

Abstract
Mutagens can be carcinogens, and traditionally, they have been identified in vitro using the Salmonella ‘Ames’ reverse mutation assay. However, prokaryotic DNA packaging, replication and repair systems are mechanistically very different to those in the humans we inevitably seek to protect. Therefore, for many years, mammalian cell line genotoxicity assays that can detect eukaryotic mutagens as well as clastogens and aneugens have been used. The apparent lack of specificity in these largely rodent systems, due partly to their mutant p53 status, has contributed to the use of animal studies to resolve data conflicts. Recently, silencing mutations at the PIG-A locus have been demonstrated to prevent glycophosphatidylinositol (GPI) anchor synthesis and consequentially result in loss of GPI-anchored proteins from the cell’s extracellular surface. The successful exploitation of this mutant phenotype in animal studies has triggered interest in the development of an analogous in vitro PIG-A mutation screening assay. This article describes the development of a robust assay design using metabolically active human cells. The assay includes viability and cell membrane integrity assessment and conforms to the future ideas of the 21st-century toxicology testing.
Introduction
Genetic toxicology plays an essential role within hazard identification and risk assessment during the development of novel drugs as well as pesticides, herbicides, flavours, and fragrances. Throughout the early stage drug development, a substance’s ability to damage DNA through genotoxic mechanisms must be fully investigated to enable accurate and cost-effective hazard and risk assessment (1). If possible, this would be completed with more emphasis on high content, high throughput in vitro genotoxicity assessment, reducing animal usage. Short falls in in silico pharmacokinetic and dynamic modelling (2)] as well as reportedly poor in vitro specificity in carcinogenicity prediction (3) have produced a battery of in vitro and in vivo genotoxicity assays developed to identify potential mutagens, aneugens and clastogens (4,5), which could benefit from a broader revision to include 21st-century approaches.
Several regulatory-accepted in vitro mammalian cell mutation assays are available to assess chemically induced gene mutation. These use cell lines derived from mice (L5178Y) and hamster (CHO, AS52 and V79), which are often p53 mutant, and humans (TK6) (6). The most commonly used genetic endpoints are mutation at the thymidine kinase (TK) and hypoxanthine–guanine phosphoribosyl transferase (HPRT) loci. Whilst data from the HPRT and TK mutation tests are widely accepted in hazard and risk assessment (6), they are relatively time-consuming (3–6 weeks) and highly labour-intensive, particularly when characterising dose–response relationships, and reportedly have poor specificity (3) that can limit their utility in a screening context. However, specificity issues are being addressed by a more recent focus on p53 competent human cell lines within in vitro Organisation for Economic Cooperation and Development (OECD) guidance documents (7). To date, in vivo gene mutation experiments have been restricted largely to transgenic models (MutaMouse™ and BigBlue). As these are more expensive than inbred animals, they are only used in a regulatory setting as a study of ‘last resort’, addressing specific concerns about a potential mutagenic signal (identified in vitro) and/or mechanism.
Recently, a novel gene mutation assay based on the endogenous phosphatidylinositol–glycan biosynthesis class-A (PIG-A) gene (8), located on the p arm of the X-chromosome (9) was developed in rodents (10). PIG-A encodes an enzyme critical to the synthesis of glycophosphatidylinositol (GPI) anchor molecules (11,12). Specifically, PIG-A is essential in the production of a catalytic subunit of the N-acetylglucosamine transferase complex (13), and combined with numerous other PIG-gene products, pig-b, pig-c etc., it contributes to the synthesis of the final branched glycan structure of the anchor. This eventually resides on the external surface of the cellular membrane, extending into the extracellular space, tethering cell-specific and conserved surface antigens (14). Whilst silencing mutations in any of these genes may prevent GPI anchor synthesis (15,16), a mutational silencing event within PIG-A is believed to be the most common cause of GPI anchor synthesis disruption, because it is X-linked (17), and a single mutation can result in a deficiency of GPI-anchored cell surface antigens. Hence, the GPI anchor-deficient phenotype is generally attributed to PIG-A mutation (18). The mutant genotype (PIG-A−) is also a characteristic of patients with paroxysmal–nocturnal–haemoglobinuria (PNH) (19), and diagnostic methods have been developed to detect and quantify cells carrying presumptive mutations at the PIG-A locus using flow cytometry (FCM) (20). The phenotype is reported to be growth neutral in vivo (21), an important factor in mutagenesis studies as it avoids mutational bias. Mutant frequency (f) and mutation rate (µ) at the PIG-A locus can be measured indirectly, using FCM, recording the loss of expression of specific GPI-anchored cellular antigens following mutagen exposure (20,22). The assay potentially has great transferability between mammalian species, due to the highly conserved nature of GPI-anchor synthesis (23).
The development of the rodent erythrocytic in vivo Pig-a gene mutation assay has gathered significant momentum, benefitting from extensive coordinated ring trials (24–27), methods to support assay transfer across mammalian species (21,23,28–34) and high throughput optimisation (29). In addition, there has been some progress in demonstrating the mechanistic basis of the assay (32,35,36), and efforts are going on to further characterise the assay in terms of genotoxic mechanisms (37,38) and chemical space. It is hoped that these activities will support the development of an OECD guideline in due course. Following recent European Union reforms to limit and/or ban animal testing (39), especially relevant to the cosmetics and consumer industries (40,41), there has been increased focus on replacing animal testing with novel approaches to quantify genotoxic hazard for human risk assessment purposes. Innovative technologies are being developed to enable high throughout, high content screening whilst retaining a high level of sensitivity (42–45).
As part of these efforts, our laboratories have focused efforts on the development of an in vitro PIG-A assay design that could be used within a screening or a regulatory setting and serve as a predictive counterpart to the in vivo assay described above. TK6 was initially proposed as the cell line of choice based on the following criteria: (i) a non-adherent cell line of haemopoetic origin and short cell cycle time to allow short experimental duration; (ii) a cell line with a proven track record of high sensitivity and specificity in genotoxicity assays; and (iii) a cell line of human origin (with functional p53) to improve risk assessment, species specificity, and with the potential for future in vitro to human extrapolation. This decision was further supported by a recent review of cell lines used in genotoxicity testing, which had preferentially highlighted the benefits of TK6 cells (46–48). Other laboratories have described the development of an analogous in vitro PIG-O assay in DT40 chicken cells (49) and an analogous in vitro PIG-A assay in TK6 cells (42). These assays theoretically provide the means for more extensive mechanistic validation (38), though for wide acceptance, the phenotypic data will need DNA sequence data to unambiguously link phenotype to genotype.
This study reports development of a PIG-A mutation assay in human B-lymphoblastoid cell lines. The aim was to establish a robust assay platform that takes into account the potential limitations of the system. Initially, enrichment techniques were evaluated to enable the assessment of GPI anchor surface proteins CD59 and CD55 (see supplementary data, available at Mutagenesis Online) as potential reporters of phenotypic PIG-A mutation. The need for this reflects early findings that the prevalence of the loss of GPI anchor phenotype in TK6 cells was very much higher than the expected mutation frequency. In addition, aerolysin sensitivity was used to validate PIG-A phenotype and was also explored as a potential basis for mutant selection, useful in tying phenotype to genotype. Dose–response data were generated throughout and were used to investigate the sensitivity of the current PIG-A assay design to both genotoxic and non-genotoxic compounds, with confocal microscopy utilised to provide additional molecular confidence in the phenotype. Throughout development, the assay design incorporated viability assessments, comprehensive gating strategies as well as a marker for non-GPI–associated cell surface protein markers, a potential source of misleading results. During the course of the study, the alternative, metabolically active human MCL-5 cells were found to provide a lower background spontaneous mutant prevalence. These assay design advancements with the associated flow cytometric gating strategies suggest development of an improved assay platform and the basis for further optimisation and validation.
Materials and methods
Cell culture
The TK6 cell line was obtained from the American Type Culture Collection (ATCC; http://www.atcc.org/). Low passage TK6 cells were obtained from Masamitsu Homna (NIH, Japan). AHH-1 and MCL-5 cell lines were purchased from Gentest Corporation (now BD Biosciences, Oxford, UK). TK6, AHH-1 and MCL-5 cells were cultured in 89% RPMI 1640, 1% l-glutamine (Gibco®, Paisley, UK) and 10% horse serum (BioSer, Sussex, UK): MCL-5 culture media were supplemented with 50 mg/mL Hygromycin B (Merck, Hoddesdon, UK). Low passage TK6 growth medium consisted of 89% RPMI 1640, 1% penicillin–streptomycin (Gibco®, Paisley, UK) and 10% heat-inactivated horse serum (cat no. 26050-088, Gibco). All cultures were kept within a controlled carbon dioxide (CO2; 5 ± 1%) incubator maintained at 37.5°C. Prior to use, cryovials of human lymphoblastoid TK6, low passage TK6, MCL-5 and AHH-1 cells were removed from cryopreservation, thawed quickly and cultured under standard conditions. All cell lines were monitored regularly to detect signs of contamination (microscopy) or morphological abnormalities and subcultured once prior to the experimental start date to ensure reproducible growth dynamics. Stock cell cultures were vitally maintained between 105 and 106 cells/mL in 75 cm2 culture flasks by routine subculturing, with an exception of the TK6 cell line, which was able to grow consistently from a 4 × 105 cells/mL seeding concentration. Populations depleted of PIG-A mutant cells were cryopreserved and stored in liquid nitrogen for a period of up to 6 months. To revive cells from cryogenic storage, they were thawed and cultured for a period of at least 72 h prior to use. Where appropriate, the use of fluorescently labelled isotype control antibody samples were utilised in order to determine the nature of cell line-specific antibody binding, thereby reporting accuracy.
Enrichment strategies to optimise CD59 expression within TK6 cells
Dynabead® magnetic bead retrieval
A mouse anti-human CD59 antibody (cat no. 555761, BD Pharmingen™, San Jose, CA, USA) was biotinylated (DSB-X™ Biotin Protein Labelling Kit, cat no. D-20655, Molecular Probes, Life™ Technologies, Paisley, UK) and used with the Dynabeads® Flow Comp™ Flexi Kit (cat no. 11061D, Molecular Probes, Life™ Technologies, Paisley, UK) to select cells expressing CD59, i.e. the cells with a PIG-A wild-type phenotype. Briefly, 5 × 107 TK6 cells were resuspended in 500 µL of 2–8°C isolation buffer and 25 µL of DSB-X™ biotin-labelled antibody, mixed well and incubated at 2–8°C (protected from light) for 10 min. Cells were then washed (2 mL cold isolation buffer), collected by centrifugation (350 × g for 8 min) and the supernatants aspirated prior to resuspension in 1 mL cold isolation buffer and the addition of 75 µL Flow Comp™ Dynabeads®. Following mixing, the suspension was incubated for 15 min (2–8°C, rolling and tilting), then transferred into a 2-mL microcentrifuge tube and placed on a magnetic rack (Qiagen) for 2 min. Following removal of the supernatant, the tube was then detached from the magnet and the pellet of bead-bound cells was washed, followed by resuspension in 1 mL of cold isolation buffer and then re-pelleted (twice). The bead-bound cells were then resuspended in 1 mL of Flow Comp™ release buffer for 10 min (25°C under rolling and tilting) and then placed on the magnetic rack for 2 min. The supernatant containing the bead-free cells was transferred into a new tube and placed on the magnetic rack for an additional 2 min to remove any residual Dynabeads®. Finally, the supernatant was centrifuged (350 × g for 8 min), and the resulting enriched population of cells expressing CD59 was transferred to a 25 cm2 cell flask for culture. Quality assurance (QA) FCM based on the PIG-A assay was used to determine the level of CD59 enrichment in the cell culture (106 cells were removed and stained, see Determination of PIG-A mutant frequency using the CD59 phenotypic marker following exposure to the potent mutagen EMS, to determine the frequency of CD59-positive cells, i.e. a PIG-A wild-type phenotype).
Clonal expansion
Cultures were established from cryopreserved TK6 cell stocks and analysed using FCM to determine the proportion of cells expressing the CD59 antigen, by means of the anti-human CD59-R-PE–labelled antibody. >10 × 106 cells were then harvested by centrifugation (250 × g for 7 min) and resuspended to a final concentration of 200 cells/mL prior to clonal expansion as follows: 100 μL cellular samples were transferred into each well of six identical 96 well plates (Greiner–Bio-One, Gloucestershire, UK), and following 72 h incubation, each well was inverted microscopically assessed for colony formation. From these plates, a total of 144 deemed well-established colonies were transferred into six 24-well plates, with the addition of fresh growth medium (~300 µL), and the cells were cultured for a further 72 h. The 10 largest colonies (excess of ~1000 cells) were transferred into separate sterile 25 cm2 flasks and cultured to confluency (~1–7 × 105 cells/mL) within pre-warmed culture media. Samples were subsequently analysed for CD59 expression by FCM, and the cultures with the resulting highest frequency of cells expressing CD59 were prepared for long-term cryopreservation.
Fluorescence-activated cell sorting
A fresh vial of TK6 cells homologous to the cultures used for the clonal expansion experimentation, subcultured from the original TK6 cell stock, were analysed for CD59 surface antigen expression using anti-human CD59-R-PE labelling as well as human leucocyte antigen–antigen DR (HLA-DR) presence utilising an anti HLA-DR fluorescein isothiocyanate (FITC) antibody and subsequent FCM. Approximately 20 000 cells that were CD59-positive and displayed uniform HLA-DR fluorescence were isolated using fluorescence-activated cell sorting (FACS; sorting on a FACSAria 1, BD Biosciences, UK). The recovered cells were pooled into 5 mL sorting tubes (BD Biosciences, Hertfordshire, UK) within culture medium, pelleted and cultured in fresh culture media within 25 cm2 culture flasks prior to long-term cryopreservation. Once more QA FCM analysis was performed to confirm the frequency of CD59-positive cells, i.e. a PIG-A wild-type phenotype following enrichment and prior to preservation.
Determination of PIG-A mutant frequency using the CD59 phenotypic marker following exposure to the potent mutagen EMS
For this purpose, 2 × 106 clonally enriched TK6 cells for CD59-positive expression (see Enrichment strategies to optimise CD59 expression within TK6 cells–Clonal expansion) were thawed and cultured under standard conditions. Following sufficient growth, 105 cells/mL were seeded into 12.5 cm2 culture flasks and treated with either vehicle [dimethyl sulphoxide (DMSO) 1% v/v] or ethylmethane sulfonate (EMS; 0–10 µg/mL) for 24 h at 37°C, 5% CO2. Following treatment, the cells were washed [10 mL phosphate-buffered saline (PBS)] and cell density determined (Beckman Coulter Z1 Particle Counter). Then, 106 cells were removed, washed (10 mL PBS) and centrifuged (250 × g for 7 min) prior to resuspension in 50 µL of fresh RPMI-based culture medium (see Cell culture). Simultaneously, parental-treated cell cultures were subcultured to 105 cells/mL and incubated for a further 24 h to enable subsequent daily PIG-A mutation assessments, only if suitable population growth has occurred to enable this (>2 × 105 cells/mL). The resuspended cellular samples were then incubated with 20 µL (0.1 mg/mL) of anti-human CD59 PE antibody for 20–30 min at 2–8°C (protected from light). Immediately following this, the cells were harvested by centrifugation (250 × g for 7 min), the supernatant was carefully removed and the pellet washed in 10 mL PBS. This was followed by re-pelleting and further resuspension in 1 mL PBS prior to being directly transferred into 5 mL BD Flow Tubes (protected from light). A portion of the cells was analysed by FCM (BD FACS Aria I) daily for four additional experimental days following the determination of spontaneous background mutant frequencies on Day 0. Mutant phenotype gating was subsequently undertaken following the use of an unstained instrument calibration standard in order to establish an autofluorescence level within the channel and hence set the detector voltages correctly to enable a fluorescence threshold value of negativity. Once established, subsequent stained samples were able to be accurately assessed for the reporting fluorophore of GPI-anchored protein (AP) status.
Investigations of FCM defined PIG-A phenotypes within TK6 cell populations
Aerolysin permeabilisation
First, 2 µM pro-aerolysin (AeroHead Scientific, Saskatoon, Canada) was activated with 2 µg/mL trypsin (diluted in 25 µg/mL Tris pH 8 buffer) in a 1:1 solution to produce a 250 nM aerolysin solution. Then, parental TK6 PIG-A wild type and PIG-A mutant populations (isolated by FACS) were adjusted to 2 × 105 cells/mL in RPMI 1640 and treated with a range of aerolysin concentrations (15.625–250 nM) in RPMI 1640. After 48 h, 150 µL cell cultures were removed from each well and viability assessed using FCM and propidium iodide (PI) DNA staining: viable cells are PI-negative, and permeabilised non-viable cells are PI-positive; PI was detected by increased fluorescence in the FL3-H detector on the FACSCalibur (Cell Quest Pro version 5.2.1).
Cell viability assessment
First, 106 untreated TK6 cells, clonally enriched for the CD59 surface marker, were resuspended in 50 µL of fresh culture medium and then incubated with 20 µL (0.1 mg/mL) of anti-human CD59 R-PE antibody for 20–30 min at 2–8°C (protected from light). Immediately following this, cells were harvested by centrifugation (250 × g for 7 min), the supernatant was removed and the pellet washed in 2 mL PBS to remove residual unbound antibody. Simultaneous to the washing steps, 1X Annexin V-binding buffer was prepared in deionised water (dH20) and placed on ice to cool. Following a second 2mL PBS wash, cells were resuspended in 2 mL 1X Annexin V-binding buffer, centrifuged and resuspended in an additional 100 µL of the 1X Annexin V-binding buffer. Then, 5 μL of Alexa Fluor® 488 Annexin V conjugate was added to the 100 µL cell suspension and incubated at room temperature for 15 min, protected from light. Post incubation, 2 mL 1X Annexin-binding buffer was added to each sample, centrifuged (250 × g for 7 min) and resuspended in a further 200 µL of 1X Annexin-binding buffer. Finally, 5 µL (0.25 μg) of 7-aminoactinomycin D (7-AAD) solution was added to each sample, incubated for a minimum 15 min on ice and subsequently diluted via the addition of 1–2 mL of PBS and transferred directly into 5 mL BD Flow Tubes, protected from light, and analysed within 4 h of initial antibody treatment.
GPI-AP candidate trials in different human cell lines
Fluorescent aerolysin
Prior to use, 1 mL of PBS was added to 25 μg of powdered (lyophilised) fluorescent aerolysin (FLAER); this stock solution was further diluted 1 in 10 (dH20) and 50 μL volumes aliquotted into 0.5 mL microcentrifuge tubes for long-term storage (wrapped aluminium foil, −20°C). Samples for each cell line, containing 106 cells in suspension, were transferred into sterile 15 mL plastic disposable tubes, centrifuged (250 × g for 7 min) and resuspended in 100 μL of fresh pre-warmed culture medium (see Cell culture). Cell pellets were carefully redispersed and treated with 5 μL of FLAER (in triplicate) at 3 concentrations; 1×, 0.5× and 0.1× in 2% bovine serum albumin (BSA)/PBS. Each tube was incubated for 15 min at room temperature and protected from direct light at all times. Post incubation, samples were centrifuged, washed with 10 mL PBS and resuspended within a further 1 mL of PBS. All samples were placed on ice and protected from direct light to maintain the stability of the suspensions and the fluorophores used, prior to flow cytometer acquisition.
CD48–B-Lymphocyte Activation Marker 1 and CD55/59 staining protocol
Samples for each cell line (106 cells) were pelleted and resuspended in fresh medium (100 µL) and fluorescently labelled with either 20 μL of mouse anti-human anti-CD48 R-PE antibody (cat no. 552855, BD Biosciences, Hertfordshire, UK) or with 20 μL of mouse anti-human anti-CD55 R-PE antibody and 20 μL of mouse anti-human anti-CD59 R-PE antibody (room temperature, protected from direct light, 30 min). The cells were pelleted (250 × g for 7 min), washed in 2 mL PBS and resuspended in a further 1 mL PBS and placed on ice, protected from light, prior to flow cytometer analysis.
Viability assessment of MCL-5 Cells to avoid potential Artefacts
First, 100 μL of 1 μM Staurosporine solution was added to six separate MCL-5 cellular suspensions (4 × 106 cells in 10 mL) and incubated for either 30 min, 1 h, 2 h, 3 h, 4 h or 8 h at 37°C. Cells were pelleted (250 × g for 7 min), washed twice in 10 mL PBS and resuspended in a further 10 mL of fresh medium. Following each treatment, cellular densities were determined, and three further cellular samples were constructed each containing 106 cells. The samples were then stained with anti-human anti-CD55 R-PE, CD59 R-PE, 7-AAD and Annexin V in an analogous protocol to that as described above.
Determination of PIG-A mutant frequency using the CD59 phenotypic marker, viability assessment and HLA-DR staining within MCL-5 cells
MCL-5 cellular cultures were established from previously cryopreserved parental stocks. Cultures were exposed to either EMS (0–161.1 µM), N-methyl-N-nitrosourea (MNU; 0–80.5 µM), N-ethyl-N-nitrosourea (ENU; 0–322.2 µM) or 2,4 dinitrophenol (2,4 DNP; 0–325.9 µM) with DMSO being utilised and as a vehicle control for 24 h. Following treatment, cultures were washed (10 mL PBS), counted, seeded (for subsequently daily analysis) and 106 cellular samples stained ready for initial flow cytometric analysis; 20 µL anti-human anti-CD59 R-PE, 40 μL HLA-DR FITC (cat no. 555871, BD Biosciences, Hertfordshire, UK) and 10 μL 7-AAD were used in an analogous fashion to previous protocols. Flow cytometric acquisition (FACSCalibur (Cell Quest Pro version 5.2.1)) was performed within an hour of sample preparation, and samples were stored on ice protected from direct light beforehand. The experiment was carried out daily for four additional days following the determination of the spontaneous background mutant frequencies on Day 0.
Tandem GPI-AP confocal expression analysis in MCL-5 cells
Untreated MCL-5 cells (106) were stained with 20 µL anti-human anti-CD55 and/or 20 µL CD59 R-PE fluorescent antibodies for 30 min, washed twice in 3 mL pre-warmed (37°C) PBS and resuspended and fixed in 1 mL of 4% PBS-buffered paraformaldehyde for 15 min at 37°C, before being washed twice more with 3 mL PBS. Coverslips, precision 0.17 mm thickness, (cat no. 474030–9000, Carl-Zeiss, Hertfordshire, UK) were placed in a 0.1-mL aliquot of 1mg/mL aqueous poly-l-lysine (Sigma-Aldrich, Gillingham, UK) on parafilm, incubated for 10 min at room temperature, washed thoroughly using dH20 and left to air-dry. Coverslips were then incubated in excess 1% BSA (Sigma-Aldrich, Gillingham, UK) PBS for at least 1 h, washed and placed into 6-well plates (Greiner Bio-One, Hertfordshire, UK). Three millilitre of stained cell suspension, at a suitable concentration to provide sufficient coverage ~105–106 cells/mL, was added to each well to enable cellular adhesion and incubated for 10 min at room temperature, protected from light. Following incubation, each coverslip was microscopically inspected to ensure that attachment was successful before being further washed in 3 mL PBS and carefully touch dried. Coverslips were then mounted within 1,4-diazabicyclo[2,2,2]octane (DABCO) (cat no. D27802, Sigma Aldrich, Gillingham, UK); 45 mL glycerol (G55-16 100 mL, Sigma Aldrich, Gillingham, UK), 2.5 mL Tris-hydrochloric acid buffer (pH 8) and 2.5 mL dH20. Polished glass slides (cat no. 11562203, Fisher Scientific, Loughborough, UK) with autoclave tape were used to construct posts, which loaded coverslips were laid down onto 50 μL DABCO, centrally between the two posts. Confocal microscopy was carried out using an LSM-710 laser scanning confocal microscope and supplied ZEN software version 2009 in conjunction with either a 40× or 63× oil immersion DIC objective (Carl Zeiss, Cambridge, UK).
Gating strategy investigations
First,106 untreated MCL-5 cells were stained with 20 µL anti-CD55/CD59 R-PE antibodies and washed (3 mL PBS) prior to flow cytometric analysis, as described previously. Samples were run/tested in triplicate utilising the different proposed gating strategies; typically FSC-A and SSC-A scatter plots were utilised to define a typically ‘single-cell’ population, from these outputs, i.e. 2-D histograms and 2-D density scatter plots were constructed utilising the FL-W measure of the fluorophore channel (PE) as well as the FSC-W, as proposed measures of particle size. Gates were drawn, which encompassed at least 90% of the parental population, to further isolate particles defined ‘single cell’ in nature and finalised outputs were commented in terms of numbers/percentage of single cell CD55/59-positive phenotypic PIG-A mutant events, as described above. All these parameters were managed using the FACS Diva™ 6 software, and doublet discrimination plots were removed to enable easier visualisation of results.
In addition, MCL-5 cellular cultures were established from previously cryopreserved parental stocks. Cultures were exposed to MNU (0–1 µg/mL) with DMSO being utilised and as a vehicle control for 24 h. Following treatment, cultures were washed (10 mL PBS), densities defined, seeded (for subsequently daily analysis) and 106 cellular samples stained ready for initial flow cytometric analysis; anti-human anti-CD59 and anti CD55 R-PE, Annexin V and 7-AAD were used in an analogous fashion to previous protocols. Flow cytometric acquisition was undertaken on a BD FACSAria 1 (FACSDiva™ 6) and performed within an hour of sample preparation, and samples were stored on ice protected from direct light beforehand.
Results and Discussion
Enrichment strategies to optimise CD59 expression within TK6 cells
Immunofluorescence antibodies to a number of GPI-anchored surface antigens (CD48 and CD59, see Supplementary figure 1, available at Mutagenesis Online) revealed that the parental TK6 cell line comprises a heterogeneous population, with a significant minority of cells GPI-anchored protein deficient. Mutagenicity is measured as an increase over the spontaneous (pre-existing) background of mutant cells, so a high background poses a significant barrier to the development of an assay to detect rare mutation events. Consequently, strategies to deplete parental TK6 cultures of the presumptive, pre-existing mutant populations are required (50). The regulatory accepted MLA and HPRT in vitro mammalian gene mutation assays have analogous requirement to purify the pretest cell cultures of mutants and subsequently enrich for wild-type cells. However, a different strategy was required for analysis of PIG-A; three approaches were assessed, immunomagnetic bead separation (Dynabead®), which is already used to increase statistical power in the in vivo Pig-a assay (29), clonal expansion and FACS. Each method was compared for its ability to produce near-homogenous cell populations with low phenotypic PIG-A mutant prevalence.
All three enrichment methods reproducibly demonstrated >90% homogeneous GPI-AP (CD59)-positive populations (Table 1). FACS produced the optimum results in terms of average CD59 wild-type enrichment, 95.3% ± 3.0, with Dynabead® and Clonal expansion yielding 89.1% ± 6.5 and 70.8% ± 25.3 CD59-positive cell cultures respectively. In terms of reproducibility, FACS and Dynabead® were shown to be the most robust methodologies, with clonal expansion resulting in a much greater range of purities post enrichment as emphasised by the elevated spread of data around the mean. Clonal expansion did however result in the optimum individual enrichment purity, with 99.8% of the subsequent population positive for CD59 expression.
Table 1.
Phenotypic PIG-A assessment of TK6 populations following enrichment
| Sample ID | Number of total events | CD59-positive (%) | CD59-negative (%) | SD | |
|---|---|---|---|---|---|
| Dynabead | 1a | 8216 | 84.1 | 15.9 | |
| 1b | 8461 | 84.3 | 15.7 | ||
| 1c | 8379 | 84.5 | 15.5 | ||
| 2 | 11388 | 96.5 | 3.5 | ||
| 3 | 10000 | 95.9 | 4.1 | ||
| Average | 9288.8 | 89.1 | 10.9 | 6.5 | |
| Clonal Expansion | 1 | 9427 | 99.8 | 0.2 | |
| 2 | 10170 | 84.5 | 15.5 | ||
| 3 | 9767 | 54.7 | 45.3 | ||
| 4 | 9836 | 99.7 | 0.3 | ||
| 5 | 10080 | 28.0 | 72.0 | ||
| 6 | 9679 | 61.8 | 38.2 | ||
| 7 | 9708 | 54.4 | 45.6 | ||
| 8 | 10443 | 83.8 | 16.2 | ||
| Average | 9888.8 | 70.8 | 29.2 | 25.3 | |
| FACS | 1a | 9480 | 95.5 | 4.5 | |
| 1b | 9467 | 96.2 | 3.8 | ||
| 1c | 9337 | 95.6 | 4.4 | ||
| 2a | 9867 | 97.7 | 2.3 | ||
| 2b | 10179 | 95.9 | 4.1 | ||
| 2c | 10194 | 87.6 | 12.4 | ||
| 3a | 10900 | 96.1 | 3.9 | ||
| 3b | 9919 | 96.5 | 3.5 | ||
| 3c | 9787 | 96.3 | 3.7 | ||
| Average | 9903.3 | 95.3 | 4.7 | 3.0 |
Anti-CD59 R-PE antibody utilised in conjunction with flow cytometry to assess GPI-AP percentage expression within Dynabead®, Clonal Expansion and FACS-enriched populations of cells (~10 000 single cellular events captured).
Within the three trialled methodologies, Dynabead® and FACS were demonstrated to be highly robust, reproducible methods; however, they lacked the refinement to achieve the prerequisite sensitivity. In the case of Dynabeads®, the technology favours the use of antigens with larger molecular weights and FACS, especially prevalent within older cell sorters, is highly dependent upon operator ability, machine usage and cellular viability. Even though clonal expansion was demonstrated the most inconsistent in terms of enrichment efficiency, the method generated the optimum enrichment yield (99.8%)—the lowest background phenotypic PIG-A mutant frequency (~2000 × 106) and was therefore used for preliminary proof of principle experiments.
Determination of PIG-A mutant frequency using the CD59 phenotypic marker following exposure to the potent mutagen EMS
In order to assess the sensitivity of the proposed assay design, the performance of CD59 as a phenotypic reporter for PIG-A mutation was evaluated in clonally enriched TK6 cell cultures (TK6CD59+) exposed to a low-dose range of EMS (Figure 1).
Figure 1.

Human lymphoblastoid cells (TK6) following 24 h EMS exposure, CD59 antigen utilised as phenotypic PIG-A marker. Day 1–4 frequencies of PIG-A mutant TK6 cells (clonally selected) following 24 h low-dose EMS exposure—CD59 antigen utilised as reporter of mutation (Dunnett’s test; *P < 0.05; n = 3, error bars ±1 SD; 0.1% PIG-A mutants = 1 mutant × 10–3 cells).
A statistically significant increase in the frequency of cells negative for CD59 staining at Day 4 of analysis was observed with a 6-fold increase compared with concurrent controls (P < 0.01, Dunnett’s test). Prior to Day 4 analysis, there were no statistically significant increases in CD59-negative cells at any dose, providing evidence for a time-dependent induction of the mutant phenotype, apparently consistent with mutation induction. The lowest observed genotoxic effect level was defined as 10 µg/mL, with minimal cytotoxicity observed [~80% relative population doubling (RPD)] at the top dose.
Mathematical modelling (PROAST38.9 software) was used to further characterise the phenotypic PIG-A mutant dose response with EMS. Benchmark dose BMD10 and BMDL10 values (51) of 0.673 and 0.503 µg/mL, respectively, were observed with comparable BMD values for EMS to those reported by Doak et al. (52) in the HPRT cell mutation assay (53). However, because the spontaneous phenotypic mutant frequency (~0.2%) in controls was high, additional studies were conducted to further investigate the relationship between the absence of extracellular GPI-AP staining and PIG-A mutation (Supplementary Figures 2 and 3, available at Mutagenesis Online).
Investigations of FCM defined PIG-A phenotypes within TK6 cell populations
Aerolysin permeabilisation has previously been reported as a means to confirm a Pig-a mutant phenotype (15,17,54). Aerolysin is a bacterial toxin secreted by Aeromonas hydrophila, which causes GPI-anchor–dependent cell lysis (54–56). Therefore, aerolysin is a GPI-anchor–specific toxin that can be used to demonstrate PIG-A mutant cell resistance.
When the parental CD59 heterogeneous TK6 cell line was treated with aerolysin and stained with the vital dye, PI, the viable cell population (PI-negative) is reflected in the frequency of cells that are negative for CD59 staining (Figure 2A). Aerolysin treatment of FACS-enriched CD59-positive cell cultures (i.e. presumptive PIG-A wild type) were predominantly PI-positive (Figure 2B), consistent with aerolysin permeabilisation at the GPI-anchor sites causing complete cell death. In contrast, aerolysin treatment of cell cultures derived from CD59-negative cells (i.e. presumptive PIG-A mutants) displayed negligible PI staining (Figure 2C), consistent with the absence of GPI-anchor sites expected of the PIG-A mutant phenotype.
Figure 2.

FCM analysis of human lymphoblastoid cells (TK6) following aerolysin and PI treatment. Flow cytometric outputs following consecutive aerolysin and PI treatment on parental TK6 strain (A) and FACS enriched CD59 positive (B) and negative populations (C).
From these studies, aerolysin treatment can also provide a means to directly select GPI-AP deficient cell clones and in turn, sufficient biological material for genomic sequencing of presumed PIG-A mutants (15,32,49,57).
Lack of cellular membrane integrity, characterised by the initial loss of membrane asymmetry, blebbing and the eventual formation of pyknotic bodies, is typically observed during mid-late stage apoptosis and subsequent cell death (58–60). Due to the extracellular localisation of GPI-AP, potential membrane alterations/inversion overserved during loss of membrane integrity could induce a misleading PIG-A mutant phenotype and hence, misleading conclusions regarding mutagenicity.
To further characterise putative phenotypic PIG-A mutants, cells were stained with Annexin V Alexa Fluor 488 and 7-AAD to determine cell membrane integrity and apoptosis status (phosphatidyl serine presence). Of the TK6CD59+ cells (97.6%), 10.4% were Annexin V-positive (apoptotic) and 2.1% were 7-AAD-positive (lack of cellular membrane integrity). The small (~2%) CD59-negative cell population (i.e. phenotypic PIG-A mutants) showed much higher levels of apoptosis or loss of cellular membrane integrity (36.4 and 39.2%, respectively (Figure 3). Additional sampling, i.e. extended event capture, n = 500 000 events, confirmed this observation (Supplementary Figure 4, available at Mutagenesis Online).
Figure 3.

Preliminary validation of presumed phenotypic PIG-A mutant TK6 cells using flow cytometry. Viability assessment of clonally enriched CD59-R-PE–stained untreated TK6 cells (A) PIG-A mutant phenotype assessment, (B—I, II) viability dot plot, (C—I, II) apoptosis measure (Annexin V) and (D—I, II) loss of membrane integrity (7-AAD; ~10 000 single cellular events), PIG-A mutant phenotype and PIG-A wild-type cytograms denoted by I and II, respectively. P gates defined from maximum auto fluorescence observed following instrument calibration standard analysis within the respective channel.
These data suggest an uneven distribution of viable cells within the CD59-positive and CD59-negative cell populations, i.e. between the putative phenotypic PIG-A wild-type and mutant populations. Moreover, the high prevalence of non-viable cells in the CD59-negative cell population challenges the previous assumptions made about the frequency of presumptive PIG-A mutants. This could result in artefact, leading to elevated background frequencies of putative PIG-A mutants and a masking consequence if not fully removed from analysis; the overall effect could greatly decrease assay sensitivity (61).
GPI-AP candidate trials in different human cell lines
The establishment of a promising assay design within TK6 cells led to the identification of a potential misinterpretation of phenotypic PIG-A mutant frequencies. In an attempt to alleviate the viability issue and hence, remove potential preparative stages that could influence viability, additional cell line and reporter phenotypic combinations were assessed in order to identify an unenriched low prevalence of spontaneous PIG-A mutants. The variability, in terms of surface antigen expression, between the different candidate human B-lymphoblastoid cell lines being considered for assay development were evaluated. Immunofluorescence staining was used to assess the expression of several well-characterised cellular surface antigens or the GPI-anchor status of the cell type, i.e. FLAER, B-lymphocyte activation marker [(BLAST-1)/(CD48)], protectin (CD59) and decay-accelerating factor (DAF/CD55).
FLAER is a fluorescently modified version of inactive aerolysin, retaining high affinity for GPI-AP moiety binding. Due to its comprehensive binding, FLAER was investigated for its use as a generic reporter for PIG-A mutation detection. CD48 was chosen for its GPI-AP characteristics and routine use as a B-lymphocyte activation marker (BLAST-1).
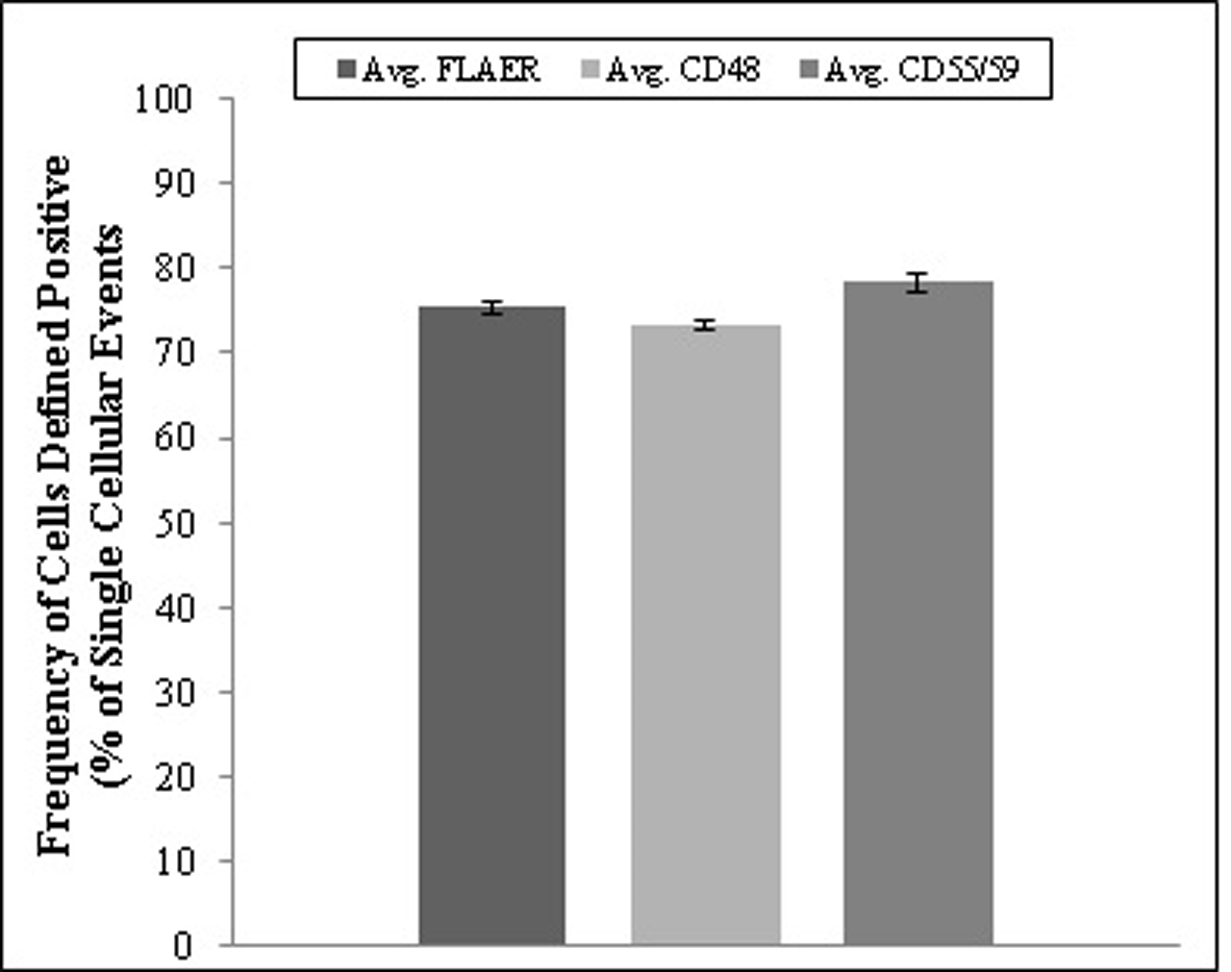
Low passage TK6 cells were shown to stain poorly for all four cell surface antigen candidates, with only 5.9% cells positive for FLAER (at top concentration 2.5 µg/mL), 2.2% cells positive for CD48 expression and 62.6% cells positive for CD55 and CD59 expression (Figure 4). The AHH-1 cell line showed higher levels of staining, i.e. 75.2% cells were positive for FLAER staining, 83.5% cells positive for CD48 staining and 99.2% cells positive for CD55 and CD59 expression (Figure 4). MCL-5 exhibited the most comprehensive staining for all fout candidates, i.e. 93.5% cells positive for FLAER, 92.1% cells positive for CD48 and 99.8% cells positive for CD55 and CD59 (Figure 4).
Figure 4.

Average cellular surface marker expression within AHH-1, MCL-5 and low passage TK6 cells. FLAER–Alexa Fluor 488™ conjugate, CD48 phycoerythrin conjugate and CD55/59 phycoerythrin conjugate fluorescent dyes utilised to estimate surface marker expression. (A) AHH-1, (B) MCL-5 and (C) low passage TK6 cells (~10 000 single cellular events; n = 3, error bars ±1 SD).
The background levels of CD55 and CD59 expression in MCL-5 cells were similar to those of the original TK6 cell lines used following enrichment (TK6CD59+/CD55+). Given these fundamental properties and the potential for artefact induction following a loss in viability during enrichment, MCL-5 cells were chosen for the continuing development of the in vitro PIG-A assay.
Viability assessment of MCL-5 Cells to avoid potential artefacts
P53 status and, hence, apoptosis functionality has been a debated topic in regard to the cell lines utilised within in vitro genetic toxicology testing (62,63). Therefore, prior to PIG-A assessment, a more detailed viability assessment was undertaken to establish apoptosis capability and provide evidence for p53 functionality. In previous TK6 cell experiments, a high prevalence of non-viable cells (apoptotic or necrotic) had been observed in the CD59-negative cell populations, which could confound an accurate estimate of the PIG-A mutant phenotype. Therefore, the impact of apoptosis on cell surface antigen expression in MCL-5 cells was assessed using cells treated with the apoptosis inducer staurosporine.
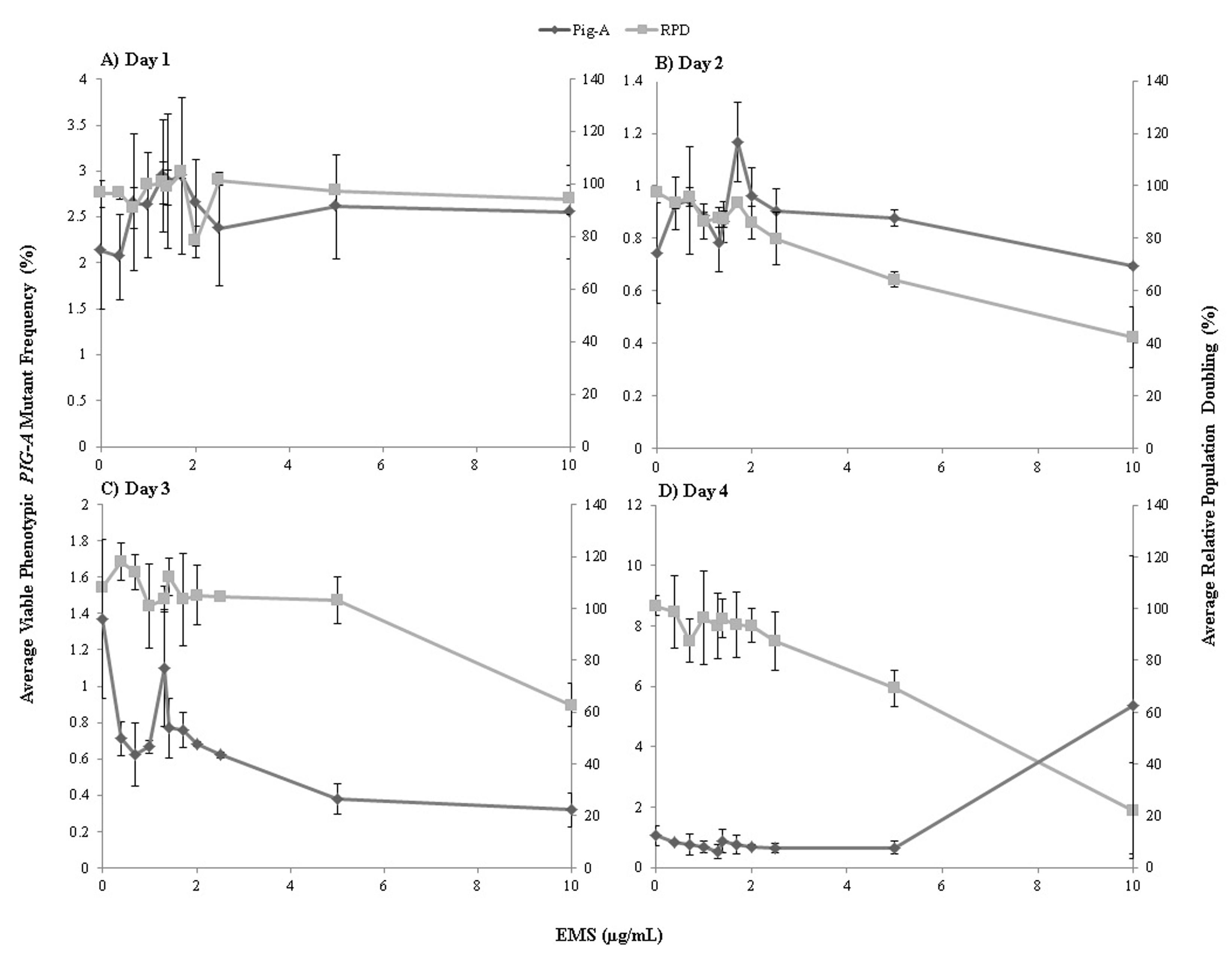
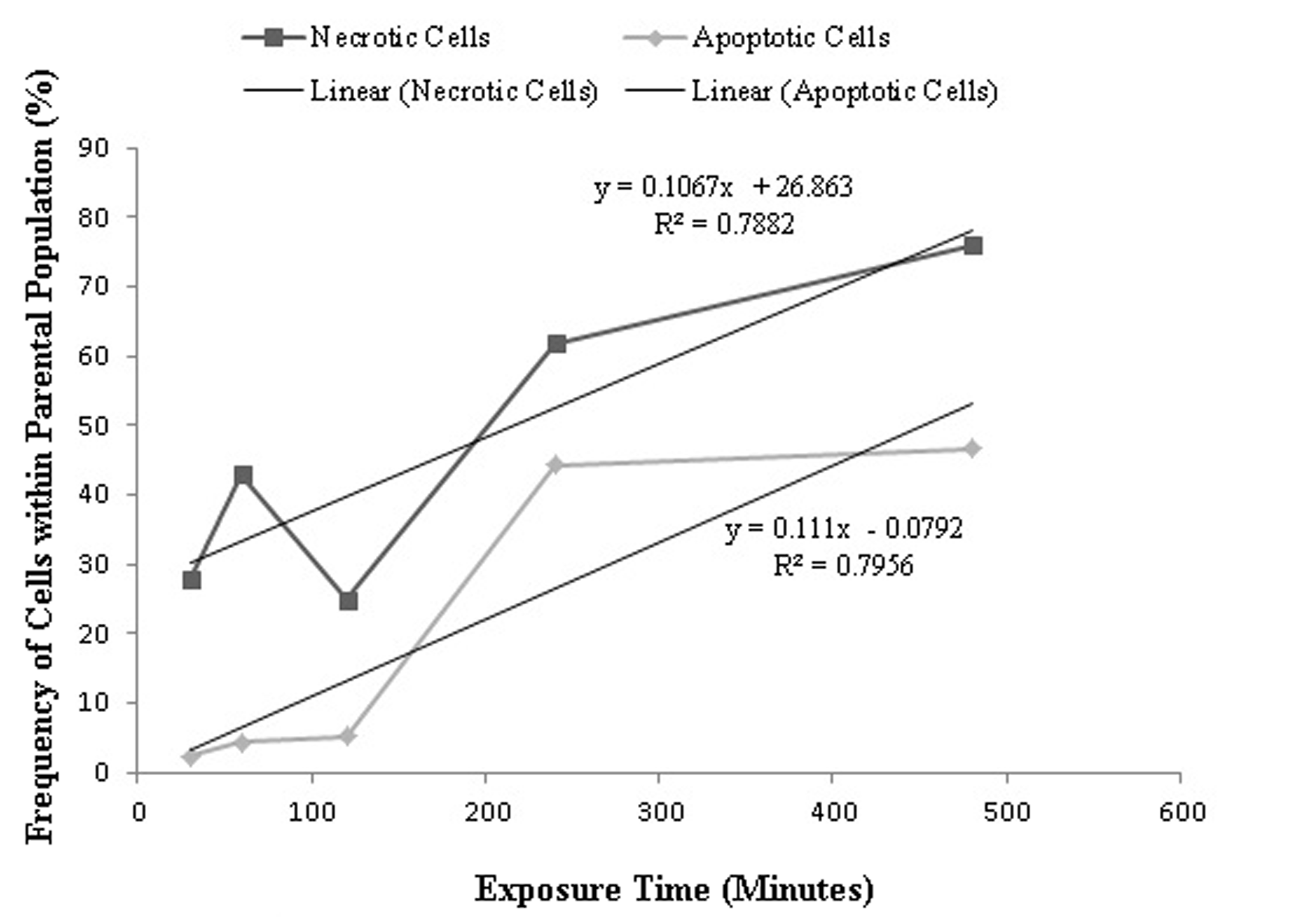
Cell viability was assessed at various time points using flow cytometric light scatter properties together with GPI-AP labelling and Annexin V and 7-AAD staining. Following staurosporine treatment, there was a dose- and time-dependent increase in the frequency of apoptotic cells and loss of cellular membrane integrity (Supplementary Figure 5, available at Mutagenesis Online); although at the earlier stages there was no significant change in cell morphology by FCM (determined by forward and side scatter profiles). Significantly, a corresponding decrease in GPI-AP expression (Figure 5) was also observed, with top doses seemingly inducing phenotypic PIG-A mutant events (Table 2).
Figure 5.

Tracking phenotypic PIG-A status through apoptosis and necrosis. Phenotypic PIG-A mutant frequency data for MCL-5 cells undergoing (A) apoptosis and (B) necrosis following a time course acute 1 µM staurosporine exposure: (1) 30 min, (2) 60 min, (3) 120 min, (4) 240 min and (5) 480 min. Relative PE detector signals utilised as the measure for surface GPI-AP expression (>3000 events analysed in each sample). ‘PIG-A WT’ gate present defines threshold for phenotypic PIG-A mutant status.
Table 2.
Tracking phenotypic PIG-A status through apoptosis and necrosis
| Sample identification | Necrotic cells | Apoptotic cells | ||||
|---|---|---|---|---|---|---|
| Percentage of dead cells | CD55/59- positive (%) | CD55/59- negative (%) | Percentage of viable cells | CD55/59- positive (%) | CD55/59- negative (%) | |
| 30 min | 27.8 | 100.0 | 0 | 2.3 | 100.0 | 0 |
| 60 min | 43.0 | 100.0 | 0 | 4.4 | 100.0 | 0 |
| 120 min | 24.9 | 100.0 | 0 | 5.2 | 100.0 | 0 |
| 240 min | 61.8 | 100.0 | 0 | 44.2 | 99.9 | 0.061 |
| 480 min | 76.0 | 98.7 | 1.3 | 46.7 | 99.9 | 0.108 |
Phenotypic PIG-A mutant frequency data for MCL-5 cells undergoing (A) apoptosis and (B) necrosis following a time course acute 1 µM staurosporine exposure (>3000 events analysed in each sample).
Since the previous observations in TK6 cells were also seen with MCL-5 cells, there is clearly the potential for cell viability artefacts to confound the PIG-A mutation phenotype assessment. It is therefore critical that viability dyes (both apoptosis and cell death) and accurate/reproducible FCM gating are applied in the development and assessment of in vitro PIG-A assays.
Determination of PIG-A mutant frequency using the CD59 phenotypic marker, viability assessment and HLA-DR staining within MCL-5 cells
To evaluate the possible benefit in phenotypic PIG-A mutation detection accuracy, transmembranous membrane protein marker staining was trialled to more accurately exclude non-viable events from FCM analysis. HLA-DR was chosen as a trial membrane marker for its comprehensive expression within the cell line utilised and to provide additional confidence in phenotypic identity. To investigate the utility of MCL-5 cells in the in vitro PIG-A assay, cell cultures were exposed to three alkylating agents, as positive mutagens, i.e. EMS, MNU or ENU and as a negative control, 2,4 DNP. Following 24 h treatment, cells were harvested and subject to daily modified cell staining including anti-HLA-DR (membrane integrity), 7-AAD and anti-CD59 to determine PIG-A phenotype (Figure 6).
Figure 6.

Trialled usage of HLA-DR staining to validate single marker PIG-A dose response data within MCL-5 Cells. Day 4, low passage MCL-5 average relative phenotypic PIG-A mutant frequency following 24 h (A) MNU (B) 2,4-DNP, (C) EMS and (D) ENU exposure, CD59 antigen utilised as reporter for mutation, inclusion of HLD-DR and 7-AAD staining to ensure cellular membrane integrity (Dunnett’s/Dunns test; *P < 0.05; n = 3; error Bars ±1 SD) PIG-A mutant data relative to the percentage of phenotypic PIG-A mutants observed within the concurrent control.
Acute MNU exposure resulted in a dose-dependent increase in the frequency of viable CD59-negative MCL-5 cells, i.e. mutant PIG-A phenotype, from 1.01% to 6.43% at the top concentration tested. Although there was a strong positive correlation between dose, cytotoxicity and phenotypic PIG-A mutants, none of the doses were statistically significant (P > 0.05). EMS exposure resulted in a comparable dose-dependent increase, with the top dose of 161 µM (equivalent to ~20 µg/mL) eliciting a statistically significant increase (P < 0.05) with a corresponding increase in cytotoxicity [8% relative increase in cell count (RICC)]. ENU exposure did not increase the number of CD59 negatively stained cells over background, i.e. the relative average phenotypic mutant frequency (P > 0.05). However, data did appear to be affected by variance between replicates, and cytotoxicity again was observed to be elevated. Acute 2,4 DNP exposure resulted in a non-linear Day 4 dose response, with no evidence for any correlation between dose and increase in relative PIG-A mutant frequency (P > 0.05). Cytotoxicity again was demonstrated to be highly dose dependent, with top doses eliciting significantly increased cytotoxicity, however, with no corresponding increments of PIG-A mutant frequency (as expected).
The data demonstrated the expected EMS and MNU results in terms of potency, MNU was observed to induce the larger fold increase in CD59-negative cells, over the concurrent controls. However, ENU did not induce the expected dose response most likely due to excessive variation within replicates. Utilising significantly shorter assay durations (4–5 days) than those recently described in analogous publications (8–14 days) (50,64) may contribute to the reduced observed genotoxic effect of ENU, possibly preventing an adequate time period for mutant phenotype manifestation. However, like 2,4 DNP, ENU showed little evidence for a positive correlation between cytotoxicity and elevated numbers of CD59 negatively stained cells, i.e. phenotypic PIG-A mutant frequency. This suggests that the inclusion of membrane assurance markers lessened the potential induction of phenotypic PIG-A mutant mimicking events. 2,4 DNP, a non-genotoxicant, was demonstrated to not increase phenotypic PIG-A mutant frequency across the entire dose range, even at doses that elicited severe cytotoxicity.
Tandem GPI-AP confocal expression analysis in MCL-5 cells
Limited physiological data, in regard to the Pig-a mechanism, is available to describe the nature of extracellular surface protein localisation and density on the surface of human lymphoblastoid cells. Within the scope of the PIG-A assay, mutagenesis is indirectly measured through the presence or the absence of GPI-AP, and therefore, mechanistic biological evidence at the cellular level could be used to provide increased integrity. Therefore, the localisation, density and coverage of initially single and sequentially tandem GPI-AP markers were investigated on the surface of MCL-5 cells using high-resolution confocal image analysis.
The proteins CD58, CD55 and CD59 were identified as possible cell surface biomarkers for the PIG-A phenotypic mutation. CD58 is a well-characterised GPI-AP, integral in strengthening the interaction between T cells and macrophages (65) and is expressed at intermediate/high levels on most subsets of normal peripheral blood leucocytes (66). CD59 (protectin) and CD55 (DAF) are used routinely for clinical diagnostic screening of PNH, associated with PIG-A mutation (19), and CD59 is also used in the in vivo Pig-a assay in rodents (29,32,34,67). However, CD58 was later demonstrated to have a transmembranous single-pass isoform (65) and therefore, CD59 and CD55 were selected for further evaluation in the development of this in vitro assay design.
Results indicated that both CD55 and CD59 immunofluorescence staining was associated comprehensively with the extracellular membrane localisation (Figure 7 AI and BI) as indicated by a distinctive halo image, reflecting the presence of peripheral cell staining. Utilsing 3-D reconstructed Z-stacked images, single GPI-AP expression was clearly defined as punctate in nature and consisted mainly of dense pockets of cell membrane expression (Figure 7 AII and BII). CD59 surface antigen expression was estimated to be less comprehensive than CD55. There was no clear association/correlation between staining density and distinct morphological features in the membrane following the construction of a composite image utilising transmitted light (Figure 7 CI). However, tandem labelling with the combined CD55/59 generated significantly greater staining and surface coverage (Figure 7 CII); and although the antibodies were still localised to densely staining regions, a much larger proportion of the extracellular membrane was visible. The punctate nature of staining was identified as a potenital limiting factor when utilsing specific excitatory sources and subsquent fluorescent image capture sysytems.
Figure 7.

GPI-AP surface expression analysis within MCL-5 cells. Utilising either or both anti-CD55 R-PE/CD59 R-PE treated 4% PFA fixed MCL-5 cells, confocal microscopy images were generated under the 63X oil immersion objective. High resolution images were produced and displayed within a 3-D reconstruction following 1024 × 1024, z-stack capture utilising the laser scanning confocal microscope. Detected, R-PE signal was retrospectively artificially coloured for image clarity and cohesion (scale bar 50 µM).
Utilsing CD55/59 tandem labelling to determine PIG-A phenotype facilitates greater confidence in the definition of presumptive PIG-A mutant cells. The unlikely probability of losing both surface antigen signals through a non-mutagenic mechanism during analysis significantly reduces the likelihood of misleading results when combined with appropriate cell viability labelling. In addition, due to the punctate nature of GPI-AP association with the surface membrane, and the nature of laser light excitation and recording, specific laser platforms could suffer from a bias due to the plane of light excitation and hence surface marker exposure.
Gating strategy investigations
Assay refinement and cell line alterations have led to the development of an in vitro PIG-A assay design utilising: (i) a membrane assurance marker (HLA-DR); (ii) viability markers (Annexin V and 7-AAD) and (iii), tandem antibody reporting markers (CD55/59). However, gating strategy also contributes significantly to the performance of the analysis. The use of alternative properties of the recorded light signal, other than solely the average or maximum fluorescence value generated, was investigated to more accurately define a viable single-cell population in which to assess PIG-A phenotype. Fluorescence width measurements (time of flight) have been recently incorporated into FCM analysis as an additional variable to be used to estimate cell size and therefore, offer a potential supplementary layer of cell characterisation during analysis.
Following the trialling of three gating strategies (Table 3), each utilising a different combination of fluorescence area and width measurements to define the single-cell population; it was apparent that the strategy had significant implications on the final presumptive PIG-A mutant frequency. Utilising pulse width measurements, analogous to that observed within a typical doublet discriminant plot, as an additional means to classify particles as single cell in nature could significantly reduce the number of events falsely being defined as single cell and therefore more accurately define the phenotypic PIG-A mutant frequency. Utilising a retrospective analysis on singlet Day 3, 24-h MNU exposure data within MCL-5 cells (Figure 8), more realistic mutant frequency data (~100 phenotypic mutants per 1 × 106 cells scored) was generated following the implementation of the refined gating strategy. However, pulse width measurements in combination with area measures for the fluorophore reporting phenotypic PIG-A status can have a biasing effect on mutant gating, when restrictive region gates were applied to the fluorescence area axis. This gating combination underestimated the number of defined phenotypic PIG-A mutant cells, unless gating counter measures were used.
Table 3.
Summary of presumed phenotypic PIG-A status of parental untreated MCL-5 cells, pre-enrichment utilising different gating strategies.
| Sample ID | Number of recorded events | Single-cell events | Trialled gated events | Number of viable cells | CD55/59-positive (%) |
|---|---|---|---|---|---|
| Instrument calibration standard | 43 856 | 20 575 | 17 574 | 17 573 | 0 |
| Original gating | 48 629 | 20 569 | n/a | 17 577 | 85.45 |
| Trialled gating 1 | |||||
| FSC-A vs PE-W 1 | 48 629 | 20 391 | 18 056 | 17 874 | 99.99 |
| FSC-A vs PE-W 2 | 59 135 | 20 495 | 17 983 | 17 750 | 99.99 |
| FSC-A vs PE-W 3 | 48 190 | 20 418 | 18 171 | 18 019 | 100.00 |
| Average | 51984.7 | 20 434.7 | 18 070 | 17 881 | 99.99 |
| Trialled gating 2 | |||||
| PE-A vs PE-W 1 | 48 629 | 20 841 | 18 584 | 18 407 | 100.00 |
| PE-A vs PE-W 2 | 59 135 | 20 882 | 18 384 | 18 140 | 100.00 |
| PE-A vs PE-W 3 | 48 190 | 20 748 | 18 431 | 18 245 | 100.00 |
| Average | 51984.7 | 20823.7 | 18466.3 | 18 264 | 100.00 |
Tandem surface antigen markers CD55 and CD59 were utilised as indirect reporters of PIG-A mutation in order to assess phenotypic PIG-A mutant status following incorporation of pulse width measurement gating strategies (>20 000 defined single cellular events were captured).
Figure 8.

Establishment of a robust in vitro PIG-A assay gating strategy. Utilising viability dye incorporation (Annexin V and 7-AAD) and tandem markers (CD55 and CD59) the proposed robust reproducible gating strategy (B) was directly compared against traditional flow cytometry gating (A). Retrospectively, Day 3, 24 h MNU (0–1 µg/mL) exposure data within MCL-5 cells was assessed using both methods in order to estimate phenotypic PIG-A mutant frequency and hence, dose response.
FCM analysis can be subjective without the use of standardised protocols. Therefore, identifying reproducible methods for establishing thresholds and utilising unbiased gating approaches are fundamental in establishing comprehensive test systems with confidence and accuracy. Utilising pulse width measures in combination with a robust threshold gating strategy could significantly reduce the number of falsely identified putative phenotypic PIG-A mutant events (Figure 8). Doublet discrimination and correct pulse width measures more accurately define the viable single-cell population and therefore can prevent the inclusion of doublets, underestimating/overestimating phenotypic mutant frequency, or debris from inducing false positives. Standardising the gating approaches and further refining assay design has been shown to reliably reduce artificially high baseline mutant frequencies (~5–50 × 10−6 mutants) and has brought the perceived assay sensitivity more in line with currently validated in vitro gene mutation endpoints (MLA/HRPT), enabling future low-dose investigations and potential quantitative dose–response modelling.
Conclusions
During this study, the proposed in vitro PIG-A assay design underwent a number of revisions and refinements following identification of potential confounding factors. The assay design, established within the metabolically active, genetically modified, human MCL-5 cell line, has demonstrated proofs of principle and the potential focus for future work. Toxicity was demonstrated to be a continuous problem associated with the misidentification of the putative PIG-A deficient phenotype; however, the studies reported here suggest that these issues are effectively addressed through the inclusion of membrane integrity markers such as HLA-DR or alternatively CD19 (50) (highlighted by the 2,4-DNP and ENU data sets; Figure 6) in combination with adequate apoptosis and cell death markers.
Toxicity measures are used to define the doses tested and relevance of results within any mutation assay. A measure utilised within the in vitro PIG-A assay design which does not account for clonal expansion/cloning efficiency could miscalculate toxicity. RICC, RPD and relative cell counts are all measures that may not be strictly suitable outside of intended usage within the MN assay based on the duration of the assay. Shifting the toxicity measure more in line with analogous test systems, such as HPRT and/or MLA, could potentially alleviate toxicity misinterpretation, seemingly more apparent at the initial stages of phenotype assessment and could facilitate minimal experimental duration.
The genotype/phenotype relationship validation is still ongoing, recent publications reporting novel PIG-L gene mutations, following loss of heterozygosity deletions within the TK6 cell line, which also contribute to the GPI-AP deficient phenotype in addition to PIG-A mutations (38,64), have heightened the need for cell line-specific identification of additional genes active within the current phenotypic PIG-A scoring mechanism. Validatory sequencing experiments should not utilise any inadvertently biasing means of loss of phenotypic mutant selection if population statistics are to be generated and focus on whole genome sequence analysis or at least multiple PIG genes from the accurately gated loss of phenotype population.
In conclusion, this study suggests that a stand-alone PIG-A assay within MCL-5 cells is more promising than a TK6 cell-based assay. Further refinement, followed by a comprehensive assessment of specificity, sensitivity and transferability could lead to the establishment of a high content, high-throughput assay system with the potential for use within a screening as well as a regulatory hazard and risk assessment environment. All data from which conclusions were drawn is contained within the article or clearly referenced.
Supplementary data
Supplementary Figures 1 to 5 are available at Mutagenesis Online.
Funding
Ph.D. work of B.R. was supported by the Engineering and Physical Sciences Research Council–GlaxoSmithKline case award [EP/J502248/1]. Work carried out at Gentronix Ltd was funded by Gentronix Ltd.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
These results were the product of collaboration between laboratories at Swansea University (including Neil Webb and Leanne Stannard) and Gentronix Ltd (including Nick Billinton, Jodie Allsup and Sabrina Khaliq), under the direction of G.E.J. and R.M.W., respectively.
Conflict of interest statement: G.E.J. consults with industry, R.M.W. founder of Gentronix Ltd and M.T. employee of Gentronix Ltd.
References
- 1. Cimino M. C. (2006)Comparative overview of current international strategies and guidelines for genetic toxicology testing for regulatory purposes. Environ. Mol. Mutagen., 47, 362–390. [DOI] [PubMed] [Google Scholar]
- 2. Ellis P. Fowler P. Booth E. Kidd D. Howe J. Doherty A. and Scott A (2014)Where will genetic toxicology testing be in 30 years’ time? Summary report of the 25th Industrial Genotoxicity Group Meeting, Royal Society of Medicine, London, November 9, 2011. Mutagenesis, 29, 73–77. [DOI] [PubMed] [Google Scholar]
- 3. Kirkland D. (2011)Improvements in the reliability of in vitro genotoxicity testing. Expert Opin. Drug Metab. Toxicol., 7, 1513–1520. [DOI] [PubMed] [Google Scholar]
- 4. FDA(2012)Guidance for Industry: S2(R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. http://www.fda.gov/downloads/Drugs/Guidances/ucm074931.pdf (accessed November 3, 2016). [PubMed] [Google Scholar]
- 5.OECD (2015) OECD Guidance Document on Revisions to OECD Genetic Toxicology Test Guidelines. https://www.oecd.org/chemicalsafety/testing/Genetic%20Toxicology%20Guidance%20Document%20Aug%2031%202015.pdf (accessed November 3, 2016). [Google Scholar]
- 6. OECD(1997)Test No. 476: In vitro Mammalian Cell Gene Mutation Test. http://www.oecd-ilibrary.org/environment/test-no-476-in-vitro-mammalian-cell-gene-mutation-test_9789264071322-en (accessed November 3, 2016). [Google Scholar]
- 7. OECD(2014)OECD Guideline for Testing of Chemicals - In Vitro Mammalian Cell Gene Mutation Assays Using the Thymidine Kinase Gene. [Google Scholar]
- 8. Miyata T. Takeda J. Iida Y. Yamada N. Inoue N. Takahashi M. Maeda K. Kitani T. and Kinoshita T (1993)The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science, 259, 1318–1320. [DOI] [PubMed] [Google Scholar]
- 9. Bessler M. Hillmen P. Longo L. Luzzatto L. and Mason P. J (1994)Genomic organization of the X-linked gene (PIG-A) that is mutated in paroxysmal nocturnal haemoglobinuria and of a related autosomal pseudogene mapped to 12q21. Hum. Mol. Genet., 3, 751–757. [DOI] [PubMed] [Google Scholar]
- 10. Bryce S. M. Bemis J. C. and Dertinger S. D (2008)In vivo mutation assay based on the endogenous Pig-a locus. Environ. Mol. Mutagen., 49, 256–264. [DOI] [PubMed] [Google Scholar]
- 11. Brodsky R. A. Mukhina G. L. Li S. Nelson K. L. Chiurazzi P. L. Buckley J. T. and Borowitz M. J (2000)Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am. J. Clin. Pathol., 114, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishimura J. Murakami Y. and Kinoshita T (1999)Paroxysmal nocturnal hemoglobinuria: An acquired genetic disease. Am. J. Hematol., 62, 175–182. [DOI] [PubMed] [Google Scholar]
- 13. Kawagoe K. Takeda J. Endo Y. and Kinoshita T (1994)Molecular cloning of murine pig-a, a gene for GPI-anchor biosynthesis, and demonstration of interspecies conservation of its structure, function, and genetic locus. Genomics, 23, 566–574. [DOI] [PubMed] [Google Scholar]
- 14. Ferguson M. A. and Williams A. F (1988)Cell-surface anchoring of proteins via glycosyl-phosphatidylinositol structures. Annu. Rev. Biochem., 57, 285–320. [DOI] [PubMed] [Google Scholar]
- 15. Hu R. Mukhina G. L. Piantadosi S. Barber J. P. Jones R. J. and Brodsky R. A (2005)PIG-A mutations in normal hematopoiesis. Blood, 105, 3848–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu R. Mukhina G. L. Lee S. H. Jones R. J. Englund P. T. Brown P. Sharkis S. J. Buckley J. T. and Brodsky R. A (2009)Silencing of genes required for glycosylphosphatidylinositol anchor biosynthesis in Burkitt lymphoma. Exp. Hematol., 37, 423–434.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dobrovolsky V. N. Miura D. Heflich R. H. and Dertinger S. D (2010)The in vivo pig-a gene mutation assay, a potential tool for regulatory safety assessment. Environ. Mol. Mutagen., 51, 825–835. [DOI] [PubMed] [Google Scholar]
- 18. Wilcox L. A. Ezzell J. L. Bernshaw N. J. and Parker C. J (1991)Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood, 78, 820–829. [PubMed] [Google Scholar]
- 19. Takeda J. Miyata T. Kawagoe K. Iida Y. Endo Y. Fujita T. Takahashi M. Kitani T. and Kinoshita T (1993)Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell, 73, 703–711. [DOI] [PubMed] [Google Scholar]
- 20. Araten D. J. Nafa K. Pakdeesuwan K. and Luzzatto L (1999)Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc. Natl. Acad. Sci. USA, 96, 5209–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peruzzi B. Araten D. J. Notaro R. and Luzzatto L (2010)The use of PIG-A as a sentinel gene for the study of the somatic mutation rate and of mutagenic agents in vivo. Mutat. Res., 705, 3–10. [DOI] [PubMed] [Google Scholar]
- 22. Chen R. Eshleman J. R. Brodsky R. A. and Medof M. E (2001)Glycophosphatidylinositol-anchored protein deficiency as a marker of mutator phenotypes in cancer. Cancer Res., 61, 654–658. [PubMed] [Google Scholar]
- 23. Dobrovolsky V. N. Elespuru R. K. Bigger C. A. Robison T. W. and Heflich R. H (2011)Monitoring humans for somatic mutation in the endogenous PIG-a gene using red blood cells. Environ. Mol. Mutagen., 52, 784–794. [DOI] [PubMed] [Google Scholar]
- 24. Cammerer Z. Bhalli J. A. Cao X., et al. (2011)Report on stage III Pig-a mutation assays using N-ethyl-N-nitrosourea-comparison with other in vivo genotoxicity endpoints. Environ. Mol. Mutagen., 52, 721–730. [DOI] [PubMed] [Google Scholar]
- 25. Dertinger S. D. Phonethepswath S. Weller P., et al. (2011)International Pig-a gene mutation assay trial: evaluation of transferability across 14 laboratories. Environ. Mol. Mutagen., 52, 690–698. [DOI] [PubMed] [Google Scholar]
- 26. Lynch A. M. Giddings A. Custer L. Gleason C. Henwood A. Aylott M. and Kenny J (2011)International Pig-a gene mutation assay trial (stage III): results with N-methyl-N-nitrosourea. Environ. Mol. Mutagen., 52, 699–710. [DOI] [PubMed] [Google Scholar]
- 27. Johnson G. E. Yamamoto M. Suzuki Y., et al. (in press) Measuring reproducibility of dose response data for the Pig-a assay using covariate benchmark dose analysis. Mutat. Res. http://www.sciencedirect.com/science/article/pii/S1383571816301012 (accessed November 3, 2016). [DOI] [PubMed] [Google Scholar]
- 28. Dobrovolsky V. N. Boctor S. Y. Twaddle N. C. Doerge D. R. Bishop M. E. Manjanatha M. G. Kimoto T. Miura D. Heflich R. H. and Ferguson S. A (2010)Flow cytometric detection of Pig-A mutant red blood cells using an erythroid-specific antibody: application of the method for evaluating the in vivo genotoxicity of methylphenidate in adolescent rats. Environ. Mol. Mutagen., 51, 138–145. [DOI] [PubMed] [Google Scholar]
- 29. Dertinger S. D. Bryce S. M. Phonethepswath S. and Avlasevich S. L (2011)When pigs fly: immunomagnetic separation facilitates rapid determination of Pig-a mutant frequency by flow cytometric analysis. Mutat. Res., 721, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kimoto T. Chikura S. Suzuki K., et al. (2011)Further development of the rat Pig-a mutation assay: measuring rat Pig-a mutant bone marrow erythroids and a high throughput assay for mutant peripheral blood reticulocytes. Environ. Mol. Mutagen., 52, 774–783. [DOI] [PubMed] [Google Scholar]
- 31. Kimoto T. Suzuki K. Kobayashi X. M. Dobrovolsky V. N. Heflich R. H. Miura D. and Kasahara Y (2011)Manifestation of Pig-a mutant bone marrow erythroids and peripheral blood erythrocytes in mice treated with N-ethyl-N-nitrosourea: direct sequencing of Pig-a cDNA from bone marrow cells negative for GPI-anchored protein expression. Mutat. Res., 723, 36–42. [DOI] [PubMed] [Google Scholar]
- 32. Dobrovolsky V. N. Shaddock J. G. Mittelstaedt R. A. Miura D. and Heflich R. H (2013)Detection of in vivo mutation in the Hprt and Pig-a genes of rat lymphocytes. Methods Mol. Biol., 1044, 79–95. [DOI] [PubMed] [Google Scholar]
- 33. Rondelli T. Berardi M. Peruzzi B. Boni L. Caporale R. Dolara P. Notaro R. and Luzzatto L (2013)The frequency of granulocytes with spontaneous somatic mutations: a wide distribution in a normal human population. PLoS One, 8, e54046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dertinger S. D. Avlasevich S. L. Bemis J. C. Chen Y. and MacGregor J.T (2014)Human erythrocyte PIG-A assay: an easily monitored index of gene mutation requiring low volume blood samples. Environ. Mol. Mutagen, 56, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobrovolsky V. N. Revollo J. Pearce M. G. Pacheco-Martinez M. M. and Lin H (2015)CD48-deficient T-lymphocytes from DMBA-treated rats have de novo mutations in the endogenous Pig-a gene. Environ. Mol. Mutagen., 56, 674–683. [DOI] [PubMed] [Google Scholar]
- 36. Revollo J. Pearce M. G. Petibone D. M. Mittelstaedt R. A. and Dobrovolsky V. N (2015)Confirmation of Pig-a mutation in flow cytometry-identified CD48-deficient T-lymphocytes from F344 rats. Mutagenesis, 30, 315–324. [DOI] [PubMed] [Google Scholar]
- 37. Luzzatto L. (2013)PNH from mutations of another PIG gene. Blood, 122, 1099–1100. [DOI] [PubMed] [Google Scholar]
- 38. Nicklas J. A. Carter E. W. and Albertini R. J (2015)Both PIGA and PIGL mutations cause GPI-a deficient isolates in the Tk6 cell line. Environ. Mol. Mutagen, 56, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.National Centre for the Replacement, Refinement and Reduction of Animals in Research (2016). The 3Rs. http://www.nc3rs.org.uk/the-3rs (accessed November 3, 2016). [Google Scholar]
- 40. Union E. (2003)7th Amendment to the Cosmetics Directive. http://ec.europa.eu/consumers/sectors/cosmetics/files/doc/antest/(2)_executive_summary_en.pdf (accessed date November 3, 2016). [Google Scholar]
- 41.European Commission (2015) Report for establishing the timetable for phasing out animal testing for the purpose of the Cosmetics Directive 2004. http://ec.europa.eu/DocsRoom/documents/13101 (accessed November 3, 2016). [Google Scholar]
- 42. Dourson M. Becker R. A. Haber L. T. Pottenger L. H. Bredfeldt T. and Fenner-Crisp P. A (2013)Advancing human health risk assessment: integrating recent advisory committee recommendations. Crit. Rev. Toxicol., 43, 467–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas R. S. Philbert M. A. Auerbach S. S., et al. (2013)Incorporating new technologies into toxicity testing and risk assessment: moving from 21st century vision to a data-driven framework. Toxicol. Sci., 136, 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mahadevan B. Snyder R. D. Waters M. D. Benz R. D. Kemper R. A. Tice R. R. and Richard A. M (2011)Genetic toxicology in the 21st century: reflections and future directions. Environ. Mol. Mutagen., 52, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. National Research Council(2007)Toxicity Testing in the 21st Century: A Vision and a Strategy. The National Academies Press, Washington, DC. [Google Scholar]
- 46. Fowler P. Smith R. Smith K. Young J. Jeffrey L. Carmichael P. Kirkland D. and Pfuhler S (2014)Reduction of misleading (‘false’) positive results in mammalian cell genotoxicity assays. III: Sensitivity of human cell types to known genotoxic agents. Mutat. Res. Genet. Toxicol. Environ. Mutagen, 767, 28–36. [DOI] [PubMed] [Google Scholar]
- 47. Fowler P. Smith K. Young J. Jeffrey L. Kirkland D. Pfuhler S. and Carmichael P (2012)Reduction of misleading (‘false’) positive results in mammalian cell genotoxicity assays. I. Choice of cell type. Mutat. Res., 742, 11–25. [DOI] [PubMed] [Google Scholar]
- 48. Fowler P. Smith R. Smith K. Young J. Jeffrey L. Kirkland D. Pfuhler S. and Carmichael P (2012)Reduction of misleading (‘false’) positive results in mammalian cell genotoxicity assays. II. Importance of accurate toxicity measurement. Mutat. Res., 747, 104–117. [DOI] [PubMed] [Google Scholar]
- 49. Nakamura J. Gul H. Tian X. Bultman S. J. and Swenberg J. A (2012)Detection of PIGO-deficient cells using proaerolysin: a valuable tool to investigate mechanisms of mutagenesis in the DT40 cell system. PLoS One, 7, e33563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kruger C. T. Hofmann M. and Hartwig A (2014)The in vitro PIG-A gene mutation assay: mutagenicity testing via flow cytometry based on the glycosylphosphatidylinositol (GPI) status of TK6 cells. Arch. Toxicol, 89, 2429–2443. [DOI] [PubMed] [Google Scholar]
- 51. Johnson G. E. Soeteman-Hernández L. G. Gollapudi B. B., et al. (2014)Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environ. Mol. Mutagen., 55, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doak S. H. Jenkins G. J. Johnson G. E. Quick E. Parry E. M. and Parry J. M (2007)Mechanistic influences for mutation induction curves after exposure to DNA-reactive carcinogens. Cancer Res., 67, 3904–3911. [DOI] [PubMed] [Google Scholar]
- 53. Gollapudi B. B. Johnson G. E. Hernandez L. G., et al. (2013)Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ. Mol. Mutagen., 54, 8–18. [DOI] [PubMed] [Google Scholar]
- 54. Peghini P. E. and Fehr J (2005)Clinical evaluation of an aerolysin-based screening test for paroxysmal nocturnal haemoglobinuria. Cytometry B. Clin. Cytom., 67, 13–18. [DOI] [PubMed] [Google Scholar]
- 55. Parker M. W. van der Goot F. G. and Buckley J. T (1996)Aerolysin—the ins and outs of a model channel-forming toxin. Mol. Microbiol., 19, 205–212. [DOI] [PubMed] [Google Scholar]
- 56. van der Goot F. G. Pattus F. Parker M. and Buckley J. T (1994)The cytolytic toxin aerolysin: from the soluble form to the transmembrane channel. Toxicology, 87, 19–28. [DOI] [PubMed] [Google Scholar]
- 57. Savoia A. Ianzano L. Lunardi C. De Sandre G. Carotenuto M. Musto P. and Zelante L (1996)Identification of three novel mutations in the PIG-A gene in paroxysmal nocturnal haemoglobinuria (PNH) patients. Hum. Genet., 97, 45–48. [DOI] [PubMed] [Google Scholar]
- 58. Garcia-Belinchon M. Sanchez-Osuna M. Martinez-Escardo L. Granados-Colomina C. Pascual-Guiral S. Iglesias-Guimarais V. Casanelles E. Ribas J. and Yuste V. J (2015)An early and robust activation of caspases heads cells for a regulated form of necrotic-like cell death. J Biol Chem, 290, 20841–20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lang F. and Hoffmann E. K (2012)Role of ion transport in control of apoptotic cell death. Compr. Physiol., 2, 2037–2061. [DOI] [PubMed] [Google Scholar]
- 60. Elmore S. (2007)Apoptosis: a review of programmed cell death. Toxicol. Pathol., 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rees, B. J., Jenkins, G. J., Lynch, A. M. and Johnson, G. E. (2012) Development of the mammalian in-vitro Pig-A gene mutation assay. Mutagenesis, 27, 789–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guest R. D. and Parry J. M (1999)P53 integrity in the genetically engineered mammalian cell lines AHH-1 and MCL-5. Mutat. Res., 423, 39–46. [DOI] [PubMed] [Google Scholar]
- 63. Morris S. M. Manjanatha M. G. Shelton S. D. Domon O. E. McGarrity L. J. and Casciano D. A (1996)A mutation in the p53 tumor suppressor gene of AHH-1 tk+/- human lymphoblastoid cells. Mutat. Res., 356, 129–134. [DOI] [PubMed] [Google Scholar]
- 64. Krüger C. T. Fischer B. M. Armant O. Morath V. Strähle U. and Hartwig A (2016)The in vitro PIG-A gene mutation assay: glycosylphosphatidylinositol (GPI)-related genotype-to-phenotype relationship in TK6 cells. Arch. Toxicol., 90, 1729–1736. [DOI] [PubMed] [Google Scholar]
- 65. Ariel O. Levi Y. and Hollander N (2009)Signal transduction by CD58: the transmembrane isoform transmits signals outside lipid rafts independently of the GPI-anchored isoform. Cell. Signal., 21, 1100–1108. [DOI] [PubMed] [Google Scholar]
- 66. Hernández-Campo P. M. Almeida J. Sánchez M. L. Malvezzi M. and Orfao A (2006)Normal patterns of expression of glycosylphosphatidylinositol-anchored proteins on different subsets of peripheral blood cells: a frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry B. Clin. Cytom., 70, 71–81. [DOI] [PubMed] [Google Scholar]
- 67. Bryce S. M. and Dertinger S. D (2012)Rapid in vivo gene mutation assay based on the pig-a gene. Google Patents. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.