1A (green), vimentin (green), α‐smooth muscle actin (α‐SMA) (red), transforming growth factor (TGF)‐β1 (red), and connective tissue growth factor (CTGF) (red), combined with blue 4′,6‐diamidino‐2‐phenylindole (DAPI) nuclear staining. Bars=50 μm.
Figure 10.
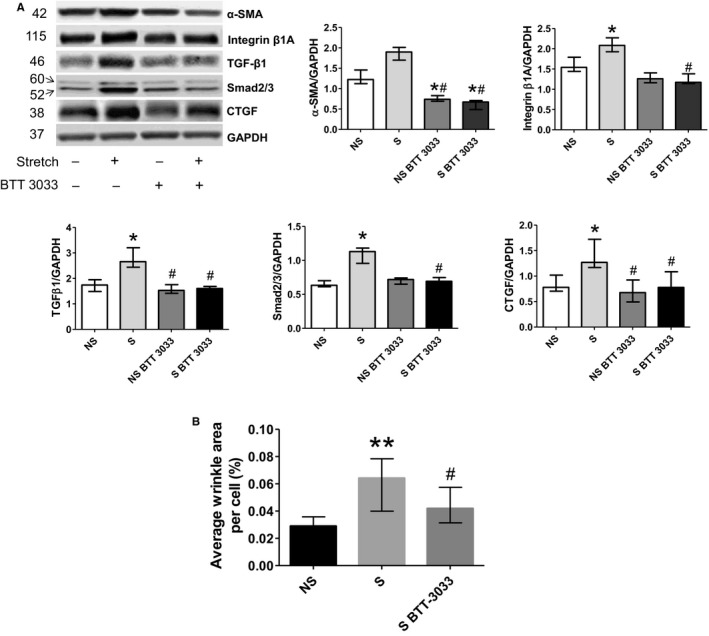
Western blot assessment of profibrotic pathway components in right ventricular (RV) cardiac fibroblast cultures. A, Cyclic mechanical stretch of cultured cardiac fibroblasts causes upregulation in the net expressions of their α‐smooth muscle actin (α‐SMA), integrin‐β1, and profibrotic signaling pathway components (transforming growth factor [TGF]‐β1), Smad 2/3, and connective tissue growth factor [CTGF]). Western blots with indicated antibodies were assessed by densitometry and normalized against the GAPDH‐positive bands (n=4). Values are expressed as medians and interquartile range. *P<0.05 vs nonstretch (NS) group; # P<0.05 vs stretch (S) group. B, Percentage of analyzed collagen fibers and average cell surface wrinkles visible area to indicate cell contraction in gel assays. Values are expressed as medians and interquartile range (n=5). **P<0.005 vs NS group; # P<0.05 vs S group.
Discussion
The mechanotransduction of RV hypertension to ECM remodeling and fibrosis and the transmission of these effects to the LV are inadequately characterized. In this study, we show that integrin‐β1A+D, in association with the fibrillar collagen binding integrins α1 and α11, appear to transduce the mechanical stress of RV hypertension to regional RV and septal HP TGF‐β1 signaling and myocardial fibrosis. In contrast, although LV geometry was substantially distorted by the hypertensive RV and LV pressures were elevated, in the experimental time frame of 6 weeks, the LV developed only mild fibrosis and maintained overall good systolic function. These results suggest the possibility of a hitherto undescribed adaptive regional mechanotransduction mechanism that may reduce LV dysfunction in the time frame produced by this experiment, through buffering effects at the septal HPs. These findings also suggest the possibility of a therapeutic window before extensive LV fibrosis and dysfunction ensue.
Regional Mechanotransduction in Isolated RV Hypertension
Given the prominent in vivo findings of RV and septal HP region fibrosis, and in cardiac fibroblast cell cultures of profibrotic signaling, and by the reduction of collagen deposition with integrin‐β1 pharmacological inhibition in vitro, our results suggest that RV hypertension and altered regional HP geometry, secondary to septal shift, induce a mechanical‐molecular wavefront of “outside in” integrin signaling that triggers regional myocardial fibrosis through the TGF‐β1 pathway in the RV and septal HPs and to a lesser degree in the LV. The marked regional activation and phosphorylation of Smad‐2/3 and upregulation of collagen‐binding integrins also suggest a positive feedback loop whereby geometrical change and mechanical wall stress induce regional Smad2/3 activation, leading to RV and HP fibrosis. Collagens may then bind to and activate collagen‐binding integrins, leading to excessive myocardial stiffness in these regions.24 Integrin‐β1A was predominantly detectable in cardiac fibroblasts and myofibroblasts. Upregulation of regional integrin‐β1A and integrin‐β1D expression was concomitant with regional changes in geometry (and hence wall stress), as seen on echocardiography, ECM remodeling, collagen deposition, and myocyte hypertrophy.21 Immunohistochemistry staining of phosphorylated Smad2/3 is congruent for activation of TGF‐β1 in these regions of remodeled myocardium. Downstream integrin signaling through molecules such as integrin‐linked kinase, FAK, and ERK25 paralleled these findings, with highest expression in the RV, followed by the septal HPs, and relatively lower expression in the LV free wall.
Our in vitro experiments further delineated this pathway, demonstrating that mechanical stretch induced aggregation of integrin‐β1 on the cell surface. This distribution change may allow outside‐in signaling, because the integrin‐β1A+D extracellular domains may allow interactions between (myo)fibroblasts and/or myocytes and the ECM to “sense” the mechanical wall stress of RV hypertension, septal shift, altered geometry, and increased fibrillar collagen, to trigger RV and septal HP biochemical signaling and ECM remodeling.26, 27 We previously identified the TGF‐β1 pathway as a central mediator of fibrosis in RV pressure load.13 TGF‐β1 binds integrin‐β1, which reciprocally activates latent TGF‐β1 and its profibrotic signaling.28, 29, 30 Consistent with our in vitro results, integrin‐β1 inhibition in fibroblasts blocks TGF‐β1 activation and prevents fibrosis.31, 32 Activated TGF‐β1 induces collagen type I production through signaling cascades (eg, Smad2/3), which are active in our models.33, 34, 35, 36 This was supported by our in vitro experiments that showed that blocking integrins decreased contractile activity of stretched fibroblasts. FAK phosphorylation with upregulation of ERK may present an interesting link in RV hypertension between integrin‐β1 upregulation and fibrosis through TGF‐β1 and endothelin‐1 signaling.13, 15, 16, 28, 29
CTGF is also upregulated by integrin‐β1, which is reciprocally upregulated by CTGF and TGF‐β1.34, 35, 36 Our in vitro experiments show that integrin‐β1 inhibition suppresses mechanical stretch‐mediated TGF‐β1 expression and collagen type I production in regionally derived fibroblasts. This reciprocal interaction and canonical inside‐out integrin signaling may be active during isolated RV hypertension. Nonetheless, CTGF remained elevated, with integrin‐β1 inhibition, suggesting that alternative pathways are active and need further delineation. CTGF also strongly upregulates elastin production, although not through integrin signaling. This suggests that therapies targeting mechanotransduction pathways to decrease collagen, while increasing elastin, may be feasible to produce a more compliant myocardium.
Despite the concomitant development of distorted LV geometry and markedly increased LV diastolic pressures, fibrosis was only mild in the LV free wall and predominantly in a perivascular distribution. Likewise, LV systolic function was maintained. Together with increased collagen I deposition at the septal HPs, we also observed increased septal HP deposition of extracellular matrix elastin. Elastin is significantly more compliant than collagen and may attenuate the increased stiffness induced by excess collagen I deposition.37, 38, 39 Likewise, expression of the stress marker NPPA was increased at the septal HPs.40 Taken together, our results suggest that the septal HP regions, where the RV and LV adjoin, while sustaining extensive stress and fibrosis, may also act as more compliant, elastin‐rich buffer zones that attenuate LV free‐wall injury. This possible adaptive mechanism has not been previously described and may act to maintain LV function, as we observed overall good LV function in this study. Preserved LV systolic function may also represent an early stage of pathological features, in which 6 weeks of PAB allow the aforementioned compensatory remodeling of the mechanically stressed heart that postpones fibrotic hypertrophy of the LV and ultimately heart failure.
Nonetheless, impaired RV and LV diastolic dysfunction was evident by delayed relaxation (prolonged τ) and increased RV and LV end‐diastolic pressures. These are important findings because end‐diastolic pressures correlate with adverse outcomes in human cardiac disease.41 RV diastolic dysfunction may be worsened by increased RV hypertrophy and fibrosis,42, 43 which is consistent with our own previous results and other studies.44, 45, 46 Although LV diastolic impairment may result from its geometrical compression by the hypertensive RV, there may be some contribution of increased LV collagen deposition. Consequently, treatments addressing RV and LV fibrosis may be beneficial.47 In parallel to fibrosis, our in vivo and in vitro results suggest that, in response to pressure‐stress and mediated by integrin‐β1A+D signaling, the pressure‐stressed myocardium upregulates prohypertrophic factors, such as TGF‐β1 and endothelin‐1,48, 49 most prominently in the RV and septal HPs and less in the LV.50, 51 Accordingly, LV myocytes were less hypertrophied than in the RV and septal HP regions, there was less cell proliferation in the LV, and gross LV wall thickness in PAB rats was not increased compared with sham controls. Taken together, our results suggest that β1‐integrins may mediate coexistent adaptive and maladaptive mechanotransduction mechanisms and that the balance between these may determine biventricular function, as frequently occurs in biological systems. Consistent with our previous studies in animal models, we found increased LV myocyte hypertrophy and no decrease in LV mass. These findings are discrepant with those of Hardziyenka et al, who found LV myocyte atrophy and decreased LV free‐wall mass in chronic thromboembolic PH and in a rat model of LV failure.52 Although the reason for these discrepant results is not apparent, the increased LV pressures resulting from left septal shift may have led to a mild LV hypertrophic response. Our RV results are also more severe than the adaptive changes seen in other studies using a PAB model but are similar to those where PAB caused severe constriction.6, 43
Limitations
Our findings are limited to RV hypertension induced by PAB and may not automatically apply to pulmonary arterial hypertension. We did not investigate the time course of development of RV and LV fibrosis and hence ultimate development of LV fibrosis and the possibility of a therapeutic window remain conjectures. We did not demonstrate whether blocking elastin production at the septal HPs increases LV fibrosis. There was low variation in fibrosis levels between individual animals and, consequently, we could not demonstrate an association between the degree of fibrosis and the severity of RV dysfunction, an association previously found in other articles.42 We have previously demonstrated apoptosis in our models as a mechanism of myocardial dysfunction.9 This analysis was not repeated in these experiments. We did not directly measure regional myocardial compliance. Histological study of fibrosis and integrin signaling pathways in human tissue from patients with PH would increase the clinical translation of our experimental findings, and this constitutes a topic worthy of future study.
Conclusion
In conclusion, isolated RV hypertension displaces the interventricular septum, leading to geometrical changes that promote RV and septal HP integrin‐β1 and profibrotic signaling, leading to increased fibrosis in these regions. Despite LV geometrical distortion and markedly increased end‐diastolic pressures, this mechanical‐molecular remodeling was attenuated in the LV in association with increased septal HP elastin deposition, suggesting a possible mechanotransduction adaptive mechanism. Future studies should investigate whether these results allow for a therapeutic window before more extensive development of LV fibrosis and dysfunction.
Sources of Funding
This work was supported by a grant from the Heart and Stroke Foundation of Canada (G‐16‐00014082).
Disclosures
Dr Connelly is a recipient of a New Investigator Award from the Canadian Institutes of Health Research and an Early Researcher Award from the Ministry of Ontario.
(J Am Heart Assoc. 2018;7:e007928 DOI: 10.1161/JAHA.117.007928.)
References
- 1. Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–S66. [DOI] [PubMed] [Google Scholar]
- 2. Belenkie I, Horne SG, Dani R, Smith ER, Tyberg JV. Effects of aortic constriction during experimental acute right ventricular pressure loading: further insights into diastolic and systolic ventricular interaction. Circulation. 1995;92:546–554. [DOI] [PubMed] [Google Scholar]
- 3. Mitchell JR, Whitelaw WA, Sas R, Smith ER, Tyberg JV, Belenkie I. RV filling modulates LV function by direct ventricular interaction during mechanical ventilation. Am J Physiol Heart Circ Physiol. 2005;289:H549–H557. [DOI] [PubMed] [Google Scholar]
- 4. Damiano RJ Jr, La Follette P Jr, Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261:H1514–H1524. [DOI] [PubMed] [Google Scholar]
- 5. Hirata M, Ousaka D, Arai S, Okuyama M, Tarui S, Kobayashi J, Kasahara S, Sano S. Novel model of pulmonary artery banding leading to right heart failure in rats. Biomed Res Int. 2015;2015:753210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. [DOI] [PubMed] [Google Scholar]
- 7. Sanz J, Dellegrottaglie S, Kariisa M, Sulica R, Poon M, O'Donnell TP, Mehta D, Fuster V, Rajagopalan S. Prevalence and correlates of septal delayed contrast enhancement in patients with pulmonary hypertension. Am J Cardiol. 2007;100:731–735. [DOI] [PubMed] [Google Scholar]
- 8. Freed BH, Gomberg‐Maitland M, Chandra S, Mor‐Avi V, Rich S, Archer SL, Jamison EB Jr, Lang RM, Patel AR. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson. 2012;14:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nielsen EA, Okumura K, Sun M, Hjortdal VE, Redington AN, Friedberg MK. Regional septal hinge‐point injury contributes to adverse biventricular interactions in pulmonary hypertension. Physiol Rep. 2017;5:e13332 DOI: 10.14814/phy2.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henderson NC, Sheppard D. Integrin‐mediated regulation of TGFbeta in fibrosis. Biochim Biophys Acta. 2013;1832:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. [DOI] [PubMed] [Google Scholar]
- 12. Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M, Schwartz MA. Integrins in mechanotransduction. Curr Opin Cell Biol. 2013;25:613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Apitz C, Honjo O, Friedberg MK, Assad RS, Van Arsdell G, Humpl T, Redington AN. Beneficial effects of vasopressors on right ventricular function in experimental acute right ventricular failure in a rabbit model. Thorac Cardiovasc Surg. 2012;60:17–23. [DOI] [PubMed] [Google Scholar]
- 14. Ruwhof C, van der Laarse A. Mechanical stress‐induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc Res. 2000;47:23–37. [DOI] [PubMed] [Google Scholar]
- 15. Friedberg MK, Cho MY, Li J, Assad RS, Sun M, Rohailla S, Honjo O, Apitz C, Redington AN. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor‐beta1 signaling and is abrogated by angiotensin receptor blockade. Am J Respir Cell Mol Biol. 2013;49:1019–1028. [DOI] [PubMed] [Google Scholar]
- 16. Nielsen EA, Sun M, Honjo O, Hjortdal VE, Redington AN, Friedberg MK. Dual endothelin receptor blockade abrogates right ventricular remodeling and biventricular fibrosis in isolated elevated right ventricular afterload. PLoS One. 2016;11:e0146767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blyth KG, Groenning BA, Martin TN, Foster JE, Mark PB, Dargie HJ, Peacock AJ. Contrast enhanced‐cardiovascular magnetic resonance imaging in patients with pulmonary hypertension. Eur Heart J. 2005;26:1993–1999. [DOI] [PubMed] [Google Scholar]
- 18. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110:3221–3228. [DOI] [PubMed] [Google Scholar]
- 20. Sun M, Chen M, Liu Y, Fukuoka M, Zhou K, Li G, Dawood F, Gramolini A, Liu PP. Cathepsin‐L contributes to cardiac repair and remodelling post‐infarction. Cardiovasc Res. 2011;89:374–383. [DOI] [PubMed] [Google Scholar]
- 21. Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107:1046–1052. [DOI] [PubMed] [Google Scholar]
- 22. Modarressi A, Pietramaggiori G, Godbout C, Vigato E, Pittet B, Hinz B. Hypoxia impairs skin myofibroblast differentiation and function. J Invest Dermatol. 2010;130:2818–2827. [DOI] [PubMed] [Google Scholar]
- 23. Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha‐smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Talior‐Volodarsky I, Arora PD, Wang Y, Zeltz C, Connelly KA, Gullberg D, McCulloch CA. Glycated collagen induces alpha11 integrin expression through TGF‐beta2 and Smad3. J Cell Physiol. 2015;230:327–336. [DOI] [PubMed] [Google Scholar]
- 25. Chen C, Li R, Ross RS, Manso AM. Integrins and integrin‐related proteins in cardiac fibrosis. J Mol Cell Cardiol. 2016;93:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsueh WA, Law RE, Do YS. Integrins, adhesion, and cardiac remodeling. Hypertension. 1998;31:176–180. [DOI] [PubMed] [Google Scholar]
- 27. MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res. 2000;46:257–263. [DOI] [PubMed] [Google Scholar]
- 28. Schultz‐Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, Murphy‐Ullrich JE. Regulation of transforming growth factor‐beta activation by discrete sequences of thrombospondin 1. J Biol Chem. 1995;270:7304–7310. [DOI] [PubMed] [Google Scholar]
- 29. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. [DOI] [PubMed] [Google Scholar]
- 30. Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, Caldarone CA, Hinz B. Integrins αvβ5 and αvβ3 promote latent TGF‐beta1 activation by human cardiac fibroblast contraction. Cardiovasc Res. 2014;102:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy‐Hulbert A, Adams RH, Sheppard D. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu S, Xu SW, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, Leask A. Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J Cell Sci. 2010;123:3674–3682. [DOI] [PubMed] [Google Scholar]
- 33. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. [DOI] [PubMed] [Google Scholar]
- 34. Hu X, Li N, Tao K, Fang X, Liu J, Wang Y, Wang H, Shi J, Ji P, Cai W, Bai X, Zhu X, Han J, Hu D. Effects of integrin ανβ3 on differentiation and collagen synthesis induced by connective tissue growth factor in human hypertrophic scar fibroblasts. Int J Mol Med. 2014;34:1323–1334. [DOI] [PubMed] [Google Scholar]
- 35. Sonnylal S, Shi‐Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, Leask A, Abraham D, Bou‐Gharios G, de Crombrugghe B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010;62:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee CH, Shah B, Moioli EK, Mao JJ. CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J Clin Invest. 2010;120:3340–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodgers UR, Weiss AS. Cellular interactions with elastin. Pathol Biol. 2005;53:390–398. [DOI] [PubMed] [Google Scholar]
- 38. Steed MM, Tyagi SC. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011;15:1927–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henderson BC, Sen U, Reynolds C, Moshal KS, Ovechkin A, Tyagi N, Kartha GK, Rodriguez WE, Tyagi SC. Reversal of systemic hypertension‐associated cardiac remodeling in chronic pressure overload myocardium by ciglitazone. Int J Biol Sci. 2007;3:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sergeeva IA, Christoffels VM. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim Biophys Acta. 2013;1832:2403–2413. [DOI] [PubMed] [Google Scholar]
- 41. Burkett DA, Slorach C, Patel SS, Redington AN, Ivy DD, Mertens L, Younoszai AK, Friedberg MK. Impact of pulmonary hemodynamics and ventricular interdependence on left ventricular diastolic function in children with pulmonary hypertension. Circ Cardiovasc Imaging. 2016;9:e004612 DOI: 10.1161/CIRCIMAGING.116.004612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rain S, Andersen S, Najafi A, Gammelgaard Schultz J, da Silva Goncalves Bos D, Handoko ML, Bogaard HJ, Vonk‐Noordegraaf A, Andersen A, van der Velden J, Ottenheijm CA, de Man FS. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail. 2016;9:e002636. doi: 10.1161/circheartfailure.115.002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mendes‐Ferreira P, Santos‐Ribeiro D, Adao R, Maia‐Rocha C, Mendes‐Ferreira M, Sousa‐Mendes C, Leite‐Moreira AF, Bras‐Silva C. Distinct right ventricle remodeling in response to pressure overload in the rat. Am J Physiol Heart Circ Physiol. 2016;311:H85–H95. [DOI] [PubMed] [Google Scholar]
- 44. Okumura K, Kato H, Honjo O, Breitling S, Kuebler WM, Sun M, Friedberg MK. Carvedilol improves biventricular fibrosis and function in experimental pulmonary hypertension. J Mol Med. 2015;93:663–674. [DOI] [PubMed] [Google Scholar]
- 45. Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. [DOI] [PubMed] [Google Scholar]
- 46. Borgdorff MA, Koop AM, Bloks VW, Dickinson MG, Steendijk P, Sillje HH, van Wiechen MP, Berger RM, Bartelds B. Clinical symptoms of right ventricular failure in experimental chronic pressure load are associated with progressive diastolic dysfunction. J Mol Cell Cardiol. 2015;79:244–253. [DOI] [PubMed] [Google Scholar]
- 47. Handoko ML, de Man FS, Allaart CP, Paulus WJ, Westerhof N, Vonk‐Noordegraaf A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: lessons from the left heart. Eur Respir Rev. 2010;19:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ayca B, Sahin I, Kucuk SH, Akin F, Kafadar D, Avsar M, Avci II, Gungor B, Okuyan E, Dinckal MH. Increased transforming growth factor‐beta levels associated with cardiac adverse events in hypertrophic cardiomyopathy. Clin Cardiol. 2015;38:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Olson AK, Ledee D, Iwamoto K, Kajimoto M, O'Kelly Priddy C, Isern N, Portman MA. C‐Myc induced compensated cardiac hypertrophy increases free fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol. 2013;55:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. [DOI] [PubMed] [Google Scholar]
- 51. Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGF‐beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hardziyenka M, Campian ME, Reesink HJ, Surie S, Bouma BJ, Groenink M, Klemens CA, Beekman L, Remme CA, Bresser P, Tan HL. Right ventricular failure following chronic pressure overload is associated with reduction in left ventricular mass: evidence for atrophic remodeling. J Am Coll Cardiol. 2011;57:921–928. [DOI] [PubMed] [Google Scholar]