

Abstract
DHX15 plays a role in leukaemogenesis and leukaemia relapse. However, the mechanism underlying the transcriptional regulation of DHX15 in ALL has not been elucidated. Our present study aimed to explore the functional promoter region of DHX15 and to investigate the transcription factors controlling the transcription of this gene. A luciferase assay performed with several truncated constructs identified a 501‐bp region as the core promoter region of DHX15. Site‐directed mutagenesis, electrophoretic mobility shift and chromatin immunoprecipitation assays showed that ETS1 and SP1 occupied the DHX15 promoter. Furthermore, knockdown of ETS1 and SP1 resulted in suppression of DHX15, whereas the overexpression of these genes led to up‐regulation of DHX15. Interestingly, in samples obtained from patients with ALL at diagnosis, both ETS1 and SP1 correlated positively with DHX15 expression. Additionally, differences in methylation of the DHX15 core promoter region were not observed between the patients and controls. In conclusion, we identified the core promoter region of DHX15 and demonstrated that ETS1 and SP1 regulated DHX15 expression in ALL.
Keywords: DHX15, ETS1, promoter, SP1
1. INTRODUCTION
Acute lymphoblastic leukaemia (ALL) is a heterogeneous haematological malignancy of the bone marrow in which lymphoblasts are overproduced and accumulate.1 The aetiology of ALL is unclear; however, aberrant gene expression due to chromosomal translocations, epigenetic abnormalities, activating mutations, hyperdiploidy and altered transcription factors2 significantly contributes to this disease. Aberrant expression of SALL4, 3 CXCR4, 4 HOX11, 5 TAL1, 5 LYL1, 5 LMO1 5 and LMO2 5 has been implicated in the pathophysiology of ALL. Currently, the primary treatment options for ALL are chemotherapy, radiotherapy and immunotherapy. Patient outcomes have markedly improved; however, the prognosis of ALL is worse in adults than in children. Approximately 50% of adult patients with ALL relapse after the initial treatment. Progress in understanding the pathophysiology of ALL will help advance treatment. Thus, identifying the novel potential targets responsible for ALL pathogenesis may lead to the discovery of novel therapies.
Human RNA helicases (HRHs) are a large family of enzymes that participate in RNA processing. Mutations, aberrant expression and chromosome abnormalities in RNA helicases have been identified in haematological malignancies. DDX3X mutations have been identified in chronic lymphocytic leukaemia6 and Burkitt's lymphomas,7, 8 although the exact significance of these mutations is not clear. The DDX10 gene fuses with the NUP98 gene to form the chimeric gene NUP98‐DDX10, 9 which is involved in de novo or secondary myeloid malignancies10, 11 as well as imatinib resistance.12 The multiplication and self‐renewal of primary human CD34+ cells can also be highly accelerated by NUP98‐DDX10.13 Dysregulation of DDX32 expression has been demonstrated in lymphoid neoplasms,14, 15 suggesting that this gene may contribute to carcinogenesis. These limited studies reveal that some members of the human RNA helicase family may play diverse biological roles in haematologic malignancies.
The DHX15 gene (alias PRP43) is a member of the DEAH‐box family and is located on the minus strand of chromosome 4 (4p15.3).16 Recent evidence has suggested that DHX15 may contribute to carcinogenesis, and overexpression of DHX15 has been observed in lung adenocarcinoma samples.17, 18 Semiquantitative RT‐PCR analysis showed up‐regulation of DHX15 in breast cancer cells. RNAi‐mediated DHX15 suppression inhibited the proliferation of MCF‐7 and T47D human breast cancer cells.19 The DHX15 p.R222G mutation has been identified in de novo or relapsed acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS) patients and has been implicated as a potential new AML driver gene.20, 21, 22, 23 Using real‐time qRT‐PCR and Western blotting, we observed higher DHX15 mRNA and protein levels, respectively, in human ALL samples than in normal bone marrow (BM) cells. Knockdown of DHX15 in Jurkat cells leads to impaired cell proliferation and increased apoptosis.24 The TSS of the DHX15 gene was identified in a previous study.16 However, the transcriptional regulatory mechanism of DHX15 in ALL remains unknown. Our present study explored the putative promoter region of the DHX15 gene to characterize the transcriptional regulatory mechanisms of DHX15 expression in ALL. The results revealed a 501‐bp functional promoter region of DHX15 that harboured binding sites for ETS1 and SP1, which regulate DHX15 expression.
2. MATERIALS AND METHODS
2.1. Cell lines and patient samples
The human T cell leukaemia Jurkat and BCP‐ALL NALM6 cell lines were grown in RPMI 1640 medium containing 10% (v/v) foetal bovine serum (FBS) (Haoyang, Tianjin, China) in a humidified 5% carbon dioxide incubator. Peripheral blood was collected from 121 newly diagnosed patients with ALL (78 males and 43 females; median age 32 [15‐81] years) at Fujian Medical University Union Hospital and from 6 healthy donors (2 males and 4 females; median age 30 [20‐45] years) following the ethical guidelines of our institution and in accordance with the Declaration of Helsinki. All patient samples were obtained prior to the initiation of any therapy. Informed consent was obtained for the procurement and analysis of these specimens.
2.2. Plasmid constructs and luciferase reporter assay
Fragments of the DHX15 promoter truncated at the 5′ end with common sequences at the 3′ end were amplified using PCR. These PCR products were cloned into the XhoI and HindIII sites in a luciferase reporter vector (pGL4.10 [luc]2) (Promega, Madison, WI, USA) lacking a promoter. Positive clones were confirmed by Sanger sequencing. Overlap extension PCR (OE‐PCR)25 was used to mutate the ETS1‐ and SP1‐binding sites. The pGL4.10‐345 plasmid was utilized as a template for the first round of OE‐PCR. The presence of the expected mutations in the plasmids was confirmed by Sanger sequencing. All fragments were amplified using the primers listed in Table S1. The Jurkat and NALM6 cells were transiently transfected using the Amaxa Cell Line Nucleofector Kit V (Lonza). A total of 2 × 106 cells were resuspended in 100 μL of Cell Line Nucleofector Solution V and mixed with 4 μg of the promoter constructs and 400 ng of the internal control (pRL‐TK). Nucleofection was performed as previously described.26 At 24 hour post‐transfection, the cells were lysed using passive lysis buffer and analysed for luciferase activity with the reagents provided in the Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The SpectraMax i3x Multi‐Mode detection platform (Molecular Devices, CA, USA) was used to measure the luciferase activity. Firefly luciferase activity was normalized to Renilla luciferase activity and shown as relative luciferase units to reflect the promoter activity.
2.3. ETS1 and SP1 constructs
The full‐length cDNA sequence for human SP1 was obtained by PCR using primers (Table S1) with NheI and HindIII sites. Then, the product was cloned into the pcDNA3.1(‐) vector (Invitrogen, Carlsbad, CA, USA) digested with the same restriction enzymes. Positive clones were validated by sequencing. The pEnter/ETS1 plasmid overexpressing human ETS1 was purchased from VigeneBio (Shandong, China). The expression or empty vectors were transfected into Jurkat and NALM6 cells using the Amaxa Nucleofector.
2.4. In silico analysis of the DHX15 gene core promoter
In silico analysis of the DHX15 promoter region between positions −345 and +156 upstream of the transcriptional start site (TSS) was performed with Matinspector (http://www.genomatix.de), Promo Alggen (http://alggen.lsi.upc.es/), TFbind (http://tfbind.hgc.gp/) and MethPrimer (http://urogene.org).
2.5. Electrophoretic mobility shift assay (EMSA)
The EMSA was conducted using a chemiluminescence EMSA kit according to the manufacturer's instructions (Beyotime, Jiangsu, China). Briefly, nuclear extracts were prepared from Jurkat and NALM6 cells using the Nuclear and Cytoplasmic Protein Extraction Kit following the manufacturer's instructions (Beyotime). The 5′‐labelled biotin probes corresponding to the putative ETS1 and SP1 sites were synthesized and annealed (Beyotime, Table S1). For regular EMSA, 20 μg of nuclear extract was incubated with the biotinylated probes at 25°C for 30 minutes. To confirm the binding specificity, a 100‐fold excess of unlabelled competitive probe (either a cold probe or a mutated cold probe) was used. To further determine the binding specificity, 4 μg of an antibody that recognized ETS1 or SP1 was also added to the reaction mixture and incubated at 25°C for 30 minutes according to the supershift assay protocol.
2.6. Chromatin immunoprecipitation assay (ChIP)
The ChIP assay was performed with the EZ‐Magna CHIP™ A kit (Millipore, MA, USA) according to the manufacturer's instructions. Briefly, two million Jurkat and NALM6 cells were harvested, crosslinked, lysed and sonicated (7‐seconds bursts with a 50‐seconds rest period at 20% power using the VibraCell™ sonicator). A 1% aliquot of the sample was used as the input DNA control. The remaining cell lysates were incubated with an RNA POL II (Millipore), ETS1 (CST), SP1 (CST) or IgG antibody overnight with rotation. Then, the samples were precipitated using protein A magnetic beads (Millipore). The chromatin‐protein complexes were washed and eluted, and the cross‐linking was subsequently reversed. After purification of the precipitated DNA, the samples were analysed using PCR. The PCR amplifications were performed with 30 cycles at 95°C for 30 seconds, 60°C for 30 seconds and 72°C for 30 seconds. An unrelated region of GAPDH was amplified to determine the binding specificity. The DNA primers used for the PCR are listed in Table S1.
2.7. RNA interference
Hollenhorst et al27 previously described the small interfering RNA (siRNA) target for ETS1, and Lars Andresen et al28 described the siRNA target for SP1. All siRNA duplexes, including a negative control without homology to known human genes, were chemically synthesized by the GenePharma Company (Shanghai, China). The RNAi knockdown experiments were performed with Jurkat and NALM6 cells transiently transfected with the siRNAs using the DharmaFECT 4 transfection reagent (Dharmacon, Inc.) following the manufacturer's instructions. Total RNA and proteins were extracted at 48 hour post‐transfection and analysed using quantitative real‐time PCR (qPCR) and Western blotting, respectively.
2.8. Quantitative PCR
Total RNA was extracted using TRIzol (Invitrogen). The concentration and purity of the RNA were determined using a spectrophotometer (NanoDrop 1000, Thermo Scientific). A total of 2 μg of RNA was reverse‐transcribed into cDNA using the PrimeScript RT Reagent Kit (Takara, Dalian, China) and the ABI2720 thermocycler (Applied Biosystems, USA). qPCR was performed with the ABI7500 real‐time PCR system (Applied Biosystems) and FastStart Universal SYBR Green Master Mix (Roche). The primers used for quantification are listed in Table S1. The mean triplicate cT values of each cDNA sample were calculated and then normalized to GAPDH. The relative gene expression levels were obtained using the 2−ΔΔCt method as previously described.29
2.9. Western blotting
RIPA buffer supplemented with the ProteinSafe Protease Inhibitor Cocktail (100×) (TransGenBiotek, China) was used to isolate total proteins from the Jurkat and NALM6 cells. The concentrations of the protein extracts were measured using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). The Western blotting analysis was performed in the following manner. A total of 20 μg of protein was separated with a 10% sodium dodecyl sulphate (SDS) polyacrylamide gel and then blotted onto PVDF membranes. The membranes were blocked by incubation with 5% skim milk in TBST buffer (10 mmol/L Tris‐HCl, pH 8.0, 150 mmol/L NaCl and 0.1% Tween‐20). After incubation with the anti‐ETS1 (14069, 1:1000 dilution, CST), anti‐SP1 (9389, 1:1000 dilution, CST), anti‐DHX15 (1:1500 dilution, Proteintech) or anti‐β‐actin (1:2000 dilution, TransGen) antibody, the detection was performed with HRP‐coupled secondary antibodies (Millipore). The intensity of each band was calculated using the Quantity One software (Bio‐Rad, Hercules, CA).
2.10. BSP
The EpiTect Fast Bisulfite Kit (QIAGEN) was used for bisulphite modification of the genomic DNA. The core promoter region of the DHX15 gene, which spans a 260‐nucleotide (nt) fragment with 30 CpG sites from −154 to +96 nt, was amplified through two rounds of PCR using primers (Table S1) designed with the Methyl Primer Express Software v1.0 (Applied Biosystems, Foster City, California). Briefly, the first‐round PCR reaction mixture (12.5 μL) contained 1 μL of modified genomic DNA and 6.25 μL of 2× DreamTaq Green PCR Master Mix (Fermentas, MD, USA). The PCR conditions were as follows: 95°C for 5 minutes, followed by 40 cycles of 95°C for 30 seconds, 50°C for 30 seconds and 72°C for 30 seconds with a final elongation at 72°C for 7 minutes. The second‐round amplification reaction (50 μL) contained 2 μL of the diluted first‐round products (1:100) and was conducted under the same conditions. After purification (TIANGEN PCR Purification Kit, Beijing, China), the purified PCR products were cloned into the pGEM‐T vector (Promega, Madison, WI, USA) according to the manufacturer's instructions. Ten independent clones from each specimen were sequenced (SanGon Biotech Co., Shanghai, China). The methylation level for each CpG site was calculated as the number of methylated CpGs in each site divided by the total number of clones sequenced.
2.11. Statistical analysis
The patient samples were stratified into low and high DHX15 expression groups based on the median value. The statistical analyses were performed with analysis of variance (ANOVA) and the t test in GraphPad Prism 6.02 (GraphPad Software, La Jolla, CA, USA). The Spearman's rho test was used to evaluate the correlation between DHX15 and ETS1 or SP1 expression in the clinical samples. The chi‐square test was used to evaluate the correlations between the clinical characteristics and the DHX15 expression level. P values < .05 (two‐sided) were considered significant.
3. RESULTS
3.1. The DHX15 core promoter is located within 501‐bp upstream of the transcription start site
To identify the DHX15 core promoter, we analysed the 2010‐bp region (−1854 to +156 bp) around the TSS (+1) (Figure 1A). Luciferase experiments suggested that transfection of the two constructs (pGL4.10‐1854 and pGL4.10‐345) into Jurkat cells resulted in stronger luciferase activity than transfection with pGL4.10 alone (both P < .0001) (Figure 1B). The difference in luciferase activity between these two constructs was not significant (P > .05) (Figure 1B). Additionally, we transfected the pGL4.10‐1854 and pGL4.10‐345 constructs into Jurkat and NALM6 cells. No significant difference in promoter activity was observed between the two constructs in either cell line (Figure 1C). The results suggested that the DHX15 promoter region spanning from −345 to +156 bp contained the full‐length promoter and was crucial for control of basal DHX15 expression. Thus, we selected the 501‐bp region for further study.
Figure 1.
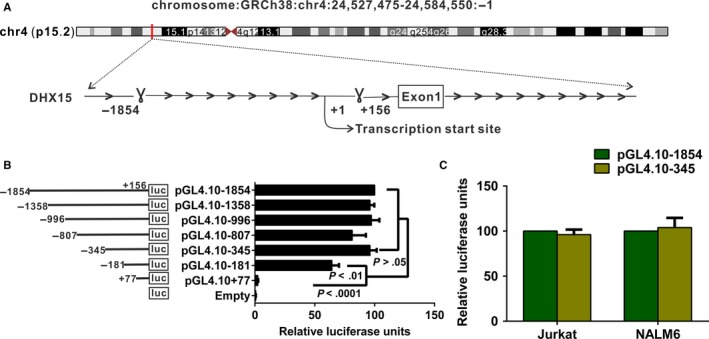
Identification of the DHX15 promoter. A, Schematic representation of the chromosome location and promoter region of the DHX15 gene. Luciferase assays with the DHX15 promoter constructs in Jurkat B and NALM6 C, cells. The results represent relative firefly/Renilla luciferase activities, with the activity of pGL4.10‐1854 considered 100%. Values shown are the means ± SDs of three independent experiments
3.2. CpG islands and functional ETS1‐ and SP1‐binding sites are present in the DHX15 promoter
Comparative analysis using three different software programs (Matinspector, Promo Alggen and TFbind) revealed putative binding sites for several transcription factors in the DHX15 core promoter region (Figure 2A). We placed emphasis on the ETS1 and SP1 transcription factor binding sites for the following reasons. The ETS1 and SP1 transcription factors drive genes that contribute to proliferation and differentiation. SP1 interacts with TATA‐binding protein‐associated factors (TAFs), which are essential for transcription.30 Additionally, SP1 has been implicated in epigenetic regulation31 and chemosensitivity32, 33 in ALL. An ETS1‐binding site was also revealed. ETS1 has been implicated in the pathogenesis of ALL.34 Site‐directed mutagenesis suggested that the ETS1 and SP1 transcription factors were essential for DHX15 promoter activity. A mutation that altered two bases of the ETS1‐binding site reduced the DHX15 promoter activity to 69% (P < .0001) (Figure 2B). Simultaneous mutations of two sites affecting the SP1‐binding site reduced the promoter activity to 62% (P < .0001) (Figure 2B). In addition, mutations in the binding sites for both ETS1 and SP1 (designated the pGL4.10‐double mutant) reduced the promoter activity to 34% (Figure 2B), indicating that these transcription factors had an additive effect. The ChIP assay revealed that RNA polymerase II bound to the DHX15 promoter (−303 to −165) (Figure 2C). CpG islands often contain potential core promoter elements,35, 36, 37 and CpG islands were identified in the DHX15 core promoter using MethPrimer (Figure 2A).38 Taken together, the results suggested that the DHX15 core promoter was located within the 501‐bp region.
Figure 2.
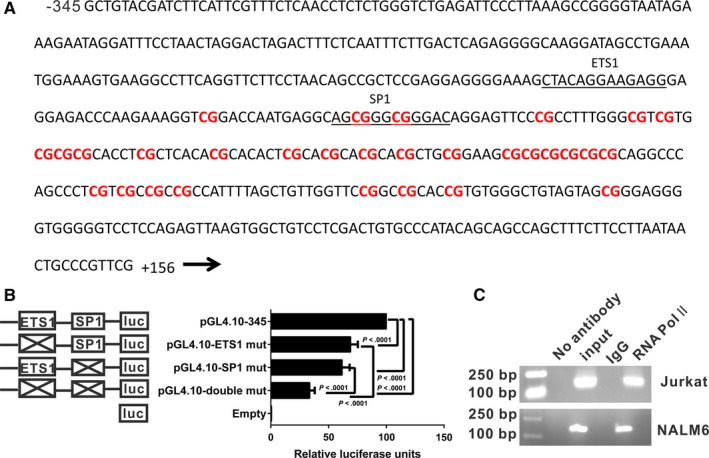
A, The genomic sequence of the DHX15 core promoter is shown. The sequence spans nucleotides −345 to +156 upstream of the DHX15 gene. The DHX15 promoter harbours binding sites for several transcription factors, which are shown underlined, based on predictions from in silico programs. Thirty CpG sites (indicated in red) are present in this region. B, Luciferase assays using constructs with mutations in the 501‐bp region with the predicted transcription binding sites in Jurkat cells. The results represent relative firefly/Renilla luciferase activities, with the activity of the WT 501‐bp region considered 100%. The values are expressed as the means ± SDs from three independent experiments. C, Jurkat and NALM6 cell chromatin was immunoprecipitated with an RNA Pol II antibody. Reactions with non‐immune IgG, no antibody and input DNA served as the negative and positive controls. After removal of the cross‐links, the immunoprecipitated DNA was PCR‐amplified using a primer flanking the basal DHX15 promoter region from −303 to −159 bp. The PCR products were subjected to agarose gel electrophoresis
3.3. ETS1 and SP1 occupy the DHX15 promoter
EMSA experiments were performed with nuclear extracts from the Jurkat and NALM6 cells. The complexity of ETS1 (Figure 3A, left) or SP1 (Figure 3A, right) binding to the DHX15 promoter was demonstrated. Complex formation between the DHX15 promoter and the ETS1 or SP1 transcription factor was reduced when unlabelled probes were added. The complexity was greatly reduced with the addition of antibodies targeting ETS1 and SP1. In addition, the complexity was unaffected in the presence of mutant probes with two base pair mutations in the binding domain. These results confirmed the binding specificity of these transcription factors for the DHX15 promoter.
Figure 3.
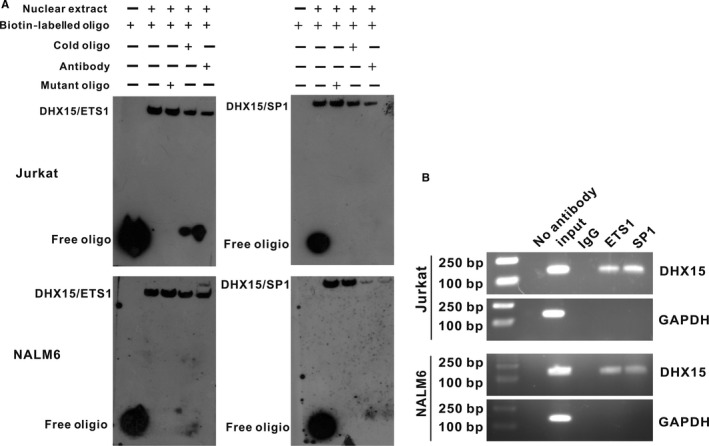
EMSA and ChIP analyses of ETS1 and SP1 binding to the DHX15 promoter. A, EMSA of ETS1 (left) and SP1 (right). The 5′‐biotin end‐labelled probe was incubated in the absence (lane 0) or presence (lane 1) of nuclear extracts from Jurkat and NALM6 cells. A cold mutated probe (lane 2) and cold probe (lane 3) were used as competitors at concentrations that were in a 100‐fold molar excess to the biotin‐labelled probe. Supershift assays were performed with 4 μg of a specific antibody against ETS1 or SP1 (lane 4). B, Equal amounts of Jurkat and NALM6 chromatin were immunoprecipitated with antibodies for ETS1 and SP1 and subsequently quantified through agarose gel electrophoresis using a primer set specific for the basal region (−181 to −36 bp). Moreover, immunoprecipitated DNA was amplified using a primer set specific to the off‐target region (GAPDH) shown in the lower panel as a negative control
To analyse recruitment of ETS1 and SP1 to the DHX15 core promoter, we performed a ChIP assay followed by PCR. A clear band was observed in the Jurkat and NALM6 cells, whereas no band was detected in the negative control (Figure 3B). The results indicated in vivo occupancy of the DHX15 core promoter by ETS1 and SP1. The ChIP and EMSA analysis results support the hypothesis that ETS1 and SP1 bind directly to the DHX15 promoter. Taken together, these results indicate that the DHX15 promoter is a eukaryotic promoter harbouring classical promoter elements, such as CpG islands and ETS1‐ and SP1‐binding sites, which together contribute to transcriptional activation of the DHX15 gene.
3.4. ETS1 and SP1 affect the DHX15 mRNA and protein levels
ETS1 and SP1 were either knocked down or overexpressed to investigate the influence of these transcription factors on the DHX15 gene transcription and protein levels in the Jurkat and NALM6 cells. siRNA‐mediated knockdown of ETS1 or SP1 reduced the DHX15 mRNA (Figure 4A, C) and, consequently, the protein levels (Figure 4E, G). The DHX15 mRNA and protein levels were further reduced by simultaneous knockdown of ETS1 and SP1. In contrast, overexpression of ETS1 or SP1 increased the DHX15 mRNA levels (Figure 4B, D), and consequently, the DHX15 protein levels (Figure 4F, H) in the Jurkat and NALM6 cells. Overexpression of ETS1 and SP1 further enhanced the DHX15 mRNA and protein levels. Taken together, the current data suggested that ETS1 and SP1 transcriptionally drove DHX15 expression through their occupancy on the DHX15 promoter.
Figure 4.
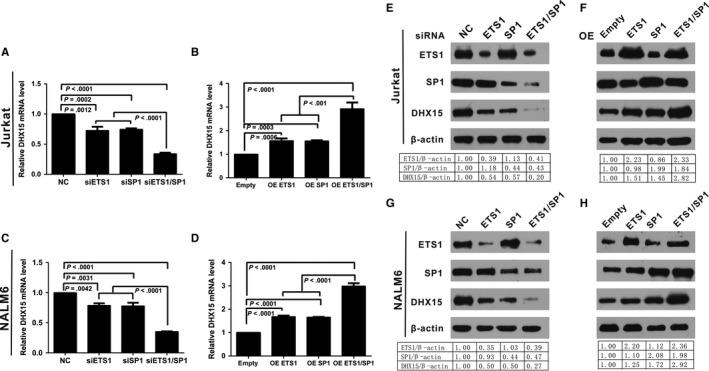
Influence of ETS1 and SP1 on DHX15 gene transcription and protein expression in Jurkat and NALM 6 cells. A, C, E, G, Knockdown of endogenous ETS1 or SP1 or ETS1 and SP1 together decreased DHX15 gene transcription and protein expression. Jurkat and NALM6 cells were transfected with 100 pmol of siRNAs targeting ETS1, SP1 or ETS1 and SP1 together or a negative control (NC). The cells were harvested 48 h after transfection, and 2 μg of the total RNA was used to detect the DHX15 mRNA level through qPCR. The relative mRNA level was obtained after comparison with the NC, which was set to 1 A, C. Western blotting analysis of total proteins with anti‐ETS1, anti‐SP1 and anti‐DHX15 antibodies was performed for the Jurkat and NALM6 cells; anti‐β‐actin served as a loading control E, G. B, D, F, H, Overexpression (OE) of ETS1 or SP1 or ETS1 and SP1 together increased DHX15 gene transcription and protein expression. Jurkat cells were transfected with 4 μg of pcDNA3.1(‐)/SP1, pEnter‐ETS1, pcDNA3.1(‐)/SP1 and pEnter‐ETS1 together or the empty control pcDNA3.1(‐) pEnter. The cells were harvested 48 h after transfection, and 2 μg of total RNA was used to detect the DHX15 mRNA level through qPCR. The relative mRNA level was obtained after comparison with the empty vector, which was set to 1 B, D. Western blotting analysis of total proteins with anti‐ETS1, anti‐SP1 and anti‐DHX15; anti‐β‐actin served as a loading control F, H
3.5. The ETS1 and SP1 levels were correlated with DHX15 expression in ALL
To assess the correlation between ETS1 and SP1 and DHX15 in a clinical setting, DHX15, ETS1 and SP1 expression was investigated using qPCR. Utilizing Spearman's rho, we observed a positive correlation between DHX15 and either ETS1 (r = .5144) or SP1 (r = .7388), indicating that these two variables were statistically dependent on one another (Figure 5A). We also investigated the biological relevance of ETS1 and SP1 for DHX15. Patients were stratified into groups with low or high DHX15 expression based on the median cut‐off value. Remarkably, we observed significant positive correlations of the transcription factor levels with DHX15; specifically, patients expressing low DHX15 levels also showed low expression levels of either ETS1 or SP1, whereas patients with high DHX15 expression demonstrated high levels of ETS1 and SP1 (both P < .0001) (Figure 5B). These data further indicated that ETS1 and SP1 regulated DHX15 expression in human ALL. Additionally, correlations of clinical characteristics and the DHX15 levels were assessed in the patients with ALL. The DHX15 expression levels were correlated with peripheral blood blasts (Table S2).
Figure 5.
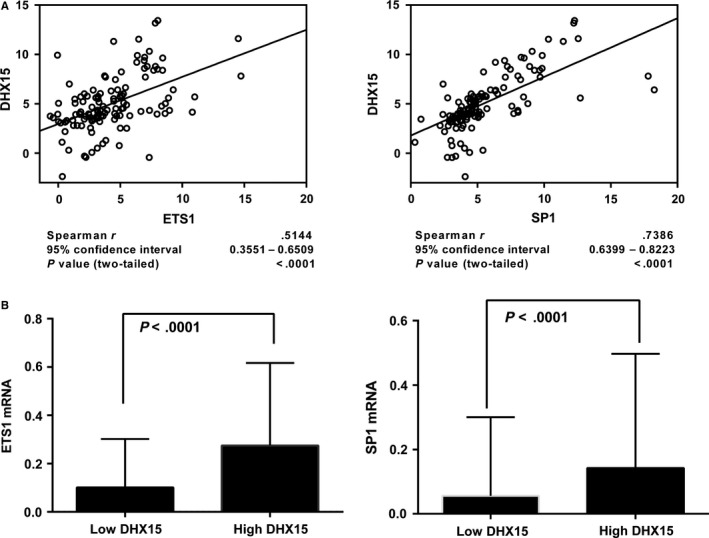
Correlation of the DHX15 levels with the ETS1 and SP1 levels in 121 ALL peripheral blood mononuclear cell (PBMC) samples. A, The qPCR results were evaluated for correlations using Spearman's correlation coefficient, and the correlation coefficient “r” was calculated. B, Box plot analyses comparing the ETS1 and SP1 levels between samples with low and high DHX15 expression levels. All qPCR results were normalized to GAPDH. The samples were divided into low and high DHX15 expression groups based on the median value
3.6. CpG methylation at the DHX15 promoter is not involved in DHX15 overexpression in ALL
The methylation status based on bisulphate sequencing of the core promoter region of DHX15 is shown as lollipop diagrams in Figure S1. Differences in DHX15 core promoter methylation were not observed between the 6 patients with ALL and the 6 healthy controls (P > .05). The CpG sites in the DHX15 gene core promoter of all samples showed a hypomethylated status.
4. DISCUSSION
Recent studies have suggested that DHX15 mutations can drive leukaemia. Higher DHX15 expression predicts a significantly worse prognosis in multiple myeloma,39 breast cancer,39 lung adenocarcinoma39 and AML.24 Transcriptional regulation is crucial for gene expression. However, the mechanism underlying the transcriptional regulation of DHX15 in ALL is unknown. Our present study provided the first characterization of a 501‐bp core promoter region in the human DHX15 gene and showed that ETS1 and SP1 directly activated DHX15 expression.
Luciferase assays of the 2010‐bp region upstream of the TSS of the human DHX15 gene identified the machinery necessary for activation of basal DHX15 expression in a 501‐bp region that exhibited full‐length construct activity. Core promoters are often enriched in CpG islands.35 The results of our in silico analysis showed the presence of CpG islands and ETS1‐ and SP1‐binding sites in this region. The core promoter is the site of action of the RNA polymerase II transcriptional machinery.35 Our ChIP assay showed that RNA polymerase II bound to the DHX15 promoter (−345 to +156). These findings strongly suggested that the core promoter region of DHX15 was located within the 501‐bp region.
The ETS1 oncogene belongs to a large family within the ETS domain family of transcription factors. ETS1 is involved in the development, differentiation and apoptosis of lymphoid cells. Elevated ETS1 expression has been demonstrated in immature T leukaemic cells40 and small lymphocytic cell lymphoma.41 Shachar Raz et al42 revealed that ETS1 was associated with antifolate resistance. ETS1 has also been implicated in T cell maturation arrest in T‐ALL.34 IKKα, which is a subunit of the IkB kinase (IKK) complex, activates nuclear factor‐kB (NF‐kB); ETS1 transactivates IKKα in EU‐4,43 which is a B‐cell precursor ALL cell line. SP1 protein expression mediates leukaemogenesis. Significantly elevated SP1 protein levels have been demonstrated in both B‐ALL and T‐ALL samples.44 A mouse model of human T‐ALL has been established by overexpressing Notch1 in bone marrow cells.45 SP1 drives Notch1 expression in T‐ALL; this finding was confirmed by the observation that SP1 down‐regulation resulted in transcriptional repression of Notch1 in T‐ALL cells.33 High hTERT expression has been observed in ALL,46 and the SP1 transcription factor plays a critical role in basal hTERT expression in Jurkat T cells47; additionally, SP1 regulates hTERT transcription in the leukaemia‐initiating cells of B‐ALL.48 These studies show that SP1 is an important mediator that exerts its effects in ALL via downstream signalling molecules.
These findings prompted an investigation of the functional relevance of the binding sites for these transcription factors on the DHX15 promoter. Site‐directed mutagenesis studies showed that ETS1 and SP1 were putative transcriptional regulators of the DHX15 promoter. The EMSA analysis results confirmed the in vitro interaction of these transcription factors with the DHX15 promoter. Using the ChIP assay, we confirmed that these interactions occurred in vivo. Overexpression and RNAi studies confirmed that the DHX15 gene was transcriptionally regulated by ETS1 and SP1. Dysregulation of DHX15 as an interacting partner with other proteins has been studied in breast cancer19 and prostate cancer.49 Here, we contribute to the understanding of the protein‐DNA interactions of the ETS1 and SP1 transcription factors with the DHX15 gene promoter.
We assessed the association of ETS1 and SP1 with DHX15 in clinical samples. A positive correlation between DHX15 and ETS1 and between DHX15 and SP1 was observed in the patients with ALL. Patients with high DHX15 expression also had high ETS1 and SP1 expression levels. These results further support the notion that ETS1 and SP1 regulate DHX15 expression. Additionally, the correlation between peripheral blood blasts and the DHX15 expression level suggests that the DHX15 expression level possibly predicts the leukaemia burden in the peripheral blood. BSP showed that the DHX15 core promoter was hypomethylated in the patients with ALL and in healthy controls. SP1 maintains the hypomethylated status of CpG islands.36 Therefore, we postulated that the presence of a SP1‐binding site in the DHX15 core promoter contributed to maintenance of the hypomethylated status of the DHX15 core promoter and active DHX15 expression.
Our present study is the first to report that the human RNA helicase DHX15 may be transcriptionally regulated in ALL by the ubiquitous transcription factors ETS1 and SP1. The two transcription factors are particularly promising therapeutic targets because these proteins mediate signals from multiple pathways. Targeting these transcription factors will likely interfere with critical signalling molecules in haematologic malignancies. The present study enhances our current understanding of the transcriptional regulation of DHX15 in ALL; however, additional studies are needed to uncover the complex transcriptional mechanisms underlying DHX15 expression.
CONFLICT OF INTEREST STATEMENT
The authors confirm that there are no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
XLC, YHC, QL, LLP, SLS and JW performed the cellular and molecular experiments. XLL, JGL and YC collected the blood samples. XLC, YHC, YL and XFL performed the data analysis. XLC, YHC and QL wrote the manuscript. SYW designed and supervised this study. SYW obtained financial support.
Supporting information
ACKNOWLEDGEMENTS
This work was financially supported by grants from the National Natural Science Foundation of China (81770163, 81270609, 81470008 and 81500139), Fujian Province Health Education Joint Research Project (WKJ‐2016‐2‐07), Special Funding of Fujian Provincial Department of Finance (Min2017‐655), the Major Science & Technology Project of Fujian Province (2012Y4012), Major Science and Technology Project of Fujian Medical University (09ZD008), Construction Project of Fujian Medical Center of Hematology (Min201704) and the National and Fujian Provincial Key Clinical Specialty Discipline Construction Program, P.R.C (2011‐1006 and 2012‐149).
Chen X‐L, Cai Y‐H, Liu Q, et al. ETS1 and SP1 drive DHX15 expression in acute lymphoblastic leukaemia. J. Cell. Mol. Med. 2018;22:2612–2621. https://doi.org/10.1111/jcmm.13525
REFERENCES
- 1. Jabbour EJ, Faderl S, Kantarjian HM. Adult acute lymphoblastic leukemia. Mayo Clin Proc. 2005;80:1517‐1527. [DOI] [PubMed] [Google Scholar]
- 2. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535‐1548. [DOI] [PubMed] [Google Scholar]
- 3. Ueno S, Lu J, He J, et al. Aberrant expression of SALL4 in acute B cell lymphoblastic leukemia: mechanism, function, and implication for a potential novel therapeutic target. Exp Hematol. 2014;42:307‐316e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crazzolara R, Kreczy A, Mann G, et al. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br J Haematol. 2001;115:545‐553. [DOI] [PubMed] [Google Scholar]
- 5. Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75‐87. [DOI] [PubMed] [Google Scholar]
- 6. Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497‐2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richter J, Schlesner M, Hoffmann S, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316‐1320. [DOI] [PubMed] [Google Scholar]
- 9. Arai Y, Hosoda F, Kobayashi H, et al. The inv(11)(p15q22) chromosome translocation of de novo and therapy‐related myeloid malignancies results in fusion of the nucleoporin gene, NUP98, with the putative RNA helicase gene, DDX10. Blood. 1997;89:3936‐3944. [PubMed] [Google Scholar]
- 10. Nakao K, Nishino M, Takeuchi K, et al. Erratum for Fusion of the nucleoporin gene, NUP98, and the putative RNA helicase gene, DZXX10, by inversion 11 (p15q22) chromosome translocation in a patient with etoposide‐related Myelodysplastic syndrome. Intern Med. 2015;54:547. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto M, Kakihana K, Kurosu T, et al. Clonal evolution with inv(11)(p15q22) and NUP98/DDX10 fusion gene in imatinib‐resistant chronic myelogenous leukemia. Cancer Genet Cytogenet. 2005;157:104‐108. [DOI] [PubMed] [Google Scholar]
- 12. Morerio C, Acquila M, Rapella A, et al. Inversion (11)(p15q22) with NUP98‐DDX10 fusion gene in pediatric acute myeloid leukemia. Cancer Genet Cytogenet. 2006;171:122‐125. [DOI] [PubMed] [Google Scholar]
- 13. Yassin ER, Abdul‐Nabi AM, Takeda A, et al. Effects of the NUP98‐DDX10 oncogene on primary human CD34 + cells: role of a conserved helicase motif. Leukemia. 2010;24:1001‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdelhaleem M. The novel helicase homologue DDX32 is down‐regulated in acute lymphoblastic leukemia. Leuk Res. 2002;26:945‐954. [DOI] [PubMed] [Google Scholar]
- 15. Alli Z, Ho M, Abdelhaleem M. Expression of DHX32 in lymphoid tissues. Exp Mol Pathol. 2005;79:219‐223. [DOI] [PubMed] [Google Scholar]
- 16. Imamura O, Sugawara M, Furuichi Y. Cloning and characterization of a putative human RNA helicase gene of the DEAH‐box protein family. Biochem Biophys Res Commun. 1997;240:335‐340. [DOI] [PubMed] [Google Scholar]
- 17. Stearman RS, Dwyer‐Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen‐induced murine model. Am J Pathol. 2005;167:1763‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu H, Ma J, Wu J, et al. Gene expression profiling analysis of lung adenocarcinoma. Braz J Med Biol Res. 2016;49:e4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin ML, Fukukawa C, Park JH, et al. Involvement of G‐patch domain containing 2 overexpression in breast carcinogenesis. Cancer Sci. 2009;100:1443‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taskesen E, Havermans M, van Lom K, et al. Two splice‐factor mutant leukemia subgroups uncovered at the boundaries of MDS and AML using combined gene expression and DNA‐methylation profiling. Blood. 2014;123:3327‐3335. [DOI] [PubMed] [Google Scholar]
- 21. Faber ZJ, Chen X, Gedman AL, et al. The genomic landscape of core‐binding factor acute myeloid leukemias. Nat Genet. 2016;48:1551‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sood R, Hansen NF, Donovan FX, et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse leukemia. Leukemia. 2016;30:501‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Farrar JE, Schuback HL, Ries RE, et al. Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res. 2016;76:2197‐2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan L, Li Y, Zhang HY, et al. DHX15 is associated with poor prognosis in acute myeloid leukemia (AML) and regulates cell apoptosis via the NF‐kB signaling pathway. Oncotarget. 2017;8:89643‐89654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR‐driven overlap extension. Nat Protoc. 2007;2:924‐932. [DOI] [PubMed] [Google Scholar]
- 26. Chicaybam L, Sodre AL, Curzio BA, et al. An efficient low cost method for gene transfer to T lymphocytes. PLoS ONE. 2013;8:e60298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hollenhorst PC, Chandler KJ, Poulsen RL, et al. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet. 2009;5:e1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andresen L, Jensen H, Pedersen MT, et al. Molecular regulation of MHC class I chain‐related protein A expression after HDAC‐inhibitor treatment of Jurkat T cells. J Immunol. 2007;179:8235‐8242. [DOI] [PubMed] [Google Scholar]
- 29. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101‐1108. [DOI] [PubMed] [Google Scholar]
- 30. Chiang CM, Roeder RG. Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Science. 1995;267:531‐536. [DOI] [PubMed] [Google Scholar]
- 31. Mulero‐Navarro S, Carvajal‐Gonzalez JM, Herranz M, et al. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis. 2006;27:1099‐1104. [DOI] [PubMed] [Google Scholar]
- 32. Gu L, Findley HW, Zhou M. MDM2 induces NF‐kappaB/p65 expression transcriptionally through Sp1‐binding sites: a novel, p53‐independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood. 2002;99:3367‐3375. [DOI] [PubMed] [Google Scholar]
- 33. Koyama D, Kikuchi J, Hiraoka N, et al. Proteasome inhibitors exert cytotoxicity and increase chemosensitivity via transcriptional repression of Notch1 in T‐cell acute lymphoblastic leukemia. Leukemia. 2014;28:1216‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dadi S, Le Noir S, Payet‐Bornet D, et al. TLX homeodomain oncogenes mediate T cell maturation arrest in T‐ALL via interaction with ETS1 and suppression of TCRalpha gene expression. Cancer Cell. 2012;21:563‐576. [DOI] [PubMed] [Google Scholar]
- 35. Butler JE, Kadonaga JT. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002;16:2583‐2592. [DOI] [PubMed] [Google Scholar]
- 36. Suzuki Y, Tsunoda T, Sese J, et al. Identification and characterization of the potential promoter regions of 1031 kinds of human genes. Genome Res. 2001;11:677‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emami KH, Navarre WW, Smale ST. Core promoter specificities of the Sp1 and VP16 transcriptional activation domains. Mol Cell Biol. 1995;15:5906‐5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427‐1431. [DOI] [PubMed] [Google Scholar]
- 39. Mizuno H, Kitada K, Nakai K, et al. PrognoScan: a new database for meta‐analysis of the prognostic value of genes. BMC Med Genomics. 2009;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sacchi N, de Klein A, Showalter SD, et al. High expression of ets‐1 gene in human thymocytes and immature T leukemic cells. Leukemia. 1988;2:12‐18. [PubMed] [Google Scholar]
- 41. Rovigatti U, Watson DK, Yunis JJ. Amplification and rearrangement of Hu‐ets‐1 in leukemia and lymphoma with involvement of 11q23. Science. 1986;232:398‐400. [DOI] [PubMed] [Google Scholar]
- 42. Sanchez‐Medal L. Iron deficiency in pregnancy and childhood. Bol Oficina Sanit Panam. 1971;70:350‐359. [PubMed] [Google Scholar]
- 43. Gu L, Zhu N, Findley HW, et al. Identification and characterization of the IKKalpha promoter: positive and negative regulation by ETS‐1 and p53, respectively. J Biol Chem. 2004;279:52141‐52149. [DOI] [PubMed] [Google Scholar]
- 44. Malik D, Kaul D, Chauhan N, et al. miR‐2909‐mediated regulation of KLF4: a novel molecular mechanism for differentiating between B‐cell and T‐cell pediatric acute lymphoblastic leukemias. Mol Cancer. 2014;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183:2283‐2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cogulu O, Kosova B, Karaca E, et al. Evaluation of telomerase mRNA (hTERT) in childhood acute leukemia. Leuk Lymphoma. 2004;45:2477‐2480. [DOI] [PubMed] [Google Scholar]
- 47. Takakura M, Kyo S, Kanaya T, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551‐557. [PubMed] [Google Scholar]
- 48. Liu S, Liu H, Qin R, et al. The cellular senescence of leukemia‐initiating cells from acute lymphoblastic leukemia is postponed by beta‐Arrestin1 binding with P300‐Sp1 to regulate hTERT transcription. Cell Death Dis. 2017;8:e2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jing Y, Nguyen MM, Wang D, et al. DHX15 promotes prostate cancer progression by stimulating Siah2‐mediated ubiquitination of androgen receptor. Oncogene. DOI: 10.1038/onc.2017.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials