

Abstract
Midazolam, a benzodiazepine derivative, is widely used for sedation and surgery. However, previous studies have demonstrated that Midazolam is associated with increased risks of congenital malformations, such as dwarfism, when used during early pregnancy. Recent studies have also demonstrated that Midazolam suppresses osteogenesis of mesenchymal stem cells (MSCs). Given that hypertrophic chondrocytes can differentiate into osteoblast and osteocytes and contribute to endochondral bone formation, the effect of Midazolam on chondrogenesis remains unclear. In this study, we applied a human MSC line, the KP cell, to serve as an in vitro model to study the effect of Midazolam on chondrogenesis. We first successfully established an in vitro chondrogenic model in a micromass culture or a 2D high‐density culture performed with TGF‐β‐driven chondrogenic induction medium. Treatment of the Midazolam dose‐dependently inhibited chondrogenesis, examined using Alcian blue‐stained glycosaminoglycans and the expression of chondrogenic markers, such as SOX9 and type II collagen. Inhibition of Midazolam by peripheral benzodiazepine receptor (PBR) antagonist PK11195 or small interfering RNA rescued the inhibitory effects of Midazolam on chondrogenesis. In addition, Midazolam suppressed transforming growth factor‐β‐induced Smad3 phosphorylation, and this inhibitory effect could be rescued using PBR antagonist PK11195. This study provides a possible explanation for Midazolam‐induced congenital malformations of the musculoskeletal system through PBR.
Keywords: chondrogenesis, mesenchymal stem cells, Midazolam, peripheral benzodiazepine receptor
1. INTRODUCTION
Benzodiazepines are widely used sedative drugs for applying regional anaesthesia.1 To obtain more specific functions to treat patients, numerous types of benzodiazepine derivatives have been synthesized. Among them, Midazolam (Dormicum®) is a short‐acting benzodiazepine derivative that has been extensively used in surgical procedures, premedication and induction for general anaesthesia. Compared with other types of anaesthetics, pharmacokinetics and the drug mechanism of benzodiazepine are well understood.2, 3 In addition to sedation, benzodiazepines possess anticonvulsant, anxiolytic amnestic, hypnosis and centrally mediated muscle relaxation properties.4, 5 They produce a sedation effect primarily through modulation of gamma‐aminobutyric acid (GABA) receptors in the central nervous system6, 7 to block nerve impulses. GABA binds with GABAA, GABAB and GABAC receptors. Among them, the GABAA receptor is of particular significance for psychopharmacology because it contains a variety of binding sites at which behaviour‐modifying drugs act to produce some or all of their effects.8 Benzodiazepines bind to benzodiazepine sites on GABA receptors and allosterically modulate the response of the channel upon GABA binding.9 The function of benzodiazepines acting on GABAA receptors is to increase the amplitude or decay time of GABA‐mediated inhibitory post‐synaptic potentials, thus increasing the inhibitory tone of GABAergic synapses to reduce the firing of neuron populations.10
In addition to the central nervous system, benzodiazepines act on peripheral tissues performed with peripheral benzodiazepine receptor (PBR) or mitochondria translocator proteins.11, 12 A PBR is distinct in its pharmacological, anatomical, structural and physiological aspects when compared with central benzodiazepine receptors.12, 13 PBR is an 18 kD protein, localize to the mammalian mitochondria membrane, and are highly conserved among various mammalian species in various types of peripheral tissue.11 It has been shown that PBR distributes ubiquitously in most types of tissues, including bone marrow stromal cells.14 Putative PBR functions are involved in numerous types of physiological processes, such as steroidogenesis, apoptosis, cell proliferation, regulation of mitochondrial membrane potential, mitochondria respiratory chains, voltage‐dependent calcium channels and microglial activation.11, 12, 15, 16, 17 PK11195 is a competitive antagonist that specifically binds to PBR18 and has been shown to reverse the inhibitory effect of Midazolam on the anxiolytic and antidepressant effects of Midazolam.19
Although numerous papers have proved the safety of benzodiazepine and its derivatives, there remain in vitro and in vivo studies indicating the toxic effects that can occur after benzodiazepine treatment.20, 21 For example, previous study has demonstrated that the incident rate of congenital malformations was 0.23% if the maternal plasma was diagnosed as benzodiazepine positive.21 Furthermore, Deck et al summarized the database of all infants born with major congenital malformations to mothers on antiepileptic drugs from Boston Medical Center from the years 2003 to 2010, including cleft lips/palate, cardiac defects and urogenital defects. Results showed that for women on benzodiazepine monotherapy during pregnancy, major congenital malformations were high and extended to 10.6%. Among their infants, the rate for cleft lip/palate was approximately 0.747%.22 Recent in vitro and in vivo animal studies have suggested that exposure to clinically relevant general anaesthetics at the peak of brain development could be detrimental to immature mammalian neurons as demonstrated by massive and widespread apoptotic neurodegeneration.23 In addition, previous studies exposing young mice to benzodiazepine derivatives, either treated alone or together with other chemicals, have resulted in high birth rates of runts, increased retarded ossification of skull bones and a variety of sternal defects.24, 25 More importantly, Midazolam has been found to inhibit osteogenesis of human bone marrow‐derived mesenchymal stem cells (hMSCs).26 However, the detailed mechanisms for benzodiazepine‐regulated bone/cartilage differentiation that associated with foetal growth retardation remain unknown.
As mentioned above, benzodiazepine and its derivatives are associated with birth defects and cleft lip/palates27 and have been linked to osteotoxicity.26 Because chondrocytes are the precursors of osteogenesis during musculoskeletal development, we sought to investigate in this study the Midazolam‐regulated chondrogenesis of human MSCs and the molecular mechanisms underlying this process.
2. MATERIALS AND METHODS
2.1. Experimental system
KP cells, an immortalized human MSC line, were established from primary human MSCs, which were transduced using a retroviral vector LXSN‐16E6E7.28, 29 KP cells display similar types of cell surface marker profiles as parental primary human MSCs do and retain the stem‐like properties. KP cells can not only renew themselves, but also differentiate into mesenchymal and nonmesenchymal cell lineages without neoplastic transformation under certain conditions.28 This type of cell line has been used in studies of stem cell biology and regenerative medicine.30 The cell line used in this study was kindly provided by Dr. Shih‐Chieh Hung (China Medical University). We also purchased primary human bone marrow‐derived MSCs from Lonza Walksville Inc., which were maintained in growth medium: DMEM low glucose containing 10% foetal bovine serum (GIBCO with selected lot), 100 units/mL penicillin and 100 μg/mL streptomycin (both from GIBCO) at 37°C with a humidified 5% CO2 atmosphere and medium was changed twice a week. Only early passages (passages 3‐6) of primary hMSCs were used in this experiment. To induce chondrogenic differentiation, KP cells or hMSCs were treated with chondrogenic induction medium (DMEM low glucose, 0.1 μmol/L Dexamethasone, 50 μmol/L L‐ascorbic acid‐2‐phosphate, 1× Insulin‐Transferrin‐Selenium, 40 μg/mL L‐proline, 100× L‐glutamine, 5 ng/mL transforming growth factor‐β3).31, 32 For the pelleted culture, 3 × 105 cells per pellet were placed in a 15‐ml centrifuge tube and centrifuged to produce a pellet. The medium was changed every 3 days. For high‐density cultures, cells were seeded at 3 × 104 cells/cm2 overnight and the medium was replaced with chondrogenic induction medium the following day.31 The chondrogenic differentiation was accessed after 7 or 14 days of treatment.31, 32
2.2. Alcian blue staining and quantification
Cells in the pellet culture or at high densities were rinsed with PBS and fixed with 4% paraformaldehyde for 10 minutes. After fixation, cells were rinsed twice with PBS and then stained with 1% Alcian blue for 30 minutes and rinsed in PBS until the blue colour was removed from the negative control cells. The amount of Alcian blue‐stained glycosaminoglycan (GAG) was quantified as per Woods et al, with modification, by dissolving the stained GAG in 6 mol/L guanidine hydrochloride overnight at 4°C. The absorbance of dissolved stained GAG was then quantified using a spectrophotometer at OD 620 nm.33
2.3. Western blot analysis
After treatment, cells were rinsed twice using chilled PBS and then lysed and collected using a radioimmunoprecipitation assay buffer (Thermo) plus protease and phosphatase inhibitor cocktails (Thermo). After sonication, cell lysate was centrifuged at 15 000 × g for 30 minutes at 4°C and supernatant was collected in an eppendorf tube and stored at −80°C. Protein concentration was assessed using a bicinchoninic acid protein assay (Bio‐Rad) as per the manufacturer's instructions. Fifteen to thirty μg of protein was resolved using SDS‐PAGE, followed by electro‐transferred onto a methanol‐soaked polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was blocked with 5% milk or 2% bovine serum albumin (for phosphorylated protein) in Tris‐Buffered Saline‐Tween 20 (TBST) and then incubated with primary antibody overnight at 4°C. The membrane was then rinsed with TBST and the immunocomplex on membrane was detected using horse‐radish peroxidase‐conjugated secondary antibody, and the final immunocomplexes were visualized using fluorography with an enhanced chemiluminescence reagent (GE Healthcare Life Sciences).
2.4. Immunostaining and immunofluorescence
Cells in micropellet cultures were fixed with 4% paraformaldehyde, washed in PBS and then sectioned and frozen (LEICA CM 1950). The 10‐μm‐thick sections were rinsed twice in PBS, followed by soaking in SuperBlock blocking buffer (Thermo) for 1 hour at room temperature, and then incubated with primary antibody overnight at 4°C. The sections were then rinsed twice with PBS and stained with secondary antibody conjugated with Alexa 594 (Thermo). Images were taken using an Olympus epifluorescence microscope. For high‐density cultures, cells were rinsed twice with PBS and then fixed with 4% paraformaldehyde, soaked in SuperBlock blocking buffer (Thermo) for 1 hour at room temperature and then incubated with primary antibodies overnight at 4°C. The sections were then rinsed twice with PBS and stained with secondary antibody conjugated Alexa 594 (Thermo). Images were taken using an Olympus epifluorescence microscope or confocal microscope (Olympus FV‐1000).
2.5. Transfection of small interfere RNA
Small interference RNA (siRNA) (ON‐TARGETplus™ siRNA Smartpool) specifically against human PBR was purchased from GE Dharmacon RNAi Technologies, and the transfection of siRNA (5 nmol/L) was performed using a Lipofectamine 2000 transfection reagent (Thermo Fisher Sci) as per manufacturer instructions. siRNA with a scrambled RNA sequence (siN) served as a transfection control. The protein levels of PBR in MSCs after siRNA transfection were detected using Western blot analysis.
2.6. Statistics
All quantified results are shown in mean ± SEM of three to four independent experiments. Statistical analyses were performed using ANOVA followed by Tukey's test for significant difference. Significance was accepted when P < .05.
3. RESULTS
3.1. Establishment of chondrogenic differentiation
We first established an in vitro chondrogenic model using either micropellet or high cell density cultures. KP cells were treated with or without transforming growth factor‐β3 (TGF‐β) containing chondrogenic induction medium for 14 days; the chondrogenic differentiation was examined using Alcian blue staining to stain GAGs or by immunostaining of type II collagen. Results demonstrated that without chondrogenic induction, the morphology of the micropellets showed incomplete structures and fractures in the centre of pellets (C), whereas pellets treated with chondrogenic induction medium showed a condensed morphology (CHON) (Figure 1A). In control pellets, cells were lightly stained using Alcian blue, whereas chondrogenic induction medium‐treated micropellets (CHON) were strongly stained (Figure 1A). In addition to the expression of GAG, we also examined the expression of type II collagen in micropellets performed with immunostaining. The results showed that the staining of type II collagen was not clear in control cells, but chondrogenic induction medium‐treated micropellets showed high staining of type II collagen (Figure 1B). Similar results could also be found in cells seeded at a high seeding density (3 × 104 cells/cm2) where Alcian blue and type II collagen were able to be stained in chondrogenic induction medium‐treated cells (CHON) (Figure 1C), but not in control cells (C). These results suggest that in vitro chondrogenic differentiation was successfully established and can be used in further studies.
Figure 1.

Establishment of chondrogenesis by micropellet and high‐density culture of mesenchymal stem cells. The KP cells were trypsinized and cell number was counted. About 3 × 105 cells were used and centrifuged at 150 × g to make a micropellet. For chondrogenic induction, the micropellets were treated with or without chondrogenic induction medium for 14 d. The chondrogenic differentiation was accessed using (A) Alcian blue staining and (B) type II collagen immunofluorescence staining. (C) Chondrogenic differentiation of KP cells at high‐density culture (3 × 104 cells/cm2) for 14 d in the presence or absence of chondrogenic induction medium. Chondrogenesis was assessed using Alcian blue staining and immunofluorescence staining of type II collagen. Scale bar: (A) 400 μm, (B, C) 200 μm. The nuclei were stained with DAPI
3.2. Midazolam inhibits chondrogenic differentiation
To test for Midazolam‐inhibited hMSC chondrogenesis, KP cells either in micropellets or in high‐density cultures were treated without (C) or with TGF‐β‐containing chondrogenic induction medium (CHON) in the presence of different doses of Midazolam (1, 10 and 20 μmol/L, denoted as CHON + MDZ1, CHON + MDZ10 and CHON + MDZ20, respectively). The Midazolam only group at dose of 10 μmol/L (MDZ10) was used as a negative control. The chondrogenic differentiation was analysed using Alcian blue, immunostaining of type II collagen as previously described. TGF‐β‐containing chondrogenic induction medium significantly increased Alcian blue content in micropellet cultures, and this chondrogenic induction was dose‐dependently inhibited by the treatment of Midazolam (Figure 2A,B). Similar results from the immunostaining of type II collagen can be observed in Figure 2C. At high cell density cultures, the chondrogenic differentiation was inhibited by Midazolam dose‐dependently (Figure S1). Early studies have indicated that SOX trios (SOX5, SOX6 and SOX9) are crucial for chondrogenesis.34, 35 To further characterize Midazolam‐inhibited protein levels of chondrogenic markers, we performed Western blot analysis to examine the protein levels of SOX5, SOX6, SOX9 and type II collagen. KP cells were seeded at density of 3 × 104 cells/cm2 and were treated with or without chondrogenic differentiation medium in the presence of various doses of Midazolam (1, 10 and 20 μmol/L) for 7 days. The chondrogenic induction successfully increased the protein levels of SOX9 and type II collagen, whereas Midazolam significantly inhibited protein levels of SOX9 and type II collagen (Figure 2D‐F). Interestingly, the chondrogenic induction did not affect the protein levels of SOX5/SOX6 and treatment of Midazolam did not alter the protein levels SOX5/SOX6. These results demonstrate that Midazolam‐inhibited chondrogenic differentiation is mediated by the inhibition of the expression of chondrogenic‐associated proteins.
Figure 2.
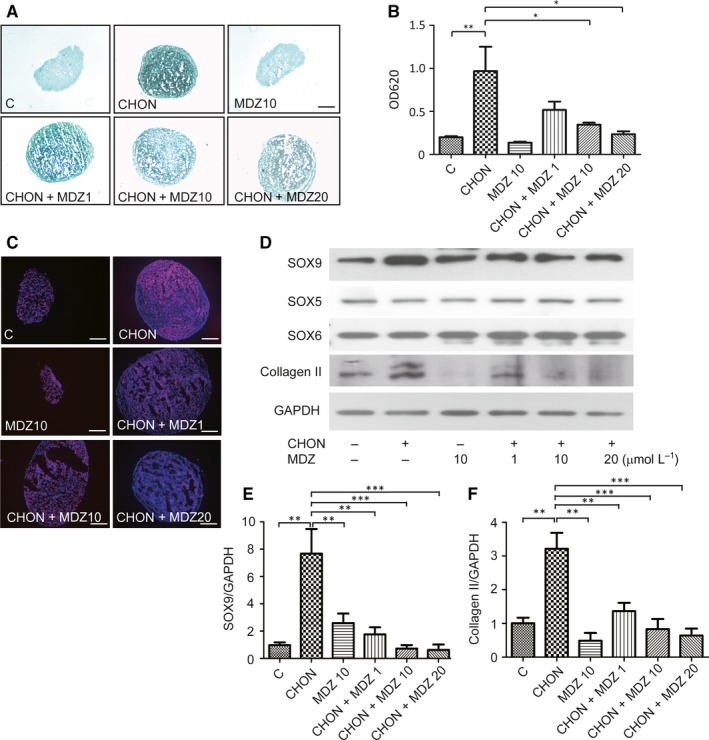
Midazolam dose‐dependently inhibits chondrogenic differentiation. KP cells in micropellets were treated without (C) or with chondrogenic induction medium (CHON) and in the presence of various concentrations of Midazolam (1, 10 and 20 μmol/L, denoted as CHON + MDZ 1, CHON + MDZ 10 and CHON + MDZ 20, respectively) for 14 d. Midazolam only at concentration of 10 μmol/L (MDZ10) was used as a negative control. Control pellets (C) were treated with serum‐free medium plus insulin‐transferrin‐selenium. Chondrogenesis was evaluated using (A, B) Alcian blue staining and (C) immunofluorescence staining of type II collagen. (B) Alcian blue was quantified by dissolving it in guanidine hydrochloride, after which the optical density of the samples was read by a spectrophotometer at OD620 nm. Quantification results are normalized with untreated control. Scale bar: (A) 400 μm; (C) 200 μm. (D) KP cells were seeded at a high density (3 × 104 cells/cm2) and treated with or without chondrogenic induction medium in the presence of various doses of Midazolam for 7 d. Protein levels of chondrogenic marker SOX5, SOX6, SOX9 and type II collagen were examined using Western blot analysis. The protein level of GAPDH served as an internal control. Quantification of SOX9 (D) and type II collagen (E) results are presented as mean ± SEM of three independent experiments (**P ≦ .01, ***P ≦ .005)
3.3. Midazolam inhibits chondrogenic differentiation of primary mesenchymal stem cells
Given that KP cells have been transfected using exogenous genes to maintain MSC stemness,28, 29 whether the incorporated genes produce different responses in these cells remain unclear. To test the inhibitory effects of Midazolam in chondrogenesis, we performed primary hMSCs to evaluate the results that were obtained from the KP MSC cell line. Primary hMSCs were seeded at a high density (3 × 104 cells/cm2) with or without chondrogenic induction medium in the presence of Midazolam (0, 1, 10 and 20 μmol/L) at the indicated time, and chondrogenic differentiation was evaluated using Alcian blue staining and immunostaining of type II collagen. Similar to the results that were obtained from KP MSCs, chondrogenic induction medium successfully induced an increase in Alcian blue staining, whereas Midazolam inhibited Alcian blue staining dose‐dependently (Figure 3A,B). In addition, chondrogenic induction‐induced type II collagen was dose‐dependently inhibited by the treatment of Midazolam (Figure 3C). Similar was confirmed using micropellet culture of primary hMSCs (Figure S2). Together, these results demonstrate that Midazolam‐inhibited chondrogenic differentiation is reproducible in MSCs from different sources.
Figure 3.

Midazolam inhibits chondrogenic differentiation in primary mesenchymal stem cells. Primary mesenchymal stem cells were seeded at 3 × 104 cells/cm2 and treated without (C) or with chondrogenic induction medium (CHON) in the presence of various concentrations of Midazolam (1, 10 and 20 μmol/L, denoted as CHON+ CHON + MDZ 1, CHON + MDZ 10 and CHON + MDZ 20, respectively) for 7 d. Midazolam only at concentration of 10 μmol/L (MDZ10) was used as a negative control. Chondrogenesis was evaluated using (A) Alcian blue staining and (B) Alcian blue was quantified by dissolving it in guanidine hydrochloride, after which the optical density of the samples was read by a spectrophotometer at OD620 nm. Quantification results are normalized with untreated control. Results are presented as mean ± SEM of three independent experiments (*P ≦ .05, **P ≦ .01, ***P ≦ .005). (C) Immunostaining of type II collagen. Scale bar: (A) 200 μm; (C) 50 μm
Because the cell type that we used was taken from peripheral tissue, we sought to understand whether Midazolam‐inhibited chondrogenesis was mediated by PBR. We first examined PBR inhibitor PK11195 on the effect of Midazolam on chondrogenesis. Primary hMSCs were seeded at a high density (3 × 104 cells/cm2) with or without chondrogenic induction medium in the presence of Midazolam (10 μmol/L) and co‐treated with PK11195 (0, 0.1, 1 and 10 μmol/L) for 7 days. The chondrogenic differentiation was accessed using Alcian blue staining and immunostaining of type II collagen. We found that treatment of PK11195 using a 10 μmol/L dose significantly improved the Midazolam‐inhibited effect on chondrogenic differentiation (Figure 4A,B). Similar results were obtained using immunostaining with type II collagen (Figure 4C).
Figure 4.

Peripheral benzodiazepine receptor antagonist PK11195 rescues Midazolam‐induced suppression of chondrogenesis. Primary mesenchymal stem cells were seeded at 3 × 104 cells/cm2 and treated without or with chondrogenic induction medium in the presence of Midazolam (10 μmol/L, MDZ10, CHON + MDZ, respectively) or co‐treated with PK11195 (0.1, 1 and 10 μmol/L, denoted as PK0.1, PK1 and PK10, respectively) for 7 d. Cells were stained with Alcian blue (A, B) or immunoblotted with type II collagen (C). (B) Quantification of Alcian blue and these quantification results are normalized with untreated control. Representative images are selected from at least three independent experiments. Results are presented as mean ± SEM of three independent experiments (*P ≦ .05, **P ≦ .01). Scale bar: 50 μm
Our results indicate that treatment of cells with a higher dose of PK11195 (20 μmol/L) resulted in a decrease in cell number, which represented an off‐target effect of PK11195 (data not shown). We therefore performed siRNA specific to PBR to elucidate the effect of PBR in our model. The control (siRNA with a scrambled sequence, siN, 5 nmol/L) or siPBR (5 nmol/L) was transfected into primary hMSCs. Protein levels of PBR after siRNA transfection were determined using Western blot analysis, and the results showed the successful knockdown by siPBR of the endogenous PBR protein levels (Figure 5A) without altering cell numbers (data not shown). The transfected primary MSCs were then seeded at a high density (3 × 104 cells/cm2) in the presence of chondrogenic medium with or without Midazolam (10 μmol/L) for 7 days. The chondrogenic differentiation was determined using Alcian blue staining (Figure 5B,C) and immunostaining of type II collagen (Figure 5D). Transfection of scrambled siRNA did not rescue Midazolam‐suppressed Alcian blue and type II collagen content during chondrogenesis. In addition, knockdown of PBR (siPBR) rescued the Midazolam‐inhibited chondrogenic differentiation (Figure 5). Similar results were found using Western blot to detect the protein levels of SOX9 and type II collagen (Figure 5E‐G). However, knockdown of PBR did not affect the protein levels of SOX5/SOX6. These results suggest that inhibition of PBR by either PK11195 or siRNA rescues Midazolam‐suppressed chondrogenic differentiation, which indicates that PBR mediates Midazolam‐inhibited chondrogenic differentiation.
Figure 5.

Knockdown of peripheral benzodiazepine receptor (PBR) by siRNA rescues Midazolam‐induced suppression of chondrogenesis. The specific small interference RNA against human PBR (siPBR) was transfected to knockdown PBR expression in primary mesenchymal stem cells (MSCs). A scrambled siRNA (siN) served as a negative control. (A) The protein level of PBRs in transfected cells was accessed using Western blot analysis. Cells were then treated without or with chondrogenic induction medium in the absence or presence of Midazolam (10 μmol/L, MDZ, CHON + MDZ, respectively) for 7 d. Cells were then fixed and stained with (B, C) Alcian blue or (D) immunostained with type II collagen. The nuclei were stained with DAPI. C, Quantification of Alcian blue and these quantification results are normalized with untreated control. (E‐G) Primary MSCs, transfected with either siN or siPBR, were seeded at high cell density and then treated with or without chondrogenic induction medium or co‐treated with or without Midazolam (10 μmol/L) for 7 d. Protein levels of SOX5, SOX6, SOX9 and type II collagen were analysed using Western blot analysis. Levels of vinculin served as an internal control (upper panel). Quantification results of (F) SOX9 and (G) type II collagen are shown. Quantification results are presented as mean ± SEM of three independent experiments (*P ≦ .05, ***P ≦ .005)
To further characterize that Midazolam inhibits chondrogenesis via inhibition of TGF‐β signalling, we assessed the phosphorylation of Smad3, one of the major downstream signalling molecules of TGF‐β.36 We found that administration of Midazolam significantly suppressed TGF‐β‐induced Smad3 phosphorylation dose‐dependently (Figure 6A). Interestingly, if cells were co‐treated with PBR antagonist PK11195, the Midazolam‐induced suppression of Smad3 phosphorylation could be rescued (Figure 6B).
Figure 6.
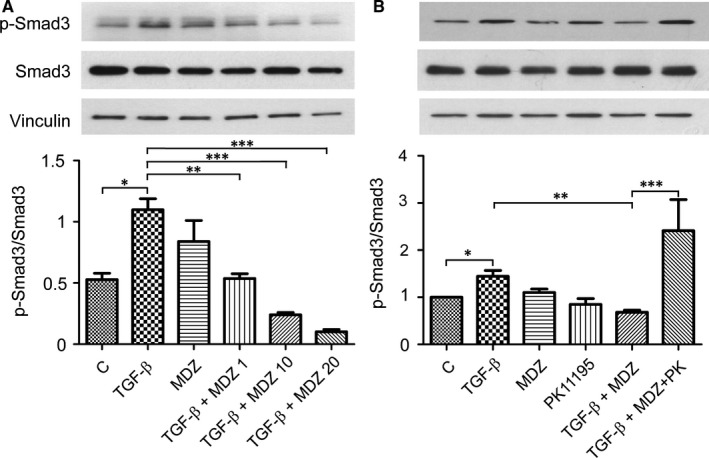
Midazolam inhibits transforming growth factor (TGF)‐β‐induced Smad3 phosphorylation. KP cells were seeded at 3 × 104 cells/cm2, serum starved for 16 h and then treated with TGF‐β (5 ng/mL) in the presence of various doses of Midazolam (0, 1, 10 and 20 μmol/L) for 1 h. (A) Levels of Smad3 phosphorylation; total Smad3 and vinculin were analysed using Western blot analysis. Quantification results are presented as mean ± SEM of three independent experiments (*P ≦ .05, **P ≦ .01, ***P ≦ .005). (B) KP cells were seeded at 3 × 104 cells/cm2, serum starved for 16 h and then treated with TGF‐β (5 ng/mL) in the presence of Midazolam (10 μmol/L, MDZ) or co‐treated with PK11195 (10 μmol/L, PK). Levels of Smad3 phosphorylation; total Smad3 and vinculin were analysed using Western blot analysis. Quantification results are presented as mean ± SEM of three independent experiments (*P ≦ .05, **P ≦ .01, ***P ≦ .005)
In summary, TGF‐β induces Smad3 phosphorylation. Phosphorylated Smad2/3 can interact with Smad4 and translocate into the nucleus to trigger chondrogenic‐related gene transcription. Midazolam‐induced suppression of chondrogenesis may be mediated by the interaction with PBR, which in turn ameliorates TGF‐β‐induced Smad signalling.
4. DISCUSSION
The present study demonstrates the role of Midazolam in chondrogenesis inhibition. However, other recent studies have demonstrated that chondrocytes are the precursors of osteogenesis,37, 38 but because they have only demonstrated the possible osteotoxicity of benzodiazepine,26 the role of Midazolam in chondrogenic differentiation remains unclear. We were interested in whether Midazolam‐induced congenital musculoskeletal defects were caused by the inhibition of chondrogenic differentiation. We first demonstrated benzodiazepine derivative Midazolam‐inhibited chondrogenic differentiation via PBR in human MSCs. Inhibition of PBR by either PK11195 or siRNA specific to PBR rescued the inhibitory effect of Midazolam. More importantly, Midazolam‐inhibited TGF‐β‐induced SMAD3 phosphorylation was mediated by PBRs. The results provided here indicate that PBR plays a crucial role in Midazolam‐inhibited chondrogenesis, which may correlate with congenital malformation in pregnant women who have been treated with benzodiazepine for anxiety or insomnia.
Although benzodiazepine derivatives may cause musculoskeletal defects, the molecular mechanisms of how these benzodiazepine derivatives result in congenital defects remain unknown. To confirm the inhibitory effect of Midazolam, we applied TGF‐β‐containing chondrogenic induction medium31, 32 in human MSCs. We used several differentiation markers to indicate chondrogenesis, including SOX9, type II collagen and an increased amount of GAGs, which could be stained using Alcian blue. SOX9 is a transcriptional factor that plays a key role in the regulation of chondrogenesis, and multiple signalling pathways are known to regulate the expression and activity of SOX9 during chondrogenesis39, 40, 41, 42. Activated SOX9 in turn regulates chondrogenic‐dependent markers, such as type II collagen43, 44 and aggrecan.45 In addition, SOX9, together with SOX5 and SOX6 to form SOX trios, and expression of these trios lead to chondrogenesis.34, 35 In current study, we found that Midazolam inhibits protein levels of SOX9 but not SOX5 and SOX6 in chondrogenic induction. One possible reason is that SOX9 is found to be one of the TGF‐β/Smad3 downstream effectors46 to regulate chondrogenesis. The down‐regulation of SOX9 may result in disruption of the formation of SOX trio, which in turn inhibits the transcription of chondrogenesis‐related genes. On the other hand, we have discovered that Midazolam suppressed chondrogenic differentiation markers at the range of 1‐20 μmol/L in human MSCs. Interestingly, the plasma concentrations of benzodiazepine derivatives from clinical samples ranged from 0.1 to 50 μmol/L.47, 48 These results suggest that administration of Midazolam at clinical dosages suppresses chondrogenic differentiation by inhibiting the levels of chondrogenic‐dependent markers.
We also investigated how Midazolam acts on MSCs to suppress their chondrogenic differentiation. Because MSCs were isolated from peripheral tissues, we aimed for the PBR. Early reports showed that PBR is expressed ubiquitously in most types of tissues, including bone marrow14 and is required for adipogenic differentiation and MSC proliferation.49 It is also known that PBR expresses in murine osteoblasts and osteoclasts, suggesting that PBR may be involved in bone homeostasis.50 To confirm that PBR mediated inhibitory effects of Midazolam, we used a specific antagonist, PK11195, which has been widely used to study PBR functions.18, 51 We demonstrated that Midazolam‐induced suppression of chondrogenesis was blocked by PK11195 as shown by chondrogenic markers, such as SOX9 and type II collagen, and by the expression of GAG. This suggests that Midazolam has an inhibitory function against MSCs differentiating into chondrocytes through PBR. However, several studies have indicated that certain types of GABA receptor subunits can be detected in murine MSCs, chondrogenic cells, growth plate chondrocytes, cartilage and osteoblasts.52, 53, 54 It is uncertain whether these subunits are able to assemble to become a functional GABA receptor and whether the GABA receptor can be expressed in hMSCs, which in turn mediate the effect of Midazolam on chondrogenesis. Together, these results suggest that Midazolam acts on hMSCs through PBRs to inhibit chondrogenesis.
At higher doses of PK11195 treatment (higher than 20 μmol/L), we observed cell loss (data not shown), which may have been damaging to our conclusion. To rule out this possible off‐target effect of PK11195, we used siRNA specific to PBRs to evaluate the function of PBR on the actions of Midazolam in chondrogenesis. Twenty‐four hours after transfection of siRNA, the protein level of PBRs was significantly decreased compared with the scrambled control. Midazolam‐induced suppression of chondrogenesis was markedly rescued with siRNA‐down‐regulated PBR in hMSCs. This result further confirmed the role of PBR in mediating the inhibitory effect of Midazolam in chondrogenesis.
The major signalling pathway used to induce chondrogenesis in this study was TGF‐β signalling. TGF‐β, a pleiotropic cytokine, is involved in the regulation of cell division and suppression of immune response.55 Upon binding of TGF‐β to its receptor, the receptor complex recruits and phosphorylates the carboxy terminus of receptor‐regulated Smad proteins (R‐Smads: Smad2 and Smad3).36, 56 Phosphorylated Smads can interact with Smad4 to become a complex, and this complex translocates into the nucleus and turns on gene transcription.55 Smad3 is able to activate SOX9 via recruitment of CREB‐binding protein/p300.46 In addition, TGF‐β/Smad signalling is essential for osteocartilage development, bone homeostasis and MSC fate decision.57 Disruption of TGF‐β/Smad signalling in vivo resulted in serious problems in the musculoskeletal system.58 Midazolam has been found to inhibit protein phosphorylation. For example, a previous report indicated that Midazolam inhibits platelet activation by inhibiting protein phosphorylation of protein kinase C.59 In mouse hippocampal slices, Midazolam was found to inhibit extracellular signal‐related kinase 1/2 activity, and this inhibition was likely mediated via N‐methyl‐d‐aspartate receptors, phospholipase C and PKC‐dependent signalling.60 We therefore proposed that the chondrogenic inhibitory effect of Midazolam was caused by the inhibition of Smad 3 phosphorylation. Indeed, Midazolam can dose‐dependently inhibit Smad3 phosphorylation (Figure 6) and this Smad3 phosphorylation inhibition was mediated by PBR, as proved when administration of PK11195 reversed the inhibitory effect of Midazolam. We therefore speculate that the inhibitory effect of Midazolam on TGF‐β‐induced Smad 3 phosphorylation is mediated by binding to PBR. Several studies have demonstrated that in most types of tissue, PBR is primarily located in the outer membrane of mitochondria.13 In addition to the outer mitochondrial membrane, PBR was also located in various subcellular regions. Mukherjee and Das first reported that although the amount of [3H]‐labelled R05‐4864 (another PBR ligand) binding sites was highest in the mitochondrial fraction, the nuclear and cytosolic fractions also contained significant amounts of these binding sites.61 A study conducted by Kuhlmann and Guilarte discovered that after neural injury, PBR localization was in cytosol or perinuclear areas in microglia cells and macrophages.62 In addition, subcellular localization of PBR was also found in cytosol or in the nuclei of different cell types (ie MA‐10, MCF‐7 and MDA‐MB‐231), and these nuclear localized PBR was implicated to correlate with cell proliferation and nuclear cholesterol transport.63 This cytosolic localization of PBR supports our speculation that Midazolam can bind with cytosolic PBR to regulate TGF‐β‐induced Smad3 phosphorylation, which also occurs in cytosol. Further study of how Midazolam binds to cytosolic PBRs and exerts its function should be investigated.
Taken together, our findings suggest that Midazolam inhibits chondrogenesis via PBR to inhibit TGF‐β signalling, which in turn suppresses chondrogenesis. This partially explains why this sedative drug can result in congenital malformation of newborns if a pregnant woman is treated with benzodiazepine for anxiety and insomnia. Thus, the future application of these types of drugs must be more thoroughly considered. Further studies to elucidate how these benzodiazepine derivatives interact with their receptors and cause various outcomes require more sophisticated investigation.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
YCC, KCW, ECS and YKW initiated the project; YCC and KCW designed the experiments and YCC conducted experiments of the project; BMH provided reagents; YCC, KCW, ECS and YKW analysed the results; YCC, KCW, ECS and YKW wrote manuscript; BMH edited the manuscript; YKW and ECS supervised the progress. All authors read and confirmed the final draft of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Ms. Chun‐Yi Hsu, Yung‐Shan Hsu and Yu‐Ting Wu for their excellent technical support and administrative effort on the project. This work was supported mainly by grant from Ministry of Science and Technology, Taiwan (MOST 105‐2320‐B‐006‐030) to YKW. This work was also supported, at least in part by the grant from An Nan Hospital‐China Medical University (ANHRF104‐01) to ECS, Chang Gung Memorial Hospital (Chia Yi, CMRPG6F0291) to KCW and grant from Academia Sinica (2396‐106‐0100) to YKW.
Chen Y‐C, Wu K‐C, Huang B‐M, So EC, Wang Y‐K. Midazolam inhibits chondrogenesis via peripheral benzodiazepine receptor in human mesenchymal stem cells. J Cell Mol Med. 2018;22:2896–2907. https://doi.org/10.1111/jcmm.13584
Yung‐Ching Chen and King‐Chuen Wu contributed to the manuscript equally.
Contributor Information
Edmund Cheung So, Email: edmunsotw@gmail.com.
Yang‐Kao Wang, Email: humwang@mail.ncku.edu.tw.
REFERENCES
- 1. Tobias JD, Leder M. Procedural sedation: a review of sedative agents, monitoring, and management of complications. Saudi J Anaesth. 2011;5:395‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mohler H, Fritschy JM, Rudolp U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2‐8. [DOI] [PubMed] [Google Scholar]
- 3. Olkkola KT, Ahonen J. Midazolam and other benzodiazepine. Handb Exp Pharmacol. 2008;182:335‐360. [DOI] [PubMed] [Google Scholar]
- 4. Swinyard EA, Castellion AW. Anticonvulsant properties of some benzodiazepines. J Pharmacol Exp Ther. 1966;151:369‐375. [PubMed] [Google Scholar]
- 5. Tallman JF, Paul SM, Skolnick P, Gallager DW. Receptors for the age of anxiety: pharmacology of the benzodiazepines. Science. 1980;207:274‐281. [DOI] [PubMed] [Google Scholar]
- 6. Bickford PC, Breiderick L. Benzodiazepine modulation of GABAergic responses is intact in the cerebellum of aged F344 rats. Neurosci Lett. 2000;291:187‐190. [DOI] [PubMed] [Google Scholar]
- 7. Carmel I, Fares FA, Leschiner S, Scherübl H, Weisinger G, Gavish M. Peripheral‐type benzodiazepine receptors in the regulation of proliferation of MCF‐7 human breast carcinoma cell line. Biochem Pharmacol. 1999;58:273‐278. [DOI] [PubMed] [Google Scholar]
- 8. Sieghart W. Structure and pharmacology of gamma‐aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181‐234. [PubMed] [Google Scholar]
- 9. Atack JR. The benzodiazepine binding site of GABA(A) receptors as a target for the development of novel anxiolytics. Expert Opin Investig Drugs. 2005;14:601‐618. [DOI] [PubMed] [Google Scholar]
- 10. Greenfield LJ Jr. Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure. 2013;22:589‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li F, Liu J, Liu N, Kuhn LA, Garavito RM, Ferguson‐Miller S. Translocator protein 18 kDa (TSPO): an old protein with new functions? Biochemistry. 2016;55:2821‐2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18 kDa): new nomenclature for the peripheral‐type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402‐409. [DOI] [PubMed] [Google Scholar]
- 13. Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40:475‐486. [DOI] [PubMed] [Google Scholar]
- 14. Kumar A, Muzik O, Chugani D, Chakraborty P, Chugani HT. PET‐derived biodistribution and dosimetry of the benzodiazepine receptor‐binding radioligand (11)C‐(R)‐PK11195 in children and adults. J Nucl Med. 2010;51:139‐144. [DOI] [PubMed] [Google Scholar]
- 15. Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ‐aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338‐351. [PubMed] [Google Scholar]
- 16. Lin R, Angelin A, Da Settimo F, et al. Genetic analysis of dTSPO, an outer mitochondria membrane protein, reveals its function in apoptosis, longevity, and ab42‐induced neurodegeneration. Aging Cell. 2014;13:507‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang M, Wang X, Zhao L, et al. Macroglia‐microglia interactions via TSPO signaling regulates microglial activation in mouse retina. J Neurosci. 2014;34:3793‐3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walter RB, Pirga JL, Cronk MR, Mayer S, Appelbaum FR, Banker DE. PK11195, a peripheral benzodiazepine receptor (pBR) ligand, broadly blocks drug efflux to chemosensitize leukemia and myeloma cells by a pBR‐independent, direct transporter‐modulating mechanism. Blood. 2005;106:3584‐3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qiu Z, Li M, He J, et al. Translocator protein mediates the anxiolytic and antidepressant effects of midazolam. Pharmacol Biochem Behav. 2015;139:77‐83. [DOI] [PubMed] [Google Scholar]
- 20. Dolovich LR, Addis A, Vaillancourt JM, Power JD, Koren G, Einarson TR. Benzodiazepine use in pregnancy and major malformations or oral cleft: meta‐analysis of cohort and case‐control studies. BMJ. 1998;317:839‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laegreid L, Olegård R, Conradi N, Hagberg G, Wahlström J, Abrahamsson L. Congenital malformations and maternal consumption of benzodiazepines: a case‐control study. Dev Med Child Neurol. 1990;32:432‐441. [DOI] [PubMed] [Google Scholar]
- 22. Deck GM, Nadkarni N, Montouris GD, Lovett A. Congenital malformations in infants exposed to antiepileptic medications in utero at Boston Medical Center from 2003 to 2010. Epilepsy Behav. 2015;51:166‐169. [DOI] [PubMed] [Google Scholar]
- 23. Nikizad H, Yon JH, Carter LB, Jevtovic‐Todorovic V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann N Y Acad Sci. 2007;1122:69‐82. [DOI] [PubMed] [Google Scholar]
- 24. Buttar HS, Moffatt JH. Pre‐ and postnatal development of rats following concomitant intrauterine exposure to propoxyphene and chlordiazepoxide. Neurobehav Toxicol Tetratol. 1983;5:549‐556. [PubMed] [Google Scholar]
- 25. Mehanny SZ, Abdel‐Rahman MS, Ahmed YY. Teratogenic effect of cocaine and diazepam in CF1 mice. Teratology. 1991;43:11‐17. [DOI] [PubMed] [Google Scholar]
- 26. Zhang T, Shao H, Xu K, Kuang LT, Chen RF, Xiu HH. Midazolam suppresses osteogenic differentiation of human bone marrow‐derived mesenchymal stem cells. Eur Rev Med Pharmacol Sci. 2014;18:1411‐1418. [PubMed] [Google Scholar]
- 27. Laganà AS, Triolo O, D'Amico V, et al. Management of women with epilepsy: from preconception to post‐partum. Arch Gynecol Obstet. 2016;293:493‐503. [DOI] [PubMed] [Google Scholar]
- 28. Hung SC, Yang DM, Chang CF, et al. Immortalization without neoplastic transformation of human mesenchymal stem cells by transduction with HPV16 E6/E7 genes. Int J Cancer. 2004;110:313‐319. [DOI] [PubMed] [Google Scholar]
- 29. Tsai CC, Chen CL, Liu HC, et al. Overexpression of hTERT increases stem‐like properties and decreases spontaneous differentiation in human mesenchymal stem cell lines. J Biomed Sci. 2010;17:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen WH, Lai MT, Wu ATH, et al. In vitro stage‐specific chondrogenesis of mesenchymal stem cells committed to chondrocytes. Arthritis Rheum. 2009;60:450‐459. [DOI] [PubMed] [Google Scholar]
- 31. Gao L, McBeath R, Chen CS. Stem cell shape regulates a chondrogenic versus myogenic fate through Rac1 and N‐cadherin. Stem Cells. 2010;28:564‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143‐147. [DOI] [PubMed] [Google Scholar]
- 33. Woods A, Wang G, Beier F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem. 2005;280:11626‐11634. [DOI] [PubMed] [Google Scholar]
- 34. Ikeda T, Kamekura S, Mabuchi A, et al. The combination of SOX5, SOX6 and SOX9 (the SOX trio) provides signals sufficient for the induction of permanent cartilage. Arthritis Rheum. 2004;50:3561‐3573. [DOI] [PubMed] [Google Scholar]
- 35. Kim HJ, Im GI. Electroporation‐mediated transfer of SOX trio genes (SOX‐5, SOX‐6, and SOX‐9) to enhance the chondrogenesis of mesenchymal stem cells. Stem Cells Dev. 2011;20:2103‐2114. [DOI] [PubMed] [Google Scholar]
- 36. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature. 2003;425:577‐584. [DOI] [PubMed] [Google Scholar]
- 37. Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci USA. 2014;111:12097‐12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10:e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813‐2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85‐89. [DOI] [PubMed] [Google Scholar]
- 41. Lefebvre V, Dvir‐Ginzberg M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect Tissue Res. 2017;58:2‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leung VY, Gao B, Leung KK, et al. SOX9 governs differentiation stage‐specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 2011;7:e1002356 https://doi.org/10.1371/journal.pgen.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bell DM, Leung KK, Wheatley SC, et al. SOX9 directly regulates the type II collagen gene. Nat Genet. 1997;16:174‐178. [DOI] [PubMed] [Google Scholar]
- 44. Ng LJ, Wheatley S, Muscat GE, et al. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108‐121. [DOI] [PubMed] [Google Scholar]
- 45. Sekiya I, Tsuji K, Koopman P, et al. SOX9 enhances aggrecan gene promoter/enhancer activity and is up‐regulated by retinoic acid in a cartilage‐derived cell line, TC6. J Biol Chem. 2000;15:10738‐10744. [DOI] [PubMed] [Google Scholar]
- 46. Furumatsu T, Tsuda M, Taniguchi N, Tajima Y, Asahara H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB‐binding protein/p300 recruitment. J Biol Chem. 2005;280:8343‐8350. [DOI] [PubMed] [Google Scholar]
- 47. Bowling AC, DeLorenzo RJ. Micromolar affinity benzodiazepine receptors: identification and characterization in central nervous system. Science. 1982;216:1247‐1250. [DOI] [PubMed] [Google Scholar]
- 48. Wilms H, Claasen J, Rohl C, Sievers J, Deuschl G, Lucius R. Involvement of benzodiazepine receptors in neuroinflammatory and neurodegenerative diseases: evidence from activated microglial cells in vitro. Neurobiol Dis. 2003;14:417‐424. [DOI] [PubMed] [Google Scholar]
- 49. Lee DH, Kang SK, Lee RH, et al. Effects of peripheral benzodiazepine receptor ligands on proliferation and differentiation of human mesenchymal stem cells. J Cell Physiol. 2004;198:91‐99. [DOI] [PubMed] [Google Scholar]
- 50. Kam WW, Meikle SR, Zhou H, et al. The 18 kDa translocator protein (peripheral benzodiazepine receptor) expression in the bone of normal, osteoprotegerin or low calcium diet treated mice. PLoS ONE. 2012;7:e30623 https://doi.org/10.1371/journal.pone.0030623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bono F, Lamarche I, Prabonnaud V, Le Fur G, Herbert JM. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem Biophys Res Commun. 1999;265:457‐461. [DOI] [PubMed] [Google Scholar]
- 52. Mizuta K, Osawa Y, Mizuta F, Xu D, Emala CW. Functional expression of GABAB receptors in airway epithelium. Am J Respir Cell Mol Biol. 2006;39:296‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takahata Y, Hinoi E, Takarada T, Nakamura Y, Ogawa S, Yoneda Y. Positive regulation by gamma‐aminobutyric acid B receptor subunit‐1 of chondrogenesis through acceleration of nuclear translocation of activating transcription factor‐4. J Biol Chem. 2012;287:33293‐33303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tamayama T, Maemura K, Kanbara K, Gallager DW. Expression of GABA(A) and GABA(B) receptors in rat growth plate chondrocytes: activation of the GABA receptors promotes proliferation of mouse chondrogenic ATDC5 cells. Mol Cell Biochem. 2005;273:117‐126. [DOI] [PubMed] [Google Scholar]
- 55. Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF‐beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284:F243‐F252. [DOI] [PubMed] [Google Scholar]
- 56. Cleary MA, van Osch GJ, Brama PA, Hellingman CA, Narcisi R. FGF, TGFb and Wnt crosstalk: embryonic to in vitro cartilage development from mesenchymal stem cells. J Tissue Eng Regen Med. 2015;9:332‐342. [DOI] [PubMed] [Google Scholar]
- 57. Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF‐beta signaling in bone remodeling. J Clin Invest. 2014;124:466‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang X, Ma Y, You T, et al. Roles of TGF‐β/Smad signaling pathway in pathogenesis and development of gluteal muscle contracture. Connect Tissue Res. 2015;56:9‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sheu JR, Hsiao G, Luk HN, et al. Mechanisms involveed in the antiplatelet activity of Midazolam in human platelet. Anesthesiology. 2002;96:651‐658. [DOI] [PubMed] [Google Scholar]
- 60. Gao H, Han M, Zhang L, et al. Anesthetics inhibit extracellular signal‐related kinase 1/2 phosphorylation via NMDA receptor, phosphorlipase C and protein kinase C in mouse hippocampal slices. Neurochem Intl. 2017;103:36‐44. [DOI] [PubMed] [Google Scholar]
- 61. Mukherjee S, Das SK. Subcellular distribution of “peripheral type” biding sites for [3 h] r05‐4864 in guinea pig lung. J Biol Chem. 1989;264:16713‐16718. [PubMed] [Google Scholar]
- 62. Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptor after trimethyltin neurotoxicity. J Neurochem. 2000;74:1694‐1704. [DOI] [PubMed] [Google Scholar]
- 63. Hardwick M, Fertikh D, Culty M, Li H, Vidic B, Papadopoulos V. Peripheral‐type benzodiazepine receptor (PBR) in human breast cancer. Cancer Res. 1999;59:831‐842. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials