

Abstract
Methemoglobinemia is a rare overlooked differential diagnosis in patients presented with cyanosis and dyspnea unrelated to cardiopulmonary causes. Our patient is 29 year old Indian non-smoker male, his story started 6 months prior to presentation to our center when he had generalized fatigue and discoloration of hands. He presented with persistent polycythemia with elevated hemoglobin level. The patient was misdiagnosed in another center as polycythemia and treated with Imatinib. The diagnosis of PV was revisited and ruled out in view of negative JAK2, normal erythropoietin level and absence of features of panmyelosis. Clinical cyanosis and lowoxygen saturation in the presence of normal arterial oxygen tension was highly suggestive of methemoglobinemia. Arterial blood gas revealed a methemoglobin level of 38% (normal: 0-1.5%). Cytochrome B5 reductase (Methemoglobin reductase B) was deficient at level of <2.6 U/g Hb) (normal: 6.6-13.3), consistent with methemoglobin reductase (cytochrome b5) deficiency and hence the diagnosis of congenital methemoglobinemia was established. The role of Imatinib in provoking methemoglobinemia is questionable and association between Imatinib and methemoglobinemia never described before. In our case, there were no other offending drugs in aggravating the patients’ symptoms and cyanosis. The patient started on Vitamin C 500 mg once daily for which he responded well with less cyanosis and significant reduction of methemoglobin level. Congenital methemoglobinemia is a rare underreported hemoglobin disease and often clinically missed. Upon extensive review of English literature for cases of congenital methemoglobinemia due to deficiency of cytochrome b5 reductase, we found 23 cases diagnosed as type I (including the case reported here). 17 cases (~74%) of type I and 6 cases (27%) of type II. There is male predominance 73% versus 26% in females. Almost half of reported cases 12 cases (52%) are Indian, 2 Japanese, 3 English, 2 Arabic, one case Spanish and one case Italian. For type I, the median calculated age is 31 years with cyanosis and shortness of breath being the most common sign and symptoms. For type II: Six cases were reported in English literature, all in pediatric age group with median calculated age at presentation is 6 years with neurologic manifestations and mental retardation are the most common type II associated symptoms. Due to lack of systematic epidemiological studies, congenital methemoglobinemia is under diagnosed as it is under investigated and usually overlooked especially when presenting in adulthood and in absence of obvious acquired agents.
Key words: hemoglobin, congenital, methemoglobinemia
Introduction
Methemoglobinemia is a rare overlooked differential diagnosis in patients presented with cyanosis and dyspnea unrelated to cardiopulmonary causes. Methemoglobinemia is usually asymptomatic, even when methemoglobin (metHb) levels are as high as 40% of the total hemoglobin (Hb) value.1
Hereditary congenital methemoglobinemia due to deficiency of nicotinamide adenine dinucleotide (NADH) cytochrome b5 reductase enzyme is even extremely rare recessively inherited disorder that is not well documented in literature.
Here, we report a rare case of methemoglobinemia presented with persistent polycythemia in absence of cardiopulmonary causes led to false diagnosis of polycythemia vera for which the patient needlessly undergone bone marrow examination and received Imatinib.
Case Report
Our patient is 29 year old Indian nonsmoker male, his story started 6 months prior to presentation to our center when he had generalized fatigue and discoloration of hands (Figure 1). His CBC revealed high red cell count 6.7 (4.5-5.5 × 106/uL), elevated hemoglobin at 20 (13-17 g/dL) with normal leukocyte and platelets counts and unremarkable peripheral smear.
Figure 1.
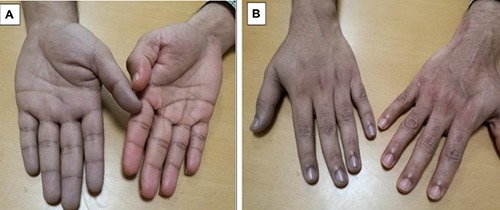
Peripheral cyanosis of patient’s hand at diagnosis compared to his brothers’ palm: anterior (A) and posterior view (B).
Based on his persistent polycythemia, the patient was evaluated in other center and diagnosed as polycythemia vera (PV), bone marrow examination revealed erythroid hyperplasia, however with no panmyelosis. Although molecular studies revealed negative JAK-2 mutation; the patient was treated with Imatinib (Indian version) based on the assumed diagnosis of PV.
In our institute, the patient was re-evaluated; where was found to have dusky discoloration (cyanosis) of the face and both upper limbs of long standing history back to nearly age of thirteen but became more evident in last one year after treatment with Imatinib. He had no history of chest pain, syncope or palpitations with no history of medications intake or exposure to oxidant chemicals. There was no history of growth or developmental retardation or neurological symptoms and no hepatosplenomegaly.
Oxygen saturation by pulse oximetry showed saturation of 92%, while the patient showed no evidence of respiratory distress and his clinical examination was unremarkable.
The diagnosis of PV was revisited and ruled out in view of negative JAK2, normal erythropoietin level and absence of features of panmyelosis in the bone marrow biopsy, which was repeated in our center four months after stopping treatment with Imatinib.
The patients’ clinical picture including cyanosis in absence of any evidence of cardiopulmonary disease; raised the possibility of an alternative diagnosis including rare hemoglobinopathies such as hemoglobin M disease. Hemolysis work up including hemoglobinopathies screening by high performance liquid chromatography (HPLC) and manual hemoglobin electrophoresis revealed normal hemoglobin pattern.
Arterial blood gas showed the following results: pH 7.380 (7.35-7.45), PC O2: 38 mmHg (35-45 mmHg), Normal PO2 at: 96-101 mmHg (83-108), normal arterial O2 saturation at 100% (95-99.0%), low HCO3 21.8 (23-29 mmol/L) and low oxygen saturation (O2 Hb): at 62.4% (94-98%).
Clinical cyanosis and low oxygen saturation in the presence of normal arterial oxygen tension was highly suggestive of methemoglobinemia. Arterial blood gas (ABG) to check for methemoglobinemia performed and revealed a methemoglobin level of 38% (normal: 0-1.5%). Cytochrome B5 reductase (Methemoglobin reductase B) was measured spectrophotometrically at Mayo Clinic Medical Laboratories; and came deficient at level of <2.6 U/g Hb) (normal: 6.6-13.3), consistent with methemoglobin reductase (cytochrome b5) deficiency and hence the diagnosis of congenital methemoglobinemia was established. Karyotyping was done to screen for chromosomal abnormalities and came normal. Other family members were asymptomatic with no family history of cyanosis or hemoglobin disorders. Family screening for methemoglobinemia cannot be conducted.
Due to lack of oral tablets of methylene blue, the patient started on Vitamin C 500 mg once daily. After one month of treatment, the cyanosis became clinically less evident with significant reduction of methemoglobin concentration measured by ABG (methemoglobin levels 19.2 %).
Discussion
Methemoglobin results from oxidation of ferrous iron to ferric iron within the heme-moiety of hemoglobin.1 In healthy individuals, less than 1% of hemoglobin is present in the oxidized form (methemoglobin), which has limited ability to carry oxygen, however with increased oxygen affinity at the remaining binding sites.2 This results in decrease of oxygen delivery to tissue leading to hypoxemia and lactic acidosis.
Acquired methemoglobinemia caused by oxidizing agents and usually presents in older age groups is not uncommon; however, congenital deficiency of the methemoglobin reductase enzyme is extremely rare that only a few cases are reported in the English medical literature around the world.
That is why the incidence and clinical characteristics of congenital Methemoglobinemia is basically unknown, due to underreporting and of these cases.
One possible hint to the diagnosis of methemoglobinemia is the presence of a saturation gap. This occurs when there is a difference between the SO2 that has been measured by means of pulse oximetry (the lower value) and the SO2 that has been calculated by means of arterial blood-gas analysis. Typically, this saturation gap is greater than 5% in cases of methemoglobinemia. 3 The failure of very high oxygen saturation to correct cyanosis is very indicative of methemoglobinemia.
Although methemoglobin at level of 20% indicates enzyme deficiency, patients with congenital disease develop physiological compensatory mechanisms and can tolerate elevated levels of methemoglobin (up to 40%) without symptoms. Levels of >70% are usually incompatible with life.1 These adaptation mechanisms include changes in the concentration of 2,3- Diphosphoglycerate and pH, synthesis of globin chains, and secondary polycythemia.4,5
Compensatory elevation of hemoglobin concentration is observed in patients with recessive hereditary methemoglobinemia, because of the methemoglobinemiainduced left shift in the oxyhemoglobin dissociation curve.6 Congenital methemoglobinemia is further classified into two main types with one due to methemoglobin reductase enzyme deficiency and the other due to an abnormal oxygen affinity hemoglobin termed hemoglobin M.7
Methemoglobin reductase enzyme deficiency is either type I or type II; type I: Cytochrome b5 reductase deficiency, demonstrable only in the erythrocytes, presents as uncomplicated, benign methemoglobinemia, and associated with a normal life expectancy with only fatigue and dyspnea being the most commonly reported symptoms.
Hereditary type II methemoglobinemia, is global (affects both red and white blood cells) and is associated with severe neurologic dysfunction and reduced life expectancy.8 The enzyme’s activity is less than 20% of normal.
Type II congenital methemoglobinemia constitutes approximately 10% of all cases of congenital methemoglobinemia and usually causes death within the first few years of life.9
There is a significant delay in the onset of symptoms in a patient with type I hereditary methemoglobinemia, as most studies indicate that patients are asymptomatic in infancy and childhood. There is a single reported case wherein the onset of symptoms in the patient started at the age of 8 years.10 With such late presentations, treating physicians would usually think of acquired methemoglobinemia, investigating exposure to an oxidative stress as the most likely cause of the findings.
Clinical cyanosis is diagnostically challenging as causes are multiple; especially in absence of cardiopulmonary causes. In the case reported here, the cyanosis was originally overlooked and hematology work-up was misdirected towards investigating polycythemia. In heterozygous cytochrome b5 reductase deficiency, methemoglobin reductase activity is low, and the patient will have a lower threshold for acquired methemoglobinemia in response to exogenous oxidative stress.
The role of Imatinib in provoking methemoglobinemia is questionable and association between Imatinib and methemoglobinemia never described before. In our case, there were no other offending drugs in aggravating the patients’ symptoms and cyanosis.
When congenital methemoglobinemia is suspected, enzyme activity in all immediate family members should be evaluated. As a result of autosomal recessive transmission, In heterozygous deficiency, methemoglobin reductase activity is low and hence heterozygotes will have a lower threshold for acquired methemoglobinemia in response to exogenous oxidative stress. However, the level of enzyme activity is not low enough to produce clinical disease under normal circumstances.11
This category of rare hemoglobin disease is underreported and often clinically missed, In 1943, the first description of familial idiopathic methemoglobinemia in the United Kingdom was reported in 2 members of one family.12
Upon extensive review of English literature for cases diagnosed as congenital Methemoglobinemia due to deficiency of cytochrome b5 reductase, we found 23 cases diagnosed as type I (including the case reported here). The details of the cases are listed in Table 1.10-25
Table 1.
Clinical characteristics of reported cases of Congenital Methemoglobinemia due to (NADH)-cyto b5 Reductase deficiency reported in English literature.
| Author [ref] | Age/Sex Nationality | Hb (gm/dL) | Presentation | Type | MetHb level | Family study | (NADH)-cyto b5 reductase | Management (MB), mg/kg (vit C) | MetHb after treatment (%) | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| Londhei [10] | 18/M Indian | 19 | Cyanosis Headache Bilateral ptosis | I | 37 | Pos (sister) | 14 (30-35) | Oral Vit C thrice/day Aspirin Phlebotomy | 18 | Cyanosis improved |
| Shonola [11] | 24 days/M | - | Mild central cyanosis | I | 26 | Pos | 4.2 (10.1-19.4) | Oral MB (1.5) Vit C (5) | 4.8 | Cyanosis improved |
| Takishe [13] | 38/F japanese | - | Cyanosis Convulsion | I | 33 | Neg | N/M | N/M | N/M | N/M |
| Yasmin [14] | 21/MIndian | - | Mild cyanosis exertional dyspnea | I | 29 | Neg | Deficient N/M | N/M | - | Mild cyanosis persisted |
| Canturk [15] | 20/M | - | Cyanosis Bronchial asthma symptoms | I | 40 | Pos | N/M | MB | 8-9 | MetHb decreased |
| Badawi [16] | 16/ M Arabic | 17 | Cyanosis fatigue dyspnea on exertion | I | 40 | Pos | <2.6 (6.6-13.3) | Oral Vit C | N/M | Cyanosis and symptoms persisted |
| Badawi [16] | 12/M Arabic | - | No symptoms | I | 25 | Brother | - | - | - | - |
| Ramanamurty [17] | 55/M Indian | 15 | Weakness Syncope Cyanosis | I | 30 | Neg | 16 (35.0±5.00) | N/A | - | N/A |
| Dhiraj [18] | 33/M Indian | 18 | Incidental cyanosis | I | 20-30 | Neg | N/A | N/A | N/A | N/A |
| Gerli [19] | 64/M Italian | - | Markedcyanosis | I | 36 | Pos | 5% normal value | Oral Vit C | 25 | N/A |
| Prabhakar [20] | 1 month/M Indian | 14 | Central cyanosis | I | 29 2.8 (n:<1.5%) | Pos | 9.36 (30-40) | MB | - | Good response to MB therapy |
| Prabhakar [20] | 32 y/F Mother Indian | 14 | REPL | I | 7.5 (n: <1.5%) | Pos | 20 | Not needed | - | - |
| Prabhakar [20] | 34/ M Father Indian | 15 | No symptoms | I | 2.8 | Pos | 24 | Not needed | - | - |
| Melanie [21] | F/English Mother | - | - | I | Pos | <0.1 I | - | - | - | |
| Melanie [21] | English/ M Father | - | - | I | Pos | 6.37 | - | - | - | |
| Melanie [21] | English/ M | - | - | I | Pos | 7.75 I (11.51-29.9) | - | - | ||
| Vives [22] | 2 year/F Spanish | - | Neurological symptoms | II | Pos | - | - | - | - | |
| Yawata [23] | M Japanese | - | Mental retardation skeletal anomalies | II | - | RBC: (0.3-4) PLT (13-27), lympho cytes (18-31), fibrobl ast (50) | - | - | - | |
| Prabhakar [24] | 5/F Indian | 11 | Cyanosis cerebral palsy, microcephaly generalized hypertonia | II | 51 | Pos | 6.0 | MB (0.5) followed by vit C 500 twice a day | - | Re-developed Cyanosis when vit C stopped |
| Prabhakar [24] | 13/M Indian | 9 | - | II | 13 | Pos | 6.21 | - | - | - |
| Prabhakar [24] | 8/M Indian | 14 | - | II | 42 | Pos | 9.60 | - | - | - |
| Nellicka [25] | 1/F Indian | - | - | II | 24 | - | - | Vit C high dose | - | Symptoms improved |
Methylene blue (MB); ascorbic acid (vit C); not mentioned (N/M); recurrent early pregnancy loss (REPL).
Seventeen cases (~74%) of type I and 6 cases (27%) of type II. There is male predominance, 73% versus 26% in females. Almost half of reported cases 12 cases (52%) are Indian, 2 Japanese, 3 English, 2 Arabic, one case Spanish and one case Italian.
For type I, the median calculated age is 31 years with cyanosis and shortness of breath being the most common sign and symptoms.
For type II: Only six cases were reported in English literature, all in pediatric age group with median calculated age at presentation is 6 years with neurologic manifestations and mental retardation are type II associated symptoms.
The modalities of treatment vary between: Methylene blue (MB) alone, MB with Ascorbic acid or Ascorbic acid alone. Ascorbic acid either alone or in combination with MB was used in seven cases within this category with no response in one case,5 while the other six cases (including our case) responded well.
Conclusions
Due to lack of systematic epidemiological studies, congenital methemoglobinemia is under diagnosed as it is under investigated and usually overlooked especially when presenting in adulthood and in absence of obvious acquired agents.
In the present report, we highpoint this clinically hidden case of methemoglobinemia which is misdiagnosed as polycythemia vera and led to needlessly patient’s exposure to invasive bone marrow procedures and even therapy with Imatinib.
References
- 1.Jaffe ER, Hultquist DE, et al, eds. The Metabolic and Molecular Basis of b5 reductase deficiency and enzymopenic hereditary methemoglobinemia. In: Inherited Disease. 7th ed. New York, NY: McGraw-Hill; 1995. pp 2267-2280. [Google Scholar]
- 2.Baraka AS, Ayoub CM, Kaddoum RN, et al. Severe oxyhemoglobin desaturation during induction of anesthesia in a patient with congenital methemoglobinemia. Anesthesiology 2001;95: 1296-7. [DOI] [PubMed] [Google Scholar]
- 3.Barker SJ, Tremper KK, Hyatt J. Effects of methemoglobinemia on pulse oximetry and mixed venous oximetry. Anesthesiology 1989;70:112-7. [DOI] [PubMed] [Google Scholar]
- 4.Kern K, Langevin PB, Dunn BM. Methemoglobinemia after topical anesthesia with lidocaine and benzocaine for a difficult intubation. J Clin Anesth 2000;12:167-72. [DOI] [PubMed] [Google Scholar]
- 5.Maurtua MA, Emmerling L, Ebrahim Z. Anesthetic management of a patient with congenital methemoglobinemia. J Clin Anesth 2004;16:455-7. [DOI] [PubMed] [Google Scholar]
- 6.Fermo E, Bianchi P, Vercellati C, et al. Recessive hereditary methemoglobinemia: Two novel mutations in the NADH- cytochrome b5 reductase gene. Blood Cells Mol Dis 2008;41:50-5. [DOI] [PubMed] [Google Scholar]
- 7.Miller DR.. Hemoglobinopathies in children. Massachusetts: PSG Publishing; 1980 [Google Scholar]
- 8.Melanie J. Percy1 and Terry R. Lappin. Recessive congenital methaemoglobinaemia: cytochrome b5 reductase deficiency. Br J Hematol 2008;141:298-308 [DOI] [PubMed] [Google Scholar]
- 9.Hirono H. Lipids of myelin, white matter and graymatter in a case of generalized deficiency of cytochrome b5 reductase in congenital methemoglobinemia with mental retardation. Lipids 1980;15:272-5. [DOI] [PubMed] [Google Scholar]
- 10.Londhey V, Khadilkar K, Gad J, et al. Congenital methaemoglobinaemia: A rare cause of cyanosis in an adult patient. J Assoc Physicians India 2014;62:269-71. [PubMed] [Google Scholar]
- 11.Shonola S, Da-Silva MD. congenital methemoglobinemia: a rare cause of cyanosis in an adult patient. J Assoc Physicians India 2014;62:269-71. [PubMed] [Google Scholar]
- 12.Deeny J, Murdock ET, Rogan JJ. Familial idopathic methaemoglobinaemia. Brit Med J 1943;721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13 Kitao T, Sugita Y, Yoneyama Y, Hattori K. Methemoglobin reductase (cytochrome b5 reductase) deficiency in congenital methemoglobinemia. Blood 1974;44:879-84. [PubMed] [Google Scholar]
- 14.Hamirani YS, Franklin W, Grifka RG, Stainback RF. Methemoglobinemia in a young man. Tex Heart Inst J 2008; 35:76-7. [PMC free article] [PubMed] [Google Scholar]
- 15.Tasci C, Nevruz O, Candir N, Bilgic H. A methemoglobinemia case who was previously diagnosed and treated as asthma. Respir Med Case Rep 2012;6:11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badawi MA, Badawi MA, Wali SO, Alsaggaf RZ. Hereditary methemoglobinemia manifesting in adolescence. J Appl Hematol 2016;7:108-10. [Google Scholar]
- 17.Ramanamurthy SV. Methemoglobinemia: a reappraisal with an Indian perspective. Available from: www.apiindia.org/medicine_update_2013/chap77.pdf . [Google Scholar]
- 18.Trivedi DJ, Joshiraj B, Bidkar V, Rao R. Methemoglobinemia: living with dormant devil. Indian J Clin Biochem 2017;32:248-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerli GC, Beretta L, Bianchi M, et al. Methemoglobinemia; a description of a case of NADH methemoglobulin reductase deficiency. Minerva Med 1981; 72:1925-30. [PubMed] [Google Scholar]
- 20.Kedar P, Warang P, Ghosh K, Colah R. Recessive congenital methemoglobinemia due to NADH-cytochrome b5 reductase deficiency associated with recurrent early pregnancy loss (REPL) in an Indian family. Ann Hematol 2012;91:1985-6. [DOI] [PubMed] [Google Scholar]
- 21.Percy MJ, Gillespie MJS, Savage G, et al. Familial idiopathic methemoglobinemia revisited: original cases reveal 2 novel mutations in NADHcytochrome b5 reductase. Blood 2002;100:10. [DOI] [PubMed] [Google Scholar]
- 22.Vives-Corrons JL, Pujades A, Vela E, et al. Congenital methemoglobin-reductase (cytochrome b5 reductase) deficiency associated with mental retardation in a Spanish girl. Acta Haematol 1978;59:348-53. [DOI] [PubMed] [Google Scholar]
- 23.Yawata Y, Ding L, Tanishima K, Tomoda A. New variant of cytochrome b5 reductase deficiency (b5Rkurashiki) in red cells, platelets, lymphocytes, and cultured fibroblasts with congenital methemoglobinemia, mental and neurological retardation, and skeletal anomalies. Am J Hematol 1992;40:299-305 [DOI] [PubMed] [Google Scholar]
- 24.Kedar PS1, Nadkarni AH, Phanasgoankar S, et al. Congenital methemoglobinemia caused by Hb- MRatnagiri ((beta-63CAT—>TAT, His—>Tyr) in an Indian family. Am J Hematol 2005;79:168-70. [DOI] [PubMed] [Google Scholar]
- 25.Nellicka S, John A, Abraham V. Congenital methemoglobinemia Type 2. A rare case report. Pediatric Oncall J 2017;14:19. [Google Scholar]