

Abstract
Glioblastoma (GBM) is the most common and most lethal primary brain tumor in adults, causing roughly 14,000 deaths each year in the U.S. alone. Median survival following diagnosis is less than 15 months with maximal surgical resection, radiation, and temozolomide chemotherapy. The challenges inherent in developing more effective GBM treatments have become increasingly clear, and include its unyielding invasiveness, its resistance to standard treatments, its genetic complexity and molecular adaptability, and subpopulations of GBM cells with phenotypic similarities to normal stem cells, herein referred to as glioblastoma stem cells (GSCs). Because GSCs are required for tumor growth and progression, differentiation-based therapy represents a viable treatment modality for these incurable neoplasms.
The following protocol describes a collection of procedures to establish a high throughput screening platform aimed at the identification of small molecules that promote GSC astroglial differentiation. At the core of the system is a glial fibrillary acidic protein (GFAP) differentiation reporter-construct. The protocol contains the following general procedures: (1) establishing GSC differentiation reporter lines; (2) testing/validating the relevance of the reporter to GSC self-renewal/clonogenic capacity; and (3) high-capacity flow-cytometry based drug screening.
The screening platform provides a straightforward and inexpensive approach to identify small molecules that promote GSCs differentiation. Furthermore, utilization of libraries of FDA-approved drugs holds the potential for the identification of agents that can be repurposed more rapidly. Also, therapies that promote cancer stem cell differentiation are expected to work synergistically with current "standard of care" therapies that have been shown to target and eliminate primarily more differentiated cancer cells.
Keywords: Cancer Biology, Issue 131, Stem Cells, Glioma, Astrocytic Differentiation, Lentivirus, Reporter System, Drug Screen, Differentiation Therapy, Acetyl Choline, Calcium Signaling
Introduction
Recent studies have shown that tumors contain a small population of cells, termed cancer stem cells (CSCs) or tumor-initiating cells, which are responsible for tumor progression, metastasis, and resistance to chemo- and radio-therapies 1,2. The presence of cancer stem cells and their more differentiated progenies within tumors is considered an important factor promoting intratumoral heterogeneity and thus represents a major hurdle in treating cancers3. Tumor cell hierarchy, provided by the cancer stem cell theory, has inspired the development of new strategies to treat cancers 4. One approach for targeting cancer stem cells is to identify and inhibit signaling pathways that are known to be required during embryonic development of the affected organ. Indeed, we and others have previously published multiple papers describing the ongoing requirement for the neural stem cell-relevant signaling pathways Sonic Hedgehog and Notch in glioblastoma5,6,7. This work has helped in solidifying the rationale for several GBM clinical trials. A second approach for targeting cancer stem cells is to promote their differentiation. This approach has received a lot of support due to the favorable results from preclinical and clinical studies in treating acute promyelocytic leukemia with retinoic acids (ATRA, a vitamin-A analog). Here ATRA was found to remove the maturation block and promote cancer cell differentiation8. More recently, Piccirillo and colleagues have elegantly shown that BMP-4 promotes GSC differentiation into astrocytes with significant anti-GBM effects in vitro and in vivo9.
The rationale for the current study is based on a "reversed engineering" approach for targeting GSCs. Given the vast heterogeneity present in GBM and with poor differentiation being one of the hallmarks of cancer, we asked if we could promote a more favorable phenotype - differentiation into an astrocyte-like state. Here, we do not have prior knowledge of the signaling pathways that maintain GSCs in a given tumor specimen but rather aim to achieve a desired phenotype (e.g. GFAP positivity).
This report describes the procedures used to establish GSC differentiation reporter-lines from the transduction of GSC-enriched cultures to GSC clonal selection. The glioblastoma neurosphere lines used were established at the laboratory of Professor Angelo Vescovi from patients with a diagnosis of primary glioblastoma at Hospital San Raffaele - Milano, Italy. These lines have been extensively studied in several publications 6,10,11,12,13,14. It is highly recommended that individuals who are interested in implementing these techniques in their laboratory determine the relevance of the reporter to cancer stem cell self-renewal capacity in the cells they plan to study (this is true for any reporter). A detailed protocol for one of the in vitro clonogenic assays accepted in the field is provided to accomplish this15,16. Finally, a detailed protocol describing the utilization of the differentiation reporter-lines in a flow-cytometry based drug screen is provided at the end. Of note, similarly to the astroglial differentiation system described here, we have successfully established and validated GSC reporter lines integrating an MAP2:GFP (neuronal differentiation) reporter. Therefore, the methodologies describe in this paper may be applied to study cellular differentiation into various cell lineages.
Some of the figures in this report can be found in a recent publication: "Atracurium Besylate and other neuromuscular blocking agents promote astroglial differentiation and deplete glioblastoma stem cells18.
Protocol
NOTE: Astroglial and neuronal lentivirus reporter systems were purchased as pre-packaged, concentrated lentiviral preparations. Basic knowledge of flow cytometry technique is required. Also, for a full use of this protocol the user will need access to a flow cytometer with high throughput capacity (accepts 96-well plates as sample source).
1. Lentiviral Transcriptional Reporter System
NOTE: In all flow cytometric analyses, use parental, non-transduced, cells or vector-transduced (non-fluorescent) cells for establishing baseline fluorescence. Also, note that in all steps where mechanical trituration is called for, be gentle. Harsh trituration can kill a significant number of the fragile GSCs and influence flow cytometry results.
Plate 1x106 cells in 2 mL of complete neural stem cell growth medium in a 6-multiwell plate.
Transduce cells by adding lentivirus reporter at a multiplicity of infection (moi) equal to 5.
Add 2 µL of polybrene for a final concentration of 8 µg/mL.
Incubate the cells at 37 °C and 5% CO2 overnight.
The next day, replace growth medium to remove unbound virus. Place the plate back in the incubator at 37 °C and 5% CO2 and incubate for 24 h.
Harvest 0.5 mL of the total volume; spin down cells at 360 x g for 5 min at room temperature in a 15- mL conical tube and remove supernatant by aspiration. Add 0.5 mL of fresh neural stem cell growth medium to the remaining cells and return the plate to the incubator for continued expansion.
Add 200 µL of dissociation reagent and incubate for 5 min at 37 °C.
Dissociate cells by gently pipetting up and down. NOTE: Harsh trituration may result in significant killing of GSCs, complete dissociation is usually achieved after 20 - 30 times.
To minimize cell attachment or aggregation, add 800 µL of Hank's Balanced Salt Solution (HBSS) for a final volume of 1 mL.
Transfer 200 µL of each cell suspension to a well of a 96-multiwell plate.
For this procedure, use a benchtop flow cytometer with 96-well plate capability equipped with a blue laser for excitation and with the capability of detecting green fluorescent protein. Perform flow cytometric analysis with at least 10,000 viable cells for each acquisition.
Determine the percentage of GFP-positive cells by flow cytometry.
2. Subclone Selection, Expansion, and Validation
Plate cells in 100 µL of neural stem cell medium at a density of 0.7 cells per well of a 96-multiwell plate.
Culture clones for 11 days at 37 °C and 5% CO2. This step is highly cell type dependent and will likely require an adjustment based on the cell line used. As a general rule, a sphere diameter ≥ 100 µm is a good indication of the presence of clonogenic GSCs in the neurospheres.
Using a fluorescent microscope equipped with a FITC filter, mark the wells that contain a single neurosphere where ~1-5% of the cells are GFP-positive. NOTE: Determining the exact percentage of GFP-positive cells is not too critical at this point. Each of the subclones will later be evaluated by flow cytometry. This step is performed to reduce the total number of clones to be analyzed with emphasis on neurospheres that originate by undifferentiated, GFAP-negative, GSC.
Expand selected reporter clones until there are a sufficient number of cells for analysis by flow cytometry. A sub-confluent well of a 6-multiwell plate containing ≤ 1.5x105 cells/mL should provide a sufficient number of cells. NOTE: A detailed procedure for the isolation and expansion of GSCs is available 17.
3. Determination of GFAP:GFP Expression by Flow Cytometry
Harvest an aliquot of cells (0.5 mL) from each reporter clone and non-transduced controls to determine the exact percentage of GFP-positive cells. Considering reporter clones that contain ~1 - 5% GFP-positive cells.
Spin cells at 360 x g for 5 min at room temperature and remove supernatant by aspiration.
To each pellet, add 200 µL of cell dissociation reagent and incubate the tubes in a water bath set to 37 °C.
Triturate cells gently to achieve a single cell suspension (usually between 20 to 30 times).
To minimize cell attachment or aggregation, add 800 µL HBSS to a final volume of 1 mL.
Transfer 200 µL per well of a 96-multiwell plate from each cell suspension.
For this procedure, use a benchtop flow cytometer with 96-well plate capability. Perform flow cytometric analysis with at least 10,000 viable cells for each acquisition.
4. ELDA Self-Renewal Assay to Assess Clonogenic Capacity
NOTE: For controls, both parental, non-transduced, as well as GFP-expressing lentivirus-transduced cells, should be used to determine the relative clonogenic potential of the GSC differentiation reporter lines to the original GSC cultures from which they were derived.
Dissociate differentiation-reporter subclones into single-cell suspensions as described above.
Plate cells in 96-multiwell plates in 100 µL of complete neural stem cell growth media at cell densities ranging between 5 and 500 cells per well.
Incubate cells for 9 to 11 days at 37 °C and 5% CO2.
Score positive wells by direct visualization of neurospheres under a light microscope. A well should be considered "positive" if it contains at least a single large neurosphere.
Plug the data: total wells analyzed and the number of positive wells using the ELDA online interface available at http://bioinf.wehi.edu.au/software/elda/index.html.
5. Drug Library Dilution Preparation
Remove library plates from storage at -80 °C, cover with aluminum foil (protect light-sensitive compounds). Thaw for approximately 30 min to an hour at room temperature.
Use a 12-channel multichannel pipettor to dilute the library compounds to 0.2 mM in complete neural stem cell medium. Dilute DMSO to 10% in growth medium NOTE: The final concentration of DMSO should be 0.1% when added to cells. A higher concentration of DMSO in the culture may result in toxicity. For this reason, it is recommended to test sensitivity to DMSO in the cell line used, before treatment.
Use DMSO as vehicle control to treat cells found in the left and right columns of each plate (Columns 1 and 12; Wells A through H).
Cover the diluted library plates with aluminum foil and return the original library plates to the -80 °C freezer for long-term storage.
6. Drug Screen
NOTE: DMSO-treated cells should be used to set the baseline fluorescence and adjust gating.
Plate 5x103 cells in a 96-multiwell plate in 99 µL of complete neural stem cell growth medium.
Using a 12-channel multichannel pipettor, treat cells with diluted library compounds at a final concentration of 2 µM (1 µL of 0.2 mM drug into 99 µL of cell suspension) or with DMSO (control). Incubate the plates for 72 h at 37 °C and 5% CO2.
Using a 12-channel multichannel pipettor add 150 µL of cell dissociation reagent to each well and incubate for 20 min at 37 °C.
Dissociate cells by gently triturating with a 12-channel multichannel pipettor until single cell suspension is achieved (usually between 20 to 30 times). The amount of time that is required to dissociate neurospheres completely will vary between GSC lines. While the recommended cell dissociation reagent (see materials) is safe and incubation for up to 45 minutes had no effect on viability in multiple GSC lines tested, verify for each GSC line to be used in screening. NOTE: The final volume in each well should be roughly 250 µL (100 µL of cell suspension + 150 µL of cell dissociation reagent).
Determine the percentage of cells expressing the GFAP:GFP reporter by flow cytometry. Use standard gating strategy. First, plot forward and side scatters to get a general sense of cell size and viability. Then place a gate on the viable single-cell population. This is the population for which green fluorescence (eGFP) will be determined. Any compound that results in an increase in the percentage of GFP-positive cells by over three standard deviations over control (DMSO-treated) is considered as a positive "hit." This threshold should be adjusted according to the specific application and the desired stringency. NOTE: For the high throughput screen is recommended using a single GSC reporter subclone. Following hit identification, each hit should be validated against additional reporter subclones from the same GSC line. To increase the confidence for true hits is also recommend testing compounds against reporter subclones isolated from different GSC neurospheres lines.
Representative Results
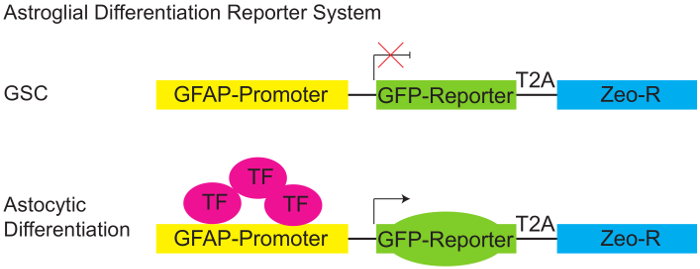
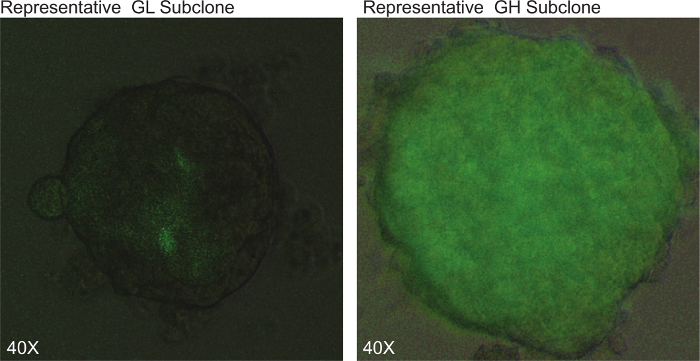
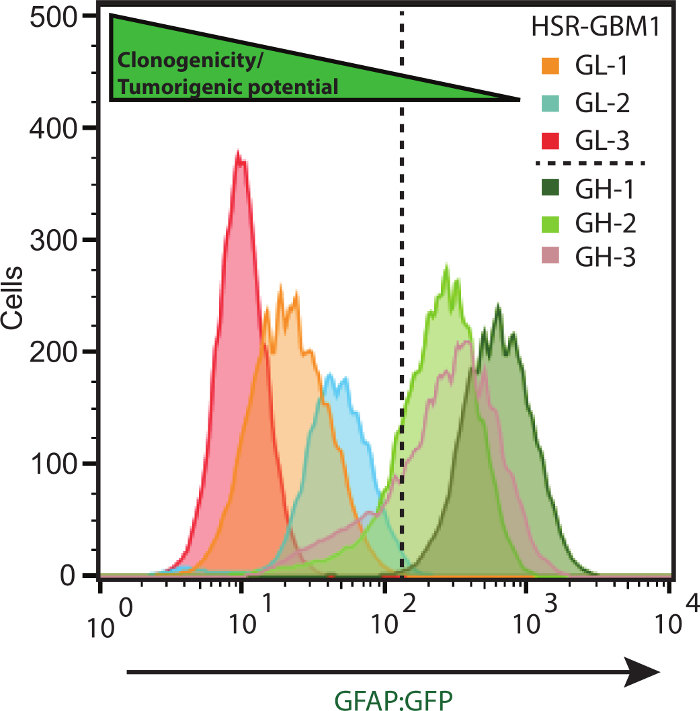
Three independent patient-derived neurospheres lines were transduced with the lentivirus astroglial reporter encoding for a green-fluorescent protein (GFP) fused in-frame with a Zeocin resistance cassette and driven by the human GFAP promoter element (Figure 1). Next individual clones were isolated by plating 0.7 cells per well in a 96 well plate (Figure 2), this was followed by flow cytometric determination of the percentage of cells expressing GFP (Figure 3). Neurosphere clones, derived from single cells, containing ≤5% GFP-positive cells are referred to as GL (GFAP Low) while clones containing ≥75% GFP positive cells are referred to as GH (GFAP High) and are considered to be more differentiated as compared to the GL subclones (Figure 4).
To identify agents and pathways which may control astroglial differentiation in GSCs, a small-molecule drug screen using two NIH Clinical Collection libraries was performed. Cells were treated for 72 hours with 727 library agents from the NIH clinical collection I and II, set at a concentration of 2 µM or an equal volume of DMSO as a control. The effect of these agents was tested on cell viability in all our patient-derived GBM neurosphere lines at concentrations ranging from 0.2 µM to 20 µM, before the drug screening. The concentration used in this protocol allowed us to identify compounds that were able to induce astroglial differentiation and at the same time, it minimized potentially off-target toxic effects due to higher drug concentrations.
Following incubation, we determined the percentage of cells expressing the GFAP-GFP reporter by flow cytometry. Baselines for viability and percentage of GFP-positive cells were determined in at least three wells for each library plate, and a positive hit was determined as an increase in the percentage of GFP-positive cells of three standard deviations over baseline (DMSO) and a minimum threshold of 25% GFP positive cells. We identified 12 drugs that induced sufficient increase in the GFP-positive population (Table 1).
Figure 1: Astroglial Differentiation Reporter System. Schematic diagram of pGreenZeo GFAP:GFP reporter. GSCs do not express the appropriate combination of transcription factors necessary to activate the glial fibrillary acidic protein (GFAP) promoter and therefore will not express GFP (top panel). However, when the appropriate transcription factors are present (e.g. when cells acquire an astroglial fate - differentiate) the GFAP promoter becomes active, and the cells will express the GFP reporter (bottom panel). (GSC - Glioma stem cell, T2A-protein linker, Zeo-R - Zeocin resistance gene, TF - transcription factor). Please click here to view a larger version of this figure.
Figure 2: Astroglial Differentiation - Subclone selection. Representative pictures of HSR-GBM1 GSC subclones expressing low levels (GL) or high levels (GH) of the GFAP:GFP reporter using fluorescence microscopy (40X magnification is shown). Please click here to view a larger version of this figure.
Figure 3: Flow Cytometry for the Determination of Green Fluorescence. HSR-GBM1 patient-derived neurosphere line was transduced with GFAP:GFP reporter lentivirus and multiple subclones were selected based on GFP expression in the neurosphere initiating-cell and confirmed by flow cytometry. These clones were named either GL (GFAP Low) or GH (GFAP High). Please click here to view a larger version of this figure.
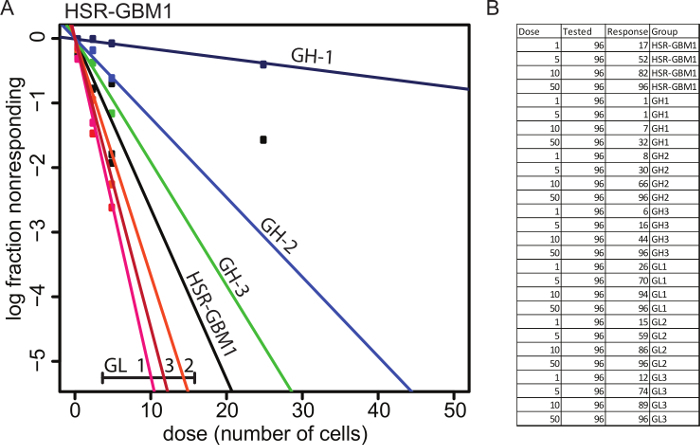
Figure 4: Functional characterization of HSR-GBM1 GFAP:GFP subclones. GL subclones are more clonogenic in vitro as indicated by increased GSC frequencies which are measured by the Extreme Limiting Dilution Analysis (ELDA). Please click here to view a larger version of this figure.
| Name | Fold increase in GFP + Cells | Description | Blood Brain Barrier | Drug Bank Probability |
| Vinorelbine | 8.97 | Anti-mitotic chemotherapy | - | 0.88 |
| Diphenoxylate | 13.89 | Antidiarrheal | + | 0.96 |
| Lomerizine | 10.89 | Calcium channel blocker / Cerebral vasodilator | NA | NA |
| Phenprobamate | 10.73 | Anxiolytic / Muscle relaxant, centrally acting | NA | NA |
| 6-Azauridine | 15.07 | Antimetabolite / Antiviral | NA | NA |
| Irinotecan | 14.14 | Topisomerase I inhibitor | + | 0.63 |
| Atracurium Besylate | 8.16 | Nondepolarizing skeletal muscle relaxant | + | 0.93 |
| Glimepiride | 8.75 | Antidiabetic | + | 0.73 |
| Hexachlorophene | 9.83 | Antiseptic | + | 0.92 |
| Digoxin | 8.23 | Cardiotonic glycoside | - | 0.72 |
| Flecainide | 10 | Anti-arrhythmia agent | + | 0.86 |
| Nisoldipine | 5.05 | Calcium channel blocker | - | 0.95 |
Table 1: Small molecules inducing GFAP-GFP reporter expression in HSR-GBM1 GL-1 Twelve compounds were found to increase the percentage of GFP-expressing cells significantly (fold increase is shown) and met the stringent criteria of ≥3 standard deviations over the baseline, DMSO-treated cells, and with no less than 25% GFP-positive cells. The ability and probability of a given agent to cross the blood brain barrier (Drug Bank (−). Abbreviation: NA (information not available).
Discussion
While most of the previous studies of GSCs focused primarily on the markers that define them, in this study we decided to take the reverse approach. We focus primarily on the differentiated progenies generated by GSCs (e.g., cells expressing astroglial and neuronal markers). Here we demonstrate the utilization of a cell-based high throughput drug screening system, which is based on human GFAP promoter-dependent expression of GFP. All the experiments were performed utilizing patient-derived neurosphere glioblastoma cell lines. A detailed protocol describing the isolation and expansion of these lines is described in Galli et al17.
The system not only aided us in the identification of small molecules which are capable of inducing cellular differentiation of GSCs but also helped us to determine unequivocally that the expression of the astroglial differentiation marker GFAP determines the clonogenic capacity of individual GBM cells by comparing GSC frequencies. The ELDA assay was developed by Yifang Hu and Gordon K. Smyth. Readers are encouraged to read the manuscript for an in-depth understanding of the assay strengths and limitations15. The colony formation scoring step is highly cell type dependent and will likely require an adjustment based on the cell line used. Moreover, as a general rule, we use sphere diameter ≥100 µm as a good indication that the neurosphere was originated by a clonogenic GSC.
Furthermore, the drug screening system we describe here allows for the identification of novel pathways (specifically acetylcholine and calcium transport) that are required to maintain GSCs in the undifferentiated state (see Table 1). The starting drug concentration may vary based on the library and cell line utilized. Also, the time required for cellular differentiation may need adjustment. Furthermore, tumorigenic validation of drug hits requires an in vivo tumor initiation assay 18.
Potentially a minor limitation of this technique is that spontaneous differentiation inevitably occurs in any stem-cell enriched culture, and this phenomenon tends to increase with the number of passages in culture and differs between individual subclones. Indeed, we observed spontaneous differentiation in our subclones generally after passage 15. Therefore, we limited our differentiation analyses to cultures at passage numbers not exceeding five.
Therefore, perhaps the most critical point in this methodology is to keep in vitro passaging of these GSCs to the minimum and when working in vitro, to maintain culture density below 1.5x105 cells/mL. Furthermore, it is highly recommended that each drug "hit" is validated against additional reporter subclones from the same GSC line, as well as, against reporter subclones isolated from different patient-derived GSC neurospheres lines. This will increase the confidence that a true "hit" is at hand.
The versatility of the methodology described in this robust protocol strengthens the therapeutic value of a drug-induced cancer stem cell differentiation and should help in identifying new drugs as potential novel therapeutic strategies for GBM and other tumors. Finally, the assay may be optimized to be used with non-neoplastic neural stem cells, other cancer types, and with different differentiation reporters.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work has been partially supported by NIH R01CA187780.
References
- Maher EA, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15(11):1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71(12):4055–4060. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Sul J, Fine HA. Malignant gliomas: new translational therapies. Mt Sinai J Med. 2010;77(6):655–666. doi: 10.1002/msj.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar EE, Chaudhry A, Farah MH, Eberhart CG. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am J Pathol. 2007;170(1):347–355. doi: 10.2353/ajpath.2007.060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG. Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res. 2013;19(12):3224–3233. doi: 10.1158/1078-0432.CCR-12-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck KC, et al. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res. 2010;16(24):6060–6070. doi: 10.1158/1078-0432.CCR-10-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrell RP, Jr, et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid) N Engl J Med. 1991;324(20):1385–1393. doi: 10.1056/NEJM199105163242002. [DOI] [PubMed] [Google Scholar]
- Piccirillo SG, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444(7120):761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- Bar EE, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25(10):2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177(3):1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert UD, et al. CD133/CD15 defines distinct cell subpopulations with differential in vitro clonogenic activity and stem cell-related gene expression profile in in vitro propagated glioblastoma multiforme-derived cell line with a PNET-like component. Folia Neuropathol. 2012;50(4):357–368. doi: 10.5114/fn.2012.32365. [DOI] [PubMed] [Google Scholar]
- Lim KS, et al. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene. 2014;33(35):4433–4441. doi: 10.1038/onc.2013.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert UD, et al. ZEB1 Promotes Invasion in Human Fetal Neural Stem Cells and Hypoxic Glioma Neurospheres. Brain Pathol. 2014;25(6):724–732. doi: 10.1111/bpa.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1-2):70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Meyer M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015;112(3):851–856. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli R, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Spina R, Voss DM, Asnaghi L, Sloan A, Bar EE. Atracurium Besylate and other neuromuscular blocking agents promote astroglial differentiation and deplete glioblastoma stem cells. Oncotarget. 2016;7(1):459–472. doi: 10.18632/oncotarget.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]