

Abstract
The suppressor of T-cell signaling (Sts) proteins, Sts-1 and Sts-2, are homologous phosphatases that negatively regulate signaling pathways downstream of the T-cell receptor. Functional inactivation of Sts-1 and Sts-2 in a murine model leads to resistance to systemic infection by the opportunistic pathogen, C. albicans. This suggests that modulation of the host immune response by inhibiting Sts function may be a viable strategy to treat these deadly fungal pathogen infections. To better understand the molecular determinants of function and structure, we characterized the structure and steady-state kinetics of the histidine phosphatase domains of human Sts-1 (Sts-1HP) and Sts-2 (Sts-2HP). We solved the X-ray crystal structures of Sts-1HP, unliganded and in complex with sulfate to 2.5 Å and 1.9 Å, respectively, and the structure of Sts-2HP with sulfate to 2.4 Å. The steady-state kinetic analysis shows, as expected, that Sts-1HP has a significantly higher phosphatase activity than that of Sts-2HP, and that the human and mouse proteins behave similarly. In addition, comparison of the phosphatase activity of full-length Sts-1 protein to Sts-1HP reveals similar kinetics, indicating that Sts-1HP is a functional surrogate for the native protein. We also tested known phosphatase inhibitors and identified that the SHP-1 inhibitor, PHPS1, is a potent inhibitor of Sts-1 (Ki of 1.05 ± 0.15 μM). Finally, we demonstrated that human Sts-1 has robust phosphatase activity against the substrate, Zap-70, in a cell-based assay. Collectively, these data suggest that the human Sts proteins are druggable targets and provides a structural basis for future drug development efforts.
Keywords: suppressor of T-cell signaling, TULA, UBASH, histidine kinase, phosphoglycerate mutase
TOC Image
INTRODUCTION
T cells play critical roles in the recognition and elimination of foreign pathogens in the host immune system. The T cell receptor (TCR) is responsible for activating T cells, recognizing and responding to foreign antigens and inappropriate expression of endogenous proteins1. Control of T cell signaling occurs through both positive and negative regulation. Two members of the suppressor of TCR signaling (Sts) family of proteins, Sts-1 and Sts-2, have been shown to be functionally redundant negative regulators of signaling pathways downstream of the TCR2. Sts-1 and -2 share 40% sequence identity and have a multidomain structure containing an N-terminal ubiquitin-association (UBA) domain and a Src-homology 3 (SH3) domain. Both of these domains are believed to be involved in protein-protein interactions3, 4. Current models of Sts function suggest that these two domains play critical roles in localizing Sts enzymatic functions to specific intracellular regions5. In addition, both Sts-1 and Sts-2 contain a C-terminal histidine phosphatase (HP) catalytic domain with homology to phosphoglycerate mutase (PGM), part of the histidine phosphatase superfamily6–8. The C-terminal HP domain of mouse Sts-1 has been shown to have an intrinsic phosphatase activity which contributes to the ability of Sts proteins to negatively regulate signaling pathways downstream of the TCR6.
The roles of Sts-1 and -2 were discovered from analysis of T cells isolated from mice lacking Sts-1 and -22. In particular, naive Sts-1/2−/− T cells exhibit a pronounced increase in TCR-induced proliferation compared with wild-type cells. The HP domain of Sts-1 (Sts-1HP) was shown to have robust tyrosine phosphatase activity that could dephosphorylate numerous proteins, including Zap-702, 6. Zap-70 is a tyrosine kinase that plays a critical role in propagating signals initiated by TCR engagement6, 9. Hyper-phosphorylated and ubiquitinated Zap-70 was identified and characterized in Sts-1/2−/− stimulated T cells10. Moreover, Sts-knockout mice displayed significantly enhanced survival after infection with the fungal pathogen C. albicans. In a mouse model which mimics disseminated candidiasis in humans, mice were found to be profoundly resistant to infection without hyperinflammatory response that would cause tissue damage11. In addition, the mice lacking the Sts proteins were associated with a significant reduction in fungal burden11. This result underscores the role of the Sts proteins in regulating host responses to fungal pathogens and their potential as host-modulatory drug targets for antifungal therapies.
Previous structural studies of Sts-1HP and Sts-2HP have been conducted for the murine proteins6, 12–16. These structures (97% and 81% sequence identity with human Sts-1HP and Sts-2HP, respectively) revealed that these proteins have a conserved phosphoglycerate mutase fold. A comparison of the MmSts-1HP to MmSts-2HP showed a high degree of structural similarity, yet some notable structural differences in the active site6, 12, 14. Functional studies, both in vitro and in vivo, showed that Sts-1 has a much higher phosphatase activity than that of Sts-2, both on non-native substrates and the putative protein substrate, Zap-706, 11, 14, 17. The mechanism of catalysis is believed to resemble that of other histidine phosphatases, proceeding through a covalent phosphor-histidine intermediate8, 18–20.
To advance our understanding of how the human Sts proteins function and as a basis for structure-guided drug discovery, we solved the X-ray crystal structures of human Sts-1HP, both unliganded and with a sulfate occupying the active site, and of human Sts-2HP with sulfate bound. Analogous to the murine Sts proteins, the structure of the human homologs have a high degree of similarity with some subtle, yet distinct structural differences. Comparison of the steady state kinetics shows that, as with the mouse proteins, Sts-1 is a much more efficient phosphatase. In addition, we demonstrate that the intact Sts-1 protein displays similar phosphatase kinetics as the isolated Sts-1HP domain. This indicates that Sts-1HP is an effective surrogate for the full length protein when measuring phosphatase activity. We also report several potent Sts inhibitors, including the phenylhydrazonopyrazolone sulfonate PHPS1, that is known to be a cell-permeable inhibitor of the phosphatase Shp221. Finally we demonstrate that human Sts-1 dephosphorylates the protein substrate Zap-70 in a cell-based assay. Taken together, these results confirm the druggability of Sts-1 and provide a framework for structure-based drug development. In addition, these findings confirm the structural and functional similarity of the mouse and human Sts proteins, suggesting that findings in the murine model should translate well to humans.
MATERIALS AND METHODS
Cloning, expression and purification of human Sts-1HP and Sts-2HP
A cDNA fragment encoding Sts-1HP (residues 369-636) was amplified by PCR, cloned as a His-tagged protein in the pTHT vector (a modified form of pET-28 with a Tobacco Etch Virus protease site in place of the thrombin site) and expressed in Escherichia coli BL21 (DE3) strain. Bacterial cultures were grown in terrific broth supplemented with 50 mg/L kanamycin for 6 hours at 310 K, and induced with 0.3 mM isopropylthiogalactopyranoside (IPTG) at 291 K overnight. The cells were harvested by centrifugation and lysed using sonication in lysis buffer (500 mM NaCl, 5 mM imidazole and 20 mM Tris-HCl, pH 8.0). The protein was then purified on a Ni-NTA column (Qiagen) equilibrated with lysis buffer. Non-specifically bound proteins were washed away with lysis buffer supplemented with 20 mM imidazole. The protein was eluted with elution buffer (lysis buffer with 300 mM imidazole). The 6xHis-tag was removed with tobacco etch virus (TEV) protease in TEV protease buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1 mM EDTA). The N-terminal sequence remaining after cleavage, and preceding residue 369 of Sts-1HP of the crystallized protein, was QGHMASMTGGQQMGRGS. The untagged protein was concentrated to 20-24 mg/mL, buffer exchanged into storage buffer (10 mM HEPES pH 7.5, 150 mM NaCl and 5 mM BME) and stored at -80 C. The Sts-2HP protein domain (residues 393-657) was expressed and purified in the same way as Sts-1HP. The N-terminal sequence remaining after cleavage, and preceding residue 393 of Sts-2HP, was QGHMASMTGGQQMGRGS. The untagged protein was concentrated to 16-18 mg/mL and stored in buffer containing 20 mM Tris, pH 8.0, 50 mM NaCl, 10% glycerol and 5 mM DTT. For full-length human Sts-1, we cloned the gene from cDNA into the pDB-NusA vector. The protein was expressed as a fusion with the NusA protein linked by a TEV cleavage site. The cells were lysed and the protein was initially purified as detailed above for the HP domains. After the initial purification step, the NusA solubility tag was removed by treatment with TEV protease and the liberated tag was separated from the untagged protein with a second round of IMAC using Ni-NTA. The protein was further purified by size exclusion chromatography using a HiLoad 26/600 Superdex 200 (GE Life Sciences) column in 50 mM MES buffer, pH 7.5 and 150 mM NaCl.
Crystallization and data collection. Unliganded human Sts-1HP
Initial crystallization screening of human Sts-1HP was conducted at 18 C, using the hanging-drop vapor diffusion method, based on the conditions under which the mouse Sts-1HP is known to crystallize16. Rod-shaped crystals were obtained overnight when 1 μL of Sts-1HP was mixed with 1 μL reservoir solution containing 0.1M HEPES pH 7.0, 5% ethylene glycol, 0.3 M magnesium chloride and 13% PEG 8000. The crystal size and quality was improved by seeding. Prior to be being flash frozen in liquid nitrogen, the crystals were soaked in a solution containing the crystallization buffer with a final PEG 8000 concentration of 30%. Human Sts-1HP with sulfate: Initial crystallization screening of human Sts-1HP was conducted at 18 C, using the hanging-drop vapor diffusion method, based on the conditions under which the mouse Sts-1HP is known to crystallize15. Lozenge-shaped crystals were obtained overnight when Sts-1HP was mixed with the reservoir solution in a ratio of 1:1.5. The reservoir solution was consisted of 0.1 M sodium acetate pH 5.5, 0.2 M ammonium sulfate, 23% PEG 2000 MME and 2 mM DTT. These crystals were cryoprotected by soaking in the crystallization solution with added PEG 2000 MME to 30%, before being flash frozen in liquid nitrogen. Human Sts-2HP: Sparse matrix screening was conducted for Sts-2HP using the hanging-drop vapor diffusion method at 291 K. Cubic-shaped crystals were obtained after two days in a condition with equal volumes of Sts-2HP and a solution consisting of 0.1 M HEPES pH, 7.0, 22% PEG 4000, 0.2 M potassium acetate and 0.2 M lithium sulfate. Crystals of similar morphology were also obtained using a reservoir solution consisting of 0.1M Tris pH 8.0, 30% PEG 4000, 0.2 M lithium sulfate and 15 mM strontium chloride. Crystals were first soaked in a solution containing the crystallization solution with a PEG 4000 final concentration of 30% before being flash frozen in liquid nitrogen. In all three cases, data was collected at 100 K at the Advanced Photon Source on beamlines 24 ID-C and 24 ID-E. Data collection statistics are provided in Table 2.
Table 2.
Data collection and refinement statistics
| Sts-1HP unliganded | Sts-1HP sulfate | Sts-2HP sulfate | |
|---|---|---|---|
|
| |||
| Data Collection | |||
| PDB ID | 5W5G | 5VR6 | 5WDI |
| Resolution range (Å)a | 50.0 − 2.48 (2.53 − 2.48) | 50.0 − 1.87 (1.90 − 1.87) | 50.0 − 2.43 (2.47 − 2.43) |
| Wavelength (Å) | 0.97910 | 0.97910 | 0.97910 |
| Space Group | C2 | P212121 | P21212 |
| Unit Cell Dimensions | |||
| a, b, c (Å) | 116.6, 74.5, 101.7 | 62.6, 79.2, 105.1 | 77.5, 113.1, 61.0 |
| α, β, γ | 90, 100.9, 90 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Measured reflections | 64,585 | 211,226 | 73,594 |
| Unique reflections | 28,546 (1,401) | 41,518 (1,693) | 20,513 (958) |
| Mean I/σ | 11.5 (1.7) | 14.9 (4.4) | 14.1 (1.8) |
| Completeness | 93.8 (93.9) | 94.4 (77.7) | 98.3 (94.2) |
| Redundancy | 2.3 (2.1) | 5.1 (4.1) | 3.6 (3.4) |
| Rmerge | 0.12 (0.49) | 0.08 (0.20) | 0.06 (0.47) |
|
| |||
| Data Refinement | |||
|
| |||
| Resolution Range (Å) | 50.0 − 2.48 | 50.0 − 1.87 | 50.0 − 2.43 |
| Total reflections | 27,105 | 39,428 | 19,484 |
| Test set | 5% (1,441) | 5% (2,033) | 5% (996) |
| Rwork | 19.4 | 19.4 | 20.6 |
| Rfree | 24.9 | 24.1 | 25.0 |
| No. of protein atoms | 5,816 | 4,163 | 4,040 |
| No. of ligand atoms | 0 | 80 | 25 |
| No. of water atoms | 98 | 336 | 122 |
| RMSD from ideal | |||
| Bonds (Å) | 0.005 | 0.006 | 0.005 |
| Angles (°) | 0.966 | 1.1 | 1.006 |
| Mean B factor (Å2) | 61.8 | 20.6 | 38.3 |
| Ramachandran | |||
| Favored (%) | 95.82 | 97.7 | 97.3 |
| Outliers (%) | 0.26 | 0.19 | 0 |
| Clashscoreb | 1.5 (100%) | 3.2 (98%) | 1.0 (100%) |
Numbers in parentheses correspond to values for the highest resolution shell.
Value calculated by MolProbity – value in parentheses corresponds to percentile (100% is best) when compared to a representative set of structures of comparable resolution39.
Structure determination and refinement
In all cases, diffraction data was indexed, integrated and scaled using HKL-200022. Structures were solved by molecular replacement using MolRep23 with the equivalent mouse protein structures (coordinate files 2H0Q and 3D4I) as search models12, 15, 16. The models were refined using iterative rounds of manual model building with Coot24, and restrained refinement with Refmac525. Water molecules were added using Coot only after the refinement had converged. Sulfates in and around the active site were directly inserted into the corresponding difference electron density, and the model was then subjected to an additional round of refinement. The refinement statistics for the models are provided in Table 2. Note that the unstructured N-termini of the proteins (upstream of R373 in Sts-1HP, and upstream of R395 in Sts-2HP) were not ordered in the crystal structures.
Phosphatase Assays
Three commonly used spectrophotometric or fluorescent phosphatase assays were used to quantify the phosphatase activities of human Sts-1HP and Sts-2HP26–28. These assays make use of three different substrates: para-nitrophenyl phosphate (pNPP), 3-O-Methylfluorescein phosphate (OMFP) or 6,8-Difluoro-4-Methylumbelliferyl Phosphate (DifMUP). For the pNPP substrate, the reaction buffer contained 20 mM Tris, pH 7.5, 150 mM sodium chloride, 5 mM magnesium chloride and 1 mM BME. The reaction was initiated by adding the protein to the pNPP in the reaction buffer. The rate of reaction was measured continuously at 298 K by quantifying the appearance of the nitrophenol product at 405nm on a SpectraMax Plus (Molecular Devices). For the OMFP substrate, a 10X assay buffer, consisting of 300 mM Tris, pH 7.4, 750 mM NaCl and 10 mM EDTA was diluted tenfold and DTT added to a final concentration of 1 mM. OMFP is added to the assay buffer from a freshly made 10 mM stock in DMSO. The production of OMF was measured continuously at 298 K by fluorescence (excitation 485nm and emission 525nm) on a SpectraMax Gemini (Molecular Devices). For the DifMUP substrate, a 10X assay buffer, consisting of 500 mM Bis-Tris, pH 7.5, 750 mM NaCl and 20mM EDTA, was diluted tenfold and fresh DTT was added to a final concentration of 1 mM. DifMUP is added from a freshly made 10 mM stock in DMSO. The rate of reaction was measured continuously at 298 K by quantifying the production of DifMU by fluorescence (excitation 360 nm, emission 450 nm) on a SpectraMax Gemini (Molecular Devices).
Steady state kinetics
Initial velocities (v0) were determined from slopes of linear segments of progress curves using time points well before the reaction had proceeded to 10% of completion. Initial velocities were determined at varying substrate concentrations and plotted against substrate concentration. The data was fit with a rectangular hyperbola using the Michaelis Menten equation and values for Km and kcat were determined (Kaleidagraph, Synergy Software).
Analysis of Inhibition of Sts-1HP and Sts-2HP
The phosphatase assays were used to analyze the inhibition of the two enzymes. To determine the mode of inhibition double reciprocal plots of 1/v0 vs 1/[substrate] were constructed using measurements of rate as described above. As the mode of inhibition was determined to be competitive, the data was fit using the following equation:
| (1) |
The value of the inhibition constant, Ki, was determined either from the fit of the data to equation 1, or by analyzing the steady state kinetics (as detailed above) in the presence of the inhibitor and using the following equation:
| (2) |
In all cases, the determination of inhibition constants was carried out in triplicate experiments at multiple inhibitor concentrations and the final amount of DMSO added during the assay was below 2% of the total volume. The error values provided are standard error from triplicate measurements.
Zap-70 dephosphorylation assay
HEK (human embryonic kidney)-293 cells were cultured in DMEM (Dulbecco’s modified Eagles medium, Gibco/Life Technologies) supplemented with 10% FBS (Fisher Hyclone) and 100 units/ml ea. penicillin and streptomycin. Zap-70 dephosphorylation was assessed as described6. Briefly, either human or murine Sts-1 expression constructs (Flag-tagged) were co-transfected into cells using Lipofectamine 2000 transfection reagent, along with plasmids encoding Lck (lymphocyte-specific protein tyrosine kinase), Zap-70 (T7-tagged), and a CD8-ζ chain chimera. Twenty four hours after transfection, cells were lysed in buffer containing 50 mM Tris/HCl (pH 7.6), 150 mM NaCl, 5 mM EDTA, and 1% NP-50, and clarified by centrifugation for 15 min at 16,000 g. Lysates were suspended in Laemmli sample buffer, and proteins were resolved by SDS-PAGE and transferred to nitrocellulose. Levels of phosphorylated Zap-70 were determined by immunoblotting with an antibody directed at Zap-70 pTyr-493 (Cell Signaling Technologies). Levels of Zap-70 and Sts-1 were determined by immunoblotting with antibodies to T7 (Novagen) and Flag (Sigma), respectively. The experiment was carried out in triplicate and the blot shown is a representative example of typical data collected.
RESULTS AND DISCUSSION
Activity of human Sts-1HP and Sts-2HP
Knockout of the Sts proteins in mice confers a profound resistance to fungal infection without deleterious immunopathology11. This highlights the clinical relevance of the Sts proteins as potential immuno-modulatory targets for the treatment of deadly pathogen infections. The enzymatic activity of the Sts proteins is carried out by a C-terminal histidine phosphatase domain with structural homology to the phosphoglycerate mutase family6. This phosphatase domain is believed to have tyrosine phosphatase activity and can act on protein substrates such as Zap-702, 6. To better understand the molecular details of the human proteins and to provide a basis for future drug discovery efforts, we set out to characterize the activity and structure of human Sts-1HP and Sts-2HP. The histidine phosphatase domains of the human Sts proteins (Sts-1HP, and Sts-2HP) both expressed and purified well using conventional methods. To determine the activity of these proteins, we used three established phosphatase assays that employed the substrates para-nitrophenyl phosphate (pNPP), 3-O-Methylfluorescein phosphate (OMFP) or 6,8-Difluoro-4-Methylumbelliferyl Phosphate (DifMUP)26, 28. The steady-state kinetic parameters of Sts-1HP and Sts-2HP for the three phosphatase substrates are given in Table 1. As expected, the phosphatase activity of Sts-2HP is considerably slower than that of Sts-1HP. In addition to the characterization of the phosphatase domain, we determined the kinetics of the full-length Sts-1 protein (Table 1). This data illustrates two key points about the human Sts proteins. The kinetics of the human Sts proteins follow the same trends and are quite similar to what has been observed for the mouse proteins6, 12, and the kinetics of the isolated Sts-1 HP domain are reasonably similar to the native, full-length protein. These findings are important for long term drug discovery efforts aimed at targeting Sts-1. They highlight the utility of the mouse model to make physiologically relevant observations that are consistent with the activity of the human protein. Furthermore, the data illustrates that the isolated HP domain, which is considerably easier to produce and work with, is a viable surrogate for measuring the phosphatase activity of the full-length protein.
Table 1.
Kinetic constants of human Sts proteins
| Substrate/Enzyme | Km (mM) | kcat (s−1) | kcat / Km (M−1s−1) |
|---|---|---|---|
| pNPP | |||
| human Sts-1HP | 2.64 ± 0.48 | 13.62 ± 0.63 | 5.16 × 103 |
| human Sts-2HP | 5.61 ± 1.69 | 1.23 ± 0.15 | 2.19 × 102 |
| Full-length Sts-1 | 3.31 ± 0.27 | 158.6 ± 6.48 | 4.77 × 104 |
|
| |||
| OMFP | |||
|
| |||
| human Sts-1HP | 0.19 ± 0.03 | 786.1 ± 52.4 | 4.14 × 106 |
| human Sts-2HP | 1.07 ± 0.24 | 5.86 ± 0.78 | 5.48 × 103 |
| Full-length Sts-1 | ND | ND | ND |
|
| |||
| DifMUP | |||
|
| |||
| human Sts-1HP | 0.002 | 100.68 | 5.03 × 107 |
| human Sts-2HP | 0.85 ± 0.23 | 4.92 ± 0.76 | 5.79 × 103 |
| Full-length Sts-1 | ND | ND | ND |
Structures of human Sts-1HP
To characterize the phosphatase active site of Sts-1 for future structure-guided drug discovery efforts, we solved the X-ray crystal structure of human Sts-1HP, both unliganded (to 2.5 Å resolution), and with the phosphate mimic, sulfate (to 1.9 Å resolution), bound in the active site. Data collection and refinement statistics are provided in Table 2. The overall structure of human Sts-1HP protomer is shown in Figure 1A. The protein has the conserved phosphoglycerate mutase (PGM) fold and is a dimer in solution (Fig. 1B). Dimerization is mediated by multiple interactions between the intertwined C-terminal dimerization domains of the protomers. This observation is consistent with reports that the physiologically relevant state of Sts-1 is a dimer and the dimerization occurs through the HP domain of the protein29.
Figure 1.
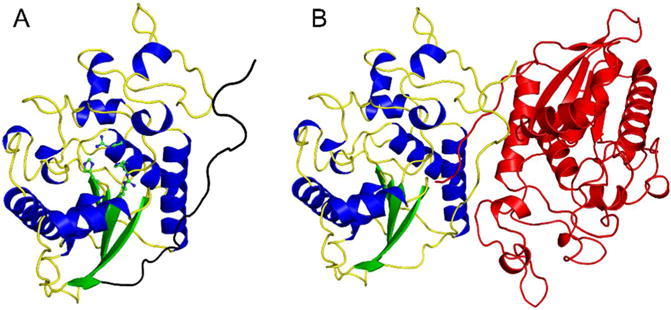
Overall structure of human Sts-1HP. The overall fold of the Sts-1HP protomer is shown (A) with the conserved active site histidine residues (H380 and H565) and arginine residues (R379 and R462) shown in ball and stick representation (carbon atoms colored green and nitrogen atoms colored blue). The dimerization domain is at the C-terminus of the protein and is colored black in this figure. The physiologically relevant dimeric form of Sts-1HP is given in (B) with one chain colored by secondary structure as in (A) and the other colored red.
The active site of Sts-1HP (Figs. 2A and B) is distinguished by the two catalytic histidine residues (H380 and H565) and two highly conserved arginine residues (R379 and R462). These residues, as well as a glutamate (Q490) and an additional arginine (R383), act to optimally position the phosphate group of the ligand for efficient catalysis (Fig. 2B). The phosphatase activity of Sts-1, like that of other PGM-family histidine phosphatases, is proposed to proceed through a two-step mechanism that involves nucleophilic attack by one of the conserved histidine residues6, 8, 18–20, 30. The active site glutamate is the likely proton donor during this attack. The second step of the reaction is a hydrolysis of the phosphor-histidine intermediate. The incoming water molecule is believed to be activated for attack by the same glutamate residue. The orientation and bond distances of the residues in the active site of sulfate-bound Sts-1HP are consistent with the proposed mechanism (Fig. 2B).
Figure 2.
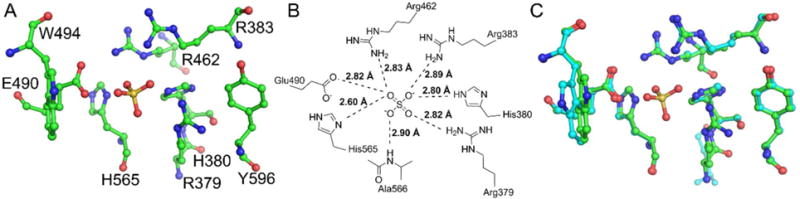
Active site of human Sts-1HP. A ball and stick diagram (A, carbon atoms colored green, nitrogen atoms colored blue, oxygen atoms colored red, and the sulfur atom is colored yellow) shows the active site residues of Sts-1HP around the phosphate binding pocket (marked by the sulfate in this structure). In addition to the conserved histidine residues and arginine residues, additional hydrogen bonding contacts are made to the sulfate by an additional arginine residue and a glutamate residue (B). The superposition of the human and murine Sts-1HP (C, overall RMSD = 0.35 Å) illustrates that the architecture of the active site is highly structurally conserved. The only observed difference is a slight change in orientation of the tryptophan (W494 in the human protein) at the edge of the active site.
As mentioned in the previous section, the degree of similarity of the mouse and human Sts-1 is an important consideration for future drug discovery efforts. In particular, a high degree of similarity suggests that results from mouse model studies are more likely to be directly translatable to humans. Comparison of the mouse and human Sts-1HP structures (Fig. 2C) shows a very high degree of structural conservation in the active site (overall RMSD = 0.35 Å). The only modest difference observed is in the orientation of a tryptophan residue (W494) near the entrance to the active site. In both the human and mouse Sts-1HP structures, however, this tryptophan has the highest B-factor of any of the active site residues. This suggests that this residue has a higher degree of conformational flexibility that the otherwise immobile active site residues, which may account for the subtle differences observed between the two structures.
Structure of human Sts-2HP
The two Sts proteins from human, Sts-1 and Sts-2, share approximately 45% overall sequence identity and 50% sequence identity in the HP domain. Despite the high degree of sequence identity, the catalytic efficiency of Sts-2HP is much lower than that of Sts-1HP for all substrates tested (Table 1). To better understand the basis for these kinetic differences we solved the X-ray crystal structure of human Sts-2HP in the presence of sulfate (data collection and refinement statistics are provided in Table 2). As expected, the overall fold of Sts-2HP is very similar to that of Sts-1HP (RMSD of 1.02 Å) with only slight differences in flexible loops and the dimerization domain observed (Fig. 3A). The active site is also highly structurally conserved, but has some subtle, yet distinct differences (Fig. 3B). While the positions of the conserved histidine and arginine residues are nearly identical in the two structures, a valine-tyrosine pair (V386 and Y596) are replaced with a glutamine-serine (Q409 and S619) in the Sts-2HP structure. This difference likely accounts for the lower rate of phosphatase activity of Sts-2. Mutation of these residues in the mouse Sts-2HP (Q372V and S582Y) increases the kcat/Km by a factor of 38 for the pNPP substrate12. The phosphatase substrates contain large aromatic groups, particularly the fluorescein and umbelliferone of the OMFP and DifMUP, respectively that could make important pi stacking interactions with the tyrosine ring of Sts-1. This likely is indicative of the differences between the native Sts-1 and Sts-2 substrates. The Sts proteins have been implicated in negatively regulating TCR signaling via the dephosphorylation of key signaling proteins downstream of the TCR2, 6. One of the putative substrates of Sts-1 is the tyrosine kinase, Zap-70, while much less is known about the possible substrates of Sts-22, 6, 17, 30. Sts-2 also shows activity against Zap-70, albeit at a much lower level than Sts-117. This, and other data, suggests that Sts-1 and Sts-2 have distinct regulatory roles. This is supported by the observations that knock-out of both Sts-1 and Sts-2 induces a greater degree of hyper-activation of signaling pathways that the loss of either protein alone2, 6, 11. The lower phosphatase activity of Sts-2 for Zap-70 and the substrates tested here does raise the possibility that the primary function of Sts-2 arises from one of the other domains of the protein and that the phosphatase activity is evolutionarily redundant. Alternative roles for Sts-2, driven by the UBA and SH3 domain interactions, have been proposed. These include the inhibition of tyrosine kinase receptor endocytosis and the promotion of caspase-independent apoptosis, proposed to be mediated by UBA or SH3 interactions with the E3 ubiquitin ligase/adaptor protein, Cbl, and with apoptosis-inducing factor, respectively29, 31. Further study is required in order to identify the physiologically relevant phosphatase substrates or binding partners of Sts-2.
Figure 3.
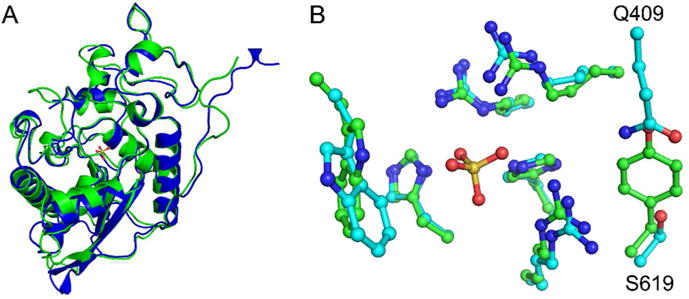
Structure of human Sts-2HP. The overall fold of human Sts-2HP is nearly identical to that of Sts-1HP. A superposition of the Sts-2HP protomer with the equivalent Sts-1HP protomer (A, Sts-1 is green and Sts-2 is blue, RMSD = 1.02 Å) shows only subtle differences in peripheral loop regions. Similarly, the active sites of the two proteins align well (B, Sts-1 has green carbon atoms, Sts-2 has cyan carbon atoms; in both structures the nitrogen atoms are blue, oxygen atoms are red and the sulfur atom is yellow) but do show some subtle, yet distinct differences. Whereas the conserved histidine and arginine residues line up well, a tyrosine and valine residue in Sts-1 (Y596 and V386) are replaced with a glutamine-serine (Q409 and S619) pair in Sts-2. It is these differences that likely account for the substrate selectivity of the Sts proteins.
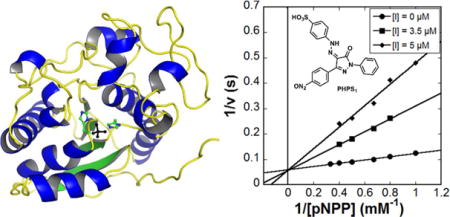
Inhibitors of Sts-1HP
Beyond the observation that phosphate and several phosphate mimics can inhibit the histidine phosphatase activity of Sts-1 and Sts-212, 13, there have been no reports of small molecule inhibitors of the Sts proteins. To test the feasibility of inhibiting Sts-1 with a druglike compound and to compare the inhibition profile with other phosphatases, we tested a number of known phosphatase inhibitors (Table 3, Fig. 4A). Of those tested, the alkaline phosphatase inhibitor tetramisole32, 33, the Ser/Thr phosphatase inhibitor glycerophosphate, and the PTP-1B/SHPTP-1 inhibitor 3,4-methyl-dephostatin34–36 did not show any observable level of inhibition of Sts-1HP phosphatase activity. The Src Homology-2 containing phosphatase (SHP2) inhibitor, PHPS1 (4-[2-[1,5-dihydro-3-(4-nitrophenyl)-5-oxo-1-phenyl-4H-pyrazol-4-ylidene]hydrazinyl]-benzenesulfonic acid)21, however, is a potent competitive inhibitor of Sts-1HP with a Ki of 1 μM (Fig. 4B, Table 3). The compound 1-(4-sulfophenyl)-3-methyl-5-pyrazolone (SMP), which is a precursor of PHPS1, also inhibits Sts-1HP (Ki of 48 μM). In addition, the phosphotyrosine analogue, sulfanilic acid, was found to be a modest inhibitor. From this data we can draw several important conclusions. The phosphatase activity of Sts-1 can clearly be modulated by small molecule inhibitors and, as expected, the inhibition profile is distinct from canonical protein tyrosine phosphates (PTPs). For example, PTP-1B is more potently inhibited by methyl-3,4-dephostatin (3.5 μM IC50) than by PHPS1 (19 μM IC50) whereas Sts-1 activity is not affected by methyl-3,4-dephostatin21, 36, 37. This data, taken together with the structure that reveals a distinct binding pocket in the active site (Fig. 4C), suggest that Sts-1 is likely a druggable target. Further in vivo studies will need to be conducted, however, to validate this conclusion.
Table 3.
Inhibition constants
| Inhibitor | Sts-1HP Ki (μM) |
|---|---|
| Orthovanadate | 4.61 ± 0.62 |
| β-glycerophosphate | NI1 |
| (−) Tetramisole | NI |
| Methyl-dephostatin | NI |
| 1-Naphthyl phosphate | NI |
| 2-Bromo-4-hydroxyacetophenone | NI |
| PHPS12 | 1.05 ± 0.15 |
| Sulfanilic acid | 480 ± 55 |
| Sulfanilamide | NI |
| SMP2 | 48.3 ± 5.61 |
NI = No observable inhibition
PHPS1: 4-[2-[1,5-dihydro-3-(4-nitrophenyl)-5-oxo-1-phenyl-4H-pyrazol-4-ylidene]hydrazinyl]-benzenesulfonic acid
SMP: 1-(4-sulfophenyl)-3-methyl-5-pyrazolone
Figure 4.
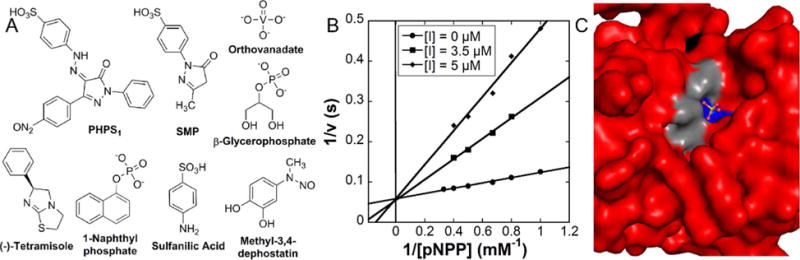
Inhibition of Sts-1HP. A set of known phosphatase inhibitors (A) were screened for their capacity to inhibit the phosphatase activity of Sts-1HP. The SHP-1 inhibitor, PHPS1, was shown to be a potent competitive inhibitor of Sts-1HP (B, Ki of 1.05 ± 0.15 μM). Shown is a Lineweaver-Burk plot of the activity of Sts-1HP (as measured by pNPP assay) with three different concentrations of the inhibitor, PHPS1. Examination of the structure of Sts-1HP shows that there is a clear binding cleft where the substrates and inhibitors bind (C, Sts-1HP shown in surface representation colored red, the conserved histidine residues are colored blue while the conserved arginine residues are colored grey).
Zap-70 Dephosphorylation by Human Sts-1
The Sts proteins have been implicated in the control of T cell activity as negative regulators of TCR signaling2, 6. Previous work on the mouse proteins demonstrated that Zap-70 is one of the protein substrates of the Sts-1 and Sts-26, 17. Our studies, detailed herein, have revealed a significant degree of structural and functional similarity between the mouse and human proteins. To confirm that human Sts-1 also targets Zap-70 as a phosphatase sub strate, we conducted an in cellulo assay measuring phosphorylation of Zap-70 in the presence and absence of Sts-1 (Fig. 5). HEK-293 cells were co-transfected with T7-tagged Zap-70, the cellular machinery to ensure Zap-70 phosphorylation (Lck and CD8-ζ chain chimera, see Materials and Methods), and either murine or human Sts-1. In the absence of any Sts-1 protein, hyper-phosphorylation of Zap-70 (Fig. 5, phosphor-Zap-70) is observed as expected. When either the mouse or human Sts-1 protein is co-expressed, the amount of phosphorylated Zap-70 is reduced to nearly nil. This data verifies that, not only are the purified proteins structurally and functionally conserved, but the human and mouse proteins behave similarly in cells. This further validates the mouse model as a viable surrogate for studies of human Sts-1.
Figure 5.

Human Sts-1HP dephosphorylates Zap-70. Using a cell-based assay (see Materials and Methods) both mouse and human Sts-1HP were tested for their ability to dephosphorylate the hyperphosphorylated substrate, Zap-70. In the absence of either Sts-1 protein, a dark band is seen for phosphorylated Zap-70 (phospho-Zap-70, top lane of the gel). When either mouse Sts-1 (m) or human Sts-1 (h) are also present in the cells, the amount of phosphorylated Zap-70 is significantly diminished without an effect on the overall expression level of the Zap-70 protein. This experiment was conducted in triplicate and the blot shown is a representative example of these data.
In summary, this work details the first structural and functional characterization of the human Sts proteins. These proteins have recently been shown to have potential as targets for immune stimulation to treat deadly pathogen infections. The studies described illustrate that the phosphatase domain of human Sts-1 has a distinct active site pocket that can be potently inhibited by a competitive inhibitor with drug-like properties21, 38. This not only serves as a proof of principle that Sts-1 can be functionally inactivated by a small molecule drug, but may provide a scaffold from which to design more potent and selective Sts inhibitors. In addition, the structural and functional similarity of the mouse and human proteins underscores the utility of the mouse model for the discovery and characterization of inhibitors of the human protein. Comparison of the human Sts-1HP and Sts-2HP structures reveal a high degree of conservation but do show some subtle distinctions. These differences are likely the key determinants of substrate specificity and may be critical features for drug discovery if selective inhibition of one Sts protein is determined to be a requirement for biological efficacy. Overall, this data suggests that the Sts proteins are druggable, they possess a well-defined binding pocket and can bind to a known drug-like molecule with high affinity. These distinct molecular features can be exploited for the development of specific and effective inhibitors of phosphatase activity. This work provides a foundation for drug discovery efforts and important tools for the structure-guided development of inhibitors of the Sts proteins.
Acknowledgments
This work was supported by Stony Brook University, the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number U01HL127522 (JF, NC), and the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH17-1-0147 (JF, NC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense. Additional support was provided by the Center for Biotechnology, a New York State Center for Advanced Technology, Stony Brook University, Cold Spring Harbor Laboratory, Brookhaven National Laboratory and Feinstein Institute for Medical Research. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
The coordinates of structures reported have been deposited in the Protein Data Bank as entries 5VR6, 5W5G, and 5WDI, for the Sts-1 sulfate bound, Sts-1 unliganded and Sts-2 sulfate bound structures, respectively.
References
- 1.Singer AL, Koretzky GA. Control of T Cell Function by Positive and Negative Regulators. Science. 2002;296:1639–1640. doi: 10.1126/science.1071551. [DOI] [PubMed] [Google Scholar]
- 2.Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. Regulation of ZAP-70 Activation and TCR Signaling by Two Related Proteins, Sts-1 and Sts-2. Immunity. 2004;20:37–46. doi: 10.1016/s1074-7613(03)00351-0. [DOI] [PubMed] [Google Scholar]
- 3.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 4.Musacchio A. How SH3 domains recognize proline. Advances in protein chemistry. 2002;61:211–268. doi: 10.1016/s0065-3233(02)61006-x. [DOI] [PubMed] [Google Scholar]
- 5.Tsygankov AY. TULA-family proteins: a new class of cellular regulators. J Cell Physiol. 2013;228:43–49. doi: 10.1002/jcp.24128. [DOI] [PubMed] [Google Scholar]
- 6.Mikhailik A, Ford B, Keller J, Chen Y, Nassar N, Carpino N. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell. 2007;27:486–497. doi: 10.1016/j.molcel.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpino N, Kobayashi R, Zang H, Takahashi Y, Jou ST, Feng J, Nakajima H, Ihle JN. Identification, cDNA cloning, and targeted deletion of p70, a novel, ubiquitously expressed SH3 domain-containing protein. Molecular and cellular biology. 2002;22:7491–7500. doi: 10.1128/MCB.22.21.7491-7500.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rigden DJ. The histidine phosphatase superfamily: structure and function. Biochem J. 2008;409:333–348. doi: 10.1042/BJ20071097. [DOI] [PubMed] [Google Scholar]
- 9.Chu DH, Morita CT, Weiss A. The Syk family of protein tyrosine kinases in T-cell activation and development. Immunological reviews. 1998;165:167–180. doi: 10.1111/j.1600-065x.1998.tb01238.x. [DOI] [PubMed] [Google Scholar]
- 10.Carpino N, Chen Y, Nassar N, Oh HW. The Sts proteins target tyrosine phosphorylated, ubiquitinated proteins within TCR signaling pathways. Mol Immunol. 2009;46:3224–3231. doi: 10.1016/j.molimm.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naseem S, Frank D, Konopka JB, Carpino N. Protection from systemic Candida albicans infection by inactivation of the Sts phosphatases. Infect Immun. 2015;83:637–645. doi: 10.1128/IAI.02789-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Jakoncic J, Carpino N, Nassar N. Structural and functional characterization of the 2H-phosphatase domain of Sts-2 reveals an acid-dependent phosphatase activity. Biochemistry. 2009;48:1681–1690. doi: 10.1021/bi802219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Jakoncic J, Parker KA, Carpino N, Nassar N. Structures of the phosphorylated and VO(3)-bound 2H-phosphatase domain of Sts-2. Biochemistry. 2009;48:8129–8135. doi: 10.1021/bi9008648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Jakoncic J, Wang J, Zheng X, Carpino N, Nassar N. Structural and functional characterization of the c-terminal domain of the ecdysteroid phosphate phosphatase from bombyx mori reveals a new enzymatic activity. Biochemistry. 2008;47:12135–12145. doi: 10.1021/bi801318w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakoncic J, Sondgeroth B, Carpino N, Nassar N. The 1.35 A resolution structure of the phosphatase domain of the suppressor of T-cell receptor signaling protein in complex with sulfate. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:643–647. doi: 10.1107/S1744309110014259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinman H, Ford B, Keller J, Carpino N, Nassar N. Crystallization and initial crystal characterization of the C-terminal phosphoglycerate mutase homology domain of Sts-1. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:218–220. doi: 10.1107/S1744309106003320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.San Luis B, Sondgeroth B, Nassar N, Carpino N. Sts-2 is a phosphatase that negatively regulates zeta-associated protein (ZAP)-70 and T cell receptor signaling pathways. J Biol Chem. 2011;286:15943–15954. doi: 10.1074/jbc.M110.177634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bond CS, White MF, Hunter WN. High resolution structure of the phosphohistidine-activated form of Escherichia coli cofactor-dependent phosphoglycerate mutase. J Biol Chem. 2001;276:3247–3253. doi: 10.1074/jbc.M007318200. [DOI] [PubMed] [Google Scholar]
- 19.Bond CS, White MF, Hunter WN. Mechanistic implications for Escherichia coli cofactor-dependent phosphoglycerate mutase based on the high-resolution crystal structure of a vanadate complex. J Mol Biol. 2002;316:1071–1081. doi: 10.1006/jmbi.2002.5418. [DOI] [PubMed] [Google Scholar]
- 20.Zheng Q, Jiang D, Zhang W, Zhang Q, Zhao Q, Jin J, Li X, Yang H, Bartlam M, Shaw N, Zhou W, Rao Z. Mechanism of dephosphorylation of glucosyl-3-phosphoglycerate by a histidine phosphatase. J Biol Chem. 2014;289:21242–21251. doi: 10.1074/jbc.M114.569913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A. 2008;105:7275–7280. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Vagin A, Teplyakov A. Molecular replacement with MOLREP, Acta crystallographica. Section D, Biological crystallography. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics, Acta crystallographica. Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method, Acta crystallographica. Section D, Biological crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Sergienko EA. Phosphatase high-throughput screening assay design and selection. Methods Mol Biol. 2013;1053:7–25. doi: 10.1007/978-1-62703-562-0_2. [DOI] [PubMed] [Google Scholar]
- 27.Tautz L, Sergienko EA. High-throughput screening for protein tyrosine phosphatase activity modulators. Methods Mol Biol. 2013;1053:223–240. doi: 10.1007/978-1-62703-562-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tierno MB, Johnston PA, Foster C, Skoko JJ, Shinde SN, Shun TY, Lazo JS. Development and optimization of high-throughput in vitro protein phosphatase screening assays. Nat Protoc. 2007;2:1134–1144. doi: 10.1038/nprot.2007.155. [DOI] [PubMed] [Google Scholar]
- 29.Kowanetz K, Crosetto N, Haglund K, Schmidt MH, Heldin CH, Dikic I. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J Biol Chem. 2004;279:32786–32795. doi: 10.1074/jbc.M403759200. [DOI] [PubMed] [Google Scholar]
- 30.San Luis B, Nassar N, Carpino N. New insights into the catalytic mechanism of histidine phosphatases revealed by a functionally essential arginine residue within the active site of the Sts phosphatases. Biochem J. 2013;453:27–35. doi: 10.1042/BJ20121769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collingwood TS, Smirnova EV, Bogush M, Carpino N, Annan RS, Tsygankov AY. T-cell ubiquitin ligand affects cell death through a functional interaction with apoptosis-inducing factor, a key factor of caspase-independent apoptosis. J Biol Chem. 2007;282:30920–30928. doi: 10.1074/jbc.M706870200. [DOI] [PubMed] [Google Scholar]
- 32.Fedde KN, Lane CC, Whyte MP. Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch Biochem Biophys. 1988;264:400–409. doi: 10.1016/0003-9861(88)90305-0. [DOI] [PubMed] [Google Scholar]
- 33.Hsu HH, Camacho NP, Anderson HC. Further characterization of ATP-initiated calcification by matrix vesicles isolated from rachitic rat cartilage. Membrane perturbation by detergents and deposition of calcium pyrophosphate by rachitic matrix vesicles. Biochim Biophys Acta. 1999;1416:320–332. doi: 10.1016/s0005-2736(98)00235-1. [DOI] [PubMed] [Google Scholar]
- 34.Fujiwara S, Watanabe T, Nagatsu T, Gohda J, Imoto M, Umezawa K. Enhancement or induction of neurite formation by a protein tyrosine phosphatase inhibitor, 3–4-dephostatin, in growth factor-treated PC12h cells. Biochem Biophys Res Commun. 1997;238:213–217. doi: 10.1006/bbrc.1997.7174. [DOI] [PubMed] [Google Scholar]
- 35.Umezawa K. Induction of cellular differentiation and apoptosis by signal transduction inhibitors. Adv Enzyme Regul. 1997;37:393–401. doi: 10.1016/s0065-2571(96)00022-2. [DOI] [PubMed] [Google Scholar]
- 36.Umezawa K, Kawakami M, Watanabe T. Molecular design and biological activities of protein-tyrosine phosphatase inhibitors. Pharmacol Ther. 2003;99:15–24. doi: 10.1016/s0163-7258(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 37.Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med Chem. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 38.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 39.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]