

Abstract
In recent years, microRNAs (miRNAs) are attracting an increasing amount of researchers’ attention, as accumulating studies show that miRNAs play important roles in various basic biological processes and that dysregulation of miRNAs is connected with diverse human diseases, particularly cancers. However, the experimental methods to identify associations between miRNAs and diseases remain costly and laborious. In this study, we developed a computational method named Network Distance Analysis for MiRNA‐Disease Association prediction (NDAMDA) which could effectively predict potential miRNA‐disease associations. The highlight of this method was the use of not only the direct network distance between 2 miRNAs (diseases) but also their respective mean network distances to all other miRNAs (diseases) in the network. The model's reliable performance was certified by the AUC of 0.8920 in global leave‐one‐out cross‐validation (LOOCV), 0.8062 in local LOOCV and the average AUCs of 0.8935 ± 0.0009 in fivefold cross‐validation. Moreover, we applied NDAMDA to 3 different case studies to predict potential miRNAs related to breast neoplasms, lymphoma, oesophageal neoplasms, prostate neoplasms and hepatocellular carcinoma. Results showed that 86%, 72%, 86%, 86% and 84% of the top 50 predicted miRNAs were supported by experimental association evidence. Therefore, NDAMDA is a reliable method for predicting disease‐related miRNAs.
Keywords: adjusted network distance, association prediction, disease, microRNA, network integration
1. INTRODUCTION
MicroRNAs (miRNAs) are a class of non‐coding RNAs which play regulatory roles in gene expressions by binding to the complementary regions of messenger transcripts to repress their translation or regulate degradation.1, 2, 3 Asince the lin‐4 and let‐7 were discovered in Caenorhabditis elegans,4 over 30 000 mature miRNAs have been found from 206 species.5 Furthermore, accumulating evidence shows that miRNA constitutes one of the most important components of cells and is involved in nearly all biological processes, including cell growth,6 immune reaction,7 cell proliferation and differentiation,8, 9 cell development,6 cell cycle regulation,10 inflammation,11 apoptosis 12 and stress response.6, 13 It is also reported that the abnormality of miRNAs is connected with various human diseases, particularly cancers.14, 15 Identifying disease‐related miRNAs is an important biomedical research field, which benefits the understanding of disease pathogenesis at the molecular level and the design of molecular tools for disease diagnosis, treatment and prevention.
In parallel with much efforts being made to identify novel miRNAs, the research community is also interested in predicting and validating miRNAs’ associations with diseases. Using experimental methods to uncover such associations is typically costly and time‐consuming. Fortunately, taking the advantage of vast biological data for miRNAs, computational methods can be an efficient complement to experimental studies. By far, existing computational methods can be broadly divided into 2 categories: (i) those constructing networks and applying the corresponding network‐based algorithms and (ii) those utilizing machine learning.
Inspired by the idea that functionally similar miRNAs tend to be related with phenotypically similar diseases, Jiang et al16 constructed a scoring system based on a discrete hypergeometric probability distribution. This method was not particularly satisfying because 2 miRNAs may be functionally related when their target genes, instead of overlapping significantly, reside in the same cellular pathways or functional modules. Some researchers developed their methods by involving other functional elements. For example, Mork et al17 combined miRNA‐protein and protein‐disease associations and proposed a novel model named miRNA‐protein‐disease (miRPD). This method took proteins into consideration, but finding enough miRNAs and diseases associated with the same proteins for the analysis was a difficult task. Shi et al18 presented a method to identify miRNA‐disease associations based on the assumption that a disease tends to be associated with miRNAs whose target genes also have associations with this disease. They carried out a random walk analysis on a protein‐protein interaction (PPI) network, and the analysis took into account the global network distance measure and the functional links between miRNAs’ targets and disease genes. However, the methods mentioned above had two common drawbacks: the high false‐positives and false‐negatives in miRNA‐target interactions and the incompletion of the disease‐gene association network.
To overcome such drawbacks, researchers developed computational models without relying on miRNA‐target interactions. Chen et al19 put forward RWRMDA, the first global network similarity‐based model, to capture the associations between miRNAs and diseases. Although the model was based on random walk that made full use of global network information, it was not applicable to new diseases without any known related miRNAs. Chen et al20 proposed another global ranking model called WBSMDA, which utilized Gaussian interaction profile kernel similarity for diseases and miRNAs. As an upgrade to RWRMDA, WBSMDA could be implemented for diseases without any known related miRNAs. However, WBSMDA might cause bias to miRNAs with more known associated diseases and its scores needed to be integrated more reasonably. Xuan et al21 presented HDMP based on weighted k most similar neighbours and the miRNA functional similarity. For a specific disease, the relevance score of a miRNA was calculated by summing all subscores of the miRNA's k neighbours. The subscore of a neighbour was calculated by multiplying the functional similarity between the miRNA and the neighbour with the weight of the neighbour; the assignment of weight was based on the neighbour's miRNA family or cluster. The members in the same miRNA family or cluster were assigned higher weights because they were usually transcribed together and therefore were more likely to be associated with similar diseases. However, this method also had some limitations: on one hand, HDMP could not be applied to the new diseases which did not have any known related miRNAs; on the other hand, HDMP did not make full use of global network similarity information. Pasquier et al22 proposed a method named MiRAI which represented distributional information on miRNAs and diseases in a high‐dimensional vector space and reduced dimensions with the help of singular value decomposition (SVD). The association score for a miRNA‐disease pair was measured by the cosine similarity between the miRNA vector in the miRNA space and the disease vector in the disease space. However, the prediction accuracy of MiRAI was low because the model had the data sparsity problem.
Besides, several computational models had adopted machine learning methods to uncover associations between miRNAs and diseases. Under the assumption that miRNAs involved in a specific tumour phenotype will exhibit aberrant regulation of their target genes, Xu et al23 introduced an approach based on the miRNA‐target‐dysregulated network (MTDN) to prioritize disease‐related miRNAs. MTDN was constructed by assembling all significant miRNA‐target pairs which were identified by miRNA expression profiles in tumour and non‐tumour tissues. For each miRNA in MTDN, 4 topological features were computed and changes in miRNA expression were captured. Then, a support vector machine (SVM) classifier was built to identify positive miRNA‐disease associations. Nevertheless, negative associations needed for training the model were hard to obtain and the prediction of supervised classifier such as SVM could be inaccurate. To address this problem, Chen et al24 proposed a semi‐supervised method named RLSMDA. It was developed under the framework of regularized least squares and the basic hypothesis that functionally related miRNAs tend to be related to phenotypically similar diseases. Compared with previous methods, RLSMDA could identify related miRNAs for diseases without any known associated miRNAs. Furthermore, only positive disease‐miRNA association samples were needed to train RLSMDA, and therefore, the model overcame the difficulties in obtaining negative samples faced by several previous studies. But the room for improvement was how to choose the best parameters. Similar to the process of random work, Chen et al25 presented another iterative model named HGIMDA to find the optimal solutions based on global network similarity information. A heterogeneous graph was constructed from various disease similarity measures, diverse miRNA similarity measures and the known miRNA‐disease associations. To calculate the association score between a miRNA and a disease, an iterative process was carried out on the heterogeneous graph, summarizing all paths between the miRNA and the disease with the length equal to 3. Xuan et al26 developed MIDP to predict potential miRNA candidates for the diseases with known related miRNAs and MIDPE for the diseases without any known related miRNAs. It is worth mentioning that the negative effect of noisy data could be decreased by restarting the walk. Chen et al27 further raised RBMMMDA which was the first computational approach for multiple types of miRNA‐disease association prediction. Based on this model, we could obtain not only new miRNA‐disease associations but also their corresponding association types. Recently, Li et al28 raised MCMDA based on the observation that the miRNA‐disease association matrix was low‐rank. They filled the candidate samples without known associations with zero and then iteratively updated them with the predictive scores.
As mentioned above, the existing methods have different limitations. For example, miRNA‐target interactions and disease‐genes associations used in some methods are inaccurate or incomplete. Furthermore, many methods could not be applied to disease without any known related miRNAs and many methods were constructed without optimal parameter. Therefore, new effective computational methods are in urgent need. Based on the assumption that functional similar miRNAs tend to be associated with similar diseases and vice versa, we developed the model of Network Distance Analysis for MiRNA‐Disease Association prediction (NDAMDA). MiRNA‐disease associations, miRNA functional similarity network, disease semantic similarity network and Gaussian interaction profile kernel similarity network were integrated in NDAMDA to uncover the potential disease‐miRNA associations. To evaluate the effectiveness of NDAMDA, global and local leave‐one‐out cross‐validation (LOOCV) as well as fivefold cross‐validation was introduced. The AUCs of global and local LOOCV were respectively 0.8920 and 0.8062, and the model obtained the average AUC of 0.8935 ± 0.0009 in fivefold cross‐validation. Besides, we accessed NDAMDA in case studies of breast neoplasms, lymphoma and oesophageal neoplasms with the validation databases of dbDEMC 29 and miR2Disease.30 As a result, 43, 36 and 43 of the top 50 candidate miRNAs for these 3 diseases were respectively confirmed by experimental discoveries in recent years. We further evaluated the applicability of our method to the diseases without any known related miRNAs. Prostate neoplasms was taken as the investigated diseases, and its known associated miRNAs for the investigated disease were removed from the training dataset. We found that 43 of the top 50 candidate miRNAs for prostate neoplasms were verified by experimental discoveries. Finally, we obtained 42 confirmed miRNAs in the top 50 candidate miRNAs for hepatocellular carcinoma based on the previous version of HMDD, further suggesting that this model have a good performance on different input dataset.
2. MATERIALS AND METHODS
2.1. Human miRNA‐disease associations
We downloaded the latest data of human miRNA‐disease from the HMDD database v2.0,31 which included 5430 experimentally verified human miRNA‐diseases associations, and it involved 383 diseases and 495 miRNAs. Here, we introduced matrix Y ∈ R nm × nd to express those associations in a mathematic way and entity equalled 1 if miRNA was confirmed to be related to disease , and otherwise 0. In addition, we used nm and nd to denote the number of miRNAs and diseases, respectively.
2.2. MiRNA functional similarity
Based on the assumption that functionally similar miRNAs tend to be associated with phenotypically similar diseases, Wang et al32 have calculated the miRNA functional similarity score and we downloaded them from http://www.cuilab.cn/files/images/cuilab/misim.zip. We kept the scores in the matrix FS, where the entity represented the functional similarity between miRNA and .
2.3. Disease semantic similarity model 1
We described each disease as a directed acyclic graph (DAG) with the help of the disease MeSH descriptors downloaded from the National Library of Medicine (http://www.nlm.nih.gov).33 Taking disease as an example, we used to represent it, where was the node set consisted of node D itself and its ancestor nodes, was the corresponding edge set composed of the direct edges from parent nodes to child nodes. Therefore, summing all the contributions from ancestor diseases and disease itself, we could calculate the semantic value of disease as follows:
| (1) |
| (2) |
where ∆ was the semantic contribution factor. Their own contribution to the semantic value of disease was defined as 1; the contribution decreased as the distance between and other diseases increased. Therefore, disease terms in the same layer had the same contribution to the semantic value of disease . We reasoned that 2 diseases sharing larger part of their DAGs were considered to have greater semantic similarity. Here, we defined semantic similarity between and as follows:
| (3) |
2.4. Disease semantic similarity model 2
The disease semantic similarity model was unsatisfying in considering that 2 diseases which located in the same layer of DAG() might appear in different number of disease DAGs. It is obvious that the one appeared more commonly was less specific. Therefore, we developed disease semantic similarity model 2 to complement the old one. We defined the contribution of disease t in DAG() to the semantic value of disease α as follows:
| (4) |
Based on the assumption that 2 diseases sharing larger part of their DAGs are considered to have stronger semantic similarity, we summed all the contributions from ancestor diseases and itself to determine the semantic value DV of disease in the similar way as model 1.
| (5) |
where was the node set in DAG . The disease semantic similarity matrix SS2 was given by
| (6) |
where and were the semantic value of and , respectively.
2.5. Gaussian interaction profile kernel similarity for diseases
Using the topologic information of known miRNA‐disease association network, we proposed Gaussian interaction profile kernel similarity for diseases based on the assumption that functional similar miRNAs tend to be associated with similar diseases. Here, we used the vector IP to represent the interaction profiles of diseases, and IP was calculated based on the associated information between the disease and each miRNA, that is, the ith row of the adjacency matrix Y. Then, Gaussian kernel similarity between disease and was defined based on their interaction profiles as follows:
| (7) |
where parameter was used to control the kernel bandwidth and calculated as follows.
| (8) |
where was the original adjustment coefficient and was the Gaussian interaction profile kernel similarity between disease and .
2.6. Gaussian interaction profile kernel similarity for miRNAs
Gaussian interaction profile kernel similarity matrix of miRNA could be calculated in a similar way:
| (9) |
| (10) |
was the Gaussian interaction profile kernel similarity between miRNA and .
2.7. Integrated similarity for miRNAs and diseases
Here, integrated miRNA similarity matrix S m and integrated disease similarity matrix S d were constructed based on miRNA functional similarity, disease semantic similarity and Gaussian interaction profile kernel similarity. For miRNA pairs and disease pairs that did not have similarity, we used KSm and KSd to respectively represent the similarity between them. In addition, we used FS to represent the similarity for miRNA pairs that had functional similarity; we used the average of SS1 and SS2 to represent the similarity for disease pairs that had semantic similarity.
| (11) |
| (12) |
2.8. NDAMDA
We developed NDAMDA which was constituted by 3 steps: (I) network distance computation and adjustment (II) calculation of the confidence (III) score conversion (See Figure 1).
Figure 1.
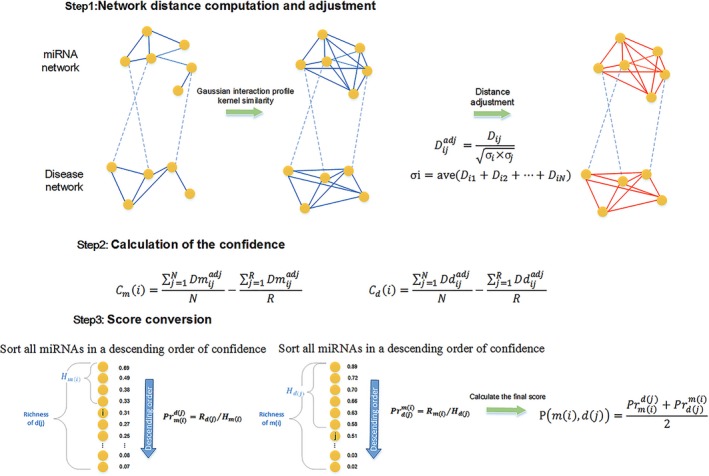
The flow chart of NDAMDA includes 3 steps: (I) network distance computation and adjustment (II) calculation of the confidence (III) score conversion. For details about each step, refer to the Materials and Method section
2.9. Network distance computation and adjustment for miRNAs
We could obtain the similarities from our previous work between two miRNAs directly, for example, we could extract functional similarity between and as , then, the raw network distance between two miRNAs with a link in the network was defined as D = 1/FS, such that a smaller D (shorter distance) would correspond to a higher functional similarity. To those miRNAs without direct links, we used Gaussian interaction profile kernel similarity to fill it. In summary, the raw distance was determined as D = 1/S m. To develop a comprehensive network, we considered both the distance between two miRNAs and their respective mean network distances to all other miRNAs, and the adjusted distance was defined as where σi and σj were the mean distance for and respectively, in raw network.
2.10. Network distance computation and adjustment for diseases
Similar to computation and adjustment for miRNAs, the scores obtained from disease semantic similarity model 1 and disease semantic similarity model 2 were used to construct the raw network of diseases, and distance between two diseases was defined as D = 1/S d after incorporating Gaussian interaction profile kernel similarity to enhance our network. We calculated the adjusted distance as:, where σi and σj were the mean distance for and respectively, in raw network.
2.11. Calculation of the confidence in miRNAs
We reasoned that, for a specific disease in the network, a related miRNA was closer to other related miRNAs than random miRNAs. Therefore, we introduced the confidence in miRNA as follows:
| (13) |
where R was richness of the given disease indicating the total number of known related miRNAs, and was the adjusted network distance between and . It could be concluded that a larger would suggest that the investigated miRNA had relatively shorter distance (stronger functional interaction) to known related miRNAs than to random miRNAs; the miRNA was therefore more likely to be associated with the investigated disease.
2.12. Calculation of the confidence to pick the diseases regulated by specific miRNA
Similarly, given a specific miRNA, we reasoned that a regulated disease deserves a stronger integrated similarity than with random diseases. Here, we introduced the confidence in , , defined as follows:
| (14) |
A larger could suggest that the disease under investigation was more likely to be associated with the given miRNA.
2.13. Score conversion
For a given disease, the confidence in specific miRNA could be compared with each other, with higher confidence indicating higher probability to be an associated miRNA. However, they could not be directly compared across diseases, because the richness varied greatly from disease to disease. For example, disease A had 20 known related miRNAs and the investigated miRNA was ranked 205th and disease B had 200 known related miRNAs and the second miRNA we investigated was ranked 205th as well. It was obvious that the second pair was more likely to associate with each other and was more likely to be an associated miRNA. Similarly, it was unreasonable to compare confidence across miRNAs. Therefore, a score conversion procedure would be needed to convert the confidence into probabilities. For a given disease, we firstly sorted all miRNAs in a descending order of confidence calculated by NDAMDA. Then, at each C m(i), we computed the corresponding precision defined as Precision = R/H, where R and H were the richness of given disease (total number of known related miRNAs) and the number of all miRNAs with higher or equal rank to C m(i), respectively. In a similar manner, score conversion for a given miRNA was applied on after sorting all diseases in a descending order.
Finally, we integrated two converted scores by averaging them, yielding the final score:
where was the final score between miRNA and disease . was the precision of when given disease and was the precision of when given miRNA .
3. RESULT
3.1. Performance evaluation
We implemented local and global LOOCV to evaluate the prediction accuracy of NDAMDA and 6 previous computational models: WBSMDA,20 RLSMDA,24 MCMDA,28 HDMP,21 RWRMDA 19 and MiRAI.22 In LOOCV, each known association was used as the validation sample and the remaining known associations were regarded as the training samples. The miRNA‐disease pairs without any known association evidence were considered as candidate samples. The known miRNA‐disease associations were obtained from the HMDD v2.0 database.31 The association scores of all miRNA‐disease pairs would be returned by NDAMDA. In global LOOCV, the score of the validation sample was compared with all the candidate samples, while in local LOOCV, the score was compared with candidate samples for the investigated disease.
In fivefold cross‐validation, the known miRNA‐disease associations were randomly partitioned into 5 equally sized subsets. Each subset was retained as the validation set in turn, and the remaining 4 subsets were used as the training set. Still, the miRNA‐disease pairs without known association evidence were regarded as the candidate samples. Then, the score of each sample in the validation set was ranked against the scores of all the candidate samples. This procedure was repeated 100 times to better estimate the mean and variance of NDAMDA's prediction accuracy. This repetition reduced the error in performance estimation as the result of fivefold cross‐validation depended on how the associations were partitioned.
In both LOOCV and fivefold cross‐validation, the model would be deemed to make a correct prediction for a validation sample, if its rank exceeded a given threshold. Furthermore, we drew receiver operating characteristics (ROC) curve by plotting the true‐positive rate (TPR) against the false‐positive rate (FPR) at various thresholds. The true‐positive rate is also known as sensitivity which represents the percentage of the validation samples ranked higher than the threshold. The false‐positive is calculated as (1‐specificity), where specificity denotes the percentage of candidate miRNA‐disease pairs ranked lower than the threshold. We calculated the area under the ROC curve (AUC) to evaluate the prediction ability of NDAMDA. AUC = 1 indicates the model has perfect prediction performance; AUC = 0.5 implies the model has random performance.
The performance comparison in local and global LOOCV is shown in Figure 2. RWRMDA was not included in global LOOCV, because it was a local method, unable to uncover potentially associated miRNAs for all diseases simultaneously. In addition, global LOOCV was not applicable to for MiRAI, because the association score for a miRNA‐disease pair calculated by the model was highly positively correlated with the number of known associated miRNAs for the disease. This means that the association scores for different diseases were not comparable. The cross‐validation results showed that in global LOOCV, the AUCs of NDAMDA, MCMDA, RLSMDA, HDMP, WBSMDA were 0.8920, 0.8749, 0.8426, 0.8366 and 0.8030, respectively; in local LOOCV, NDAMDA, MCMDA, RLSMDA, HDMP, WBSMDA, RWRMDA and MiRAI achieved AUCs of 0.8062, 0.7718, 0.6953, 0.7702, 0.8031, 0.7891 and 0.6299, respectively. In addition, the average AUC of NDAMDA (0.8935 ± 0.0009) exceeded the average AUCs of MCMDA (0.8767 ± 0.0011), RLSMDA (0.8569 ± 0.0020), HDMP (0.8342 ± 0.0010) and WBSMDA (0.8185 ± 0.0009), indicating the superior performance of NDAMDA.
Figure 2.

AUC of global LOOCV (left) compared with HGIMDA, RLSMDA, HDMP and WBSMDA; AUC of local LOOCV (right) compared with MCMDA, RLSMDA, HDMP, WBSMDA and MiRAI. As a result, NDAMDA achieved AUCs of 0.8920 and 0.8062 in the global and local LOOCV, which exceed all the previous classical models. LOOCV, leave‐one‐out cross‐validation
3.2. Case studies
To demonstrate the sound prediction accuracy of our method, we further carried out 3 types of case studies on 5 important diseases. In the first type of case studies, the top 10 and top 50 predicted miRNAs for the investigated diseases were validated by another two miRNA‐disease databases, namely dbDEMC 29 and miR2Disease.30
Breast cancer is the most commonly diagnosed in females. With more than 1 million new incidences every year, breast cancer is ranked as the second most frequent cancer type when considering both sexes together.34 Incidence rates are high in most of the developed areas, and more than half of the cases are in industrialized countries.35 It is the leading cause of death in females aged 20‐59.36 With the rapid development of high‐throughput sequencing technologies, researchers have identified plenty of miRNAs associated with breast cancer. For example, higher levels of circulating miR‐122 specifically predicted metastatic recurrence in patients with stage II‐III breast cancer.37 Besides, miR‐155 was up‐regulated greater than twofold in breast cancer compared with normal adjacent tissue (NAT),38 while a decreased level of serum miR‐155 was found after surgery and 4 cycles of chemotherapy.39 Here, we implemented NDAMDA to identify potentially related miRNAs for brest neoplasms. As a result, 9 of the top 10 and 42 of the top 50 predicted miRNAs were verified by experimental literatures from dbDEMC and miR2Disease (See Table 1).
Table 1.
The top 50 predicted miRNAs associated with breast neoplasms by sorting the association probabilities calculated by NDAMDA
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa‐mir‐16 | dbdemc | hsa‐mir‐181 | Unconfirmed |
| hsa‐mir‐1247 | Unconfirmed | hsa‐mir‐181c | Dbdemc |
| hsa‐mir‐345 | dbdemc | hsa‐mir‐100 | Dbdemc |
| hsa‐mir‐143 | dbdemc;miR2Disease | hsa‐let‐7c | Dbdemc |
| hsa‐mir‐215 | dbdemc | hsa‐mir‐1302 | Unconfirmed |
| hsa‐mir‐150 | dbdemc | hsa‐mir‐107 | Dbdemc |
| hsa‐mir‐15a | dbdemc | hsa‐mir‐503 | Dbdemc |
| hsa‐mir‐15b | dbdemc | hsa‐mir‐483 | Dbdemc |
| hsa‐mir‐10b | dbdemc;miR2Disease | hsa‐mir‐33a | Unconfirmed |
| hsa‐mir‐141 | dbdemc;miR2Disease | hsa‐mir‐422a | Dbdemc |
| hsa‐mir‐21 | dbdemc;miR2Disease | hsa‐mir‐200c | dbdemc;miR2Disease |
| hsa‐mir‐198 | dbdemc | hsa‐mir‐20a | miR2Disease |
| hsa‐mir‐590 | dbdemc | hsa‐mir‐133a | Dbdemc |
| hsa‐mir‐200a | dbdemc;miR2!Disease | hsa‐mir‐498 | Dbdemc |
| hsa‐mir‐29a | dbdemc | hsa‐mir‐145 | dbdemc;miR2Disease |
| hsa‐mir‐26b | dbdemc | hsa‐mir‐200b | dbdemc;miR2Disease |
| hsa‐mir‐675 | Unconfirmed | hsa‐let‐7d | dbdemc;miR2Disease |
| hsa‐mir‐221 | dbdemc;miR2Disease | hsa‐let‐7b | Dbdemc |
| hsa‐mir‐765 | dbdemc | hsa‐mir‐942 | Unconfirmed |
| hsa‐let‐7a | dbdemc;miR2Disease | hsa‐mir‐518a | Dbdemc |
| hsa‐mir‐29b | dbdemc;miR2Disease | hsa‐mir‐181b | dbdemc;miR2Disease |
| hsa‐mir‐17 | miR2Disease | hsa‐mir‐99b | Dbdemc |
| hsa‐mir‐195 | dbdemc;miR2Disease | hsa‐mir‐125a | dbdemc;miR2Disease |
| hsa‐mir‐1 | dbdemc | hsa‐mir‐27b | Dbdemc |
| hsa‐mir‐103b | Unconfirmed | hsa‐let‐7 g | Dbdemc |
The first column records top 1‐25 related miRNAs. The second column records the top 26‐50 related miRNAs.
Oesophageal cancer is the eighth most common cancer worldwide (accounting for about 500 000 new cases every year) and the sixth most common cause of death by cancers (with 400 000 deaths each year).34 Moreover, cancer in the oesophagus is usually 3‐4 times more common among males than females and has a very low 5‐year survival rate: only 16% in the United States and 10% in Europe.35 Squamous cell carcinoma and adenocarcinoma, the two main types of oesophageal cancer, are mainly caused by overweight, obesity and chronic gastro‐oesophageal reflux disease (GERD).40 Researchers have identified several miRNAs associated with oesophagus cancer. For instance, using real‐time RT‐PCR, a research group studied miRNA‐21 levels in serum samples from patients with oesophageal squamous cell carcinomas (ESCC); they found that the patients’ serum concentration of miRNA‐21 was remarkably higher than that of healthy controls and that a significant reduction in the concentration was observed in patients when applied surgery or chemotherapy.41, 42 Another research group noticed that the expression of exosomal miR‐21 was up‐regulated in serum from patients with ESCC compared with serum from patients with benign diseases.43 Among the top 10 and 50 potential oesophageal cancer‐related miRNAs, respectively, 8 and 43 miRNA‐disease‐predicted associations were supported by database evidence (See Table 2).
Table 2.
The top 50 predicted miRNAs associated with oesophageal neoplasms by sorting the association probabilities calculated by NDAMDA
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa‐mir‐146a | dbdemc | hsa‐mir‐765 | Dbdemc |
| hsa‐mir‐133b | dbdemc | hsa‐mir‐29b | Dbdemc |
| hsa‐mir‐1247 | Unconfirmed | hsa‐mir‐195 | Dbdemc |
| hsa‐mir‐205 | dbdemc;miR2Disease | hsa‐let‐7a | Dbdemc |
| hsa‐mir‐152 | dbdemc | hsa‐mir‐17 | Dbdemc |
| hsa‐mir‐345 | dbdemc | hsa‐mir‐1 | Dbdemc |
| hsa‐mir‐143 | dbdemc | hsa‐mir‐181 | Unconfirmed |
| hsa‐mir‐215 | dbdemc | hsa‐mir‐103b | Unconfirmed |
| hsa‐mir‐148b | dbdemc | hsa‐mir‐181c | dbdemc |
| hsa‐mir‐449b | Unconfirmed | hsa‐mir‐100 | dbdemc |
| hsa‐mir‐15a | dbdemc | hsa‐mir‐1302 | Unconfirmed |
| hsa‐mir‐150 | dbdemc | hsa‐mir‐483 | dbdemc |
| hsa‐mir‐140 | dbdemc | hsa‐let‐7c | dbdemc |
| hsa‐mir‐10b | dbdemc | hsa‐mir‐20a | dbdemc |
| hsa‐mir‐1972 | Unconfirmed | hsa‐mir‐422a | dbdemc |
| hsa‐mir‐15b | dbdemc | hsa‐mir‐145 | dbdemc |
| hsa‐mir‐21 | dbdemc;miR2 Disease | hsa‐mir‐200b | dbdemc |
| hsa‐mir‐198 | dbdemc | hsa‐mir‐107 | dbdemc;miR2Disease |
| hsa‐mir‐141 | dbdemc | hsa‐let‐7b | dbdemc |
| hsa‐mir‐590 | dbdemc | hsa‐mir‐133a | dbdemc |
| hsa‐mir‐200a | dbdemc | hsa‐mir‐200c | dbdemc |
| hsa‐mir‐29a | dbdemc | hsa‐let‐7d | dbdemc |
| hsa‐mir‐26b | dbdemc | hsa‐mir‐498 | dbdemc |
| hsa‐mir‐675 | Unconfirmed | hsa‐mir‐503 | dbdemc |
| hsa‐mir‐221 | dbdemc | hsa‐mir‐181b | dbdemc |
The first column records top 1‐25 related miRNAs. The second column records the top 26‐50 related miRNAs.
Lymphoma cancer begins in cells of the immune system and can be divided into two main categories: Hodgkin lymphoma and non‐Hodgkin lymphoma, which accounts for 90 per cent of all lymphomas.44, 45 Hodgkin lymphoma can be identified by the presence of a type of cell called the Reed‐Sternberg cell, and non‐Hodgkin consists of a large, multiple group of cancers of immune system cells.46, 47 Recently, researcher found that the expression of miR‐21 in plasma of patient with lymphoma group significantly correlated with their serum LDH level and the higher expressions of miR‐21, miR‐155 and miR‐210 in plasma of patients with lymphoma were significantly higher.48 It was also reported that miR‐203, miR‐218, miR‐181a, miR19a and miR17 were found to be associated with lymphoma: the former 3 miRNAs functioned as tumour suppressors, and the latter two were found to up‐regulate oncogenes for lymphoma.49 This finding coincided with the generally accepted idea that canine lymphoma is a common spontaneous tumour with great similarities to human lymphoma.50 Similarly, we used dbDEMC and miR2Disease to validate the potentially associated miRNAs for lymphoma, and 9 of the top 10 and 36 of the top 50 candidate miRNAs were examined by two databases (See Table 3).
Table 3.
The top 50 predicted miRNAs associated with lymphoma by sorting the association probabilities calculated by NDAMDA
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa‐mir‐1247 | Unconfirmed | hsa‐mir‐133a | dbdemc |
| hsa‐mir‐215 | dbdemc | hsa‐mir‐151 | miR2Disease |
| hsa‐mir‐15b | dbdemc | hsa‐mir‐376c | Unconfirmed |
| hsa‐mir‐10b | dbdemc | hsa‐mir‐181d | dbdemc |
| hsa‐mir‐21 | dbdemc;miR2Disease | hsa‐mir‐23a | dbdemc |
| hsa‐mir‐200a | dbdemc | hsa‐mir‐659 | Unconfirmed |
| hsa‐let‐7a | dbdemc | hsa‐let‐7d | dbdemc |
| hsa‐mir‐26b | dbdemc | hsa‐mir‐422a | dbdemc |
| hsa‐mir‐221 | dbdemc;miR2Disease | hsa‐mir‐10a | dbdemc;miR2Disease |
| hsa‐mir‐17 | dbdemc;miR2Disease | hsa‐mir‐483 | Unconfirmed |
| hsa‐mir‐33a | dbdemc | hsa‐mir‐149 | dbdemc;miR2Disease |
| hsa‐mir‐195 | dbdemc | hsa‐mir‐193b | Unconfirmed |
| hsa‐mir‐503 | dbdemc | hsa‐mir‐301b | Unconfirmed |
| hsa‐mir‐130a | dbdemc | hsa‐mir‐1323 | Unconfirmed |
| hsa‐mir‐103b | Unconfirmed | hsa‐let‐7b | dbdemc |
| hsa‐mir‐1 | dbdemc | hsa‐mir‐20a | dbdemc;miR2Disease |
| hsa‐mir‐181c | dbdemc | hsa‐mir‐26a | dbdemc |
| hsa‐mir‐107 | dbdemc | hsa‐let‐7 g | dbdemc |
| hsa‐let‐7c | dbdemc | hsa‐mir‐31 | dbdemc |
| hsa‐mir‐29b | dbdemc | hsa‐mir‐181b | dbdemc |
| hsa‐mir‐498 | Unconfirmed | hsa‐mir‐410 | Unconfirmed |
| hsa‐mir‐200c | dbdemc | hsa‐mir‐125a | dbdemc |
| hsa‐mir‐1302 | Unconfirmed | hsa‐mir‐200b | dbdemc |
| hsa‐mir‐942 | Unconfirmed | hsa‐mir‐204 | dbdemc |
| hsa‐mir‐518a | Unconfirmed | hsa‐mir‐433 | Unconfirmed |
The first column records top 1‐25 related miRNAs. The second column records the top 26‐50 related miRNAs.
In addition to the above 3 cancers, we used NDAMDA to prioritize candidate miRNAs for all diseases in HMDD v2.0 and the results are included in Table S1.
To assess the ability of NDAMDA in predicting potentially related miRNAs for diseases without any known associated miRNAs, we carried out another case study on prostate cancer. Its associated miRNAs were removed from the training set, and the rest known miRNA‐disease associations were used to train NDAMDA. In this manner, potentially related miRNAs for prostate cancer were uncovered only using the information of other diseases‐related miRNAs and the similarity measures. Subsequently, the top 50 prediction outcomes were confirmed with HMDD v2.0, dbDEMC and miR2Disease. As shown in Table 4, 8 of the top 10, 18 of the top 20 and 43 of the top 50 candidate miRNAs were verified.
Table 4.
The top 50 predicted miRNAs associated with prostate neoplasms by sorting the association probabilities calculated by NDAMDA
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa‐mir‐21 | dbdemc;miR2Disease | hsa‐mir‐31 | dbdemc;miR2Disease |
| hsa‐mir‐155 | dbdemc | hsa‐mir‐199a | dbdemc;miR2Disease |
| hsa‐mir‐146a | miR2Disease | hsa‐mir‐9 | dbdemc |
| hsa‐mir‐125b | dbdemc;miR2Disease;HMDD | hsa‐mir‐181a | dbdemc;miR2Disease |
| hsa‐mir‐17 | miR2Disease | hsa‐mir‐133a | dbdemc |
| hsa‐mir‐20a | miR2Disease | hsa‐mir‐210 | miR2Disease |
| hsa‐mir‐34a | dbdemc;miR2Disease | hsa‐let‐7b | dbdemc;miR2Disease |
| hsa‐mir‐145 | dbdemc;miR2Disease;HMDD | hsa‐mir‐200a | dbdemc |
| hsa‐mir‐221 | dbdemc;miR2Disease | hsa‐mir‐200c | dbdemc |
| hsa‐mir‐18a | Unconfirmed | hsa‐mir‐181b | dbdemc;miR2Disease |
| hsa‐mir‐16 | dbdemc;miR2Disease | hsa‐mir‐142 | Unconfirmed |
| hsa‐mir‐92a | Unconfirmed | hsa‐mir‐150 | dbdemc |
| hsa‐mir‐126 | dbdemc;miR2Disease | hsa‐mir‐34c | dbdemc |
| hsa‐mir‐19b | dbdemc;miR2Disease | hsa‐let‐7c | dbdemc;miR2Disease |
| hsa‐mir‐15a | dbdemc;miR2Disease | hsa‐mir‐146b | Unconfirmed |
| hsa‐mir‐19a | dbdemc | hsa‐mir‐122 | Unconfirmed |
| hsa‐mir‐29a | dbdemc;miR2Disease | hsa‐mir‐106b | dbdemc |
| hsa‐mir‐1 | dbdemc | hsa‐mir‐182 | dbdemc;miR2Disease |
| hsa‐mir‐222 | dbdemc;miR2Disease | hsa‐let‐7d | dbdemc;miR2Disease |
| hsa‐mir‐143 | dbdemc;miR2Disease | hsa‐mir‐141 | miR2Disease |
| hsa‐mir‐29b | dbdemc;miR2Disease | hsa‐let‐7e | dbdemc |
| hsa‐let‐7a | dbdemc;miR2Disease | hsa‐mir‐133b | dbdemc |
| hsa‐mir‐200b | Unconfirmed | hsa‐mir‐214 | dbdemc;miR2Disease |
| hsa‐mir‐223 | dbdemc;miR2Disease | hsa‐mir‐203 | Unconfirmed |
| hsa‐mir‐29c | dbdemc | hsa‐mir‐30a | miR2Disease |
The disease's associated miRNAs were removed from the training set, and the rest known miRNA‐disease associations were used to train NDAMDA. Subsequently, the top 50 prediction outcomes were confirmed with HMDD v2.0, dbDEMC and miR2Disease. The first column records top 1‐25 related miRNAs. The second column records the top 26‐50 related miRNAs.
In the final case study, we fitted our model with the miRNA‐disease association dataset from HMDD v1.0, the old version of the HMDD database. This case study was meant to demonstrate NDAMDA's robust prediction ability to various datasets. Hepatocellular carcinoma was chosen as the investigated disease; its potentially associated miRNAs were identified by NDAMDA and validated against HMDD v2.0, dbDEMC and miR2Disease. As a result, 42 of the top 50 candidates were confirmed by experimental evidence from the databases (See Table 5). Taking the 1st candidate miR‐155 as an example, it inhibited HBV infection in human hepatoma cells through enhancing innate antiviral immunity; the ectopic expression of miR‐155 up‐regulated the expression of several IFN‐inducible antiviral genes in human hepatoma cells.51
Table 5.
The top 50 predicted miRNAs associated with hepatocellular carcinoma by sorting the association probabilities calculated by NDAMDA
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa‐mir‐155 | miR2Disease;HMDD | hsa‐mir‐133a | miR2Disease |
| hsa‐mir‐16 | miR2Disease;HMDD | hsa‐mir‐150 | miR2Disease;HMDD |
| hsa‐mir‐208 | Unconfirmed | hsa‐mir‐24 | miR2Disease;HMDD |
| hsa‐let‐7a | miR2Disease;HMDD | hsa‐mir‐132 | miR2Disease |
| hsa‐mir‐15a | miR2Disease;HMDD | hsa‐mir‐141 | miR2Disease;HMDD |
| hsa‐mir‐598 | Unconfirmed | hsa‐mir‐9 | miR2Disease |
| hsa‐mir‐539 | Unconfirmed | hsa‐mir‐29c | HMDD |
| hsa‐mir‐550 | Unconfirmed | hsa‐mir‐15b | HMDD |
| hsa‐mir‐652 | Unconfirmed | hsa‐mir‐181a | miR2Disease;HMDD |
| hsa‐let‐7b | miR2Disease;HMDD | hsa‐mir‐210 | HMDD |
| hsa‐mir‐328 | miR2Disease | hsa‐mir‐30c | miR2Disease;HMDD |
| hsa‐let‐7c | miR2Disease;HMDD | hsa‐mir‐107 | miR2Disease;HMDD |
| hsa‐let‐7i | HMDD | hsa‐mir‐194 | miR2Disease |
| hsa‐let‐7d | miR2Disease;HMDD | hsa‐mir‐30d | HMDD |
| hsa‐mir‐411 | Unconfirmed | hsa‐mir‐373 | HMDD |
| hsa‐mir‐29b | HMDD | hsa‐mir‐205 | miR2Disease;HMDD |
| hsa‐mir‐143 | miR2Disease | hsa‐mir‐30a | miR2Disease;HMDD |
| hsa‐mir‐181b | miR2Disease;HMDD | hsa‐mir‐200c | HMDD |
| hsa‐mir‐126 | miR2Disease;HMDD | hsa‐mir‐25 | miR2Disease;HMDD |
| hsa‐let‐7 g | miR2Disease;HMDD | hsa‐mir‐196a | HMDD |
| hsa‐let‐7f | miR2Disease;HMDD | hsa‐mir‐191 | HMDD |
| hsa‐mir‐146b | HMDD | hsa‐mir‐32 | Unconfirmed |
| hsa‐mir‐106b | miR2Disease;HMDD | hsa‐mir‐93 | miR2Disease;HMDD |
| hsa‐mir‐29a | HMDD | hsa‐mir‐451 | Unconfirmed |
| hsa‐mir‐214 | miR2Disease;HMDD | hsa‐mir‐34c | HMDD |
The model was fitted by the miRNA‐disease association dataset from HMDD v1.0, the old version of the HMDD database. Potentially associated miRNAs for the disease were validated against HMDD v2.0, dbDEMC and miR2Disease. The first column records top 1‐25 related miRNAs. The second column records the top 26‐50 related miRNAs.
4. DISCUSSIONS
The experimental methods for identifying disease‐miRNA associations are expensive and time‐consuming. Encouragingly, plenty of computational methods for predicting disease‐related miRNAs have been proposed in recent years. To predict potentially related miRNAs for diseases at a higher accuracy than previous methods, we developed a network analysis‐based method named NDAMDA for prioritizing potentially disease‐related miRNAs. The model achieved sound prediction performance throughout global and local LOOCV, fivefold cross‐validation and 3 types of case studies on 5 major human diseases. Therefore, NDAMDA would be a useful resource for researches to discover associations between diseases and miRNAs.
In our work, the case studies were based on cancers. The hallmarks of cancer are one of the most widely acknowledged organizing principles for research on cancer, and currently, ten hallmarks have been identified to represent the acquired capabilities that distinguish cancer from normal tissue.52 These hallmarks are (1) self‐sufficiency in growth signals; (2) insensitivity to antigrowth signals; (3) evading apoptosis; (4) limitless replicative potential; (5) sustained angiogenesis; (6) tissue invasion and metastasis; (7) abnormal metabolic pathways; (8) evading the immune system; (9) chromosome abnormalities and unstable DNA; and (10) inflammation.52 Association between cancer hallmarks and genes has been indicated by the literatures.52, 53, 54 For example, in our work, miR‐155 obtained the highest score in the fifth case study on hepatocellular carcinoma; according to the data from NanoString's Hallmarks of Cancer Panel collection (https://www.nanostring.com/), two of the miRNA's gene targets, MUS81 and FLT1, have been found to be associated with Hallmark (9) and Hallmark (5), respectively. Other examples include miR‐16, miR‐1247 and miR‐21, which had the highest scores in the first, third and fourth case studies, respectively. APP, ATG12 and ATF2 are the common targets for these 3 miRNAs and have been identified to be associated with Hallmark (10). In future work, we would consider to involve the information of cancer hallmark‐gene associations in our analysis and examine whether this information could enhance the accuracy of our algorithm.
The reliable performance of NDAMDA could be attributed to several factors as follows. Firstly, heterogeneous datasets (disease‐miRNA associations from HMDD, miRNA functional similarity, disease semantic similarity and Gaussian interaction profile kernel similarity for diseases and miRNAs) were integrated to construct the informative network for prediction. Secondly, we used the adjusted network distance and the algorithm for calculating the confidence in a specific miRNA and the confidence in a specific disease. Finally, we used a score conversion procedure that considered the variation in the number of related miRNAs for different diseases.
Yet, there still exist limitations in NDAMDA. Firstly, more known miRNA‐disease associations are necessary for building a more accurate adjacency network and improving the performance of NDAMDA. Secondly, the model might cause bias to miRNAs with more known related diseases, as it was based on the assumption that the functional similar miRNAs are more likely to be connected with similar diseases. Thirdly, NDAMDA might be not applicable to the diseases whose associated miRNAs tend to distribute randomly in the network, and how to integrate two scores to calculate the final score in a more reasonable way should be studied in future. Finally, although NDAMDA exhibited a commendable predictive performance with the currently available 5430 associations between 495 miRNAs and 383 diseases from HMDD v2.0, this association dataset was still limited; it contained a large amount of unlabelled data and only a very small amount (2.86%) of labelled data, which negatively affected the prediction accuracy. As experimental research continues, more miRNA‐disease associations were expected to be biologically verified in future. With an improved association dataset, our model would be able to uncover disease‐related miRNAs at an even higher accuracy.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
Supporting information
ACKNOWLEDGEMENTS
XC was supported by National Natural Science Foundation of China under Grant Nos. 61772531 and 11631014.
Chen X, Wang L‐Y, Huang L. NDAMDA: Network distance analysis for MiRNA‐disease association prediction. J Cell Mol Med. 2018;22:2884–2895. https://doi.org/10.1111/jcmm.13583
Xing Chen and Le‐Yi Wang wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
REFERENCES
- 1. Lefebvre L, Sol D. Brains, lifestyles and cognition: are there general trends? Brain Behav Evol. 2008;72:135‐144. [DOI] [PubMed] [Google Scholar]
- 2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281‐297. [DOI] [PubMed] [Google Scholar]
- 3. Vonk J, Shackelford TK. The Oxford handbook of comparative evolutionary psychology In: Nathan PE, ed. Oxford Library of Psychology. New York: Oxford University Press; 2012:574. [Google Scholar]
- 4. Pasquinelli AE, Ruvkun G. Control of developmental timing by microRNAs and their targets. Annu Rev Cell Dev Biol. 2002;18:495‐513. [DOI] [PubMed] [Google Scholar]
- 5. Kozomara A, Griffiths‐Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68‐D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673‐676. [DOI] [PubMed] [Google Scholar]
- 7. Taganov KD, Boldin MP, Chang K‐J, Baltimore D. NF‐κB‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci. 2006;103:12481‐12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J‐F, Mandel EM, Thomson JM, et al. The role of microRNA‐1 and microRNA‐133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen C‐Z, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83‐86. [DOI] [PubMed] [Google Scholar]
- 10. Carleton M, Cleary MA, Linsley PS. MicroRNAs and cell cycle regulation. Cell Cycle. 2007;6:2127‐2132. [DOI] [PubMed] [Google Scholar]
- 11. Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581‐588. [DOI] [PubMed] [Google Scholar]
- 12. Petrocca F, Visone R, Onelli MR, et al. E2F1‐regulated microRNAs impair TGFβ‐dependent cell‐cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272‐286. [DOI] [PubMed] [Google Scholar]
- 13. Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol. 2013;774:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065‐7070. [DOI] [PubMed] [Google Scholar]
- 16. Jiang Q, Hao Y, Wang G, et al. Prioritization of disease microRNAs through a human phenome‐microRNAome network. BMC Syst Biol. 2010;4:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mork S, Pletscher‐Frankild S, Palleja Caro A, Gorodkin J, Jensen LJ. Protein‐driven inference of miRNA‐disease associations. Bioinformatics. 2014;30:392‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi H, Xu J, Zhang G, et al. Walking the interactome to identify human miRNA‐disease associations through the functional link between miRNA targets and disease genes. BMC Syst Biol. 2013;7:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen X, Liu M‐X, Yan G‐Y. RWRMDA: predicting novel human microRNA–disease associations. Mol BioSyst. 2012;8:2792‐2798. [DOI] [PubMed] [Google Scholar]
- 20. Chen X, Yan CC, Zhang X, et al. WBSMDA: within and between score for MiRNA‐disease association prediction. Sci Rep. 2016;6:21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xuan P, Han K, Guo M, et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE. 2013;8:e70204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pasquier C, Gardes J. Prediction of miRNA‐disease associations with a vector space model. Sci Rep. 2016;6:27036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu J, Li C‐X, Lv J‐Y, et al. Prioritizing candidate disease miRNAs by topological features in the miRNA target–dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011;10:1857‐1866. [DOI] [PubMed] [Google Scholar]
- 24. Chen X, Yan G‐Y. Semi‐supervised learning for potential human microRNA‐disease associations inference. Sci Rep. 2014;4:5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Yan CC, Zhang X, You Z‐H, Huang Y‐A, Yan G‐Y. HGIMDA: heterogeneous graph inference for miRNA‐disease association prediction. Oncotarget. 2016;7:65257‐65269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xuan P, Han K, Guo Y, et al. Prediction of potential disease‐associated microRNAs based on random walk. Bioinformatics. 2015;31:1805‐1815. [DOI] [PubMed] [Google Scholar]
- 27. Chen X, Yan CC, Zhang X, et al. RBMMMDA: predicting multiple types of disease‐microRNA associations. Sci Rep. 2015;5:13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li JQ, Rong ZH, Chen X, Yan GY, You ZH. MCMDA: matrix completion for MiRNA‐disease association prediction. Oncotarget. 2017;8:21187‐21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Z, Ren F, Liu C, et al. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genom. 2010;11:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98‐D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y, Qiu C, Tu J, et al. HMDD, v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070‐D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA‐associated diseases. Bioinformatics. 2010;26:1644‐1650. [DOI] [PubMed] [Google Scholar]
- 33. Lipscomb CE. Medical subject headings (MeSH). Bull Med Libr Assoc. 2000;88:265. [PMC free article] [PubMed] [Google Scholar]
- 34. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975‐2014, featuring survival. J Natl Cancer Inst 2017;109: https://doi.org/10.1093/jnci/djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74‐108. [DOI] [PubMed] [Google Scholar]
- 36. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5‐29. [DOI] [PubMed] [Google Scholar]
- 37. Wu X, Somlo G, Yu Y, et al. De novo sequencing of circulating miRNAs identifies novel markers predicting clinical outcome of locally advanced breast cancer. J Transl Med. 2012;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J, Mao Q, Liu Y, Hao X, Zhang S, Zhang J. Analysis of miR‐205 and miR‐155 expression in the blood of breast cancer patients. Chin J Cancer Res 2013;25:46‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun Y, Wang M, Lin G, et al. Serum microRNA‐155 as a potential biomarker to track disease in breast cancer. PLoS ONE. 2012;7:e47003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 41. Cai EH, Gao YX, Wei ZZ, Chen WY, Yu P, Li K. Serum miR‐21 expression in human esophageal squamous cell carcinomas. Asian Pac J Cancer Prev. 2012;13:1563‐1567. [DOI] [PubMed] [Google Scholar]
- 42. Kurashige J, Kamohara H, Watanabe M, et al. Serum microRNA‐21 is a novel biomarker in patients with esophageal squamous cell carcinoma. J Surg Oncol. 2012;106:188‐192. [DOI] [PubMed] [Google Scholar]
- 43. Tanaka Y, Kamohara H, Kinoshita K, et al. Clinical impact of serum exosomal microRNA‐21 as a clinical biomarker in human esophageal squamous cell carcinoma. Cancer. 2013;119:1159‐1167. [DOI] [PubMed] [Google Scholar]
- 44. Gall EA, Mallory TB. Malignant lymphoma: a clinico‐pathologic survey of 618 cases. Am J Pathol. 1942;18:381‐429. [PMC free article] [PubMed] [Google Scholar]
- 45. Le Count ER. Lymphoma, a benign tumor representing a lymph gland in structure. J Exp Med. 1899;4:559‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non‐hodgkin lymphoma. Lancet. 2017;390:298‐310. [DOI] [PubMed] [Google Scholar]
- 47. Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin's disease associated antigen Ki‐1 in reactive and neoplastic lymphoid tissue: evidence that Reed‐Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848‐858. [PubMed] [Google Scholar]
- 48. Ge TT, Liang Y, Fu R, et al. [Expressions of miR‐21, miR‐155 and miR‐210 in plasma of patients with lymphoma and its clinical significance]. Zhongguo shi yan xue ye xue za zhi. 2012;20:305‐309. [PubMed] [Google Scholar]
- 49. Uhl E, Krimer P, Schliekelman P, Tompkins SM, Suter S. Identification of altered MicroRNA expression in canine lymphoid cell lines and cases of B‐ and T‐Cell lymphomas. Genes Chromosom Cancer. 2011;50:950‐967. [DOI] [PubMed] [Google Scholar]
- 50. Khanna C, Lindblad‐Toh K, Vail D, et al. The dog as a cancer model. Nat Biotechnol. 2006;24:1065‐1066. [DOI] [PubMed] [Google Scholar]
- 51. Su C, Hou Z, Zhang C, Tian Z, Zhang J. Ectopic expression of microRNA‐155 enhances innate antiviral immunity against HBV infection in human hepatoma cells. Virol J. 2011;8:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang E, Zaman N, McGee S, Milanese JS, Masoudi‐Nejad A, O'Connor‐McCourt M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin Cancer Biol. 2015;30:4‐12. [DOI] [PubMed] [Google Scholar]
- 53. Gao S, Tibiche C, Zou J, et al. Identification and construction of combinatory cancer hallmark‐based gene signature sets to predict recurrence and chemotherapy benefit in stage II colorectal cancer. JAMA Oncol. 2016;2:37‐45. [DOI] [PubMed] [Google Scholar]
- 54. Fu C, Li J, Wang E. Signaling network analysis of ubiquitin‐mediated proteins suggests correlations between the 26S proteasome and tumor progression. Mol BioSyst. 2009;5:1809‐1816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials