

Abstract
Glioblastoma multiforme (GBM), the most common and aggressive primary brain tumor, has a high mortality rate despite extensive efforts to develop new treatments. GBM exhibits both intra- and intertumor heterogeneity, lending to resistance and eventual tumor recurrence. Large-scale genomic and proteomic analysis of GBM tumors has uncovered potential drug targets. Effective and “druggable” targets must be validated to embark on a robust medicinal chemistry campaign culminating in the discovery of clinical candidates. Here, we review recent developments in GBM drug discovery and delivery. To identify GBM drug targets, we performed extensive bioinformatics analysis using data from The Cancer Genome Atlas project. We discovered 20 genes, BOC, CLEC4GP1, ELOVL6, EREG, ESR2, FDCSP, FURIN, FUT8-AS1, GZMB, IRX3, LITAF, NDEL1, NKX3-1, PODNL1, PTPRN, QSOX1, SEMA4F, TH, VEGFC, and C20orf166AS1 that are overexpressed in a subpopulation of GBM patients and correlate with poor survival outcomes. Importantly, nine of these genes exhibit higher expression in GBM versus low-grade glioma and may be involved in disease progression. In this review, we discuss these proteins in the context of GBM disease progression. We also conducted computational multi-parameter optimization to assess the blood-brain barrier (BBB) permeability of small molecules in clinical trials for GBM treatment. Drug delivery in the context of GBM is particularly challenging because the BBB hinders small molecule transport. Therefore, we discuss novel drug delivery methods, including nanoparticles and prodrugs. Given the aggressive nature of GBM and the complexity of targeting the central nervous system, effective treatment options are a major unmet medical need. Identification and validation of biomarkers and drug targets associated with GBM disease progression present an exciting opportunity to improve treatment of this devastating disease.
I. Introduction
Glioblastoma multiforme (GBM) is the most malignant primary central nervous system tumor, and the prognosis for patients is often bleak. Currently, there are no curative treatment options for GBM, and despite rigorous therapeutic research, the survival rate of patients diagnosed with GBM remains low. Median overall survival is 15–23 months and 5-year survival is less than 6%, which is the lowest long-term survival rate of malignant brain tumors (Ostrom et al., 2016). An estimated 79,270 new cases of primary brain and other central nervous system (CNS) tumors were expected to be diagnosed in 2017 (Ostrom et al., 2016). To improve therapeutic options, studies to identify and validate single protein targets are underway. However, in most cases, targeted compounds that perform well in preclinical studies have failed expensive Phase III clinical trials in humans. Ultimately, several major factors are responsible for drug failure, including poor pharmacokinetic properties, emergence of resistance pathways, complex intratumoral heterogeneity, and suboptimal clinical trial design. Thus, there is a desperate need for an efficient approach to identify and vet potential drugs at the preclinical stage to prevent late stage failure. Genomic- and proteomic-scale analysis can identify proteins and pathways involved in the development of chemotherapeutic resistance mechanisms responsible for recurrent disease.
With the advent of TCGA (The Cancer Genome Atlas) consortium and resources, genomic analysis of cancers is at the forefront of drug discovery. Additionally, proteomics is gaining widespread use in drug discovery efforts. Quantitative proteomics can measure the expression and, in some cases, posttranslational modification status of up to and over 8000 proteins in the cell at any given time. The advent of novel proteomic techniques in the last decade, in tandem with the resources allocated to address the lack of a cure for GBM, will accelerate the discovery of a treatment and shed light on the feasibility of precision medicine.
The target and mechanism of action of many Food and Drug Administration (FDA)-approved drugs are not fully established. Of the approximately 1600 FDA-approved drugs with known targets, most belong to four classes: GPCRs (33%), ion channels (18%), nuclear receptors (16%), and kinases (3%) (Supplemental Fig. 1) (Santos et al., 2017). This suggests uncharted proteomic space exists in which novel drug targets may be identified. Of the six drugs approved for the treatment of GBM, three act as DNA alkylators, two are kinase inhibitors, and one is a tubulin inhibitor. Burgeoning research efforts in novel treatment areas, including alternating electric field therapy (tumor-treating fields), immunotherapy, and antibody-drug conjugates are improving patient outcomes. Much of the challenge in developing a GBM therapy lies in reaching therapeutic concentrations at the target site. Few drug molecules cross the blood-brain barrier (BBB), and those that do may be exported via efflux pumps. Therefore, valid target selection, permeability, and drug pharmacokinetics are important considerations in GBM drug design.
In this review, we highlight the importance of genomic and proteomic research on identifying novel biomarkers and drug targets for GBM treatment. Additionally, we demonstrate a genomic approach to drug discovery and uncover novel potential drug targets by performing bioinformatics analysis of TCGA data. Although further validation is necessary and increased expression of some of these targets may be a response to oncogenic stress, this approach provides a list of proteins that, if inhibited alone or in combination with other targets, could effectively treat GBM. Furthermore, we address the challenges faced in the drug discovery and delivery process and discuss potential solutions to those problems. In particular, we focus on the challenge of BBB permeability, nanocarrier design, and the application of computational methods to aid compound optimization. In recent years, major clinical trials for small molecule treatment of GBM have failed because the compounds did not reach effective concentrations in the brain (i.e., gefitinib and erlotinib) (Agarwal et al., 2010; de Vries et al., 2012). Thus, an understanding of BBB function and physiology is crucial for the development of efficacious small molecule treatment strategies and the avoidance of failed expensive clinical trials. The lack of effective treatment options for GBM emphasizes the unmet need for successful target inhibition and drug delivery strategies.
II. Current Treatment Options for Glioblastoma
Upon diagnosis, GBM treatment includes maximal surgical resection, followed by temozolomide and radiation (Stupp et al., 2005). Due to the invasive nature of GBM, surgical resection rarely eliminates all tumor cells, and postsurgical treatment is usually necessary to prevent recurrence. Treatment varies based on the age of the patient and stage of the disease. Depending on the overall health of the patient and disease status, they may also be enrolled in relevant ongoing clinical trials.
The standard-of-care temozolomide is a DNA-alkylating agent discovered in the 1970s and approved in 2005 by the FDA to treat newly diagnosed brain tumors. The first clinical trial with temozolomide was conducted in 1993, and, of the 10 patients who received adjuvant temozolomide, five patients showed significant clinical and radiographic improvement (O’Reilly et al., 1993). The success of this initial study prompted further successful studies of temozolomide treatment in GBM patients. In these studies, subsets of patients were more responsive to temozolomide treatment than others. Responsive patients had methyl-guanine-methyltransferase (MGMT) genes with methylated promoters and showed higher survival rates than patients with hypomethylated MGMT genes (Hegi et al., 2005). MGMT is a DNA repair enzyme that repairs the N7 and O6 positions of guanine alkylated by temozolomide. Although MGMT depletion does not seem to be an effective treatment strategy (Quinn et al., 2009; Robinson et al., 2010), MGMT gene methylation status nevertheless remains an important biomarker for GBM prognosis. Although temozolomide is part of the standard chemotherapeutic regimen for GBM, it presents unwanted toxicity and does not eliminate the disease. As an alternative approach, targeted therapies may limit unwanted toxicity and more effectively block tumor proliferation.
A promising targeted treatment is the anti–vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab. Bevacizumab was first approved by the FDA in 2004 to treat metastatic colorectal cancer. Since then, it has been approved for several different types of cancer, including GBM in 2009. Angiogenesis is a key survival feature of many cancers as tumors rely on nutrients from the vasculature to proliferate. VEGF is a broad mediator of tumor neovascularization, and VEGF expression is linked with GBM tumorgenicity (Cheng et al., 1996). Bevacizumab was first tested in 21 patients with malignant glioma in 2004. Patients were treated with bevacizumab at 5 mg/kg and irinotecan at 125 mg/m2 every 2 weeks, producing a significant 43% response rate (Stark-Vance, 2005). However, the Phase III “Avaglio” trial, conducted on 921 patients with newly diagnosed GBM, resulted in no overall survival benefit in bevacizumab-treated versus placebo-treated patients (median overall survival of 16.8 months for bevacizumab-treated patients and 16.7 months for placebo-treated patients) (Chinot et al., 2014). A second Phase III trial, the RTOG 0825 trial, produced similar results. Out of 637 patients receiving either 10 mg/kg bevacizumab every 2 weeks or placebo, there was no significant difference in overall survival between the two groups (median overall survival of 15.7 months for bevacizumab-treated patients vs. 16.1 months for the placebo group) (Gilbert et al., 2014). Therefore, bevacizumab treatment is an option reserved for patients with recurrent GBM.
Almost all GBM tumors that respond to first-line therapy recur. There is no standard approach for a successful treatment of recurrent GBM. Second-line treatment may take several directions, depending on factors such as tumor size and location, previous treatments, age, and time from initial diagnosis. Treatment can include surgical resection, reirradiation, nitrosoureas, temozolomide rechallenge, bevacizumab, or tyrosine kinase inhibitors (Tosoni et al., 2016). Even with these treatments, median overall survival after recurrence is 6.2 months (Gorlia et al., 2012). In a Phase II study that led to conditional FDA approval, the longest median progression-free survival (5.6 months) was seen with a combination of bevacizumab and irinotecan (Friedman et al., 2009), while longest overall survival (12 months) resulted from lomustine + bevacizumab (Taal et al., 2014). Current clinical trials involving small molecules for GBM treatment are summarized in Table 1; clinical trials involving biologics and other treatment strategies are reported in Supplemental Table 1.
TABLE 1.
List of clinical trials for glioblastoma treatment registered on www.clinicaltrials.gov
Small molecules.
| No. | Drug | Purpose | P | DR | NPE | AG | NCT number | Status |
|---|---|---|---|---|---|---|---|---|
| 1 | Abemaciclib | To evaluate the efficacy of abemaciclib in recurrent GBM | 2 | O | 47 | A,S | NCT02981940 | Recruiting |
| 2 | ACP-196 | To evaluate the efficacy and safety of ACP-196 in patients with recurrent GBM who have progressed after 1 or 2 prior systemic treatment regimens | 1/2 | O | 72 | A,S | NCT02586857 | Recruiting |
| 3 | Afatinib | To determine the maximum safe dose of afatinib that can be administered to people with brain cancer | 1 | O | 24 | A,S | NCT02423525 | Recruiting |
| 4 | Aldoxorubicin | To determine the efficacy and safety of aldoxorubicin in patients with GBM | 2 | i.v. | 28 | A,S | NCT02014844 | Completed (Groves et al., 2016) |
| 5 | Alisertib | To study the side effects and best dose of alisertib when combined with fractionated stereotactic radiosurgery in treating patients with high-grade gliomas | 1 | O | 24 | A,S | NCT02186509 | Active, not recruiting |
| 6 | AMG-232 | To study the side effects and dosage of MDM2 inhibitor AMG-232 in patients with newly diagnosed or recurrent GBM | 1 | O | 92 | A,S | NCT03107780 | Not yet recruiting |
| 7 | Ascorbate | To evaluate high-dose ascorbate in combination with standard of care treatment of GBM | 2 | i.v. | 90 | A,S | NCT02344355 | Recruiting |
| 8 | Atorvastatin | To explore the efficacy and safety of atorvastatin in combination with RT+TMZ in patients with newly diagnosed GBM | 2 | O | 32 | A,S | NCT02029573 | Completed |
| 9 | Axitinib | To test the efficacy of axitinib alone or in combination with lomustine for patients with recurrent GBM | 2 | O | 52 | A,S | NCT01562197 | Completed (Duerinck et al., 2016) |
| 10 | Axitinib + Avelumab | To determine the efficacy of axitinib + avelumab to treat patients with recurrent GBM | 2 | O | 52 | A,S | NCT03291314 | Recruiting |
| 11 | AZD1390 | To test the safety and tolerability of AZD1390 in combination with radiation therapy for the treatment of brain tumors | 1 | i.v. | 132 | A,S | NCT03423628 | Not yet recruiting |
| 12 | BAL101553 | To assess side effects and best dose of BAL101553 + radiation therapy in patients with newly diagnosed GBM | 1 | O | 30 | A,S | NCT03250299 | Recruiting |
| 13 | BBI608 (napabucasin) | To test the efficacy of BBI608 in combination with TMZ in patients with recurrent or progressive GBM | 1/2 | O | 60 | A,S | NCT02315534 | Recruiting |
| 14 | Belinostat | To determine the efficacy of belinostat in patients with newly diagnosed GBM and to determine the feasibility of adding magnetic resonance spectroscopic imaging to improve patient outcomes | 2 | i.v. | 87 | A,S | NCT02137759 | Recruiting |
| 15 | Bevacizumab + Nimustine | To determine the efficacy and feasibility of bevacizumab and nimustine treatment in patients with recurrent GBM | 2 | i.v. | 40 | A,S | NCT02698280 | Recruiting |
| 16 | BGB-290 | To assess the combination of BGB-290 and TMZ in patients with newly diagnosed or recurrent GBM | 1/2 | O | 300 | A,S | NCT03150862 | Recruiting |
| 17 | BGJ398 | To determine the efficacy of BGJ398 in patients with recurrent resectable or unresectable GBM | 2 | O | 24 | A,S | NCT01975701 | Active, not recruiting |
| 18 | BLZ945 | To characterize the safety, tolerability, pharmacokinetics, pharmacodynamics, and antitumor activity of BLZ945 against GBM | 2 | O | 151 | A,S | NCT02829723 | Recruiting |
| 19 | Buparlisib | To test the efficacy of buparlisib plus carboplatin or lomustine in patients with recurrent GBM | 1/2 | O | 35 | A,S | NCT01934361 | Completed |
| 20 | Cabazitaxel | To assess the efficacy of cabazitaxel on GBM | 2 | IF | 24 | A,S | NCT01866449 | Active, not recruiting |
| 21 | Cabozantinib | To study the feasibility and efficacy of cabozantinib for recurrent or refractory GBM | 2 | O | 10 | C,A | NCT02885324 | Recruiting |
| 22 | Capecitabine | To test the efficacy of capecitabine + bevacizumab in patients with recurrent GBM | 1 | O | 12 | A,S | NCT02669173 | Recruiting |
| 23 | Cediranib + Olaparib | To evaluate to efficacy of cediranib + olaparib in patients with recurrent GBM | 2 | O | 70 | A,S | NCT02974621 | Recruiting |
| 24 | Chlorogenic acid | To determine the pharmacokinetic characteristics of chlorogenic acid in advanced GBM | 1 | i.v. | 30 | A | NCT02728349 | Recruiting |
| 25 | Chloroquine | To assess the safety of chloroquine addition to chemoradiation in newly diagnosed GBM | 1 | O | 9 | A,S | NCT02378532 | Active, not recruiting |
| 26 | Crenolanib | To investigate crenolanib monotherapy in patients with recurrent/refractory GBM with PDGFRA gene amplification | 2 | O | 33 | A,S | NCT02626364 | Recruiting |
| 27 | Crizotinib | To assess the safety, efficacy, and safety of crizotinib in combination with RT+TMZ in patients with newly diagnosed GBM | 1 | O | 24 | A,S | NCT02270034 | Recruiting |
| 28 | Dacomitinib | To assess the efficacy and safety of dacomitinib in patients with recurrent GBM with EGFR gene amplification and/or EGFRvIII mutation. | 2 | O | 64 | A,S | NCT01520870 | Active, not recruiting |
| 29 | Dexanabinol | To determine the maximum safe dose of dexanabinol that can be administered to people with brain cancer | 1 | i.v. | 26 | A,S | NCT01654497 | Completed |
| 30 | Dimethyl fumarate | To test the safety of dimethyl fumarate in combination with RT+TMZ in patients with newly diagnosed GBM | 1 | O | 12 | A,S | NCT02337426 | Active, not recruiting |
| 31 | Disulfiram | To assess the effects of proteasome inhibition in patients with GBM | 1 | O | 20 | A,S | NCT01907165 | Active, not recruiting |
| 32 | DM-CHOC-PEN | To test the efficacy of DM-CHOC-PEN in patients with GBM | 2 | i.v. | 27 | A,S | NCT02038218 | Completed |
| 33 | Dovitinib | To determine a safe and tolerable dose of dovitinib in patients with relapsed GBM | 1 | O | 12 | A,S | NCT01972750 | Completed (Schäfer et al., 2016) |
| 34 | Dovitinib | To determine the efficacy of dovitinib on recurrent GBM | 2 | O | 33 | A,S | NCT01753713 | Completed (Ahluwalia et al., 2015) |
| 35 | Epacadostat | To determine the efficacy of epacadostat in combination with nivolumab for patients with GBM | 2 | O | 291 | A,S | NCT02327078 | Recruiting |
| 36 | Fingolimod | To evaluate the efficacy of fingolimod in patients with bevacizumab-resistant GBM | 1 | O | 5 | A,S | NCT02490930 | Completed |
| 37 | G-202 (mipsagargin) | To evaluate the activity, safety, and CNS exposure of G-202 in patients with recurrent or progressive GBM | 2 | i.v. | 26 | A,S | NCT02067156 | Completed, Publication Awaited |
| 38 | GDC-0084 | To evaluate the safety and tolerability, pharmacokinetics, pharmacodynamics and efficacy of GDC-0084 in patients with progressive or recurrent GBM | 1 | O | 29 | A,S | NCT01547546 | Completed (Wen et al., 2016) |
| 39 | HMPL-813 | To evaluate epitinib to treat GBM patients with EGFR gene amplification | 1 | O | 29 | A,S | NCT03231501 | Not yet recruiting |
| 40 | INC280 + Buparlisib | To assess the safety of the combination of INC280 and buparlisib in patients with recurrent GBM | 1/2 | O | 42 | A,S | NCT01870726 | Completed (van den Bent et al., 2017) |
| 41 | Indoximod | To assess the effect of indoximod in patients with newly diagnosed GBM | 1/2 | O | 144 | C,A,S | NCT02052648 | Recruiting |
| 42 | Ixazomib | To determine the tissue concentration of ixazomib citrate | 1 | O | 3 | A,S | NCT02630030 | Recruiting |
| 43 | JP001 | To evaluate the effect of JP001 in combination with standard chemoradiation on increasing overall survival of patients with newly diagnosed GBM | 2/3 | O | 264 | A,S | NCT03008148 | Not yet recruiting |
| 44 | Lapatinib | To test the safety and effects of a combination of lapatinib, plus RT+TMZ in patients with newly diagnosed GBM | 2 | i.v. | 70 | A,S | NCT01591577 | Recruiting |
| 45 | LB100 | To determine blood-brain barrier permeability of LB100 | 1 | i.v. | 20 | A,S | NCT03027388 | Not yet recruiting |
| 46 | LOXO-101 | To determine the efficacy of LOXO-101 in the treatment of solid tumors | 2 | O | 151 | C,A,S | NCT02576431 | Recruiting |
| 47 | LY2157299 | To test the efficacy of LY2157299 in combination with lomustine in patients with recurrent GBM | 2 | O | 180 | A,S | NCT01582269 | Active, not recruiting |
| 48 | LY2228820 | To determine an appropriate dose of LY2228820 in combination with TMZ and radiotherapy in patients with newly diagnosed GBM | 1/2 | O | 50 | A,S | NCT02364206 | Recruiting |
| 49 | Macitentan | To test the safety of macitentan in patients with newly diagnosed GBM | 1 | O | 30 | A,S | NCT02254954 | Completed |
| 50 | Marizomib | To establish the impact of marizomib on overall survival of patients with GBM | 3 | i.v. | 750 | A,S | NCT03345095 | Not yet recruiting |
| 51 | Marizomib | To determine the efficacy of marizomib in patients with newly diagnosed GBM | 1 | i.v. | 48 | A,S | NCT02903069 | Recruiting |
| 52 | Mebendazole | To determine the safety and side effects for increasing doses of mebendazole for recurrent or progressive pediatric brain tumors | 1 | O | 21 | C,A | NCT02644291 | Recruiting |
| 53 | Mibefradil | To determine the safety of mibefradil and hypofractionated re-irradiation therapy in recurrent GBM | 1 | O | 24 | A,S | NCT02202993 | Completed |
| 54 | Nabiximols | To determine the safety of nabiximols in combination with TMZ in patients with recurrent GBM | 1/2 | O | 6 | A,S | NCT01812603 | Completed (Twelves et al., 2017) |
| 55 | NVX-108 | To test the safety, tolerability, and effectiveness of NVX-108 | 1 | i.v. | 25 | A,S | NCT02189109 | Active, not recruiting |
| 56 | Olaparib | To determine efficacy of olaparib in patients with glioma, cholangiocarcinoma, or solid tumors with IDH1 or IDH2 mutations | 2 | O | 75 | A,S | NCT03212274 | Not yet recruiting |
| 57 | ONC201 | To test efficacy of ONC201 in patients with recurrent GBM | 2 | O | 30 | A,S | NCT02525692 | Recruiting |
| 58 | Ortataxel | To evaluate the efficacy of ortataxel in recurrent GBM | 2 | i.v. | 64 | A,S | NCT01989884 | Suspended |
| 59 | Palbociclib Isethionate | To test the side effects and best dose of palbociclib isethionate in treating younger patients with central nervous system tumors | 1 | O | 55 | C,A | NCT02255461 | Recruiting |
| 60 | Pazopanib | To assess pazopanib in combination with TMZ in patients with newly diagnosed GBM after surgery and RT-CT | 1/2 | O | 51 | A,S | NCT02331498 | Recruiting |
| 61 | Pembrolizumab + Vorinostat + TMZ | To test the safety and tolerability of vorinostat and pembrolizumab, in combination with TMZ and radiotherapy | 1 | O | 32 | A,S | NCT03426891 | Not yet recruiting |
| 62 | Perifosine + Torisel (Temsirolimus) | To test the effectiveness of perifosine and torisel in patients with recurrent or progressive GBM | 2 | O | 10 | A,S | NCT02238496 | Active, not recruiting |
| 63 | Plerixafor | To determine the safety of plerixafor after radiation therapy and TMZ in patients with newly diagnosed GBM | 1/2 | O | 29 | A,S | NCT01977677 | Active, not recruiting |
| 64 | PLX3397 | To test the efficacy of PLX3397 in combination with radiation therapy (RT) + TMZ in patients with newly diagnosed GBM | 1/2 | O | 65 | A,S | NCT01790503 | Active, not recruiting |
| 65 | Ponatinib | To evaluate the efficacy of ponatinib in recurrent GBM | 2 | O | 32 | A,S | NCT02478164 | Active, not recruiting |
| 66 | PQR309 | To evaluate the dual pan-PI3K and mTOR inhibitor in patients with first progression of GBM | 2 | O | >35 | A,S | NCT02850744 | Active, not recruiting |
| 67 | PT2385 | To study efficacy of HIF-2 alpha inhibitor PT2385 in patients with recurrent GBM | 2 | O | 35 | A,S | NCT03216499 | Recruiting |
| 68 | Regorafenib | To evaluate the role of regorafenib in prolonging the overall survival of GBM patients | 2 | O | 112 | A,S | NCT02926222 | Active, not recruiting |
| 69 | Ribociclib | To assess the ability of ribociclib to inhibit CDK4/CDK6/RB/E2F signaling and cell proliferation/viability in core and infiltrating tumor tissues obtained from patients with recurrent GBM | 1 | O | 20 | A,S | NCT02345824 | Recruiting |
| 70 | Sapanisertib | To determine blood-brain barrier permeability and efficacy of sapanisertib | 1 | O | 40 | A,S | NCT02133183 | Recruiting |
| 71 | Sapanisertib | To determine the best dose of sapanisertib in combination with bevacizumab in patients with recurrent GBM or advanced solid tumors | 1 | O | 23 | A,S | NCT02142803 | Recruiting |
| 72 | Selinexor | To evaluate the efficacy and safety of selinexor in patients with recurrent GBM | 2 | O | 125 | A,S | NCT01986348 | Active, not recruiting |
| 73 | Sunitinib | To determine the effectiveness of a combination of sunitinib, TMZ, and RT in newly diagnosed GBM patients harboring tumors with unmethylated MGMT promoter | 2 | O | 45 | A,S | NCT02928575 | Recruiting |
| 74 | Sunitinib | To evaluate the effect of high-dose, intermittent sunitinib in patients with recurrent GBM | 2/3 | O | 100 | A,S | NCT03025893 | Not yet recruiting |
| 75 | Tesevatinib | To assess the efficacy of tesevatinib monotherapy in recurrent GBM | 2 | O | 40 | A,S | NCT02844439 | Active, not recruiting |
| 76 | TG02 | To determine safety and efficacy of TG02 in patients with recurrent GBM and anaplastic astrocytoma | 1/2 | O | 152 | A,S | NCT02942264 | Recruiting |
| 77 | TH-302 | To determine the safety and efficacy of TH-302 in combination with bevacizumab for GBM following bevacizumab failure | 2 | O | 33 | A,S | NCT02342379 | Active, not recruiting |
| 78 | Tipifarnib | To test the safety and effectiveness of tipifarnib for newly diagnosed GBM | 1 | O | 19 | A,S | NCT02227901 | Completed |
| 79 | Tivozanib | To test the safety and effectiveness of tivozanib for recurrent GBM | 2 | O | 10 | A,S | NCT01846871 | Completed (Kalpathy-Cramer et al., 2017) |
| 80 | TPI 287 | To evaluate the safety, maximum tolerated dose (MTD), and efficacy of TPI 287 in combination with bevacizumab in patients with GBM | 2 | i.v. | 17 | A,S | NCT02047214 | Terminated |
| 81 | Trametinib | To investigate the activity of dabrafenib in combination with trametinib in children and adolescent patients with BRAF V600 mutation-positive relapsed or refractory GBM | 2 | O | 40 | C | NCT02684058 | Recruiting |
| 82 | TRC102 | To evaluate the combination of TRC102 and TMZ in patients with recurrent GBM | 2 | O | 66 | A,S | NCT02395692 | Active, not recruiting |
| 83 | Ubidecarenone | To study the side effects and dosage of ubidecarenone injectable nanosuspension in patients with recurrent GBM or gliosarcoma | 1 | i.v. | 10 | A,S | NCT03020602 | Recruiting |
| 84 | USL311 | To assess the safety and efficacy of USL311 alone and in combination with lomustine in patients with relapsed or recurrent GBM | 2 | O | 120 | A,S | NCT02765165 | Recruiting |
| 85 | VAL-083 | To investigate the efficacy of VAL-083 in patients with TMZ-bevacizumab recurrent GBM | 3 | i.v. | 180 | AS | NCT03149575 | Recruiting |
| 86 | VAL-083 | To determine the efficacy of VAL-083 on unmethylated MGMT recurrent GBM | 2 | i.v. | 48 | A,S | NCT02717962 | Recruiting |
| 87 | Veliparib | To determine efficacy of veliparib + TMZ in patients with newly diagnosed GBM | 2/3 | O | 440 | A,S | NCT02152982 | Recruiting |
| 88 | Vistusertib | To determine the efficacy of mTORC1/2 kinase inhibitor vistusertib in previously treated GBM | 1/2 | O | 52 | A,S | NCT02619864 | Recruiting |
| 89 | Vorinostat | To determine the efficacy of vorinostat + bevacizumab in patients with recurrent GBM | 2 | O | 48 | A,S | NCT01738646 | Completed (Ghiaseddin et al., 2018) |
| 90 | β-elemene | To determine the efficacy of β-elemene to maintain the health of patients with newly diagnosed malignant gliomas following standard treatment | 3 | i.v. | 100 | A,S | NCT02629757 | Recruiting |
A, adult (18–60 years old); AG, age groups; C, child (<18 years old); CDK4/6, cyclin-dependent kinase 4/6; DR, delivery route; IF, infusion; i.v., intravenous; MDM2, mouse double minute 2 homolog; mTOR, mammalian target of rapamycin complex 1; NPE, number of patients enrolled; O, oral; P, phase; S, senior (>60 years old); TMZ, temozolomide.
III. Molecular Diagnostic Signature of Glioblastoma
Glioblastoma is a grade IV glioma and the most malignant astrocytoma (Fig. 1) (Siegel et al., 2016). GBM tumors consist of a complex mixture of heterogeneous cells, complicating the search for the cell of origin. Previously, GBM was thought to originate from neural stem cells. However, studies have suggested that gliomas may differentiate directly from progenitor cells, and the type of progenitor cell each tumor originates from dictates their chemosensitivity (Persson et al., 2010). Until recently, GBM tumors have been diagnosed histologically and are characterized by increased cell density, abnormal cell types (atypia), areas of necrosis, and robust angiogenesis (Fig. 2). This histologic diagnosis hinders therapeutic approaches at personalized therapy. TCGA project improved characterization of GBM tumors with whole genome sequencing and identified key oncogenic signaling pathways to further classify tumor types. The molecular aberrations required for gliomagenesis include: mutations in the P53, retinoblastoma (RB), and receptor tyrosine kinase/Ras/phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways (Fig. 3) (Brennan et al., 2013). RB and P53 are tumor suppressors that lose function in several cancers (Weinberg, 1995; Freed-Pastor and Prives, 2012). Additionally, epithelial growth factor receptor (EGFR) expression is amplified in some GBM tumors, leading to increased cell proliferation through the receptor tyrosine kinase/Ras/PI3K/AKT signaling pathway (Huang et al., 2009). Through TCGA project, tumors were also profiled with reverse phase protein array, a high-throughput technique similar to Western blotting that detects and quantifies protein expression levels. Out of 171 antibodies, 127 correlated with transcriptomal subtype, and signaling pathway alterations were confirmed, including increased EGFR, Notch1, and Notch3 expression and activated MAPK pathway signaling (Brennan et al., 2013). Although this is a useful tool, only 171 antibodies were used in this study and therefore only 171 gene products could be profiled, providing a limited scope of potential novel drug targets. TCGA results were used by the World Health Organization to describe novel guidelines for GBM diagnosis to supplement histologic findings with the mutation status of several biomarkers of GBM, including IDH1/2, ATRX, and Histone Cluster 1 H3 Family Member A (HIST1H3A or H3F3A) (mutation at position K27M or simply H3-K27M mutation) (Reifenberger et al., 2017). The novel classification of GBM subtypes will aid patient stratification and the development of targeted therapeutics based on genetics.
Fig. 1.

Classification of brain tumors as reported from the Central Brain Tumor Registry of the United States (Ostrom et al., 2016). Numbers in parentheses indicates incidence or cases per 100,000 individuals and are age-adjusted to the 2000 United States standard population.
Fig. 2.

Common characteristics and diagnostic markers of World Health Organization grade IV glioma compared with lower-grade gliomas. Object images obtained from Servier Medical Art by Servier.
Fig. 3.

Canonical gliomagenesis mediators EGFR, P53, and retinoblastoma protein (RB1) are important for cancer signaling. EGFR is amplified or mutated to the constitutively active EGFRvIII and propagates kinase signaling cascades to promote proliferation, invasion, and angiogenesis. P53 is a tumor suppressor that is mutated in GBM, allowing B-cell lymphoma 2 (BCL2) to inhibit apoptosis. RB is another tumor suppressor that, when inactivated, releases E2F transcription factor 1 (E2F1) to activate cell cycling and growth. Percentages of aberrations of commonly mutated genes (in yellow) are reported, determined from TCGA analysis of patient samples (Brennan et al., 2013).
Molecular profiling has been used to classify GBM into four subtypes: Classic, Mesenchymal, Proneural, and Neural. Expression and aberrations of specific genes associated with each subtype have been identified (Verhaak et al., 2010). All Classic GBM tumors contain chromosome 7 amplification and chromosome 10 loss, and almost all (97%) display EGFR amplification (Verhaak et al., 2010). Mesenchymal GBM tumors show loss of NF1, contain markers of epithelial-to-mesenchymal transition (CD44 and MERTYK), and highly express genes in the tumor necrosis factor super family and NF-κB pathways. Alterations of PDGFRA and point mutations in the IDH1 gene are characteristic of Proneural subtypes. Tumors with expression of neural markers NEFL, GABRA1, SYT1, and SLC12A5 are classified as the Neural subtype. Of these subtypes, patients classified with the Proneural subtype generally had a longer overall survival, although the results were not statistically significant. Furthermore, the Proneural subtype is most common in younger patients. However, of the four subtypes, the Proneural subtype seemed the least responsive to aggressive treatment (concurrent chemo- and radiotherapy or more than three subsequent cycles of chemotherapy) (Verhaak et al., 2010).
Improvements in tumor profiling may drastically alter how GBM is treated and may improve the fidelity of new diagnoses. Furthermore, treatment of each tumor subtype may be individualized for optimal success. Although no targeted therapies have been approved for GBM yet, these diagnostic criteria may lead to more effective personalized treatments. Moreover, targeted therapies should be evaluated in a specific GBM subtype for optimal response. Further complicating the development of targeted treatments is the fact that a single cell of origin may not exist because of the cellular complexity of GBM. Conversely, multiple factors lead to the disease, and in fact, the cell of origin may not be the cell type that contains the transforming mutation. However, deciphering the cell of origin of GBM may be important to identify properly targets for drug discovery, stratify patient diagnosis, and optimize an effective treatment strategy.
IV. Characteristics of Protein Expression in Glioblastoma
Dynamic signaling pathways govern cancer cell proliferation. A major consequence of cancer signaling is an imbalance in protein expression to allow the cells to evade apoptosis, proliferate, and metastasize. Approximately 40% of GBM tumors are characterized by amplification and overexpression of EGFR, an effector of several signaling cascades that aid tumor growth, angiogenesis, migration, and metastatic spread (Brennan et al., 2013). EGFR is a receptor tyrosine kinase that, upon ligand binding, dimerizes and activates downstream signaling through the Ras/PI3K/AKT pathway. EGFR overexpression and EGFRvIII amplification may be prognostic markers that correlate with decreased overall survival of GBM patients (Shinojima et al., 2003); however, a recent meta-analysis disputes this claim (Chen et al., 2015). Nevertheless, because EGFR amplification and mutations promote glioma growth and survival, EGFR has been proposed as an attractive therapeutic target. Unfortunately, several clinical trials with EGFR inhibitors have failed, likely due to poor BBB permeability, intratumoral heterogeneity, and the difference between local versus systemic administration (Wen et al., 2014). Gliomagenesis is driven by mutations such as EGFRvIII, and those gene mutations promote tumor growth and proliferation through protein expression networks.
Large-scale proteomic research has shown that GBM tumors have increased expression of membrane proteins involved in cellular function and maintenance (P = 2.03 × 10−8), protein synthesis (P = 7.74 × 10−11), cell-to-cell signaling and interaction (P = 1.82 × 10−10), cellular movement (P = 1.34 × 10−8), and antigen presentation (P = 2.24 × 10−7) compared with normal brain tissue (Fig. 4) (Polisetty et al., 2012). More specifically, GBM tumors had increased expression of membrane proteins involved in acute phase response signaling, caveolar-mediated endocytosis signaling, and calcium signaling (Polisetty et al., 2012). To confirm these findings, we evaluated the 25 genes shown in Fig. 4 using Pharos drug target survey software (Nguyen et al., 2017). RNASeq expression of these genes was classified as medium to high in normal brain tissue and 19 out of 25 were significantly upregulated in GBM (Supplemental Table 2).
Fig. 4.

Signaling pathways involving membrane proteins upregulated in GBM as determined by LC-MS/MS and iTRAQ. Results are from proteomic analysis of human GBM tumors with Ingenuity Pathway Analysis software (Polisetty et al., 2012). Representative genes from each category are shown.
Proteomic approaches have identified proteins that are involved in chemotherapeutic resistance. For example, a study using 2D gel electrophoresis (2DGE) and mass spectrometry identified that lipocalin 2 and integrin β3 were downregulated in BCNU-resistant rat models of glioma (Suk, 2012). Furthermore, 2DGE coupled with liquid chromatography-mass spectrometry analysis identified several proteins important for the invasive properties of gliomas (Maruo et al., 2013). In particular, annexin A2 was highly expressed in an angiogenesis-dependent cell line (Maruo et al., 2013), and its overexpression further correlated with tumor aggressiveness and patient survival (Maule et al., 2016).
Although many other proteins have been found to contribute to GBM tumor growth, for this review, we will focus on targets that have been discovered through proteomic approaches and TCGA data mining. Some examples of proteins overexpressed in GBM that may represent novel drug targets that were not discovered via proteomic approaches include heat-shock protein 47 (Jiang et al., 2017b), cathepsin L (Xiong et al., 2017), glycoprotein nonmetastatic melanoma protein B (Ono et al., 2016), transcription factor 12 (Godoy et al., 2016), targeting protein for Xenopus kinesin-like protein 2 (Gu et al., 2016), and B-cell CLL/lymphoma 3 (BCL3) (Wu et al., 2016). Due to the characteristic intratumoral heterogeneity of GBM, it is likely that a single target approach will not be effective, and appropriate drug combinations will be necessary.
V. Emerging Targets in Glioblastoma
Numerous proteins are overexpressed in GBM, and abundant research has identified potential targets; however, extensive genomic and proteomic research suggests that tumor heterogeneity will likely render GBM unresponsive to single agent therapy. Of equal importance to target discovery is biomarker identification. Disease biomarkers can be used for early diagnosis and monitoring responsiveness to treatment.
A. Biomarker Identification
Biomarkers have been used successfully as tools for cancer diagnosis. Prostate cancer was one of the first to benefit significantly with the discovery of prostate specific antigen to inform early diagnosis and response to treatment. In addition, biomarkers have been discovered for ovarian, head and neck, lung, and breast cancer, among others (Petricoin et al., 2002; Varnum et al., 2003; Xiao et al., 2003–2004; Soltys et al., 2004). Gliomas are characterized in the clinic by IDH1 and IDH2 mutations and MGMT gene promoter methylation status to better inform treatment strategies; however, for GBM, proper prognostic biomarkers do not yet exist. By studying glioma tumorigenesis in detail, prognostic markers can be identified. Better prognostic markers would allow physicians to diagnose and begin treatment of GBM at early onset, possibly preventing disease progression.
Several groups have used proteomic techniques to analyze GBM and identify potential biomarkers for early diagnosis. For example, small extracellular vesicles transporting RNA and protein between cells can help clinicians diagnose and begin treatment of GBM at an earlier stage. Small extracellular vesicles in the cerebrospinal fluid carry important microRNA that could be used as biomarkers (Akers et al., 2015). In addition, the oncometabolite 2-hydroxyglutarate (2-HG) has been studied as a noninvasive biomarker in gliomas. In one study, urinary 2-HG levels were elevated in patients diagnosed with IDH1-mutant gliomas (Fathi et al., 2016). However, it is still unclear whether 2-HG levels could be used as a diagnostic measure for IDH1-mutant GBM and whether 2-HG levels could determine patient health outcome in response to chemotherapy and radiation. A computational approach was used to identify dysregulated pathways associated with short-term survival including proteins associated with gene ontology terms “protein kinase cascade” and “NF-κB pathway” (Patel et al., 2013). Despite this research, novel disease biomarkers identified with mass spectrometry-based proteomics have yet to reach the clinic (Di Meo et al., 2014).
B. Drug Discovery Targets
Genomic and proteomic techniques inform the development of precision medicine. The evolution of large-scale proteomic efforts is likely to benefit future drug discovery, and information on genomic events in GBM could lead to valuable insights about protein target candidates. Using TCGA GBM project cohort genomic analysis, we identified 20 genes with high expression that correlates with poor overall survival. These genes encode for proteins that promote the aggressive nature of GBM tumors and therefore may be important drug targets. However, further validation is necessary to confirm that the increased expression is not a response to oncogenic stress.
1. Gene Expression Associated with Reduced Patient Survival
In an effort to better understand the landscape of known and unknown GBM drug targets based on available gene expression data, we performed an analysis on 141 GBM samples from the TCGA cohort with both survival metadata and RNASeq expression data (http://cancergenome.nih.gov/). Patient sample RNASeq RSEM-normalized gene expression values and survival metadata were sourced from the TCGA GDAC Firehose (Broad Institute TCGA Genome Data Analysis Center, 2016). When multiple samples were available for a given patient, barcodes were sorted alphabetically and the first was selected for analysis.
GBM patient samples were evaluated for reduced survivability by comparing survival outcomes for patients with high and low expression of each gene (Fig. 5). Thresholding for high and low expression patient populations was evaluated using five different quantile cutoffs: 95%, 90%, 75%, 50%, and 25%. A log-rank test statistic was calculated for each cutoff to compare the survival distributions of high and low expression patient populations with the null hypothesis that there was no difference in survival curves. P-values were FDR-adjusted across all diseases, quantile cutoffs, and genes evaluated. To reduce over-fitting of a single cutoff per gene, genes for which the high expression population was associated with reduced survivability were required to have FDR-adjusted P-values ≤0.1 for at least two quantile cutoffs. Survival test statistics were calculated in R using the survival package (R Core Team, 2016).
Fig. 5.

Twenty genes were identified as associated with reduced survivability in the TCGA GBM patient cohort profiled with RNASeq expression data. Patients were stratified by high and low gene expression based on one of five expression percentile thresholds. Kaplan-Meier survival plots are shown with patients having increased expression in red and all other GBM patients shown in green. Nonadjusted P values generated using the log-rank test are shown. All P values shown survived multiple testing corrections (qValue ≤ 0.1) across all 5 percentile thresholds and 20,531 genes.
Twenty genes were identified as significantly associated with reduced survivability using the criteria described in the previous paragraph (Table 2). To further validate the statistical significance of all 20 genes, a univariate Cox proportional hazards analysis was performed using thresholds for high and low expression shown in Fig. 5. All 20 genes remained significantly associated with reduced survival (Supplemental Table 3). Additionally, a multivariate Cox proportional hazards analysis was performed taking into account clinical properties found to be significantly (Cox univariate P value <0.05) associated with reduced survivability including age, Karnofsky performance score, postoperative drug treatment, and radiation therapy (Supplemental Table 4). All but one gene (NDEL1) remained significant (P value <0.05) after controlling for significant clinical properties, but still exhibited borderline significance (P value = 0.065). It should be noted that chemotherapy and radiation treatments were received by 83% and 84% of patients, respectively and were associated with improved survival (P value = 0.00017 and 0.000001, respectively).
TABLE 2.
Gene list and descriptions from DAVID bioinformatics database (https://david.ncifcrf.gov)
| No. | Name | Full Name | Description | Reference |
|---|---|---|---|---|
| 1 | BOC | BOC cell adhesion associated, oncogene regulated | Component of a cell-surface receptor complex that mediates cell-cell interactions between muscle precursor cells | Tenzen et al. (2006) |
| 2 | CLEC4GP1 | C-type lectin domain family 4 member G pseudogene 1 | Function unknown | |
| 3 | ELOVL6 | ELOVL fatty acid elongase 6 | Fatty acid elongase specific to C12-C16 saturated and monounsaturated fatty acids | Feng et al. (2016) |
| 4 | EREG | epiregulin | May be a mediator of localized cell proliferation | Riese and Cullum (2014) |
| 5 | ESR2 | estrogen receptor 2 | Nuclear hormone receptor that binds estrogens with an affinity similar to that of ESR1 and activates expression of reporter genes containing estrogen response elements in an estrogen-dependent manner | Batistatou et al. (2004), Sareddy et al. (2016) |
| 6 | FDCSP | follicular dendritic cell secreted protein | Can bind to the surface of B-lymphoma cells, but not T-lymphoma cells, consistent with a function as a secreted mediator acting upon B-cells | Wang et al. (2010), Hou et al. (2014) |
| 7 | FURIN | furin, paired basic amino acid cleaving enzyme | Release of mature proteins from their proproteins by cleavage of -Arg-Xaa-Yaa-Arg-|-Zaa- bonds, where Xaa can be any amino acid and Yaa is Arg or Lys and regulates TGF-β bioavailability | Phillips-Mason et al. (2014) |
| 8 | FUT8-AS1 | fucosyltransferase 8 antisense RNA 1 | Fucosylation of proteins, including EGFR | Liu et al. (2011) |
| 9 | GZMB | granzyme B | This enzyme is necessary for target cell lysis in cell-mediated immune responses. It cleaves after Asp. Seems to be linked to an activation cascade of caspases (aspartate-specific cysteine proteases) responsible for apoptosis execution. It has been associated with both tumor progression and regression, in a case-dependent manner. | Medema et al. (2001), Rousalova and Krepela (2010) |
| 10 | IRX3 | iroquois homeobox 3 | Belongs to the TALE/IRO homeobox family and may have a direct functional relationship to both obesity and type 2 diabetes. IRX3 is a proneural gene important for neuronal differentiation. | Yang et al. (2010), Seol et al. (2011) |
| 11 | LITAF | lipopolysaccharide induced TNF factor | Probable role in regulating transcription of specific genes. May regulate through NF-κB1 the expression of the CCL2/MCP-1 chemokine. May play a role in TNF-alpha gene expression. | Zou et al. (2015) |
| 12 | NDEL1 | nudE neurodevelopment protein 1 like 1 | Facilitates the polymerization of neurofilaments from the individual subunits NEFH and NEFL. Required for organization of the cellular microtubule array and microtubule anchoring at the centrosome. | Hong et al. (2016) |
| 13 | NKX3-1 | NK3 homeobox 1 | Transcription factor, which binds preferentially the consensus sequence 5′-TAAGT[AG]-3′ and can behave as a transcriptional repressor. Could play an important role in regulating proliferation of glandular epithelium and in the formation of ducts in prostate. | Bhatia-Gaur et al. (1999) |
| 14 | PODNL1 | podocan like 1 | Belongs to the small leucine-rich proteoglycan (SLRP) family | Heyn et al. (2013), Yan et al. (2013), Teng and Zheng (2017) |
| 15 | PTPRN | protein tyrosine phosphatase, receptor type N | Implicated in neuroendocrine secretory processes. May be involved in processes specific for neurosecretory granules, such as their biogenesis, trafficking or regulated exocytosis or may have a general role in neuroendocrine functions. | Xie et al. (1996), Bauerschlag et al. (2011) |
| 16 | QSOX1 | quiescin sulfhydryl oxidase 1 | Catalyzes the oxidation of sulfhydryl groups in peptide and protein thiols to disulfides with the reduction of oxygen to hydrogen peroxide. May contribute to disulfide bond formation in a variety of secreted proteins. | Katchman et al. (2011) |
| 17 | SEMA4F | semaphorin 4F | Estrogen-regulated semaphorin ligand with growth cone collapse activity against retinal ganglion-cell axons | Parrinello et al. (2008) |
| 18 | TH | tyrosine hydroxylase | Plays an important role in the physiology of adrenergic neurons | Tekin et al. (2014) |
| 19 | VEGFC | vascular endothelial growth factor C | Growth factor active in angiogenesis and endothelial cell growth, stimulating proliferation and migration. Has effects on the permeability of blood vessels. May function in angiogenesis of the venous and lymphatic vascular systems during embryogenesis, and in the maintenance of differentiated lymphatic endothelium in adults. | Denicolaï et al. (2016), Dufies et al. (2017) |
| 20 | C20orf166AS1 | chromosome 20 open reading frame 166 antisense RNA 1 | Long noncoding RNA | Hu et al. (2014) |
TNF, tumor necrosis factor.
Several of the 20 significant genes (Table 2) encode proteins involved in EGFR signaling. Our results reveal novel EGFR signaling proteins that may have more prominent roles than previously thought. These proteins include proteases (FURIN, GZMB, and NDEL1), transcription factors (LITAF, IRX3, NKX3-1, and VEGFC), and receptors (ERβ, BOC, EREG, and PTPRN). Agglomerative hierarchical clustering was performed using the 20 significant genes across TCGA GBM patients, and patients were stratified based on cluster membership. One cluster group had higher average expression across the 20 genes, and this higher expression corresponded with reduced time to death and disease-free survival (Fig. 6A). Patients belonging to the cluster group with higher average expression had significantly reduced survival compared with those not included. Survival stratification significance (P = 5.59 × 10−11) was greater when evaluating by cluster group across all 20 genes compared with any of the 20 genes separately (Fig. 6B). Gene expression association with poor overall survival was further validated by applying survival test statistics to samples from three independent GBM cohorts (Freije et al., 2004; Murat et al., 2008; Yu and Fu, 2015). Eight of the 20 genes (LITAF, FURIN, VEGFC, C20orf166-AS1, ELOVL6, PODNL1, ESR2, and QSOX1) were significantly associated with reduced survivability in at least one additional GBM cohort (Supplemental Fig. 2). This additional validation supports the importance of the overexpression of these genes in the context of GBM.
Fig. 6.

(A) Hierarchical clustering was performed to identify groups of patients with similar RNASeq expression of 20 genes associated with reduced survivability in the TCGA GBM patient cohort. (B) Patients stratified using clustering dendrogram assignment into high and low expression groups showed significant differences in survival. Heatmap z-scores were calculated per gene. Agglomerative hierarchical clustering with complete linkage was performed using Euclidean and Pearson correlation distance metrics on rows and columns, respectively.
We would expect patients with more aggressive forms of brain cancer to express higher levels of the 20 genes if the tumor cells relied on expression of these genes to survive. To test this hypothesis, we downloaded normalized log2 RSEM gene expression values for GBM and low-grade glioma (LGG) patients as a combined cohort (GBMLGG) from GDAC Firehose to evaluate differences in expression. Of the 20 genes, 11 had significantly higher expression in GBM patients versus LGG patients using a Kruskal-Wallis test (P < 0.05) (Supplemental Fig. 3). Interestingly two genes, BOC and VEGFC, showed no significant difference in expression between diseases, and both have been implicated in brain tumor progression. FDCSP was not expressed in the majority of patients and could not be evaluated. The remaining six genes had significantly increased expression in LGG and indicate an interesting contrast between diseases that may warrant further investigation.
Further validation of the proteins identified in Table 2 was performed with the open-access resource Pharos (Nguyen et al., 2017) (Supplemental Table 2). The majority of the identified genes (12) had Tbio classifications while two (ESR2 and TH) had Tclin classifications and three (ELOVL6, FURIN, GZMB) were assigned a Tchem classification. All targets that were mapped to GTEx expression were classified as having high or medium expression levels in normal brain tissue. From the analysis, 21 out of 25 genes in Fig. 4 and 12 out of 20 genes in Table 2 have a known link to brain cancer. Of the 12 genes, seven are linked to GBM: BOC, ELOVL6, IRX3, LITAF, NDEL1, PTPRN, and QSOX1. Furthermore, ELOVL6 small molecule probes have been identified and could be used to validate ELOVL6 as a drug target. Given that ELOVL6, ESR2, TH, FURIN, and GZMB have probes or inhibitors identified, these proteins could be a starting point for validation of our TCGA data mining.
We expanded our analysis of the 20 genes to include 33 TCGA diseases (Fig. 7). Head and neck squamous cancer had the highest average expression of the 20 genes in the analyzed patient samples. In addition, several genes were identified that have consistently higher expression in several cancers. For example, PTPRN was highly expressed in pancreatic adenocarcinoma and the pheochromocytoma and paraganglioma cohort, and therefore, those cancer subsets may be more sensitive to targeted PTPRN therapy. Several of the genes are involved in the transcriptional regulation of EGFR, including ESR2, EREG, and VEGFC. In addition, several genes are indirectly involved in EGFR regulation, including FUT8, LITAF, FURIN, NKX3-1, and TH. Upon further validation, these transcription factors may prove to be relevant to the progression and recurrence of GBM.
Fig. 7.

Expression of 20 genes significantly associated with reduced survivability in GBM is shown across 33 TCGA diseases. Gene expression from each patient sample was converted to a z-score, and z-scores were recalculated across all diseases for each gene to show relative expression. Regions of the heatmap are circled to highlight genes with consistent higher expression (10th percentile >0.5) and previously published support for relevance to disease progression (cyan) or high expression without previously published support for disease progression (purple). Diseases are ranked by decreasing average expression and ribbon on the right is colored to indicate average expression per patient sample.
Below, we briefly summarize the 20 genes significantly associated with reduced survivability and discuss current research on the link between each gene and cancer. Further validation of each target is necessary to confirm the importance of each gene in the context of GBM. Inhibiting the activity or expression of one, or a combination, of the proteins discussed below may prove to be a viable treatment strategy for GBM.
a. BOC cell adhesion associated, oncogene regulated (BOC)
BOC is a member of the immunoglobulin/fibronectin type III repeat family and promotes myogenic differentiation. During oncogenesis, BOC promotes hedgehog pathway signaling by sustaining a feedback mechanism that enhances the concentration of Sonic hedgehog (Shh) ligand (Tenzen et al., 2006). The hedgehog signaling pathway is necessary for normal cellular processes such as embryogenesis and growth of hair follicles and taste papillae in adults (Robbins et al., 2012). In the absence of the Shh ligand, the GPCR Ptch is active, which blocks Smo signaling. When Shh ligand is present, it inactivates Ptch, allowing Smo to signal transcription of target genes. Since BOC activates hedgehog pathway signaling, it likely contributes to GBM progression and may be a potential drug target. In our analysis, increased BOC expression is strongly associated with poor overall survival (P = 1.36 × 10−6). This is the first report, to our knowledge, of BOC associated with GBM.
b. C-Type lectin domain family 4 member G pseudogene 1 (CLEC4GP1)
CLEC4G is a 32.6-kDa membrane-bound protein expressed in the liver and lymph nodes and plays a role in T-cell immune response. TCGA whole-genome sequencing revealed CLEC4G was downregulated in hepatocellular carcinoma tissue (Ho et al., 2015). As a pseudogene, CLEC4GP1 is likely a nonfunctional copy of the enzyme. Pseudogenes can arise during duplication if a mutation occurs in the DNA or with retrotransposition, in which the cDNA product of the reverse-transcribed mRNA becomes incorporated in the genome. In our analysis, increased CLEC4GP1 expression is strongly associated with poor overall survival (P = 2.63 × 10−5). CLEC4GP1 is located on chromosome 19. In one study, CLEC4GP1 mRNA expression increased in response to an mRNA-based vaccine encoding influenza A hemagglutinin from a pandemic strain (Edwards et al., 2017). Additionally, expression of CLEC4GP1 is high in samples from patients diagnosed with adenoid cystic carcinoma (Fig. 7).
c. ELOVL fatty acid elongase 6 (ELOVL6)
ELOVL6 is highly expressed in the brain, and the gene is often hypomethylated in GBM (Vyazunova et al., 2014). This enzyme performs the first and rate-limiting step of fatty acid elongation, with malonyl-CoA as a 2-carbon donor and is important for insulin sensitivity and energy metabolism (Matsuzaka and Shimano, 2009). Phospholipids containing longer acyl chains are abundant in cancer tissue, and ELOVL6 is the main enzyme responsible for fatty acid elongation in cancer (Marien et al., 2016). The gene is located on chromosome 4q25, adjacent to the EGF gene. Expression of ELOVL6 may be high because it shares an enhancer region with EGF. Enhancers perform complex functions and can activate transcription of specific genes upstream or downstream by engaging the transcriptional machinery. In acute myeloid leukemia, a novel chromosomal rearrangement was found to activate ELOVL6 and EGF (Anelli et al., 2013). ELOVL6 has been studied in the context of many cancers. Increased ELOVL6 mRNA expression was found in triple-negative breast cancer tissue (Yamashita et al., 2017). Additionally, ELOVL6 and lipid composition may be regulated by the RB-E2F transcription factor 1 pathway (Muranaka et al., 2017). An ELOVL6 inhibitor, Compound A, inhibited tumor growth in an in vivo model of squamous cell carcinoma (Marien et al., 2016), and therefore validation and pursuit of ELOVL6 inhibition in GBM is warranted.
d. Epiregulin (EREG)
EREG is a 19-kDa peptide hormone that acts as a ligand for the EGF receptor and ErbB4. When cleaved by a disintegrin and metalloproteinase (ADAM) enzyme from the transmembrane pro-peptide to an active soluble form, EREG binds EGFR family members and initiates the signaling cascade. EREG expression is upregulated in gastric (Wu et al., 2009), colon (Wu et al., 2009), lung (Sunaga et al., 2013), and head and neck (Shigeishi et al., 2008) cancers, among others. In a colon cancer xenograft model, EREG expression correlated with a positive response to the anti-EGFR monoclonal antibody cetuximab, suggesting the tumors were dependent on the EGFR signaling pathway activated by EREG (Jacobs et al., 2009). EREG transcription is regulated by insulin, Sp1, NF-κB, and AP-2 (Li et al., 2002; Ornskov et al., 2007; Orso et al., 2008). Silencing of EREG in a breast cancer cell line inhibited metastasis, angiogenesis, and tumor cell extravasation (Gupta et al., 2007). EREG is a partial agonist of EGFR dimerization and induces differentiation in breast cancer cells (Freed et al., 2017). The emerging role for EREG as a key activator of EGFR signaling driving cancer cell proliferation suggests that inhibition of EREG binding to EGFR is a potential targeted cancer treatment. In our analysis, EREG expression was associated with poor overall survival in GBM patients (P = 5.6 × 10−5). High EREG expression was also found in TCGA samples from patients diagnosed with rectal adenocarcinoma (Fig. 7). Furthermore, EREG activates the extracellular signaling-related kinase/MAPK pathway in GBM, suggesting inhibition of the EREG-EGFR interaction may be a strategy for EREG-overexpressing GBM patients (Kohsaka et al., 2014).
e. Estrogen receptor 2 (ESR2)
ESR2 encodes the gene for estrogen receptor β (ERβ), a nuclear hormone receptor for estrogen, is considered a tumor suppressor in the context of GBM and other cancers (Paruthiyil et al., 2004; Sareddy et al., 2016), and enhances chemosensitivity in NSCLC (Nikolos et al., 2018). Treatment with ERβ agonist, LY500307, is efficacious in a GBM tumor-bearing mouse model (Sareddy et al., 2016). Additionally, ERβ expression, analyzed immunohistochemically, declines as brain astrocytic tumors progress (Batistatou et al., 2004). In our analysis, increased ESR2 expression is strongly associated with poor overall survival (P = 1.68 × 10−4), which is in contrast with the tumor suppressing effects of the protein. Furthermore, expression of ESR2 is high in TCGA samples from patients diagnosed with diffuse large B-cell lymphoma (Fig. 7). The tumor-suppressing characteristics of ESRβ may prevent it from being a potential anticancer target.
f. Follicular dendritic cell secreted protein (FDCSP)
FDCSP (C4orf7) is a 9.7-kDa peptide that promotes invasion and metastasis of tumor cells. Although relatively little is known about this peptide, overexpression of FDCSP is common in tumorigenesis, especially in ovarian cancer (Wang et al., 2010). FDCSP expression has also been implicated as a marker of follicular dendritic cell sarcoma (Lorenzi et al., 2017). The position, on chromosome 4q13, and characteristics, including amino acid composition, molecular mass, and isoelectric point, suggest FDCSP may be similar to the inflammatory C-X-C chemokines, such as interleukin-8 (Marshall et al., 2002). In our analysis, increased FDCSP expression is strongly associated with poor overall survival (P = 1.24 × 10−4). FDCSP expression may be important for GBM progression.
g. Furin
Furin is a protease that activates matrix metalloproteinases, including proparathyroid hormone, transforming growth factor beta 1 precursor, proalbumin, pro-beta-secretase, membrane type-1 matrix metalloproteinase, beta subunit of pro-nerve growth factor, and von Willebrand factor. Furin is linked with tumor progression in several cancers, including head and neck squamous cell carcinoma, breast cancer, and rhabdomyosarcoma (Jaaks and Bernasconi, 2017). In astrocytoma cells, inhibition of furin decreases cell proliferation and invasiveness (Mercapide et al., 2002). Furthermore, furin promotes activation of pro-TGFβ1 and pro-TGFβ2, demonstrating a tumorigenic role in glioma-initiating cells (Ventura et al., 2017). In our analysis, increased furin expression is strongly associated with poor overall survival (P = 1.79 × 10−4). Transcription of furin is promoted by AP-1 (activator protein-1), c-Jun, and ATF-2. Proteolysis is important in cancer, and furin activates several enzymes via proteolysis that contribute to cell migration and survival, including protein kinase C (Rucci et al., 2011). Combined inhibition of furin, ADAM, calpain, and another serine protease is necessary to prevent glioma migration and slow growth mediated by protein tyrosine phosphatase μ (Phillips-Mason et al., 2014). Inhibitors of furin demonstrate antiproliferative effects and are being optimized in the context of inhibition of viral replication (Dahms et al., 2017; Małuch et al., 2017). The extensive evidence of the tumorigenic role of furin in several cancers, including brain cancer, suggests it may be a promising therapeutic target, and inhibitors of furin may improve treatment outcomes.
h. Fucosyltransferase 8 antisense RNA 1 (FUT8-AS1)
FUT8 is a 66.5-kDa enzyme located in the Golgi apparatus and extracellular space and catalyzes the transfer of fucose from GDP-fucose to N-linked type complex glycopeptides. Fucosylation is an important posttranslational glycosylation event that regulates cancer signaling processes including metastasis and epithelial-to-mesenchymal transition. The expression of FUT8-AS1 suggests FUT8 expression may be downregulated in GBM patients. In our analysis, increased FUT8-AS1 expression is strongly associated with poor overall survival (P = 8.6 × 10−5). FUT8 function has been studied in the context of several cancers. For example, knockdown of FUT8 halted growth of in vitro and in vivo models of lung cancer (Chen et al., 2013). Additionally, inhibitors have been developed that block fucosylation in models of cancer (Okeley et al., 2013). Although FUT8 expression has been studied in the context of lung, liver, colon, and other cancers, it has not been evaluated in GBM.
i. Granzyme B (GZMB)
GZMB is a serine protease in the peptidase S1 family and is involved in mediating apoptosis. This enzyme cleaves after aspartate and plays a role in the cellular caspase cascade that leads to apoptosis. GZMB is the most abundant enzyme in cytotoxic granules responsible for the clearance of tumor cells, as well as cells infected with intracellular pathogens and allogeneic cells (Choy, 2010; Cullen et al., 2010). It is also a prognostic marker in colorectal cancer (Prizment et al., 2017). In our analysis, increased GZMB expression is strongly associated with poor overall survival (P = 5.29 × 10−7). Additionally, there is high expression of GZMB in TCGA samples from patients diagnosed with diffuse large B-cell lymphoma (Fig. 7). GZMB transcription is regulated by nuclear factor of activated T cells, Ikaros, and AP-1 (Wargnier et al., 1998). GZMB gene transcription is also activated and enhanced by NF-κB, which binds approximately 10 kb downstream from the GZMB transcription start site (Huang et al., 2006), and by Janus kinase 1/signal transducer and activator of transcription signaling (Jahrsdörfer et al., 2010). The role of GZMB in apoptosis makes it an attractive anticancer target.
j. Iroquois homeobox 3 (IRX3)
IRX3 is a 5.2-kDa transcription factor in the Iroquois homeobox family of developmental factors and is involved in Shh-dependent neural patterning. IRX3 belongs to class I proteins of neural progenitor factors and is repressed by Shh signals. IRX3 contains transcription factor binding sites for ERα, Pax-5, AP-2α, AP-2β, AP-2γ, FOXD1, and C/EBP, among others. In our analysis, increased IRX3 expression is strongly associated with poor overall survival (P = 1.15 × 10−4). In addition, consistently higher expression of IRX3 is observed in TCGA samples from patients diagnosed with breast cancer (Fig. 7). IRX3 is a target gene of WHSC1L1 (Wolf-Hirschhorn syndrome candidate 1-like 1 gene, or NSD3), a known oncogene in breast cancer, and may be a regulator of WNT signaling (Yang et al., 2010). DNA methylation profiling of an oligodendroma-derived cell line revealed hypermethylation of the CpG island on an IRX3 exon, consistent with overexpression of IRX3 in tumor tissue compared with normal brain samples (Ordway et al., 2006). Although IRX transcription factors have been identified in multiple genome-wide sequencing studies in cancer, they specifically hamper the tumor-suppressing activity of the TGF-β pathway (Martorell et al., 2014). Therefore, blocking IRX3 expression or inhibiting its ability to suppress the TGF-β pathway may be an option for GBM treatment.
k. Lipopolysaccharide-induced tissue-necrosis-factor factor (LITAF)
LITAF is a lipopolysaccharide-regulated transcription factor located on chromosome 16 that regulates VEGF and plays a role in angiogenesis and inflammatory response (Tang et al., 2013). LITAF contains a small integral membrane protein of lysosome/late endosome-like domain with a YXX ϕ motif that mediates transport of membrane proteins to and from the endosome, Golgi apparatus, and lysosomes. In several cancers, LITAF induces inflammation and promotes cancer cell survival. Stimulation by lipopolysaccharide causes LITAF to translocate from the cytoplasm with its partner protein STAT6(B) to the nucleus to promote gene expression (Zou et al., 2015). In our analysis, increased LITAF expression is strongly associated with poor overall survival (P = 2.3 × 10−5). Furthermore, LITAF was in the top 30 overexpressed genes in GBM in a large-scale expression analysis study (Dreyfuss et al., 2009). Therefore, blocking the LITAF-STAT6(B) protein-protein interaction may be a viable treatment strategy. However, LITAF possesses a tumor-suppressing role in pancreatic cancer (Zhou et al., 2018b), and its expression can be induced by P53 (Tang et al., 2007). LITAF knockdown promoted tumor malignancy and growth in nude mice injected subcutaneously with prostate cancer cells (Zhou et al., 2011). Overall, LITAF plays a complex role in the progression of cancer.
l. NudE neurodevelopment protein 1 like 1 (NDEL1)
NDEL1 is a 38-kDa cytoskeletal protein that contains an N-terminal coiled coil NUDE domain and is important for the regulation of microtubule organization to promote neuronal migration. Expression of NDEL1 is highest during mitosis, and it is necessary for mitotic cell division (Toyo-Oka et al., 2005). In our analysis, increased NDEL1 expression is strongly associated with poor overall survival (P = 5.7 × 10−5), and consistently higher expression is found in acute myeloid leukemia (Fig. 7). NDEL1 has also been implicated in the development of schizophrenia via its protein-protein interactions with Disrupted-in-Schizophrenia 1 (DISC1) (Hayashi et al., 2015). NDEL1 associates with microtubules, dynein, CENPF, and ZNF365. Additionally, the NDEL1 gene contains P53, c-myc, and ARP-1 transcription factor binding sites. With an increased understanding of the role of NDEL1 in cancer migration, effective, targeted inhibitors could be developed to control tumor growth.
m. NK3 homeobox 1 (NKX3-1)
NKX3-1 is a transcription factor that negatively regulates epithelial cell growth in prostate tissue. Loss of NKX3-1 is common in prostate cancer patients (He et al., 1997; Bhatia-Gaur et al., 1999). NKX3-1 negatively regulates the PI3K-AKT pathway to suppress tumor growth, and heterozygous deletions of NKX3-1 and PTEN cause prostate adenocarcinomas in mice (Abate-Shen et al., 2003). Additionally, NKX3-1 functions as a tumor suppressor in hepatocellular carcinoma (Jiang et al., 2017a). In our analysis, increased NKX3-1 expression is strongly associated with poor overall survival (P = 1.74 × 10−4), and consistently higher expression is also found in prostate adenocarcinoma (Fig. 7). To our knowledge, NKX3-1 has not yet been studied in the context of GBM.
n. Podocan like 1 (PODNL1)
PODNL1 is an extracellular protein expressed in tibial nerves, coronary arteries, and bone marrow mesenchymal stem cells and is involved in proteinaceous extracellular matrix formation. It belongs to the small leucine-rich proteoglycan (SLRP) family of 17 genes and is a member of Class V SLRPs, residing on chromosome 19q. SLRPs also act upstream of signaling cascades, including receptor tyrosine kinases like ErbB family members (Schaefer and Iozzo, 2008). Interestingly, the Class V SLRPs bind collagen I and inhibit cell growth by inducing cyclin-dependent kinase inhibitor 1 expression (Shimizu-Hirota et al., 2004). Additionally, another SLRP family member, decorin, binds to EGFR and lowers receptor levels by caveolin-mediated internalization (Zhu et al., 2005; Seidler et al., 2006; Ma et al., 2014). High expression of PODNL1 correlates with poor prognosis in ovarian cancer (Teng and Zheng, 2017). Methylation of the PODNL1 gene may be important for phenotypic changes that occur during aging (Heyn et al., 2013), and PODNL1 expression is associated with high-grade glioma (Yan et al., 2013). Our analysis supports these findings; increased PODNL1 expression is strongly associated with poor overall survival (P = 3.61 × 10−6). Because several proteins in the SLRP family play a role in cancer progression, PODNL1 may have an important function as well.
o. Protein tyrosine phosphatase, receptor type N (PTPRN)
PTPRN (also known as islet antigen-2 or IA-2) is a gene encoding a 105.8-kDa protein in the protein tyrosine phosphatase family responsible for signaling processes related to cell growth, differentiation, and oncogenic transformation. Hypermethylation of PTPRN in ovarian cancer patients was associated with shorter survival (Bauerschlag et al., 2011). It was initially discovered as a gene differentially expressed in human pancreatic beta islet cells and is localized on the plasma membrane and in endosomes. PTPRN depletion reduced small cell lung cancer cell growth (Xu et al., 2016). Valproic acid induced the expression of PTPRN as a result of increased acetylation in the promoter region (Witt et al., 2013). Analysis of TCGA samples from patients with pancreatic adenocarcinoma, pheochromocytoma, paraganglioma, GBM, and LGG revealed consistently higher expression of PTPRN (Fig. 7). In our analysis, increased PTPRN expression is strongly associated with poor overall survival (P = 2.19 × 10−5). Although PTP family proteins have been well studied in the context of cancer, little work has been done to elucidate the role of PTPRN in brain cancer.
p. Quiescin sulfhydryl oxidase 1 (QSOX1)
QSOX1 is a flavin adenine dinucleotide-dependent 82.6-kDa enzyme that forms disulfide bonds in proteins by oxidizing sulfhydryl groups. It is found in the extracellular space, Golgi apparatus, and endoplasmic reticulum, where it functions alongside protein disulfide isomerase to fold nascent proteins (Hoober and Thorpe, 2002). QSOX1 contains one thioredoxin domain and one ERV/ALR sulfhydryl oxidase domain. In pancreatic cancer, QSOX1 expression correlates with cell migration and survival, and QSOX1-mediated migration of pancreatic ductal carcinoma cells may be activated by MMP-2 and MMP-9 (Katchman et al., 2011). Interestingly, loss of NKX3-1 expression correlates with an increase in QSOX1 expression in prostate cancer (Song et al., 2009). In our analysis, increased QSOX1 expression is strongly associated with poor overall survival (P = 1.96 × 10−8). Proteomic analysis using iTRAQ identified that QSOX1 expression was upregulated in hepatocellular carcinoma (Guo et al., 2017). Furthermore, knockdown of QSOX1 sensitizes nasopharyngeal carcinoma cells to radiation (Zhou et al., 2018). Ebselen, a covalent inhibitor of QSOX1, suppressed pancreatic tumor growth in vivo (Hanavan et al., 2015). Much work has been done to elucidate the complex role of QSOX1 in several cancers, and it clearly plays an important role in disease progression.
q. Semaphorin 4F (SEMA4F)
SEMA4F is a membrane-bound glycoprotein in the semaphorin family of receptors. Semaphorins are involved in eliciting intracellular signaling cascades and may be receptors for EGFR signaling ligands. Therefore, semaphorins are important regulators of tumor growth, angiogenesis, migration, and apoptosis (Capparuccia and Tamagnone, 2009). For example, SEMA3B was found to be a marker for poor survival in patients over 50 diagnosed with GBM (Rich et al., 2005). In contrast, SEMA4D can stimulate or inhibit breast cancer cell migration and adhesion, depending on the presence of receptor tyrosine kinases ERBB2 and MET (Swiercz et al., 2008). In our analysis, increased SEMA4F expression is strongly associated with poor overall survival (P = 3.24 × 10−9). SEMA4F is linked to the induction of prostate cancer neurogenesis (Ayala et al., 2008) and may be important for breast cancer progression (Gabrovska et al., 2011). SEMA4F knockdown was linked to Schwann cell proliferation in the development of neurofibroma downstream of the loss of NF1 tumor suppressor function (Parrinello et al., 2008). The molecular mechanisms driving the function of this signaling receptor in cancer are complex.
r. Tyrosine hydroxylase (TH)
TH, as its name suggests, hydroxylates tyrosine to form the precursor for dopamine, l-dopa, and is induced by hypoxic stress via HIF1α, common in the tumor microenvironment. TH is also a marker for neurons containing downstream products dopamine, norepinephrine, and epinephrine. In our analysis, increased TH expression is strongly associated with poor overall survival (P = 4.62 × 10−5). TH gene expression is also significantly increased in pheochromocytoma and paraganglioma (Fig. 7). To date, eight inhibitors of TH have been studied. One of the inhibitors, alpha-methyl-p-tyrosine, was used to treat pheochromocytoma; however, use was discontinued because of severe side effects. In general, inhibition of TH may rely on a small therapeutic window for safe usage, because of the crucial role of the enzyme in dopamine synthesis.
s. Vascular endothelial growth factor C (VEGFC)
VEGFC is a dimeric, secreted growth factor in the VEGF (vascular endothelial growth factor) family. The VEGF family contains five members, VEGFA, placenta growth factor, VEGFB, VEGFC, and VEGFD, and acts by binding tyrosine kinase VEGF receptors on the cell surface. VEGFC binds and activates VEGFR-2 and VEGFR-3. VEGFC is overexpressed in peripheral blood mononuclear cells and plays an important role in lymphoangiogenesis (Mandriota et al., 2001). VEGFC is also strongly overexpressed in patients with thyroid cancer (Fig. 7). Furthermore, VEGFC expression is upregulated in brain tumors including GBM and hemangioblastomas, suggesting this protein is important for tumor-associated inflammation (Jenny et al., 2006). In our analysis, increased VEGFC expression is strongly associated with poor overall survival (P = 7.08 × 10−7). Expression of VEGFC is associated with poor overall survival in GBM (P < 0.001 and P = 0.023) (Xu et al., 2013; Zhao et al., 2016). VEGFC is targeted by microRNA-144 and microRNA-186 to halt tumor growth in cervical and bladder cancer, respectively (He et al., 2017; Tao et al., 2018). High expression of this protein in GBM suggests VEGFR-3 plays a vital role in cancer proliferation, potentially as much as VEGFR-1. CS2164 is a novel multikinase inhibitor that targets VEGFR-1, VEGFR-2, VEGFR-3, PDGFR alpha, c-Kit, Aurora kinase b, and CSF-R1 and exhibited antitumor potency in mouse xenograft models of colon, lung, liver, and stomach cancer (Zhou et al., 2017). Inhibitors of VEGFR-1 or VEGFR-3, or inhibitors of the maturation of VEGFC, could be efficacious in GBM, based on the strong correlation between poor prognosis in several cancers and VEGFC expression.
t. Chromosome 20 open reading frame 166 antisense RNA 1 (C20orf166AS1)
C20orf166AS1 is an 8.5-kb- long noncoding RNA (lncRNA). C20orf166AS1 was reported as a prostate-cancer-specific lncRNA that was negatively correlated with prostate cancer (Hu et al., 2014). Analysis of TCGA samples supports these findings; C20orf166AS1 expression is consistently higher in prostate adenocarcinoma patient samples than in normal tissue (Fig. 7). In our analysis, increased C20orf166AS1 expression is strongly associated with poor overall survival (P = 3.6 × 10−7). Aside from its possible role in prostate cancer, C20orf166AS1 function has not been fully elucidated.
2. Protein Targets Identified via Proteomic Approaches
Although the application of modern proteomic approaches has yet to reach its full potential in GBM research, several important studies have identified potential drug targets. Traditionally, proteomics have been performed with 2DGE and mass spectrometry. Although useful, 2DGE has several major limitations. For example, 2DGE cannot detect low abundance proteins, proteins with a molecular mass greater than 100 kDa, or hydrophobic membrane proteins (Hanash et al., 2002). In addition, proteins with isoelectric point values outside the pH range go undetected, including important GBM proteins such as EGFR and VEGFR (Iwadate et al., 2004). Proteomic technologies have overcome these challenges with several methods, namely targeted mass spectrometry via SRM (selected reaction monitoring), iTRAQ, and SWATH-MS (sequential window acquisition of all theoretical mass spectra). Here we discuss several preclinical protein targets involved in GBM identified via proteomic approaches.
Several GBM proteomic studies have identified annexin A2 as a possible drug target (Polisetty et al., 2012; Maruo et al., 2013; Yu et al., 2016). Annexin A2 is a calcium-binding cytoskeletal protein expressed in cancer cells and is strongly correlated with tumor aggression, metastasis, and glioma patient survival (Maule et al., 2016). The protein aids the conversion of plasminogen to plasmin, a serine protease that activates metalloproteinases and degrades the extracellular matrix to promote cell metastasis (Hajjar and Krishnan, 1999). Consistent overexpression of annexin A2 emphasizes its role in various subtypes of GBM. Thus, annexin A2 may be a promising drug target. Small molecule annexin A2 inhibitors have been developed to prevent human papilloma virus (Reddy et al., 2012; Woodham et al., 2015). Further validation of annexin A2 inhibitors in models of GBM is warranted.
One study identified nine potential GBM targets by comparing microarray data sets of neural stem cells and GBM stem cells and further validating the findings with RT-PCR and Western blot (Stangeland et al., 2015). Nine overexpressed proteins, PBK, CENPA, KIF15, DEPDC1, CDC6, DLG7, KIF18A, EZH2, and HMMR, correlated with poor patient survival and are potential GBM drug targets. CENPA was further validated as a potential target in GBM-initiating cells (Behnan et al., 2016). PBK is a MAPKK involved in p38-mediated cell motility and DNA damage response (Ayllón and O’Connor, 2007) and has been validated in vivo as a GBM target (Joel et al., 2015). EZH2 has also been validated as a target in GBM, and overexpression is associated with poor prognosis (Grinshtein et al., 2016; Zhang et al., 2017). CDC6, a gene involved in the RB/E2F pathway, was associated with decreased astrocytic glioma patient survival (Ferreira et al., 2015). Additionally, HMMR was validated as a potential target for GBM stem cell inhibition (Tilghman et al., 2014). The other proteins have not been validated further in the context of GBM but may also represent potential drug targets.
Proteomic approaches may also explain potential reasons for drug or target failure. To determine why antiangiogenic therapies failed, a proteomic approach based on selected reaction monitoring was employed on patient-derived intracranial GBM xenografts in rodents (Demeure et al., 2016). Levels of tricarboxylic acid cycle enzymes such as isocitrate dehydrogenase and aldehyde dehydrogenase decrease in response to antiangiogenic therapy, suggesting the cells evade death by increasing glycolysis (Demeure et al., 2016). Additionally, a systems-based statistical analysis of a proteomic and transcriptomic signature of GBM was identified, concluding a strong link between GBM invasive properties and the TGF-β signaling pathways (Ghosh et al., 2017). Targeting these pathways may inhibit GBM proliferation; however, target validation is necessary to rule out proteins that do not drive tumor growth.
3. Target Validation
Correlation between gene expression and patient survival does not necessarily indicate the gene (or protein) is critical for tumor progression or a viable drug target. For example, tyrosine hydroxylase is required for the synthesis of dopamine, and inhibition of TH, at least by the reported inhibitors, showed significant adverse effects. Therefore, rigorous validation of the 20 genes determined from TCGA analysis is crucial to move forward and develop a viable treatment option for GBM.
Clinical trials often fail due to insufficient target validation in the preclinical stage of the drug discovery process. To validate each target appropriately, CRISPR-Cas9-mediated gene knockdown can be used to assess tumor growth in vitro and in vivo. Gene knockouts that significantly inhibit tumor growth would be pursued for druggability. High throughput small molecule binding screens of each target could be run using differential scanning fluorimetry or other binding determination methods. For targets with selective inhibitors, further validation can be performed. Although our TCGA analysis results demonstrate a potential direction for GBM drug discovery research, target validation is required before further effort is used to develop inhibitors of these targets.
C. Synthetic Lethality
GBM tumor heterogeneity will likely render single target inhibition ineffective. In general, combination therapies are necessary to halt tumor growth. A potential approach to identify synergistic interactions is to perform “synthetic lethal” screens. Synthetic lethality is the concept that a combination of two or more gene mutations or alterations is necessary for cell death, and the mutation or inhibition of only one of the genes allows tumor cells to survive (Weidle et al., 2011). Synthetic lethal combinations can be identified via several strategies. For example, large, short hairpin RNA (shRNA) libraries can be used to screen cell lines with an inhibitor, that, when in combination with certain shRNAs, causes a lethal phenotype. Synthetic lethal pairs can also be discovered computationally by mining large datasets. By using this method, the known synthetic lethal relationship between P53 and PLK1 was validated by comparing patient survival data with pairs of genes in which the expression of one of the genes was underrepresented (Szczurek et al., 2013).
Several other synthetic lethal combinations have been identified in the context of GBM. Large-scale, shRNA library screening identified that the inhibition of MYC, P38MAPK, or ERK signaling pathways may be synthetically lethal with PI3K inhibitor PX-866 (Kim et al., 2011). EGFR inhibition is synthetically lethal with pharmacological stabilization of P53 (Mai et al., 2017). P53 mutations have also sensitized GBM cells to combined p-AKT inhibition and radiation by antagonizing DNA repair (Palanichamy et al., 2018). Furthermore, IDH1-mutated gliomas are potentially more susceptible to BCL-xL inhibition than other gliomas (Karpel-Massler et al., 2017). Continued work in this area is expected to generate novel effective treatment strategies for GBM.
D. Preclinical Models of Glioblastoma
Preclinical in vivo models of GBM can recapitulate hallmarks of cancer, including tissue invasion, sustained angiogenesis, evasion of apoptosis, and cancer-specific metabolism that cannot be modeled in vitro. Robust models of GBM that mimic the human tumor microenvironment are needed to assess drug safety profiles and reduce clinical trial failure. There are three major types of preclinical GBM models: chemically induced models, xenograft models, and genetically engineered mouse models. Some of the current and state-of-the-art strategies for developing animal models of GBM will be summarized here. [This subject has been reviewed extensively by Huszthy et al. (2012), Miyai et al. (2017), and Schuhmacher and Squatrito (2017)].
GBM mouse models have evolved in an attempt to mirror human tumor characteristics and microenvironment. One of the earliest models, the chemically induced GBM tumor, is generated by treating rats with N-nitroso compounds. The spontaneity of tumor generation in this model provides insight about the underlying molecular pathways involved in chemically induced mutagenesis. However, the rat tumors generally do not model human GBM histologic characteristics and cell lines suffer from genetic drift (Huszthy et al., 2012). Xenografts of human tumor cell lines injected into immunodeficient mice have also been used. However, these models can be difficult to establish and do not factor in immune response or changes in stromal environment (Huszthy et al., 2012). Therefore, GEMMs are excellent as in vivo GBM models because of the extensive molecular characterization of the human GBM tumor genome, which confirmed key mutations that drive oncogenesis. Compounds of interest can be tested on several variations of GEMMs, including those generated via combinations of P53, PTEN, NF1, RB, and PDGF alterations (reviewed in depth in Simeonova and Huillard, 2014). GEMMs have also provided valuable insight on the cell of origin of GBM. For example, GBM tumors can form in mice with conditional tumor suppressor alleles of NF1, P53, and PTEN that are injected with cre recombinase-expressing adenovirus (Alcantara Llaguno et al., 2009). The downsides of GEMMs are that they can be costly and time consuming and do not exhibit the heterogeneity of human GBM tumors. Additional in vivo models include orthotopic models, in which GBM cells are injected intracranially, and patient-derived xenograft models, in which primary patient tumors are cultured and implanted in mice subcutaneously (William et al., 2017). Furthermore, a Human Glioblastoma Cell Culture (HGCC) open resource has been organized to promote in vitro and in vivo testing (Xie et al., 2015). The HGCC resource contains a bank of 48 GBM cell lines derived from patients for translational research use. This bank allows robust in vivo representations of GBM to promote new discoveries. Numerous in vivo models of GBM exist, but none perfectly capture the complexity of tumor biology and microenvironment.
Because each GBM tumor model has its shortcomings, there remains a need for better preclinical models for compound screening. One strategy to meet this need involves avatar mice and coclinical models of GBM (Malaney et al., 2014). The mouse avatar allows efficient testing of different treatment strategies by implanting GBM tumor tissue resected from the patient into mice with the goal of selecting a promising therapy for each individual patient (Malaney et al., 2014). Unfortunately, grafted patient-derived xenograft tumors are altered by the mouse biology and do not predict response to treatment with great accuracy (Ben-David et al., 2017). CRISPR/Cas9 technology was previously used to generate P53, PTEN, and NF1 gene deletions in mice (Zuckermann et al., 2015). Although the CRIPSR/Cas9 system represents a more convenient model for in vivo tumor development, the need for an accurate model of GBM still exists. In general, in vivo models that mimic human intratumoral heterogeneity, tumor initiation, and tumor microenvironment are needed to accurately assess in vivo efficacy of a drug.
VI. Blood-Brain Barrier
A. Characteristics of the Blood-Brain Barrier
The BBB is responsible for nutrient transport, homeostasis, and communication between the body and the brain and also prevents foreign substances from reaching the brain. Research on the BBB dates to the 1880s, when a barrier to the transport of solutes from the blood to the brain was discovered. Paul Ehlrich furthered BBB research with experiments demonstrating that passage into the brain of peripherally injected dyes was impeded. Small molecule permeability of the BBB is an important consideration for drug development. Not only does the BBB impede small molecule transport, but active BBB transporters clear foreign material that passes the protective layers. The BBB is a dynamic, flexible interface between the brain and the body.
The BBB is composed of a monolayer of endothelial, ependymal, and tanycytic cells held together by restrictive tight junctions (Fig. 8). Two types of cellular junctions halt passive diffusion and prevent leakiness between the endothelial cells: intercellular adherens junctions and paracellular tight junctions. Adherens junctions are composed of vascular endothelium, cadherin, actinin, and catenin (Vorbrodt and Dobrogowska, 2003). Tight junctions consist of three major proteins: occludin, claudin, and junction adhesion molecules. Occludins are regulated by phosphorylation of serine, tyrosine, and threonine residues. Junction adhesion molecules regulate the formation of tight junctions during the acquisition of cell polarity (Ebnet et al., 2004). Furthermore, there are several other important cytoplasmic accessory proteins including zonula occludens and cingulin. Altogether, these proteins maintain the integrity of the BBB.
Fig. 8.

The blood-brain barrier protects the brain from foreign material with a layer of endothelial cells bound by adherens junctions [i.e., vascular endothelial (VE)-cadherin] and tight junctions [i.e., junction adhesion molecules (JAMs), endothelial cell adhesion molecule (ESAM), claudins, and occludins].
Nutrients and small molecules may be transported in and out of the brain by various methods, including passive diffusion, carrier-mediated transport, endocytosis, and active transport. Small biomolecules, such as water and various lipid-soluble molecules, are transported by passive diffusion. Typically, small lipophilic compounds will diffuse through the BBB; however, these properties make compounds more likely to be P-glycoprotein (Pgp) substrates or be taken up by peripheral tissues (Banks, 2009). Thus, although lower molecular mass and ClogP values are often optimal in theory for CNS drug discovery, the determination of appropriate values is a balancing act. Carrier-mediated transport is driven by two major protein families, the solute carrier (SLC) superfamily and ATP binding cassette (ABC) transporters. The main function of these transporters is to carry essential amino acids and glucose from the blood to the brain. Carrier-mediated transport may be hijacked by drug delivery. For example, System L has a broad substrate specificity for large molecules and, therefore, can transport levodopa (Nutt et al., 1984; Christensen, 1990). Endocytosis imports nutrients such as insulin via the formation of intracellular transport vesicles (Duffy and Pardridge, 1987). Active transport requires energy in the form of ATP hydrolysis; the µ-opioid agonist fentanyl is likely taken up into the brain via active transport mechanisms (Henthorn et al., 1999). However, due to the complexity of the BBB neovascular unit, drug uptake and efflux likely proceed via multiple transport pathways.
The BBB poses several challenges for effective drug discovery. One challenge is reaching and maintaining effective CNS permeation and drug concentration. The brain uses efflux pumps at the luminal side of the BBB to recognize and remove foreign substances. In particular, ABC transporters prevent a large influx of lipophilic molecules, xenobiotics, toxic metabolites, and drugs (Fletcher et al., 2016). CNS tumors compromise the structural integrity of the BBB, causing it to be leaky at the tumor core (Chacko et al., 2013). Although this may suggest that small molecules may be more permeable at the tumor site, the BBB surrounding the proliferating cells at the tumor’s edge remains intact (van Tellingen et al., 2015). Thus, BBB physiology and compound permeability are critical considerations for the CNS drug discovery process.
BBB transporters may provide an opportunity for the pursuit of alternative drug targets. l-dopa, melphalan, baclofen, and gabapentin are examples of drugs that cross the BBB via neutral amino acid transporters. Organic cation-carnitine transporters are used by verapamil, levofloxacin, and cephaloridine (Banks, 2016). Generally, compounds that use these transporters are similar in size and shape to the endogenous substrate of the protein. Additionally, uptake and efflux transporters can be inhibited by saturating the transporters. For example, saturating the large neutral amino acid transporter with large neural amino acids competes off the excess branched chain amino acids that enter the brain and cause neurotoxicity in maple syrup urine disease. Furthermore, a recent study demonstrated that metastasizing cells may signal to break down the BBB with microRNA-181c, allowing the cells to propagate in the brain (Leinenga et al., 2016). As a whole, successful drug discovery and development will involve efficient and reliable drug delivery methods to significantly improve treatment.
B. Blood-Brain Barrier Computational Modeling for Drug Discovery
Lipinski et al. (2001) developed a groundbreaking method of screening for orally bioavailable, drug-like molecules by using physicochemical properties known as the “rule of five.” Traditional CNS drugs are biased toward targeting monoamine GPCRs, transporters, and ion channels. Therefore, an assessment of the physicochemical properties of CNS drugs would conclude that CNS drugs should be small lipophilic compounds. Generally, CNS drugs are smaller and more lipophilic than oral non-CNS drugs and have fewer hydrogen bond donors and a lower topological polar surface area (Ghose et al., 2012). However, with increasing drug discovery efforts focused on nontraditional CNS targets, understanding of CNS-penetrant compounds could expand. To ameliorate this problem, a CNS multiparameter optimization (CNS MPO) algorithm was designed by Pfizer scientists with the goal of streamlining the CNS drug discovery process (Wager et al., 2010). The CNS MPO algorithm involves six physicochemical parameters [lipophilicity (ClogP), distribution at pH = 7.4 (ClogD), molecular mass, topical polar surface area (TPSA), pKa (of the most basic center), and the number of hydrogen bond donors (HBD)] relative to CNS penetration and success. The value of each parameter is weighted (0 to 1) based on the probability of the compound crossing the BBB (0 = low probability; 1 = high probability). For example, it is well known that a compound with a ClogP value less than 0 will be less likely to cross the BBB, therefore a compound with a ClogP value less than zero would receive a 0 for the ClogP parameter. The total CNS MPO desirability score is the summation of the weighted scores based on each of the six properties, with a range from 0 to 6. An analysis of FDA-approved CNS drugs demonstrated 74% have a CNS MPO desirability score ≥4 (Wager et al., 2010). To assess the BBB permeability of novel GBM clinical candidates, we applied the algorithm to 73 of the small molecule compounds currently undergoing clinical trials related to GBM treatment.
We determined the CNS desirability score for 73 of the GBM drug candidates reported in Table 1, using the CNS MPO.v2 algorithm (Fig. 9A) (Rankovic, 2017). The CNS MPO.v2 desirability score weighs five important CNS physicochemical properties, molecular mass, lipophilicity (ClogP), number of hydrogen bond donors, topical polar surface area, and pKa (of the most basic center), from 0 to 1. These properties were calculated with ADMET Predictor Version 8. The desirability score was the summation of the weighted score of each component, with the number of HBD score doubled. The HBD value was found to correlate strongly with BBB permeability, whereas ClogD, a variable used in the original equation, was somewhat redundant to ClogP and removed (Rankovic, 2017). Interestingly, only 37% of the small molecule candidates in clinical trials currently have a score ≥4, a much lower percentage than the 74% of FDA-approved CNS drugs. This may highlight the significance of emphasizing BBB permeability in early-stage drug discovery and may explain future clinical trial failures (or highlights the use of novel drug delivery methods). The compound that scored highest, LB100 with a 5.68 out of 6, is a protein phosphatase 2A inhibitor. All scores of tested compounds are listed in Supplemental Table 5. Compounds with a desirable molecular mass (score = 1) made up 23% of the group, whereas an almost equal amount (25%) had higher-than-optimal molecular mass values, over 500 Da (Fig. 9B). Most of the compounds (48%) had a ClogP score of 1 (Fig. 9C). A large portion of the compounds had a favorable topical polar surface area (45% between 40 and 90 Å2; Fig. 9D). Several of the compounds had an appropriate number of hydrogen bond donors as well (36% with scores >0.8; Fig. 9E). The majority of the compounds (64%) had a pKa desirability score of 1 (pKa < 8 for the most basic center) (Fig. 9F). Use of this CNS MPO algorithm together with other useful tools for predicting biologic behavior of small molecules could enhance and accelerate the drug discovery process.
Fig. 9.

CNS MPO Version 2 scores were calculated for 73 of the GBM drug candidates listed in Table 1. Plots are shown for scores calculated for total CNS MPO score (A), molecular weight distribution (B), LogP value distribution (C), polar surface area value distribution (D), hydrogen bond donor total distribution (E), and pKa value (of the most basic center) distribution (F).
VII. Drug Discovery Challenges in GBM
CNS drugs typically have a lower FDA-approval rate than non-CNS drugs. Additionally, oncology drug discovery attrition rates are characteristically high, second only to the therapeutic area of woman’s health (Kola and Landis, 2004). Thus, brain tumor drug discovery is characterized by major obstacles and historical failure.
In a study of CNS drugs entered into clinical trials from 1990 to 2012, CNS drugs were 45% less likely to pass Phase III trials than non-CNS drugs, with 46% failing to show improved efficacy over placebo (Kesselheim et al., 2015). Although bevacizumab received FDA approval, other antiangiogenesis drug candidates have been less effective. The Phase III “REGAL” (Recentin in Glioblastoma Alone and with Lomustine) trial comparing cediranib and cediranib + lomustine versus placebo in patients with recurrent GBM failed to reach the primary endpoint of progression-free survival prolongation (Batchelor et al., 2013). By using a different approach, rindopepimut, a conjugate of the EGFRvIII mutation site with an immunogenic carrier protein keyhole limpet hemocyanin, demonstrated efficacy in Phase I and II trials in combination with temozolomide. Progression-free survival and median overall survival (OS) were 10–15 and 22–26 months, respectively, compared with 6 and 15 months in historical controls (Swartz et al., 2014). Unfortunately, in the Phase III study, rindopepimut failed to meet OS endpoint criteria; however, this was due to a significant outperformance of the control arm (median OS = 20.0 months) compared with the treatment arm (median OS = 20.1 months) (Weller et al., 2017). Trials with rindopepimut will continue, but this failure highlights an important obstacle faced when bringing a novel therapy to the market.
Several obstacles impede the drug discovery process for GBM treatment. Challenges include identifying an effective target at the early research stages amid the complex intratumoral molecular heterogeneity, identifying a therapy that is permeable to the BBB, and developing robust clinical trials to assess the effectiveness of the potential treatment. Furthermore, a recent study highlighted the variation in the epigenetic tumor microenvironment of in vitro and in vivo models, suggesting that research with in vitro cancer cell lines is a “therapeutic roadblock” to GBM drug discovery (Miller et al., 2017). This study identified a single gene, jumonji C-domain-containing protein 6 (JMJD6), as a potential target. JMJD6 interacts with bromodomain containing 4 (BRD4), and JMJD6 shRNA knockdown was lethal in both in vitro and in vivo models (Miller et al., 2017).
As for the pharmacokinetics of the drug, there are several important limitations to consider for any drug candidate. First, the compound must reach the tumor site without diffusing into other tissue and must reach therapeutic concentrations. For example, a retrospective pharmacokinetic analysis of lapatinib after a failed Phase I/II clinical trial revealed that therapeutic concentrations of the drug were not reached (Reardon et al., 2013). Additionally, CNS drugs must be able to cross the BBB, which means they must have appropriate lipophilicity and size. Lipophilicity is measured by the octanol-water partition coefficient of a compound (ClogP), and CNS drugs optimally have a ClogP = 2 (Hansch and Leo, 1979). The size of a compound is measured by its molecular weight and polar surface area, which are optimal below 450 g/mol and 90 Å2, respectively, for CNS drugs (Rankovic, 2015). This is a large obstacle for biologics, since EGFR antibodies cannot cross the BBB. Generally, only 0.1%–0.2% of an administered antibody crosses the BBB and reaches the tumor site (Poduslo et al., 1994). (Here, it should be noted that bevacizumab likely does not need to cross the BBB to target the VEGF receptors in the lumen of capillaries of blood vessels in the brain.) Drugs could be administered intratumorally, as with the case of DNX-2401. In a Phase I trial, DNX-2401, an oncolytic adenovirus, demonstrated antitumor activity with no dose-limiting side effects (Lang et al., 2018). Intratumoral injections, although effective, may be time-consuming, unfamiliar to oncologists, and pose biosafety concerns. Another consideration is the presence of Pgp efflux pumps that remove foreign material escaping past the BBB. Although the BBB is impaired at the tumor site, allowing for increased permeability, the dense endothelium of vasculature providing nutrients to the tumor is not compromised, and therefore most of the BBB remains intact (Bart et al., 2000). These issues should be addressed in the preclinical phase, before bringing drug candidates into clinical trials.
Retrospective analysis of EGFR inhibitors provided insight into their failure in GBM clinical trials. EGFR inhibitors are widely and effectively used in preclinical models of GBM; however, clinical trials with these inhibitors failed to detect any improvement in outcome. These tyrosine kinase inhibitors (TKI), namely erlotinib and gefitinib, likely failed clinical trials due to limited brain exposure from Pgp and ABCG2-mediated efflux (Agarwal et al., 2010; de Vries et al., 2012). Additionally, gefitinib inhibits signaling of EGFR proteins with mutations in exons 19 and 21 of the TK domain that are often absent in gliomas (Marie et al., 2005). This phenomenon suggests more rigorous preclinical research should be conducted before expensive clinical trials are initiated.
A few recent successes in TKIs, osimertinib and GDC-0084, should be noted. The third generation EGFR inhibitor osimertinib (AZD9291) has been studied for its efficacy against non-small cell lung cancer (NSCLC) and is undergoing a large Phase I/II trial to determine the maximum tolerated dose in patients with advanced NSCLC (NCT01802632). Preclinical evaluation of osimertinib demonstrated the compound is more BBB permeable than gefitinib and other TKIs (Ballard et al., 2016). Another TKI, GDC-0084, was demonstrated to cross the BBB in a first-in-human Phase I dose-escalation study in patients with high-grade glioma (Wen et al., 2016). Extensive structure-activity relationship analysis on the dual PI3K/mammalian target of rapamycin complex 1 inhibitor revealed that removal of a methyl group at the two position of the pyridine side chain of the purine-based scaffold increased cellular potency and human metabolic stability and decreased efflux ratios (Heffron et al., 2016). Since BBB permeability has been a major problem with current EGFR TKIs, osimertinib and GDC-0084 both represent exciting inhibitors that have the potential to become efficacious treatments for brain and potentially other cancers.
Although recent efforts have advanced GBM drug discovery, nonpharmacokinetic problems, including clinical trial organization, remain a large obstacle to drug development. Because GBM is an orphan disease, clinical trial participation is low, which prevents the detection of subtle differences in treatment with statistical significance. Other challenges include determination of appropriate controls, stratification according to prognostic factors, and definition of clinical endpoint (Reardon et al., 2011). In addition, it is difficult to monitor the molecular signature of a brain tumor because surgeries are expensive and risky. It will be important to establish nonimaging methods of determining drug efficacy because targeted therapies may be cytostatic. In addition, biomarkers to measure treatment response will be useful for GBM clinical trials.
VIII. Advances in Drug Delivery
A. Nanocarriers
The current standard of care for GBM, temozolomide, suffers from problems with poor drug delivery. The compound is not soluble under physiologic conditions, easily inactivated via hydrolysis in the cells, and causes harmful side effects because of off-target DNA damage (Fang et al., 2015). Furthermore, several clinical trials have failed, such as those with gefitinib and erlotinib, due to the inability of the compounds to pass the BBB. While modifications to the compounds could improve BBB permeability, such modifications may affect target binding or decrease compound potency. Thus, nanocarrier delivery has emerged as a promising approach, especially for drug transport across the BBB using methods such as nanoparticles or prodrugs (Fig. 10).
Fig. 10.

Three drug delivery strategies for crossing the blood-brain barrier. In receptor-mediated endocytosis, a drug is conjugated to a ligand that binds to a receptor on the blood-brain barrier to trigger endocytosis. Small lipophilic compounds can be taken up by passive diffusion. Carrier-mediated transport is driven by two major protein families, the solute carrier superfamily and ATP binding cassette transporters, and these transporters can be hijacked for drug delivery.
1. General Properties of Nanoparticles
In general, nanoparticles (NPs) can be classified as organic/polymeric (e.g., liposomes, dendrimers, micelles, and ferritin) or inorganic (e.g., gold NPs, quantum dots, iron oxide, and lanthanide ions) (Safari and Zarnegar, 2014). Organic NPs provide more flexibility in terms of chemistry and structure for fabrication, whereas inorganic NPs have interesting physical properties including particular optical or magnetic features (Cheng et al., 2014; Verma et al., 2014; Abadeer and Murphy, 2016).
There are several important factors influencing the BBB permeability of NPs, including composition, size, charge, and shape (Safari and Zarnegar, 2014). NP size is a fundamental characteristic that determines passive targeting and biodistribution within brain tumors. NPs within 5–100 nm had selective penetration into brain tumors, with low accumulation in healthy brain tissue, due to the enhanced permeability and retention effect (Cheng et al., 2011; Fang et al., 2015; Mo et al., 2016; Jiang et al., 2017c). Surface charge is also an important factor determining NP transport across the BBB. Neutral and low concentrations of anionic NPs can be used safely as colloidal drug carriers to the brain. In contrast, high concentration anionic NPs (zeta potential between −15 and −60 mV) and cationic NPs (zeta potential between 15 and 45 mV) compromise BBB integrity (Lockman et al., 2004). In general, most of the NP formulations for brain delivery have a moderate zeta potential (between ±1 and ±15 mV). Therefore, NP surface charge can influence toxicity and distribution profiles. In addition, drug carrier activity can be enhanced by conjugation with different types of ligands. For example, ligands can be: 1) capable of mediating protein adsorption (e.g., polysorbate) (Petri et al., 2007), 2) capable of increasing charge and hydrophobicity (e.g., amphiphilic peptides), 3) able to improve blood circulation (e.g., polyethylene glycol) (Ishida et al., 2001), and 4) able to target macromolecules on the cell membrane directly such as peptides (e.g., RGD, NGR, and pep-1 peptides), proteins (transferrin), and antibodies (e.g., nicotinic acetylcholine receptor, EGFR, IL13Rα2, and LRP1 antibodies) (Wiley et al., 2013; Shilo et al., 2014; Saraiva et al., 2016). Overall, several parameters can influence the ability of NPs to transverse the BBB. Therefore, it is important that appropriate NP systems be designed and modulated to meet the objectives of the proposed function.
2. Nanocarriers in Clinical Trials
With proper characteristics in terms of composition, size, shape, surface charge, and coating chemistry, many nanocarriers can be transported effectively across the BBB in both in vitro and in vivo models by endocytosis and/or transcytosis. Subsequently, clinical trials using nanocarriers to deliver drugs and target brain tumors have been conducted (Table 3).
TABLE 3.
Nanoparticle-based agents in preclinical and clinical trials for brain tumors (clinicaltrials.gov)
| Name | Material | Target ligand | Therapeutic agent | Size (nm) | Status |
|---|---|---|---|---|---|
| Organic/Polymer | |||||
| NL CPT-11 (Clarke et al., 2017) | Liposome | — | irinotecan | 96–101 | Phase I NCT00734682 |
| SGT-53 (Camp et al., 2013) | Liposome | transferrin | wild-type P53 gene | 90 | Phase II NCT02340156 |
| SGT-94 (Siefker-Radtke et al., 2016) | Liposome | transferrin | RB94 gene | 108 | Phase I NCT01517464 |
| 2B3-101 (Gaillard et al., 2014) | Liposome | glutathione | doxorubicin | 95 | Phase I/IIa NCT01386580 |
| Inorganic | |||||
| Cornell dots (Phillips et al., 2014) | silica nanoparticles | cyclic arginine–glycine–aspartic acid (cRGDY) peptides | — | 7 | Phase 0 NCT01266096 |
| NU-0129 | gold nanoparticles | spherical nucleic acid for BCL2L12 | — | Phase 0/1 NCT03020017 | |
| NanoTherm (Maier-Hauff et al., 2011) | iron oxide | — | — | 15 | Approved in Europe in 2013 |
There are NP-based systems that have progressed to clinical trials (Anselmo and Mitragotri, 2016). For example, CPT-11 is a nanoliposomal irinotecan without a targeting molecule that was evaluated in Phase I studies in patients with recurrent high-grade gliomas (NCT00734682). In a Phase II clinical trial, SGT-53 was combined with temozolomide to treat patients with recurrent malignant gliomas, with the aim of evaluating tumor cell death, antitumor efficacy, safety, and overall survival (NCT02340156). SGT-53, a nanocarrier developed by SynerGene Therapeutics, Inc., is composed of cationic liposomes encapsulating a plasmid of P53 tumor suppressor and conjugating antitransferrin receptor antibody. Another gene therapy that has reached clinical trials is SGT-94, which contains RB94 plasmid DNA that encodes a truncated form of the RB gene, in a liposome with an antitransferrin receptor antibody (Siefker-Radtke et al., 2016). SGT-94 was found to cross the BBB and, therefore, may be a promising delivery method for GBM treatment (Kim et al., 2014). Regarding liposome-based nanocarriers, 2B3-101 is composed of PEGylated liposomal doxorubicin displaying glutathione as a targeting ligand for glutathione transporters. 2B3–101 enhanced doxorubicin delivery to the brain extracellular space compared with free doxorubicin and completed a Phase I/IIa clinical trial (Gaillard et al., 2014). In addition to liposomal-based systems, inorganic nanocarriers such as Cornell Dots; silica NPs conjugated with cyclic arginine–glycine–aspartic acid (cRGDY) peptides; and NU-0129, small gold NPs attached to spherical nucleic acids targeted for BCL2L12, are being tested (Phillips et al., 2014). Aminosilane-coated superparamagnetic iron oxide, called NanoTherm, was approved in Europe in 2013 to treat GBM. Treatment of GBM patients with hyperthermia plus radiotherapy with NanoTherm resulted in median overall survival of 13.4 months following first recurrence compared with 6.2 months with conventional treatments in a previous study population (Maier-Hauff et al., 2011). These clinical trials demonstrate the ability of NPs to increase compound concentration near GBM tumors and potentially improve potency and efficacy.
B. Prodrugs
Due to the presence of the BBB, only a small percentage of the administered drug reaches the brain. While NPs are a viable method to overcome this issue, another attractive chemical modification-based strategy, the prodrug, has been designed to increase BBB permeability.
A prodrug is a complex formed between a drug and a chemical moiety that increases its solubility or cell permeability (Laksitorini et al., 2014). Release of the active drug is controlled at the specific organ or tissue in the body, depending on the unique cellular conditions of each location, such as pH, enzyme distribution, and transporter expression (Gupta et al., 2009; Yang et al., 2011). Prodrugs are designed to overcome various physicochemical and biopharmaceutical obstacles such as low solubility in water or lipid membranes, low target selectivity, chemical instability, and toxicity (Rautio et al., 2008).
Multiple prodrug strategies have been employed to facilitate transport into the CNS (Table 4). Lipidization of the therapeutic molecule can enhance passive diffusion by masking polar groups on the parent drug. The classic example is the acetylation of the hydroxyl group of morphine to procure heroin. Because heroin is more lipophilic than morphine, brain uptake is 100-fold greater for heroin than for morphine (Oldendorf et al., 1972). However, although increasing lipophilicity enhances transport of drugs across the BBB, it also increases uptake in other tissues, potentially causing off-target toxicity. As another prodrug strategy, chemical delivery systems (CDS) link an active drug molecule to a lipophilic carrier that is oxidized to the hydrophilic form in the brain. This process results in a sustained and enhanced brain-specific release of free drug. CDS have improved brain targeting for several molecules, including zidovudine (AZT), ganciclovir, benzylpenicillin, and estradiol. For example, attaching a redox-based CDS to estradiol prolongs half-life in the brain (Mullersman et al., 1988), although recent studies have found high quantities of estradiol still accumulate in the uterus upon treatment (Prokai-Tatrai et al., 2013). Carrier-drug conjugates have transporters within the brain capillary endothelium and deliver compounds that are fundamental to normal brain function, e.g., large neutral amino acid transporter (Gomes and Soares-da-Silva, 1999), glucose transporter (Fernández et al., 2003), sodium-dependent vitamin C transporter 2 (Manfredini et al., 2002), peptide transporters, monocarboxylic acid transporters, organic cation transporters, organic anion transporters, and concentrative nucleoside and nucleotide transporters (Bhowmik et al., 2015). Prodrugs with close structural resemblance to substrates of these transporters will be recognized by them. Therefore, these transporters have become targets for drug/prodrug design and transport across the BBB. Ligand-drug conjugates may undergo receptor-mediated transport through the BBB, e.g., insulin, transferrin, leptin, peptides, and antibodies (Friden et al., 1991; Fukuta et al., 1994; Kang et al., 2015). The activities depend on the affinity of the ligand for its receptor; high-affinity ligands prevent systemic toxicity, but can become trapped in the CNS (Moos and Morgan, 2001). In addition, researchers have synthesized active drugs conjugated to brain-targeting moieties, a linear chain tertiary amine (N,N-dimethyl amino) and cyclic tertiary amine (scopine), to achieve better brain uptake (Li et al., 2014; Wang et al., 2014). Furthermore, antibody-drug conjugates as targeted drug delivery systems are a promising novel strategy. For example, anti-EGFR antibody-drug conjugates, like ABT-414, may be useful as a monotherapy or in combination with temozolomide (for an extensive review, see Gan et al., 2017). A novel cell-based drug delivery system takes advantage of postoperative inflammatory signals such as IL-8 and CXCL1/KC to deliver paclitaxel in neutrophils to GBM tumors (Xue et al., 2017). This neutrophil-based drug delivery system effectively slows recurrent tumor growth in vivo; however, it does not enhance the suppression of primary glioma growth. Nonetheless, novel drug delivery systems have the potential to improve the efficacy of small molecules.
TABLE 4.
Prodrug strategies for the transport of drugs into the CNS
| Strategy | Example | Chemical Structure | Action |
|---|---|---|---|
| Lipidization | Heroin (Fernandez et al., 2003) |  |
Acetylation of the hydroxyl group changes the physicochemical properties of heroin to favor brain uptake. |
| Chemical delivery system | Estradiol-CDS (Estredox) (Mullersman et al., 1988) |  |
After oxidation and hydrolysis, the concentration of estradiol CDS in rat brain was elevated four to five times longer than after estradiol treatment. |
| Carrier-drug conjugates | LAT1 (Gomes and Soares-da-Silva, 1999) |  |
The conversion of dopamine into its α-amino acid, l-dopa, enables the brain to uptake dopamine via LAT1. |
| GLUT1 (Fernandez et al., 2003) | 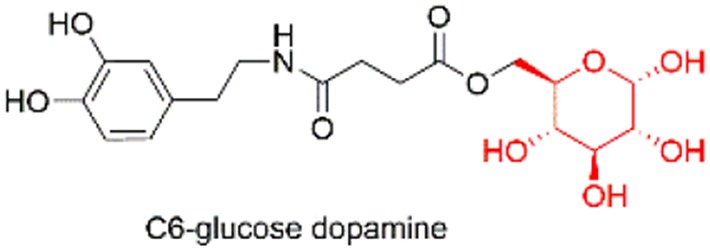 |
Dopamine linked to the C6 position of glucose had the best affinity for GLUT1. | |
| SVCT2 (Manfredini et al., 2002) | 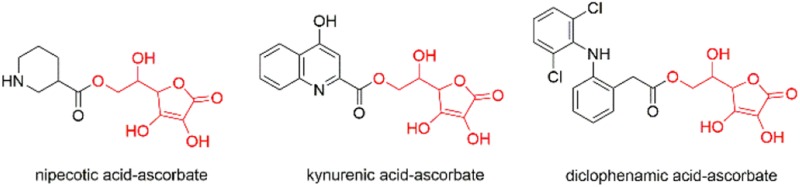 |
When nipecotic, kynurenic, and diclophenamic acids were conjugated to ascorbic acid, interaction with SVCT2 transporters improved. | |
| Ligand-drug conjugates | Insulin/transferrin (Friden et al., 1991; Fukuta et al., 1994; Wang et al., 2014) | — | CNS accumulation of methotrexate is improved by conjugating it to an antibody (OX-26), which is recognized by the transferrin receptor. |
| Targeting moiety-drug conjugates | N,N-dimethyl amino (Li et al., 2014) | 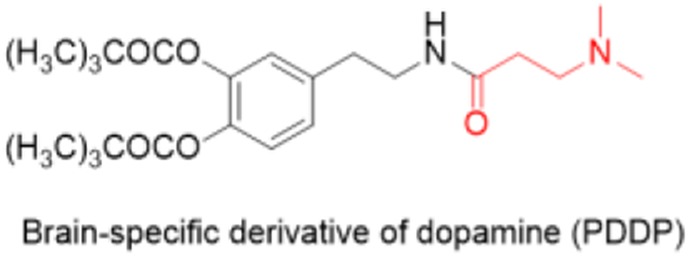 |
Conjugation with N,N-dimethyl amino significantly enhanced the brain-uptake efficiency of dexibuprofen, naproxen, 5-fluorouracil, and dopamine. |
| Scopine, cyclic tertiary amine (Wang et al., 2014) | 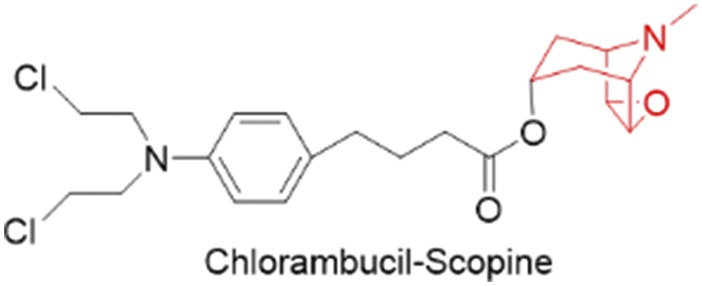 |
Chlorambucil-scopine prodrug significantly improved the cellular uptake both in vitro and in vivo. |
GLUT1, glucose transporter; LAT, large neutral amino acid transporter; SVCT2, sodium-dependent vitamin C transporter 2.
IX. Conclusions and Future Directions
Treatment of GBM is a complex and formidable, but not unsolvable, problem. The girth of available genomic information directs research strategies, allowing researchers to pursue meaningful hypotheses supported by patterns in population-level genomics. In tandem, novel proteomic tools are a valuable resource that will enhance our understanding of GBM tumor complexity. Genomic methods have already revealed a molecular fingerprint of the disease and pathways on which to focus our research efforts. Despite the emergence of more specific molecular classifications of GBM, targeted therapies to treat specific GBM subtypes are not yet realized. Numerous failed clinical trials suggest combination therapies will likely be the most promising method of GBM treatment, and emphasis should be applied to drug design and pharmacokinetic properties. With this study, we have identified 20 genes that may play important roles in GBM progression. These genes should be validated as potential targets for GBM drug discovery, as they correlate with poor overall patient survival. We have also uncovered novel transcription factors and signaling molecules involved in GBM that may regulate EGFR signaling. Targeting transcription factors and membrane proteins upstream of EGFR signaling may prove a promising therapeutic strategy for the treatment of GBM. Several genes identified in our analysis have been linked with GBM or EGFR signaling in previous studies. It should be noted that gene expression of the 20 targets could be a consequence of oncogenic stress, rather than tumor growth, and further target validation is necessary. This analysis may reorganize research priorities toward targeting receptors and proteins involved in glioma progression. In the future, both genomic and proteomic approaches will be standard tools not only to identify novel drug targets, but also to identify noninvasive biomarkers for diagnosis and treatment response.
Acknowledgments
We acknowledge Lily Zhao for thoughtful discussions and assistance with the manuscript.
Abbreviations
- AKT
protein kinase B
- BBB
blood-brain barrier
- CDS
chemical delivery system
- 2DGE
2D gel electrophoresis
- EGFR/vIII
epidermal growth factor receptor/variant III
- ERβ
estrogen receptor β
- FDA
Food and Drug Administration
- GBM
glioblastoma multiforme
- HBD
hydrogen bond donors
- 2-HG
2-hydroxyglutarate
- LGG
low-grade glioma
- MAPK
mitogen-activated protein kinase
- MPO
multiparameter optimization
- NF-κB
nuclear factor-κB
- NP
nanoparticle
- NSCLC
non-small cell lung cancer
- OS
overall survival
- Pgp
P-glycoprotein
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homolog
- RB
retinoblastoma
- TCGA
The Cancer Genome Atlas
- TGF
transforming growth factor
- TKI
tyrosine kinase inhibitors
- TPSA
topical polar surface area
- VEGF
vascular endothelial growth factor
Authorship Contributions
Participated in research design: Bankhead, Muangsin, Neamati.
Conducted experiments: Bankhead.
Performed data analysis: Shergalis, Bankhead.
Wrote or contributed to the writing of the manuscript: Shergalis, Bankhead, Luesakul, Neamati.
Footnotes
This work was supported by the National Institutes of Health, National Cancer Institute [Grant CA193690].
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.
References
- Abadeer NS, Murphy CJ. (2016) Recent progress in cancer thermal therapy using gold nanoparticles. J Phys Chem C 120:4691–4716. [Google Scholar]
- Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, Borowsky AD, Cardiff RD, Shen MM. (2003) Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res 63:3886–3890. [PubMed] [Google Scholar]
- Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia MS, Papadantonakis N, Alva Venur V, Schilero C, Peereboom DM, Stevens G, Rosenfeld S, Vogelbaum MA, Elson P, Nixon AB, et al. (2015) Phase II trial of dovitinib in recurrent glioblastoma. J Clin Oncol 33:2050. [Google Scholar]
- Akers JC, Ramakrishnan V, Kim R, Phillips S, Kaimal V, Mao Y, Hua W, Yang I, Fu CC, Nolan J, et al. (2015) miRNA contents of cerebrospinal fluid extracellular vesicles in glioblastoma patients. J Neurooncol 123:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. (2009) Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli L, Zagaria A, Coccaro N, Tota G, Impera L, Minervini CF, Pastore D, Minervini A, Casieri P, Specchia G, et al. (2013) A novel t(4;16)(q25;q23.1) associated with EGF and ELOVL6 deregulation in acute myeloid leukemia. Gene 529:144–147. [DOI] [PubMed] [Google Scholar]
- Anselmo AC, Mitragotri S. (2016) Nanoparticles in the clinic. Bioeng Transl Med 1:10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, Shine D, Kadmon D, Thompson T, Miles BJ, et al. (2008) Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res 14:7593–7603. [DOI] [PubMed] [Google Scholar]
- Ayllón V, O’Connor R. (2007) PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene 26:3451–3461. [DOI] [PubMed] [Google Scholar]
- Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, Pickup K, Jordan A, Hickey M, Grist M, et al. (2016) Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res 22:5130–5140. [DOI] [PubMed] [Google Scholar]
- Banks WA. (2009) Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 9:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA. (2016) From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 15:275–292. [DOI] [PubMed] [Google Scholar]
- Bart J, Groen HJ, Hendrikse NH, van der Graaf WT, Vaalburg W, de Vries EG. (2000) The blood-brain barrier and oncology: new insights into function and modulation. Cancer Treat Rev 26:449–462. [DOI] [PubMed] [Google Scholar]
- Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, et al. (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31:3212–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batistatou A, Stefanou D, Goussia A, Arkoumani E, Papavassiliou AG, Agnantis NJ. (2004) Estrogen receptor beta (ER beta) is expressed in brain astrocytic tumors and declines with dedifferentiation of the neoplasm. J Cancer Res Clin Oncol 130:405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerschlag DO, Ammerpohl O, Bräutigam K, Schem C, Lin Q, Weigel MT, Hilpert F, Arnold N, Maass N, Meinhold-Heerlein I, et al. (2011) Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology 80:12–20. [DOI] [PubMed] [Google Scholar]
- Behnan J, Grieg Z, Joel M, Ramsness I, Stangeland B. (2016) Gene knockdown of CENPA reduces sphere forming ability and stemness of glioblastoma initiating cells. Neuroepigenetics 7:6–18. [Google Scholar]
- Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R, et al. (2017) Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet 49:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, Norton CR, Gridley T, Cardiff RD, Cunha GR, et al. (1999) Roles for Nkx3.1 in prostate development and cancer. Genes Dev 13:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmik A, Khan R, Ghosh MK. (2015) Blood brain barrier: a challenge for effectual therapy of brain tumors. BioMed Res Int 2015:320941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng SY, Chakravarty D, Sanborn JZ, Berman SH, et al. TCGA Research Network (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center (2016) Firehose stddata__2016_01_28 run., Broad Institute of MIT Harvard, Cambridge, MA: DOI: 10.7908/C11G0KM9. [Google Scholar]
- Camp ER, Wang C, Little EC, Watson PM, Pirollo KF, Rait A, Cole DJ, Chang EH, Watson DK. (2013) Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther 20:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparuccia L, Tamagnone L. (2009) Semaphorin signaling in cancer cells and in cells of the tumor microenvironment - two sides of a coin. J Cell Sci 122:1723–1736. [DOI] [PubMed] [Google Scholar]
- Chacko AM, Li CS, Pryma DA, Brem S, Coukos G, Muzykantov V. (2013) Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: crossing the blood-brain barrier divide. Expert Opin Drug Deliv 10:907–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, Yang PC, Hsiao M, Hsu TL, Wong CH. (2013) Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci USA 110:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Xu HZ, Yao Y, Qin ZY. (2015) Prognostic value of epidermal growth factor receptor amplification and EGFRvIII in glioblastoma: meta-analysis. Acta Neurol Scand 132:310–322. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Huang HJ, Nagane M, Ji XD, Wang D, Shih CC, Arap W, Huang CM, Cavenee WK. (1996) Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA 93:8502–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome AM, Burda C, Basilion JP. (2011) Addressing brain tumors with targeted gold nanoparticles: a new gold standard for hydrophobic drug delivery? Small 7:2301–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Morshed RA, Auffinger B, Tobias AL, Lesniak MS. (2014) Multifunctional nanoparticles for brain tumor imaging and therapy. Adv Drug Deliv Rev 66:42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. (2014) Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370:709–722. [DOI] [PubMed] [Google Scholar]
- Choy JC. (2010) Granzymes and perforin in solid organ transplant rejection. Cell Death Differ 17:567–576. [DOI] [PubMed] [Google Scholar]
- Christensen HN. (1990) Role of amino-acid-transport and countertransport in nutrition and metabolism. Physiol Rev 70:43–77. [DOI] [PubMed] [Google Scholar]
- Clarke JL, Molinaro AM, Cabrera JR, DeSilva AA, Rabbitt JE, Prey J, Drummond DC, Kim J, Noble C, Fitzgerald JB, et al. (2017) A Phase 1 trial of intravenous liposomal irinotecan in patients with recurrent high-grade glioma. Cancer Chemother Pharmacol 79:603–610. [DOI] [PubMed] [Google Scholar]
- Cullen SP, Brunet M, Martin SJ. (2010) Granzymes in cancer and immunity. Cell Death Differ 17:616–623. [DOI] [PubMed] [Google Scholar]
- Dahms SO, Jiao GS, Than ME. (2017) Structural studies revealed active site distortions of human furin by a small molecule inhibitor. ACS Chem Biol 12:1211–1216. [DOI] [PubMed] [Google Scholar]
- Demeure K, Fack F, Duriez E, Tiemann K, Bernard A, Golebiewska A, Bougnaud S, Bjerkvig R, Domon B, Niclou SP. (2016) Targeted proteomics to assess the response to anti-angiogenic treatment in human glioblastoma (GBM). Mol Cell Proteomics 15:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denicolaï E, Tabouret E, Colin C, Metellus P, Nanni I, Boucard C, Tchoghandjian A, Meyronet D, Baeza-Kallee N, Chinot O, et al. (2016) Molecular heterogeneity of glioblastomas: does location matter? Oncotarget 7:902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. (2012) Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs 30:443–449. [DOI] [PubMed] [Google Scholar]
- Di Meo A, Diamandis EP, Rodriguez H, Hoofnagle AN, Ioannidis J, Lopez M. (2014) What is wrong with clinical proteomics? Clin Chem 60:1258–1266. [DOI] [PubMed] [Google Scholar]
- Dreyfuss JM, Johnson MD, Park PJ. (2009) Meta-analysis of glioblastoma multiforme versus anaplastic astrocytoma identifies robust gene markers. Mol Cancer 8:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerinck J, Du Four S, Bouttens F, Verschaeve V, Chaskis C, Andre C, Van Fraeyenhove F, D’haene N, Salmon I, Neyns B. (2016) Final results from the AXIG trial: a randomized Phase II clinical trial investigating axitinib alone or in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol 18 (Suppl 6):8. [Google Scholar]
- Duffy KR, Pardridge WM. (1987) Blood-brain-barrier transcytosis of insulin in developing rabbits. Brain Res 420:32–38. [DOI] [PubMed] [Google Scholar]
- Dufies M, Giuliano S, Ambrosetti D, Claren A, Ndiaye PD, Mastri M, Moghrabi W, Cooley LS, Ettaiche M, Chamorey E, et al. (2017) Sunitinib stimulates expression of VEGFC by tumor cells and promotes lymphangiogenesis in clear cell renal cell carcinomas. Cancer Res 77:1212–1226. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Ohno S, Vestweber D. (2004) Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci 117:19–29. [DOI] [PubMed] [Google Scholar]
- Edwards DK, Jasny E, Yoon H, Horscroft N, Schanen B, Geter T, Fotin-Mleczek M, Petsch B, Wittman V. (2017) Adjuvant effects of a sequence-engineered mRNA vaccine: translational profiling demonstrates similar human and murine innate response. J Transl Med 15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Wang K, Stephen ZR, Mu Q, Kievit FM, Chiu DT, Press OW, Zhang M. (2015) Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl Mater Interfaces 7:6674–6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi AT, Nahed BV, Wander SA, Iafrate AJ, Borger DR, Hu RL, Thabet A, Cahill DP, Perry AM, Joseph CP, et al. (2016) Elevation of urinary 2-hydroxyglutarate in IDH-mutant glioma. Oncologist 21:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YH, Chen WY, Kuo YH, Tung CL, Tsao CJ, Shiau AL, Wu CL. (2016) Elov16 is a poor prognostic predictor in breast cancer. Oncol Lett 12:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández C, Nieto O, Fontenla JA, Rivas E, de Ceballos ML, Fernández-Mayoralas A. (2003) Synthesis of glycosyl derivatives as dopamine prodrugs: interaction with glucose carrier GLUT-1. Org Biomol Chem 1:767–771. [DOI] [PubMed] [Google Scholar]
- Ferreira WA, Araújo MD, Anselmo NP, de Oliveira EH, Brito JR, Burbano RR, Harada ML, Borges Bdo N. (2015) Expression analysis of genes involved in the RB/E2F pathway in astrocytic tumors. PLoS One 10:e0137259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. (2016) ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat 26:1–9. [DOI] [PubMed] [Google Scholar]
- Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, Valley CC, Ferguson KM, Leahy DJ, Lidke DS, et al. (2017) EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell 171:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev 26:1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freije WA, Castro-Vargas FE, Fang ZX, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. (2004) Gene expression profiling of gliomas strongly predicts survival. Cancer Res 64:6503–6510. [DOI] [PubMed] [Google Scholar]
- Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B, Starzyk RM. (1991) Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc Natl Acad Sci USA 88:4771–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, et al. (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733–4740. [DOI] [PubMed] [Google Scholar]
- Fukuta M, Okada H, Iinuma S, Yanai S, Toguchi H. (1994) Insulin fragments as a carrier for peptide delivery across the blood–brain barrier. Pharm Res 11:1681–1688. [DOI] [PubMed] [Google Scholar]
- Gabrovska PN, Smith RA, Tiang T, Weinstein SR, Haupt LM, Griffiths LR. (2011) Semaphorin–plexin signalling genes associated with human breast tumourigenesis. Gene 489:63–69. [DOI] [PubMed] [Google Scholar]
- Gaillard PJ, Appeldoorn CC, Dorland R, van Kregten J, Manca F, Vugts DJ, Windhorst B, van Dongen GA, de Vries HE, Maussang D, et al. (2014) Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101). PLoS One 9:e82331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan HK, van den Bent M, Lassman AB, Reardon DA, Scott AM. (2017) Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat Rev Clin Oncol 14:695–707. [DOI] [PubMed] [Google Scholar]
- Ghiaseddin A, Reardon D, Massey W, Mannerino A, Lipp ES, Herndon JE, McSherry F, Desjardins A, Randazzo D, Friedman HS, et al. (2018) Phase II study of bevacizumab and vorinostat for patients with recurrent World Health Organization grade 4 malignant glioma. Oncologist 23:157–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. (2012) Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci 3:50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Funk CC, Caballero J, Shah N, Rouleau K, Earls JC, Soroceanu L, Foltz G, Cobbs CS, Price ND, et al. (2017) A cell-surface membrane protein signature for glioblastoma. Cell Syst 4:516–529.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy PR, Montaldi AP, Sakamoto-Hojo ET. (2016) HEB silencing induces anti-proliferative effects on U87MG cells cultured as neurospheres and monolayers. Mol Med Rep 14:5253–5260. [DOI] [PubMed] [Google Scholar]
- Gomes P, Soares-da-Silva P. (1999) L-DOPA transport properties in an immortalised cell line of rat capillary cerebral endothelial cells, RBE 4. Brain Res 829:143–150. [DOI] [PubMed] [Google Scholar]
- Gorlia T, Stupp R, Brandes AA, Rampling RR, Fumoleau P, Dittrich C, Campone MM, Twelves CC, Raymond E, Hegi ME, et al. (2012) New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group Phase I and II clinical trials. Eur J Cancer 48:1176–1184. [DOI] [PubMed] [Google Scholar]
- Grinshtein N, Rioseco CC, Marcellus R, Uehling D, Aman A, Lun XQ, Muto O, Podmore L, Lever J, Shen YQ, et al. (2016) Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget 7:59360–59376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves MD, Portnow J, Boulmay BC, Chawla SP, Dinh H, Chawla S, Wieland S, Levitt DJ. (2016) Phase 2 study of aldoxorubicin in relapsed glioblastoma. J Clin Oncol 34:2027. [Google Scholar]
- Gu JJ, Zhang JH, Chen HJ, Wang SS. (2016) TPX2 promotes glioma cell proliferation and invasion via activation of the AKT signaling pathway. Oncol Lett 12:5015–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Jing R, Zhong JH, Dong X, Li YX, Liu YK, Huang TR, Zhang CY. (2017) Identification of CD14 as a potential biomarker of hepatocellular carcinoma using iTRAQ quantitative proteomics. Oncotarget 8:62011–62028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D, Gupta SV, Lee K-D, Amidon GL. (2009) Chemical and enzymatic stability of amino acid prodrugs containing methoxy, ethoxy and propylene glycol linkers. Mol Pharm 6:1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massagué J. (2007) Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446:765–770. [DOI] [PubMed] [Google Scholar]
- Hajjar KA, Krishnan S. (1999) Annexin II: a mediator of the plasmin/plasminogen activator system. Trends Cardiovasc Med 9:128–138. [DOI] [PubMed] [Google Scholar]
- Hanash SM, Bobek MP, Rickman DS, Williams T, Rouillard JM, Kuick R, Puravs E. (2002) Integrating cancer genomics and proteomics in the post-genome era. Proteomics 2:69–75. [PubMed] [Google Scholar]
- Hanavan PD, Borges CR, Katchman BA, Faigel DO, Ho TH, Ma CT, Sergienko EA, Meurice N, Petit JL, Lake DF. (2015) Ebselen inhibits QSOX1 enzymatic activity and suppresses invasion of pancreatic and renal cancer cell lines. Oncotarget 6:18418–18428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C, Leo A. (1979) Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York. [DOI] [PubMed] [Google Scholar]
- Hayashi MA, Felicori LF, Fresqui MA, Yonamine CM. (2015) Protein-protein and peptide-protein interactions of NudE-Like 1 (Ndel1): a protein involved in schizophrenia. Curr Protein Pept Sci 16:754–767. [DOI] [PubMed] [Google Scholar]
- He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, Curtis RT, Shell BK, Bostwick DG, Tindall DJ, et al. (1997) A novel human prostate-specific, androgen-regulated homeobox gene (NKX3. 1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics 43:69–77. [DOI] [PubMed] [Google Scholar]
- He X, Ping J, Wen D. (2017) MicroRNA-186 regulates the invasion and metastasis of bladder cancer via vascular endothelial growth factor C. Exp Ther Med 14:3253–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron TP, Ndubaku CO, Salphati L, Alicke B, Cheong J, Drobnick J, Edgar K, Gould SE, Lee LB, Lesnick JD, et al. (2016) Discovery of clinical development candidate GDC-0084, a brain penetrant inhibitor of PI3K and mTOR. ACS Med Chem Lett 7:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- Henthorn TK, Liu Y, Mahapatro M, Ng KY. (1999) Active transport of fentanyl by the blood-brain barrier. J Pharmacol Exp Ther 289:1084–1089. [PubMed] [Google Scholar]
- Heyn H, Moran S, Esteller M. (2013) Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner Syndrome. Epigenetics 8:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DW, Kai AK, Ng IO. (2015) TCGA whole-transcriptome sequencing data reveals significantly dysregulated genes and signaling pathways in hepatocellular carcinoma. Front Med 9:322–330. [DOI] [PubMed] [Google Scholar]
- Hong JH, Kwak Y, Woo Y, Park C, Lee SA, Lee H, Park SJ, Suh Y, Suh BK, Goo BS, et al. (2016) Regulation of the actin cytoskeleton by the Ndel1-Tara complex is critical for cell migration. Sci Rep 6:31827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoober KL, Thorpe C. (2002) Flavin-dependent sulfhydryl oxidases in protein disulfide bond formation. Methods Enzymol 348:30–34. [DOI] [PubMed] [Google Scholar]
- Hou S, Landego I, Jayachandran N, Miller A, Gibson IW, Ambrose C, Marshall AJ. (2014) Follicular dendritic cell secreted protein FDC-SP controls IgA production. Mucosal Immunol 7:948–957. [DOI] [PubMed] [Google Scholar]
- Hu CC, Gan P, Zhang RY, Xue JX, Ran LK. (2014) Identification of prostate cancer lncRNAs by RNA-seq. Asian Pac J Cancer Prev 15:9439–9444. [DOI] [PubMed] [Google Scholar]
- Huang C, Bi E, Hu Y, Deng W, Tian Z, Dong C, Hu Y, Sun B. (2006) A novel NF-kappa B binding site controls human granzyme B gene transcription. J Immunol 176:4173–4181. [DOI] [PubMed] [Google Scholar]
- Huang PH, Xu AM, White FM. (2009) Oncogenic EGFR signaling networks in glioma. Sci Signal 2:re6. [DOI] [PubMed] [Google Scholar]
- Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen PØ, Miletic H, Thorsen F, Bjerkvig R. (2012) In vivo models of primary brain tumors: pitfalls and perspectives. Neuro-oncol 14:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida O, Maruyama K, Tanahashi H, Iwatsuru M, Sasaki K, Eriguchi M, Yanagie H. (2001) Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res 18:1042–1048. [DOI] [PubMed] [Google Scholar]
- Iwadate Y, Sakaida T, Hiwasa T, Nagai Y, Ishikura H, Takiguchi M, Yamaura A. (2004) Molecular classification and survival prediction in human gliomas based on proteome analysis. Cancer Res 64:2496–2501. [DOI] [PubMed] [Google Scholar]
- Jaaks P, Bernasconi M. (2017) The proprotein convertase furin in tumour progression. Int J Cancer 141:654–663. [DOI] [PubMed] [Google Scholar]
- Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, et al. (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27:5068–5074. [DOI] [PubMed] [Google Scholar]
- Jahrsdörfer B, Vollmer A, Blackwell SE, Maier J, Sontheimer K, Beyer T, Mandel B, Lunov O, Tron K, Nienhaus GU, et al. (2010) Granzyme B produced by human plasmacytoid dendritic cells suppresses T-cell expansion. Blood 115:1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny B, Harrison JA, Baetens D, Tille JC, Burkhardt K, Mottaz H, Kiss JZ, Dietrich PY, De Tribolet N, Pizzolato GP, et al. (2006) Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J Pathol 209:34–43. [DOI] [PubMed] [Google Scholar]
- Jiang J, Liu Z, Ge C, Chen C, Zhao F, Li H, Chen T, Yao M, Li J. (2017a) NK3 homeobox 1 (NKX3. 1) up-regulates forkhead box O1 expression in hepatocellular carcinoma and thereby suppresses tumor proliferation and invasion. J Biol Chem 292:19146–19159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Zhou T, Wang Z, Bin Q, Xia H. (2017b) HSP47 promotes glioblastoma stemlike cell survival by modulating tumor microenvironment extracellular matrix through TGF-beta pathway. ACS Chem Neurosci 8:128–134. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Wang X, Liu X, Lv W, Zhang H, Zhang M, Li X, Xin H, Xu Q. (2017c) Enhanced antiglioma efficacy of ultrahigh loading capacity paclitaxel prodrug conjugate self-assembled targeted nanoparticles. ACS Appl Mater Interfaces 9:211–217. [DOI] [PubMed] [Google Scholar]
- Joel M, Mughal AA, Grieg Z, Murrell W, Palmero S, Mikkelsen B, Fjerdingstad HB, Sandberg CJ, Behnan J, Glover JC, et al. (2015) Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo. Mol Cancer 14:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalpathy-Cramer J, Chandra V, Da X, Ou Y, Emblem KE, Muzikansky A, Cai X, Douw L, Evans JG, Dietrich J, et al. (2017) Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J Neurooncol 131:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T, Jiang M, Jiang D, Feng X, Yao J, Song Q, Chen H, Gao X, Chen J. (2015) Enhancing glioblastoma-specific penetration by functionalization of nanoparticles with an iron-mimic peptide targeting transferrin/transferrin receptor complex. Mol Pharm 12:2947–2961. [DOI] [PubMed] [Google Scholar]
- Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, Banu MA, Garcia F, Roth KA, Bruce JN, et al. (2017) Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 8:1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchman BA, Antwi K, Hostetter G, Demeure MJ, Watanabe A, Decker GA, Miller LJ, Von Hoff DD, Lake DF. (2011) Quiescin sulfhydryl oxidase 1 promotes invasion of pancreatic tumor cells mediated by matrix metalloproteinases. Mol Cancer Res 9:1621–1631. [DOI] [PubMed] [Google Scholar]
- Kesselheim AS, Hwang TJ, Franklin JM. (2015) Two decades of new drug development for central nervous system disorders. Nat Rev Drug Discov 14:815–816. [DOI] [PubMed] [Google Scholar]
- Kim SS, Rait A, Kim E, Pirollo KF, Nishida M, Farkas N, Dagata JA, Chang EH. (2014) A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 8:5494–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YW, Liu TJ, Koul D, Tiao N, Feroze AH, Wang J, Powis G, Yung WK. (2011) Identification of novel synergistic targets for rational drug combinations with PI3 kinase inhibitors using siRNA synthetic lethality screening against GBM. Neuro-oncol 13:367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka S, Hinohara K, Wang L, Nishimura T, Urushido M, Yachi K, Tsuda M, Tanino M, Kimura T, Nishihara H, et al. (2014) Epiregulin enhances tumorigenicity by activating the ERK/MAPK pathway in glioblastoma. Neuro-oncol 16:960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J. (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–715. [DOI] [PubMed] [Google Scholar]
- Laksitorini M, Prasasty VD, Kiptoo PK, Siahaan TJ. (2014) Pathways and progress in improving drug delivery through the intestinal mucosa and blood-brain barriers. Ther Deliv 5:1143–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, Prabhu SS, Rao G, Fuller GN, Aldape KD, et al. (2018) Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol DOI: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinenga G, Langton C, Nisbet R, Götz J. (2016) Ultrasound treatment of neurological diseases - current and emerging applications. Nat Rev Neurol 12:161–174. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. (2002) IKKα, IKKβ, and NEMO/IKKγ are each required for the NF-κB-mediated inflammatory response program. J Biol Chem 277:45129–45140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou Y, Qi B, Gong T, Sun X, Fu Y, Zhang Z. (2014) Brain-specific delivery of dopamine mediated by N,N-dimethyl amino group for the treatment of Parkinson’s disease. Mol Pharm 11:3174–3185. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. [DOI] [PubMed] [Google Scholar]
- Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Khoo KH, Yu CJ, Yang PC, Hsu TL, et al. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci USA 108:11332–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman PR, Koziara JM, Mumper RJ, Allen DD. (2004) Nanoparticle surface charges alter blood-brain barrier integrity and permeability. J Drug Target 12:635–641. [DOI] [PubMed] [Google Scholar]
- Lorenzi L, Döring C, Rausch T, Benes V, Lonardi S, Bugatti M, Campo E, Cabeçadas J, Simonitsch-Klupp I, Borges A, et al. (2017) Identification of novel follicular dendritic cell sarcoma markers, FDCSP and SRGN, by whole transcriptome sequencing. Oncotarget 8:16463–16472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HI, Hueng DY, Shui HA, Han JM, Wang CH, Lai YH, Cheng SY, Xiao X, Chen MT, Yang YP. (2014) Intratumoral decorin gene delivery by AAV vector inhibits brain glioblastomas and prolongs survival of animals by inducing cell differentiation. Int J Mol Sci 15:4393–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai WX, Gosa L, Daniels VW, Ta L, Tsang JE, Higgins B, Gilmore WB, Bayley NA, Harati MD, Lee JT, et al. (2017) Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat Med 23:1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier-Hauff K, Ulrich F, Nestler D, Niehoff H, Wust P, Thiesen B, Orawa H, Budach V, Jordan A. (2011) Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol 103:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaney P, Nicosia SV, Davé V. (2014) One mouse, one patient paradigm: new avatars of personalized cancer therapy. Cancer Lett 344:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małuch I, Levesque C, Kwiatkowska A, Couture F, Ly K, Desjardins R, Neugebauer WA, Prahl A, Day R. (2017) Positional scanning identifies the molecular determinants of a high affinity multi-leucine inhibitor for furin and PACE4. J Med Chem 60:2732–2744. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, et al. (2001) Vascular endothelial growth factor‐C‐mediated lymphangiogenesis promotes tumour metastasis. EMBO J 20:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredini S, Pavan B, Vertuani S, Scaglianti M, Compagnone D, Biondi C, Scatturin A, Tanganelli S, Ferraro L, Prasad P, et al. (2002) Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J Med Chem 45:559–562. [DOI] [PubMed] [Google Scholar]
- Marie Y, Carpentier AF, Omuro AMP, Sanson M, Thillet J, Hoang-Xuan K, Delattre J-Y. (2005) EGFR tyrosine kinase domain mutations in human gliomas. Neurology 64:1444–1445. [DOI] [PubMed] [Google Scholar]
- Marien E, Meister M, Muley T, del Pulgar TG, Derua R, Spraggins JM, Van de Plas R, Vanderhoydonc F, Machiels J, Binda MM, et al. (2016) Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma. Oncotarget 7:12582–12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AJ, Du QJ, Draves KE, Shikishima Y, HayGlass KT, Clark EA. (2002) FDC-SP, a novel secreted protein expressed by follicular dendritic cells. J Immunol 169:2381–2389. [DOI] [PubMed] [Google Scholar]
- Martorell Ò, Barriga FM, Merlos-Suárez A, Attolini CSO, Casanova J, Batlle E, Sancho E, Casali A. (2014) Iro/IRX transcription factors negatively regulate Dpp/TGF-beta pathway activity during intestinal tumorigenesis. EMBO Rep 15:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruo T, Ichikawa T, Kanzaki H, Inoue S, Kurozumi K, Onishi M, Yoshida K, Kambara H, Ouchida M, Shimizu K, et al. (2013) Proteomics-based analysis of invasion-related proteins in malignant gliomas. Neuropathology 33:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaka T, Shimano H. (2009) Elovl6: a new player in fatty acid metabolism and insulin sensitivity. J Mol Med (Berl) 87:379–384. [DOI] [PubMed] [Google Scholar]
- Maule F, Bresolin S, Rampazzo E, Boso D, Della Puppa A, Esposito G, Porcù E, Mitola S, Lombardi G, Accordi B, et al. (2016) Annexin 2A sustains glioblastoma cell dissemination and proliferation. Oncotarget 7:54632–54649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP, de Jong J, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, Franken K, Hahne M, Albar JP, Melief CJ, et al. (2001) Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc Natl Acad Sci USA 98:11515–11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercapide J, Lopez De Cicco R, Bassi DE, Castresana JS, Thomas G, Klein-Szanto AJ. (2002) Inhibition of furin-mediated processing results in suppression of astrocytoma cell growth and invasiveness. Clin Cancer Res 8:1740–1746. [PubMed] [Google Scholar]
- Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, Factor DC, Kim LJY, Morrow JJ, Wu Q, et al. (2017) Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 547:355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyai M, Tomita H, Soeda A, Yano H, Iwama T, Hara A. (2017) Current trends in mouse models of glioblastoma. J Neurooncol 135:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J, He L, Ma B, Chen T. (2016) Tailoring particle size of mesoporous silica nanosystem to antagonize glioblastoma and overcome blood-brain barrier. ACS Appl Mater Interfaces 8:6811–6825. [DOI] [PubMed] [Google Scholar]
- Moos T, Morgan EH. (2001) Restricted transport of anti‐transferrin receptor antibody (OX26) through the blood–brain barrier in the rat. J Neurochem 79:119–129. [DOI] [PubMed] [Google Scholar]
- Mullersman G, Derendorf H, Brewster ME, Estes KS, Bodor N. (1988) High-performance liquid chromatographic assay of a central nervous system (CNS)-directed estradiol chemical delivery system and its application after intravenous administration to rats. Pharm Res 5:172–177. [DOI] [PubMed] [Google Scholar]
- Muranaka H, Hayashi A, Minami K, Kitajima S, Kohno S, Nishimoto Y, Nagatani N, Suzuki M, Kulathunga LA, Sasaki N, et al. (2017) A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 6:e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, et al. (2008) Stem cell-related “Self-Renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 26:3015–3024. [DOI] [PubMed] [Google Scholar]
- Nguyen DT, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, Hersey A, Holmes J, Jensen LJ, Karlsson A, et al. (2017) Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res 45 (D1):D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolos F, Thomas C, Bado I, Gustafsson JÅ. (2018) ERβ sensitizes NSCLC to chemotherapy by regulating DNA damage response. Mol Cancer Res 16:233–242. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Woodward WR, Hammerstad JP, Carter JH, Anderson JL. (1984) The “on-off” phenomenon in Parkinson’s disease: relation to levodopa absorption and transport. N Engl J Med 310:483–488. [DOI] [PubMed] [Google Scholar]
- Okeley NM, Alley SC, Anderson ME, Boursalian TE, Burke PJ, Emmerton KM, Jeffrey SC, Klussman K, Law CL, Sussman D, et al. (2013) Development of orally active inhibitors of protein and cellular fucosylation. Proc Natl Acad Sci USA 110:5404–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. (1972) Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 178:984–986. [DOI] [PubMed] [Google Scholar]
- Ono Y, Chiba S, Yano H, Nakayama N, Saio M, Tsuruma K, Shimazawa M, Iwama T, Hara H. (2016) Glycoprotein nonmetastatic melanoma protein B (GPNMB) promotes the progression of brain glioblastoma via Na+/K+-ATPase. Biochem Biophys Res Commun 481:7–12. [DOI] [PubMed] [Google Scholar]
- Ordway JM, Bedell JA, Citek RW, Nunberg A, Garrido A, Kendall R, Stevens JR, Cao D, Doerge RW, Korshunova Y, et al. (2006) Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis 27:2409–2423. [DOI] [PubMed] [Google Scholar]
- O’Reilly SM, Newlands ES, Glaser MG, Brampton M, Rice-Edwards JM, Illingworth RD, Richards PG, Kennard C, Colquhoun IR, Lewis P, et al. (1993) Temozolomide: a new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur J Cancer 29A:940–942. [DOI] [PubMed] [Google Scholar]
- Ornskov D, Nexo E, Sorensen BS. (2007) Insulin induces a transcriptional activation of epiregulin, HB-EGF and amphiregulin, by a PI3K-dependent mechanism: identification of a specific insulin-responsive promoter element. Biochem Biophys Res Commun 354:885–891. [DOI] [PubMed] [Google Scholar]
- Orso F, Penna E, Cimino D, Astanina E, Maione F, Valdembri D, Giraudo E, Serini G, Sismondi P, De Bortoli M, et al. (2008) AP-2 alpha and AP-2 gamma regulate tumor progression via specific genetic programs. FASEB J 22:2702–2714. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. (2016) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro-oncol 18:v1–v75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanichamy K, Patel D, Jacob JR, Litzenberg KT, Gordon N, Acus K, Noda SE, Chakravarti A. (2018) Lack of constitutively active DNA repair sensitizes glioblastomas to Akt inhibition and induces synthetic lethality with radiation treatment in a p53-dependent manner. Mol Cancer Ther 17:336–346. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Noon LA, Harrisingh MC, Digby PW, Rosenberg LH, Cremona CA, Echave P, Flanagan AM, Parada LF, Lloyd AC. (2008) NF1 loss disrupts Schwann cell-axonal interactions: a novel role for semaphorin 4F. Genes Dev 22:3335–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. (2004) Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G(2) cell cycle arrest. Cancer Res 64:423–428. [DOI] [PubMed] [Google Scholar]
- Patel VN, Gokulrangan G, Chowdhury SA, Chen Y, Sloan AE, Koyüturk M, Barnholtz-Sloan J, Chance MR. (2013) Network signatures of survival in glioblastoma multiforme. PLOS Comput Biol 9:e1003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, et al. (2010) Non-stem cell origin for oligodendroglioma. Cancer Cell 18:669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri B, Bootz A, Khalansky A, Hekmatara T, Müller R, Uhl R, Kreuter J, Gelperina S. (2007) Chemotherapy of brain tumour using doxorubicin bound to surfactant-coated poly(butyl cyanoacrylate) nanoparticles: revisiting the role of surfactants. J Control Release 117:51–58. [DOI] [PubMed] [Google Scholar]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, et al. (2002) Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359:572–577. [DOI] [PubMed] [Google Scholar]
- Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, Humm J, Gönen M, Kalaigian H, Schöder H, et al. (2014) Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med 6:260ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Mason PJ, Craig SE, Brady-Kalnay SM. (2014) A protease storm cleaves a cell-cell adhesion molecule in cancer: multiple proteases converge to regulate ptpmu in glioma cells. J Cell Biochem 115:1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Berg CT. (1994) Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci USA 91:5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polisetty RV, Gautam P, Sharma R, Harsha HC, Nair SC, Gupta MK, Uppin MS, Challa S, Puligopu AK, Ankathi P, Purohit AK, Chandak GR, Pandey A, Sirdeshmukh R. (2012) LC-MS/MS analysis of differentially expressed glioblastoma membrane proteome reveals altered calcium signaling and other protein groups of regulatory functions. Mol Cell Proteomics 11:M111.013565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prizment AE, Vierkant RA, Smyrk TC, Tillmans LS, Nelson HH, Lynch CF, Pengo T, Thibodeau SN, Church TR, Cerhan JR, et al. (2017) Cytotoxic T cells and granzyme B associated with improved colorectal cancer survival in a prospective cohort of older women. Cancer Epidemiol Biomarkers Prev 26:622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai-Tatrai K, Szarka S, Nguyen V, Sahyouni F, Walker C, White S, Talamantes T, Prokai L. (2013) “All in the mind”? Brain-targeting chemical delivery system of 17β-estradiol (Estredox) produces significant uterotrophic side effect. Pharm Anal Acta DOI: 10.4172/2153-2435.S7-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, et al. (2009) Phase II trial of temozolomide plus O(6)-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol 27:1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. (2015) CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem 58:2584–2608. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. (2017) CNS physicochemical property space shaped by a diverse set of molecules with experimentally determined exposure in the mouse brain. J Med Chem 60:5943–5954. [DOI] [PubMed] [Google Scholar]
- Rautio J, Laine K, Gynther M, Savolainen J. (2008) Prodrug approaches for CNS delivery. AAPS J 10:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2016). R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Reardon DA, Galanis E, DeGroot JF, Cloughesy TF, Wefel JS, Lamborn KR, Lassman AB, Gilbert MR, Sampson JH, Wick W, et al. (2011) Clinical trial end points for high-grade glioma: the evolving landscape. Neuro-oncol 13:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J, McLendon RE, Suttle AB, et al. (2013) A Phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res 19:900–908. [DOI] [PubMed] [Google Scholar]
- Reddy TR, Li C, Fischer PM, Dekker LV. (2012) Three-dimensional pharmacophore design and biochemical screening identifies substituted 1,2,4-triazoles as inhibitors of the annexin A2-S100A10 protein interaction. ChemMedChem 7:1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. (2017) Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14:434. [DOI] [PubMed] [Google Scholar]
- Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, Rasheed BA, Dobra A, Dressman HK, Bigner DD, Nevins JR, et al. (2005) Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res 65:4051–4058. [DOI] [PubMed] [Google Scholar]
- Riese DJ, II, Cullum RL. (2014) Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol 28:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. (2012) The hedgehog signal transduction network. Sci Signal 5:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CG, Palomo JM, Rahmathulla G, McGraw M, Donze J, Liu L, Vogelbaum MA. (2010) Effect of alternative temozolomide schedules on glioblastoma O6-methylguanine-DNA methyltransferase activity and survival. Br J Cancer 103:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousalova I, Krepela E. (2010) Granzyme B-induced apoptosis in cancer cells and its regulation. Int J Oncol 37:1361–1378. [DOI] [PubMed] [Google Scholar]
- Rucci N, Sanità P, Angelucci A. (2011) Roles of metalloproteases in metastatic niche. Curr Mol Med 11:609–622. [DOI] [PubMed] [Google Scholar]
- Safari J, Zarnegar Z. (2014) Advanced drug delivery systems: nanotechnology of health design A review. J Saudi Chem Soc 18:85–99. [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. (2016) Nanoparticle-mediated brain drug delivery: overcoming blood–brain barrier to treat neurodegenerative diseases. J Control Release 235:34–47. [DOI] [PubMed] [Google Scholar]
- Sareddy GR, Li XN, Liu JY, Viswanadhapalli S, Garcia L, Gruslova A, Cavazos D, Garcia M, Strom AM, Gustafsson JA, et al. (2016) Selective estrogen receptor β agonist LY500307 as a novel therapeutic agent for glioblastoma. Sci Rep 6:24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer L, Iozzo RV. (2008) Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem 283:21305–21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer N, Gielen GH, Kebir S, Wieland A, Till A, Mack F, Schaub C, Tzaridis T, Reinartz R, Niessen M, et al. (2016) Phase I trial of dovitinib (TKI258) in recurrent glioblastoma. J Cancer Res Clin Oncol 142:1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher AJ, Squatrito M. (2017) Animal models in glioblastoma: use in biology and developing therapeutic strategies, in Advances in Biology and Treatment of Glioblastoma (Somasundaram K. ed) pp 219–240, Springer, Cham, Switzerland. [Google Scholar]
- Seidler DG, Goldoni S, Agnew C, Cardi C, Thakur ML, Owens RT, McQuillan DJ, Iozzo RV. (2006) Decorin protein core inhibits in vivo cancer growth and metabolism by hindering epidermal growth factor receptor function and triggering apoptosis via caspase-3 activation. J Biol Chem 281:26408–26418. [DOI] [PubMed] [Google Scholar]
- Seol MA, Chu IS, Lee MJ, Yu GR, Cui XD, Cho BH, Ahn EK, Leem SH, Kim IH, Kim DG. (2011) Genome-wide expression patterns associated with oncogenesis and sarcomatous transdifferentation of cholangiocarcinoma. BMC Cancer 11:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeishi H, Higashikawa K, Hiraoka M, Fujimoto S, Mitani Y, Ohta K, Takechi M, Kamata N. (2008) Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol Rep 19:1557–1564. [PubMed] [Google Scholar]
- Shilo M, Motiei M, Hana P, Popovtzer R. (2014) Transport of nanoparticles through the blood-brain barrier for imaging and therapeutic applications. Nanoscale 6:2146–2152. [DOI] [PubMed] [Google Scholar]
- Shimizu-Hirota R, Sasamura H, Kuroda M, Kobayashi E, Saruta T. (2004) Functional characterization of podocan, a member of a new class in the small leucine-rich repeat protein family. FEBS Lett 563:69–74. [DOI] [PubMed] [Google Scholar]
- Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, et al. (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63:6962–6970. [PubMed] [Google Scholar]
- Siefker-Radtke A, Zhang XQ, Guo CC, Shen Y, Pirollo KF, Sabir S, Leung C, Leong-Wu C, Ling CM, Chang EH, et al. (2016) A Phase l study of a tumor-targeted systemic nanodelivery system, SGT-94, in genitourinary cancers. Mol Ther 24:1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. (2016) Cancer statistics, 2016. CA Cancer J Clin 66:7–30. [DOI] [PubMed] [Google Scholar]
- Simeonova I, Huillard E. (2014) In vivo models of brain tumors: roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol Life Sci 71:4007–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys SG, Le QT, Shi GY, Tibshirani R, Giaccia AJ, Koong AC. (2004) The use of plasma surface-enhanced laser desorption/ionization time-of-flight mass spectrometry proteomic patterns for detection of head and neck squamous cell cancers. Clin Cancer Res 10:4806–4812. [DOI] [PubMed] [Google Scholar]
- Song H, Zhang B, Watson MA, Humphrey PA, Lim H, Milbrandt J. (2009) Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene 28:3307–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangeland B, Mughal AA, Grieg Z, Sandberg CJ, Joel M, Nygård S, Meling T, Murrell W, Vik Mo EO, Langmoen IA. (2015) Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 6:26192–26215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark-Vance V. (2005) Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. Neuro Oncol 7:369. [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- Suk K. (2012) Proteomic analysis of glioma chemoresistance. Curr Neuropharmacol 10:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunaga N, Kaira K, Imai H, Shimizu K, Nakano T, Shames DS, Girard L, Soh J, Sato M, Iwasaki Y, et al. (2013) Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene 32:4034–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz AM, Li QJ, Sampson JH. (2014) Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 6:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz JM, Worzfeld T, Offermanns S. (2008) ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J Biol Chem 283:1893–1901. [DOI] [PubMed] [Google Scholar]
- Szczurek E, Misra N, Vingron M. (2013) Synthetic sickness or lethality points at candidate combination therapy targets in glioblastoma. Int J Cancer 133:2123–2132. [DOI] [PubMed] [Google Scholar]
- Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D, de Vos FY, et al. (2014) Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled Phase 2 trial. Lancet Oncol 15:943–953. [DOI] [PubMed] [Google Scholar]
- Tang X, Molina M, Amar S. (2007) p53 short peptide (p53pep164) regulates lipopolysaccharide-induced tumor necrosis factor-α factor/cytokine expression. Cancer Res 67:1308–1316. [DOI] [PubMed] [Google Scholar]
- Tang X, Yang Y, Yuan H, You J, Burkatovskaya M, Amar S. (2013) Novel transcriptional regulation of VEGF in inflammatory processes. J Cell Mol Med 17:386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P, Wen H, Yang B, Zhang A, Wu X, Li Q. (2018) miR-144 inhibits growth and metastasis of cervical cancer cells by targeting VEGFA and VEGFC. Exp Ther Med 15:562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin I, Roskoski R, Carkaci-Salli N, Vrana KE. (2014) Complex molecular regulation of tyrosine hydroxylase. J Neural Transm (Vienna) 121:1451–1481. [DOI] [PubMed] [Google Scholar]
- Teng C, Zheng H. (2017) Low expression of microRNA-1908 predicts a poor prognosis for patients with ovarian cancer. Oncol Lett 14:4277–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. (2006) The cell surface membrane proteins Cdo and Boc are components and targets of the hedgehog signaling pathway and feedback network in mice. Dev Cell 10:647–656. [DOI] [PubMed] [Google Scholar]
- Tilghman J, Wu H, Sang YY, Shi XH, Guerrero-Cazares H, Quinones-Hinojosa A, Eberhart CG, Laterra J, Ying MY. (2014) HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res 74:3168–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosoni A, Franceschi E, Poggi R, Brandes AA. (2016) Relapsed glioblastoma: treatment strategies for initial and subsequent recurrences. Curr Treat Options Oncol 17:49. [DOI] [PubMed] [Google Scholar]
- Toyo-Oka K, Sasaki S, Yano Y, Mori D, Kobayashi T, Toyoshima YY, Tokuoka SM, Ishii S, Shimizu T, Muramatsu M, et al. (2005) Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum Mol Genet 14:3113–3128. [DOI] [PubMed] [Google Scholar]
- Twelves C, Short S, Wright S. (2017) A two-part safety and exploratory efficacy randomized double-blind, placebo-controlled study of a 1:1 ratio of the cannabinoids cannabidiol and delta-9-tetrahydrocannabinol (CBD:THC) plus dose-intense temozolomide in patients with recurrent glioblastoma multiforme (GBM). J Clin Oncol 35:2046. [Google Scholar]
- van den Bent M, Azaro A, Vos F, Sepulveda J, Yung WA, Wen P, Lassman A, Joerger M, Tabatabai G, Rodon J, et al. (2017) A Phase IB/II, open-label, multicenter study of capmatinib (INC280) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. Neuro Oncol 19:vi16–vi17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. (2015) Overcoming the blood–brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 19:1–12. [DOI] [PubMed] [Google Scholar]
- Varnum SM, Covington CC, Woodbury RL, Petritis K, Kangas LJ, Abdullah MS, Pounds JG, Smith RD, Zangar RC. (2003) Proteomic characterization of nipple aspirate fluid: identification of potential biomarkers of breast cancer. Breast Cancer Res Treat 80:87–97. [DOI] [PubMed] [Google Scholar]
- Ventura E, Weller M, Burghardt I. (2017) Cutting edge: ERK1 mediates the autocrine positive feedback loop of TGF-β and furin in glioma-initiating cells. J Immunol 198:4569–4574. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Cancer Genome Atlas Research Network (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma J, Lal S, Van Noorden CJ. (2014) Nanoparticles for hyperthermic therapy: synthesis strategies and applications in glioblastoma. Int J Nanomedicine 9:2863–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorbrodt AW, Dobrogowska DH. (2003) Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev 42:221–242. [DOI] [PubMed] [Google Scholar]
- Vyazunova I, Maklakova VI, Berman S, De I, Steffen MD, Hong W, Lincoln H, Morrissy AS, Taylor MD, Akagi K, et al. (2014) Sleeping Beauty mouse models identify candidate genes involved in gliomagenesis. PLoS One 9:e113489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TT, Hou X, Verhoest PR, Villalobos A. (2010) Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci 1:435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Zhou L, Li S, Wei J, Wang W, Zhou T, Liao SJ, Weng DH, Deng DR, Weng YJ, et al. (2010) C4orf7 contributes to ovarian cancer metastasis by promoting cancer cell migration and invasion. Oncol Rep 24:933–939. [PubMed] [Google Scholar]
- Wang X, Li J, Xu C, Li Y, Gong T, Sun X, Fu Y, He Q, Zhang Z. (2014) Scopine as a novel brain-targeting moiety enhances the brain uptake of chlorambucil. Bioconjug Chem 25:2046–2054. [DOI] [PubMed] [Google Scholar]
- Wargnier A, Lafaurie C, Legros-Maïda S, Bourge JF, Sigaux F, Sasportes M, Paul P. (1998) Down-regulation of human granzyme B expression by glucocorticoids - Dexamethasone inhibits binding to the Ikaros and AP-1 regulatory elements of the granzyme B promoter. J Biol Chem 273:35326–35331. [DOI] [PubMed] [Google Scholar]
- Weidle UH, Maisel D, Eick D. (2011) Synthetic lethality-based targets for discovery of new cancer therapeutics. Cancer Genomics Proteomics 8:159–171. [PubMed] [Google Scholar]
- Weinberg RA. (1995) The retinoblastoma protein and cell cycle control. Cell 81:323–330. [DOI] [PubMed] [Google Scholar]
- Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al. (2017) Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 18:1373–1385. [DOI] [PubMed] [Google Scholar]
- Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, Robins HI, Lieberman FS, Gilbert MR, Mehta MP, et al. (2014) Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American brain tumor consortium trial 04-02. Neuro-oncol 16:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PY, Cloughesy TF, Olivero A, Lu X, Mueller L, Coimbra AF, Gerstner ER, Ahnert JR. (2016) A first-in-human Phase 1 study to evaluate the brain-penetrant PI3K/mTOR inhibitor GDC-0084 in patients with progressive or recurrent high-grade glioma. J Clin Oncol 34:2012. [DOI] [PubMed] [Google Scholar]
- Wiley DT, Webster P, Gale A, Davis ME. (2013) Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci USA 110:8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William D, Mullins CS, Schneider B, Orthmann A, Lamp N, Krohn M, Hoffmann A, Classen CF, Linnebacher M. (2017) Optimized creation of glioblastoma patient derived xenografts for use in preclinical studies. J Transl Med 15:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt D, Burfeind P, von Hardenberg S, Opitz L, Salinas-Riester G, Bremmer F, Schweyer S, Thelen P, Neesen J, Kaulfuss S. (2013) Valproic acid inhibits the proliferation of cancer cells by re-expressing cyclin D2. Carcinogenesis 34:1115–1124. [DOI] [PubMed] [Google Scholar]
- Woodham AW, Taylor JR, Jimenez AI, Skeate JG, Schmidt T, Brand HE, Da Silva DM, Kast WM. (2015) Small molecule inhibitors of the annexin A2 heterotetramer prevent human papillomavirus type 16 infection. J Antimicrob Chemother 70:1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Li L, Jiang G, Zhan H, Wang N. (2016) B-cell CLL/lymphoma 3 promotes glioma cell proliferation and inhibits apoptosis through the oncogenic STAT3 pathway. Int J Oncol 49:2471–2479. [DOI] [PubMed] [Google Scholar]
- Wu WK, Tse TT, Sung JJ, Li ZJ, Yu L, Cho CH. (2009) Expression of ErbB receptors and their cognate ligands in gastric and colon cancer cell lines. Anticancer Res 29:229–234. [PubMed] [Google Scholar]
- Xiao X, Liu D, Tang Y, Guo F, Xia L, Liu J, He D. (2003–2004) Development of proteomic patterns for detecting lung cancer. Dis Markers 19:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Notkins AL, Lan MS. (1996) IA-2, a transmembrane protein tyrosine phosphatase, is expressed in human lung cancer cell lines with neuroendocrine phenotype. Cancer Res 56:2742–2744. [PubMed] [Google Scholar]
- Xie Y, Bergström T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, Segerman A, Wicher G, Niklasson M, Baskaran S, et al. (2015) The human glioblastoma cell culture resource: validated cell models representing all molecular subtypes. EBioMedicine 2:1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Ji W, Fei Y, Zhao Y, Wang L, Wang W, Han M, Tan C, Fei X, Huang Q, et al. (2017) Cathepsin L is involved in X-ray-induced invasion and migration of human glioma U251 cells. Cell Signal 29:181–191. [DOI] [PubMed] [Google Scholar]
- Xu H, Cai T, Carmona GN, Abuhatzira L, Notkins AL. (2016) Small cell lung cancer growth is inhibited by miR-342 through its effect of the target gene IA-2. J Transl Med 14:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhong Z, Yuan J, Zhang Z, Wei Q, Song W, Chen H. (2013) Collaborative overexpression of matrix metalloproteinase-1 and vascular endothelial growth factor-C predicts adverse prognosis in patients with gliomas. Cancer Epidemiol 37:697–702. [DOI] [PubMed] [Google Scholar]
- Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y, Wei Z, Wang L, Kong L, Sun H, et al. (2017) Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol 12:692–700. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Nishiumi S, Kono S, Takao S, Azuma T, Yoshida M. (2017) Differences in elongation of very long chain fatty acids and fatty acid metabolism between triple-negative and hormone receptor-positive breast cancer. BMC Cancer 17:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Zhang L, Xu T, Zhou J, Qin R, Chen C, Zou Y, Fu D, Hu G, Chen J, et al. (2013) SAMSN1 is highly expressed and associated with a poor survival in glioblastoma multiforme. PLoS One 8:e81905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Aloysius H, Inoyama D, Chen Y, Hu L. (2011) Enzyme-mediated hydrolytic activation of prodrugs. Acta Pharm Sin B 1:143–159. [Google Scholar]
- Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. (2010) Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res 70:8487–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Fu WM. (2015) Identification of differential splicing genes in gliomas using exon expression profiling. Mol Med Rep 11:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Feng L, Liu D, Zhang L, Wu B, Jiang W, Han Z, Cheng S. (2016) Quantitative proteomics reveals the novel co-expression signatures in early brain development for prognosis of glioblastoma multiforme. Oncotarget 7:14161–14171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yu XG, Chen L, Zhang Z, Feng S. (2017) EZH2 overexpression is associated with poor prognosis in patients with glioma. Oncotarget 8:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Hou C, Hou A, Zhu D. (2016) Concurrent expression of VEGF-C and neuropilin-2 is correlated with poor prognosis in glioblastoma. Tohoku J Exp Med 238:85–91. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yang Z, Tsuji T, Gong J, Xie J, Chen C, Li W, Amar S, Luo Z. (2011) LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene 30:1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chen HM, Qu S, Li L, Zhao W, Liang ZG, Yu BB, Chen KH, Lu QT, Lin GX, et al. (2018a) Reduced QSOX1 enhances radioresistance in nasopharyngeal carcinoma. Oncotarget 9:3230–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang J, Yu X, Jiang X, Shi Y, Weng Y, Kuai Y, Lei L, Ren G, Feng X, et al. (2018b) LITAF is a potential tumor suppressor in pancreatic cancer. Oncotarget 9:3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shan S, Li ZB, Xin LJ, Pan DS, Yang QJ, Liu YP, Yue XP, Liu XR, Gao JZ, et al. (2017) CS2164, a novel multi-target inhibitor against tumor angiogenesis, mitosis and chronic inflammation with anti-tumor potency. Cancer Sci 108:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JX, Goldoni S, Bix G, Owens RT, McQuillan DJ, Reed CC, Iozzo RV. (2005) Decorin evokes protracted internalization and degradation of the epidermal growth factor receptor via caveolar endocytosis. J Biol Chem 280:32468–32479. [DOI] [PubMed] [Google Scholar]
- Zou J, Guo P, Lv N, Huang D. (2015) Lipopolysaccharide-induced tumor necrosis factor-α factor enhances inflammation and is associated with cancer. Mol Med Rep 12:6399–6404. [DOI] [PubMed] [Google Scholar]
- Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DT, Tschida B, Moriarity B, et al. (2015) Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6:7391. [DOI] [PMC free article] [PubMed] [Google Scholar]