

SUMMARY
It is widely believed that inflammation associated with obesity has an important role in the development of type 2 diabetes. IκB kinase beta (IKKβ) is a crucial kinase that responds to inflammatory stimuli such as Tumor Necrosis Factor α (TNFα), by initiating a variety of intracellular signaling cascades, and is considered to be a key element in the inflammation-mediated development of insulin resistance. We show here, contrary to expectation, that IKKβ-mediated inflammation is a positive regulator of hepatic glucose homeostasis. IKKβ phosphorylates the spliced form of X-Box Binding Protein 1 (XBP1s) and increases the activity of XBP1s. We have used three experimental approaches to enhance the IKKβ activity in the liver of obese mice, and observed increased XBP1s activity, reduced ER stress, and a significant improvement in insulin sensitivity and consequently in glucose homeostasis. Our results reveal a beneficial role of IKKβ-mediated hepatic inflammation in glucose homeostasis.
ETOC blurb
Inflammatory signaling via IKKβ in the liver is beneficial for glucose homeostasis, running counter to the prevailing view that inflammation caused by obesity leads to insulin resistance
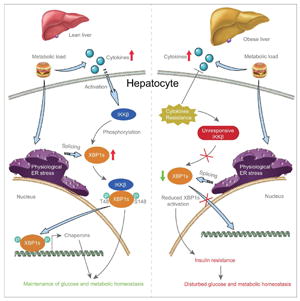
INTRODUCTION
According to projections of the World Health Organization, type 2 diabetes will affect 300 million people or more by the year 2025 (Wild et al., 2004; Xu et al., 2013). The total cost of diabetes and its complications to the global economy in 2010 was estimated at around $376 billion; this is expected to increase to $490 billion in 2030 (Hu, 2011). Despite enormous research efforts in the study of obesity and diabetes, the molecular mechanisms that lead to insulin resistance and type 2 diabetes associated with obesity is not fully understood.
A leading notion in the fields of obesity and type 2 diabetes is that obesity leads to increased inflammation in a variety of metabolically active tissues locally, initiates inflammatory stimuli such as Tumor Necrosis Factor α (TNFα) and other cytokines either locally or systemically, and that these stimuli in turn lead to the development of insulin resistance and type 2 diabetes (Gregor and Hotamisligil, 2011; Olefsky and Glass, 2010; Shoelson et al., 2006). To date no pharmacologic treatment has emerged that is based on blocking inflammation or the activity of inflammatory molecules in obesity.
Certain conditions, such as accumulation of misfolded proteins in the endoplasmic reticulum (ER) lumen, and uncontrolled nutrient and energy homeostasis, perturb ER homeostasis and lead to a condition defined as ER stress (Lee and Ozcan, 2014; Park and Ozcan, 2013; Walter and Ron, 2011). ER stress is marked by activation of a complex intracellular signaling network that is referred to as the unfolded protein response (UPR) (Gardner et al., 2013; Lee and Ozcan, 2014; Park and Ozcan, 2013; Walter and Ron, 2011).
IRE1, which is one of the main UPR signaling molecules, is activated by autophosphorylation, and it cleaves the full-length mRNA of the X-box binding protein 1 (XBP1), leading to the translation of the spliced form of XBP1 (XBP1s), a master regulator for ER homeostasis. Nuclear translocation and activity of XBP1s are substantially reduced in the liver of obese versus lean mice (Park et al., 2010). Decreased XBP1s activity plays a key role in the development of insulin resistance and type 2 diabetes in obese mice (Lee et al., 2011; Park et al., 2010; Zhou et al., 2011), and that reinstatement of XBP1s activity in the liver of obese and diabetic mice greatly increases insulin sensitivity, reduces blood glucose levels, and restores glucose homeostasis (Deng et al., 2013; Zhou et al., 2011).
Increased inflammation in obesity is also believed to contribute to the development of ER stress (Gregor and Hotamisligil, 2011; Zhang et al., 2008). However, we recently showed that the inflammatory signaling molecule p38 mitogen-activated protein kinase (p38 MAPK) - which is also activated by TNFα - in fact increases the activity of XBP1s and reduces ER stress; also that activation of p38 MAPK in the liver of obese and diabetic mice significantly enhances insulin sensitivity and improves glucose homeostasis.
IκB kinase beta (IKKβ) is one of the most important kinases that mediates the effects of general inflammatory stimuli inside the cell, and is a major downstream regulator of TNF signaling (Hacker and Karin, 2006; O’Dea and Hoffmann, 2009; Senftleben and Karin, 2002). TNF activates p38 MAPK and IKKβ simultaneously (Hacker and Karin, 2006; Karin, 2005b; O’Dea and Hoffmann, 2009). IKKβ was previously believed to cause ER stress. However, recent unexpected findings on the positive effects of TNF-p38 MAPK-XBP1s axis signaling on ER stress (Lee et al., 2011) led us to investigate whether IKKβ has any effect on XBP1s, and thus on ER stress signaling.
RESULTS
IKKβ Upregulates XBP1s Activity by Increasing Its Stability
To investigate whether IKKβ influences the activity of XBP1s, we co-expressed IKKβ and XBP1s in human embryonic kidney 293 (HEK293) cells by infecting the cells with an XBP1s-expressing adenovirus (Ad-XBP1s) and different doses of an adenovirus that expresses constitutively active IKKβ (Ad-ca-IKKβ). Overexpression of ca-IKKβ increased protein levels of XBP1s in a dose dependent manner (Figure 1A). Total IKKβ levels were up-regulated in response to Ad-ca-IKKβ infection (Figure 1A). Consequently, the nuclear levels of p65 (Figure S1A) and NF-κB target genes were also significantly up-regulated (Figures S1B).
Figure 1. IKKβ Increases XBP1s’ Protein Stability.
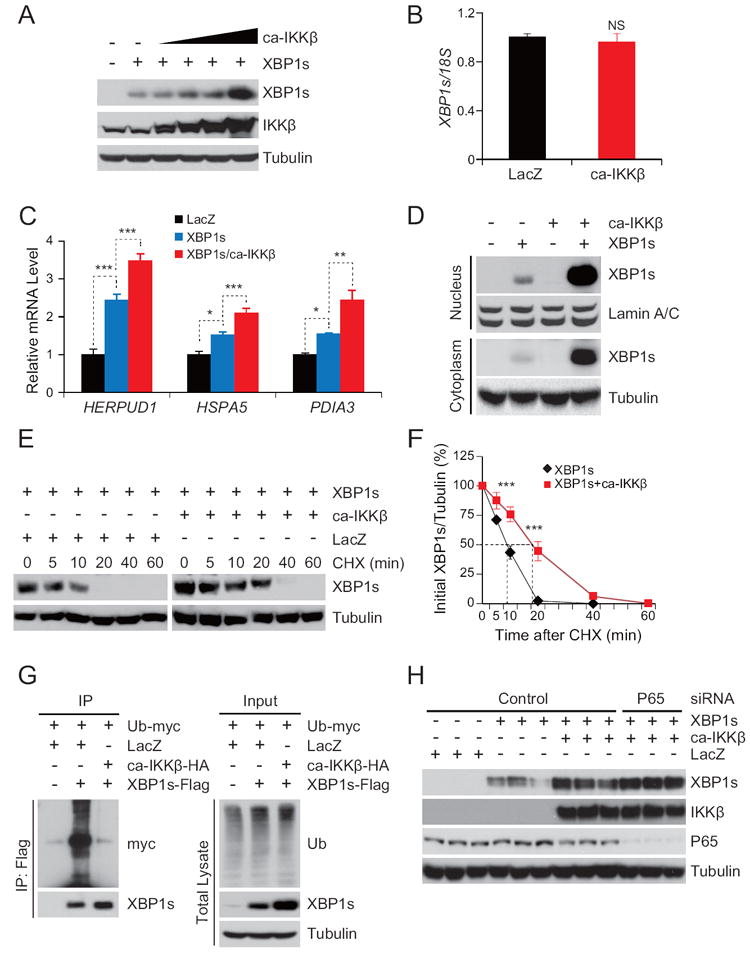
(A) XBP1s and IKKβ levels in HEK293 cells infected with XBP1s-expressing adenovirus (Ad-XBP1s) and different doses of constitutively active IKKβ expressing adenovirus (Ad-ca-IKKβ).
(B) Endogenous XBP1s mRNA levels in HEK293 cells infected with Ad-LacZ or Ad-ca-IKKβ.
(C) Gene expression levels of HERPUD1, HSPA5 and PDIA3 in HEK293 cells infected with Ad-XBP1s alone or together with Ad-ca-IKKβ. Cells infected with Ad-LacZ were used as control.
(D) Cytoplasmic and nuclear XBP1s protein levels from HEK293 cells infected with Ad-XBP1s alone or together with Ad-ca-IKKβ.
(E) HEK293 cells transfected with XBP1s alone or together with ca-IKKβ and further treated with cycloheximide (CHX, 20 μg ml-1) for the indicated time periods. XBP1s and tubulin protein levels were analyzed with immunoblotting. The blots from the cells transfected with XBP1s only were exposed for a longer time (left bands) and the blots from cells transfected with XBP1s and ca-IKKβ together were exposed for a shorter time (right bands) to achieve comparable autoradiographic signals for total XBP1s from both groups at the zero time point.
The results represented in A-E were reproduced in three independent experiments.
(F) The graph depicts the averaged ratio of the autoradiographic signals of XBP1s to tubulin levels in HEK293 cells from three independent experiments. These three independent experiments are shown in Figure 1E, Figure S1M and S1N. The dotted lines show the half-life of XBP1s protein.
(G) 293T cells were transfected with XBP1s-flag and Ub-myc with or without ca-IKKβ-HA. Ubiquitinylated XBP1s levels in HEK293 (left); The global ubiquitination levels in HEK293 cells were detected by western blotting using anti-Ub antibody (right). These results were reproduced in four independent experiments.
(H) XBP1s, IKKβ, p65 and tubulin protein levels in HEK293 cells, which were transfected with XBP1s alone or together with ca-IKKβ, after transfection with p65 and control siRNA. These results were reproduced in two independent experiments.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (B), one-way analysis of variance (ANOVA) (C), and two-way analysis of variance (ANOVA) with Bonferroni multiple-comparison analysis (F). *P < 0.05, **P < 0.01, ***P <0.001, NS: Nonsignificant.
We also performed the same experiment in four other cell lines, which all yielded similar results (Figures S1C-F). Co-expression of ca-IKKβ with human XBP1s (hXBP1s) has also increased the protein levels of hXBP1s (Figure S1G).
Next, we infected HEK293 cells with Ad-XBP1s alone or with Ad-ca-IKKβ, and analyzed the levels of endogenous XBP1s mRNA. IKKβ did not enhance endogenous XBP1s mRNA levels (Figure 1B). Analysis of mRNA levels of both exogenously and endogenously expressed XBP1s in other cells lines also did not show any alterations (Figure S1H and S1I). These results provided support that IKKβ increased protein levels of XBP1s without effecting the mRNA levels and thus without creating ER stress. Activating transcription factor-6 (ATF6) is another UPR transcription factor and cleaved form (ATF6n) is up-regulated in response to ER stress to re-establish ER homeostasis (Lee and Ozcan, 2014; Park and Ozcan, 2013). We next co-expressed ca-IKKβ with ATF6n. Unlike XBP1s, IKKβ did not alter ATF6n levels (Figure S1J). These results indicate that IKKβ has specific activity on XBP1s.
We also infected HEK293 cells with Ad-XBP1s alone, or with Ad-XBP1s plus Ad-ca-IKKβ, and analyzed level of XBP1s target genes and nuclear translocation of XBP1s. XBP1s target gene expression, including homocysteine-inducible ER stress protein (HERPUD1), heat shock protein 5 (HSPA5), and protein disulphide isomerase family A member 3 (PDIA3) was significantly increased at the presence of XBP1s (Figure 1C), which is further increased by ca-IKKβ co-expression (Figure 1C). IKKβ-expression also increased nuclear translocation of XBP1s (Figure 1D).
Next we sought to investigate whether other isoforms of IKK, IKKα and IKKγ, also has effects on XBP1s’ activity. Similar to IKKβ, IKKα also increased the XBP1s protein levels (Figure S1K). However, IKKγ did not lead to an increase in XBP1s protein levels (Figure S1L). Considering the major reported roles of IKKβ in regulation of metabolic homeostasis, we focused our research on further investigating the IKKβ.
Given that IKKβ increases the level of XBP1s protein without altering its mRNA levels (Figures 1A and 1B), we examined whether IKKβ affects the stability of the XBP1s protein. We first infected HEK293 cells with Ad-XBP1s alone, or with Ad-XBP1s plus Ad-ca-IKKβ, and then treated the cells with cycloheximide to inhibit global translation. In the absence of IKKβ expression, XBP1s started to degrade within 5 minutes of treatment, and was undetectable 20 minutes after adding the cycloheximide (Figure 1E); however, this degradation rate was significantly reduced when IKKβ and XBP1s were expressed together (Figure 1E). Densitometric analysis of western blot results from three independent experiments (Figure 1E, S1M and S1N) confirmed that XBP1s degradation was significantly inhibited by IKKβ (Figure 1F). The half-life of XBP1s was around 10 minutes in the absence of IKKβ; but when IKKβ was co-expressed with XBP1s, the half-life of XBP1s was prolonged to 20 minutes (Figure 1F). These results indicate that IKKβ increases the amount of XBP1s protein by enhancing its stability. Considering that XBP1s is recycled in a proteasome-dependent manner (Zhang et al., 2014), we also tested whether IKKβ affects the level of ubiquitination of XBP1s. A myc-tagged ubiquitin (Ub-myc)-expressing vector was transfected into cells with a flag-tagged XBP1s (XBP1s-flag), in the presence or absence of an HA-tagged constitutively active IKKβ (IKKβ-HA). We found that co-expression of IKKβ and XBP1s robustly reduced the ubiquitination of XBP1s, without affecting global ubiquitination levels in the cell (Figure 1G).
To investigate whether p65 signaling is involved in IKKβ–mediated XBP1s stabilization, we used a highly efficient p65 siRNA. (Figure 1H) and co-transfected XBP1s and IKKβ in p65 siRNA transfected cells; depletion of p65 did not block IKKβ–mediated upregulation of XBP1s protein levels (Figure 1H).
IKKβ Interacts with and Phosphorylates XBP1s
The increased protein level of XBP1s caused by IKKβ prompted us to hypothesize that IKKβ stabilizes XBP1s by direct modification, through a physical interaction. To test this hypothesis, we transfected cells ca-IKKβ-HA and XBP1s-flag expressing vectors and immunoprecipitated XBP1s from cell lysates; IKKβ was indeed co-immunoprecipitated with XBP1s (Figure 2A), which indicates that XBP1s and IKKβ interact. We also immunoprecipitated IKKβ and examined the precipitates for the presence of XBP1s. XBP1s was also co-immunoprecipitated with IKKβ (Figure 2B).
Figure 2. IKKβ Interacts with XBP1s and Phosphorylates It.
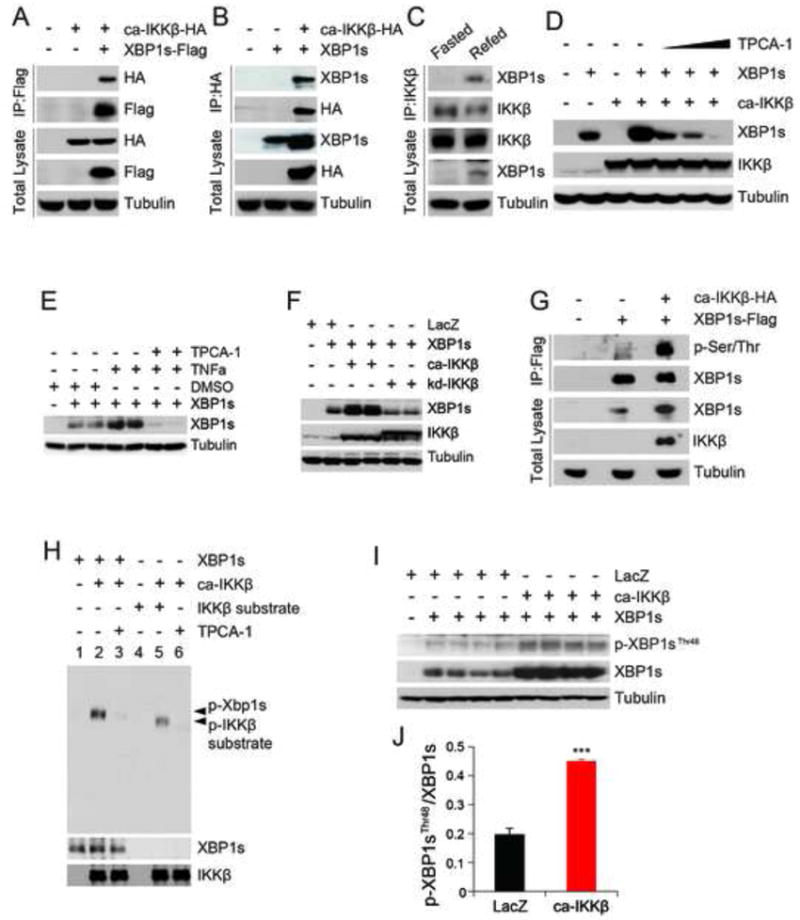
(A) Immunoblotting for HA-tagged ca-IKKβ and flag-tagged XBP1s proteins after immunoprecipitation of XBP1s from HEK293 cells.
(B) Immunoblotting for flag-tagged XBP1s and HA-tagged ca-IKKβ proteins after immunoprecipitation of IKKβ from HEK293 cells.
(C) XBP1s and IKKβ protein levels after immunoprecipitation of IKKβ from the liver lysates of 24 h fasted, or fasted for 24 hours and re-fed for 1 hour.
(D) XBP1s and tubulin protein levels in cells transfected with the indicated vectors and further treated with DMSO or TPCA-1 at doses (0.1, 1 and 10 μM).
(E) XBP1s and tubulin protein levels in HEK293 cells transfected with XBP1s and further treated with DMSO or TPCA-1 (20 μM) for 2 hours following incubating with vehicle or TNFα (100 ng ml-1) for 30 minutes.
(F) XBP1s, IKKβ and tubulin protein levels in HEK293 cells transfected with the indicated vectors.
(G) Phosphorylated XBP1s levels in HEK293 transfected with the indicated vectors. We specifically loaded three times more amounts of IP samples from XBP1s-transfected cells when compared to the XBP1s-ca-IKKβ co-transfected cells to achieve comparable total XBP1s autoradigraphical signal from each sample.
(H) Phospho-XBP1s levels after IP of XBP1s from IKKβ kinase assay.
(I) Phospho-XBP1sThr48 and total XBP1s protein levels in HEK293 cells infected with the indicated adenovirus.
(J) Densitometric quantification of ratio of Phospho-XBP1sThr48 to total XBP1s protein in Figure 2I.
Experiments in A, B, D and I were reproduced in three and in C, E, F, G and H were reproduced in two independent experiments.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test. **P < 0.01, ***P < 0.001.
We next investigated whether the interaction between XBP1s and IKKβ that we observed in vitro, exists between endogenous XBP1s and endogenous IKKβ in vivo. We have previously reported that re-feeding after fasting greatly increases XBP1s protein levels in the liver of lean mice (Park et al., 2010). To investigate whether endogenous XBP1s interacts with IKKβ during normal physiological processes, lean healthy mice were fasted for 24 hours, then re-fed for one hour, at which point liver lysates were processed for IKKβ immunoprecipitation; we found that IKKβ immunoprecipitation pulled down XBP1s (Figure 2C), which confirmed that endogenous XBP1s and endogenous IKKβ interact in the liver.
To determine whether IKKβ’s kinase activity is responsible for the increased stability and consequent enhanced protein levels of XBP1s, HEK293 cells were transfected with XBP1s-flag in the presence or absence of ca-IKKβ-HA; the cells were then treated with [(aminocarbony)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1), an IKKβ inhibitor. Inhibition of IKKβ‘s kinase activity greatly blocked the IKKβ-mediated increase in total protein levels of XBP1s (Figure 2D). To also investigate whether endogenous IKKβ can also affect XBP1s levels, we expressed XBP1s in cells and then treated with vehicle or with TNFα at the absence or presence of TPCA-1. TNFα stimulation increased XBP1s protein levels. However, pre-treatment with IKKβ inhibitor TPCA-1 blocked TNFα-induced up-regulation of XBP1s (Figure 2E). Next, we sought to investigate whether the IKKβ’s effect on XBP1s is mediated through its kinase activity. For this purpose, we compared the effects of ca-IKKβ and a mutant IKKβ that lacks kinase activity 8(kd-IKKβ). Expression of ca-IKKβ greatly upregulated XBP1s protein levels. However, kd-IKKβ, despite expression of more protein compared to ca-IKKβ, did not have any effect on XBP1s (Figure 2F). Furthermore, we also stimulated the cells with IL-1β, another cytokine that have ability to increase IKK activity. As shown in Figure S1O, stimulation of cells, which are transfected with XBP1s, led to a robust increase in XBP1s protein levels and IKKβ phosphorylation.
These findings then led us to ask whether IKKβ phosphorylates XBP1s: we transfected HEK293 cells with an XBP1s-flag in the presence or absence of IKKβ-HA, and immunoprecipitated XBP1s from the cell lysates. Use of a general anti-phosphoserine/threonine antibody revealed a robust, IKKβ-mediated increase in levels of XBP1s phosphorylation (Figure 2G).
Together, the above results indicate that elevated IKKβ activity enhances Ser/Thr phosphorylation of XBP1s. However to address whether IKKβ has a direct physical interaction with XBP1s, and whether it directly phosphorylates XBP1s, we transfected HEK293 cells with a flag-ca-IKKβ, then immunoprecipitated the ca-IKKβ protein from the cell lysates, and performed an in vitro IKKβ kinase assay, using a His-TF-XBP1s fusion protein as the substrate; an IKKβ substrate and TPCA-1 (20 μg ml-1) were used as positive and negative controls, respectively. Immunoprecipitated IKKβ successfully phosphorylated its substrate in in vitro setting (line 5, Figure 2H); but when TPCA-1 was present during the assay, the phosphorylation was blocked (line 6, Figure 2H). Analysis of XBP1s phosphorylation confirmed that XBP1s was robustly phosphorylated by IKKβ (line 2, Figure 2H) and that this phosphorylation was inhibited by TPCA-1 (line 3, Figure 2H).
To determine the sites on XBP1s that are phosphorylated by IKKβ, we performed the same in vitro kinase assay as described above with His-TF-XBP1s protein, in the presence and absence of IKKβ, then immunoprecipitated XBP1s from the reaction buffer. Subsequently, possible phosphorylation sites on XBP1s were analyzed with tandem mass spectrometry (MS/MS). Results showed that IKKβ directly phosphorylates XBP1s at Thr48 and Ser148 residues (Figures S2A and S2B).
We have previously produced an antibody that is specific for p-XBP1Thr48 (Lee et al., 2011). We next infected HEK293 cells with Ad-XBP1s alone, or with Ad-XBP1s plus Ad-ca-IKKβ; immunoblotting with p-XBP1Thr48 antibody demonstrated that XBP1sThr48 phosphorylation was indeed increased by IKKβ (Figure 2I and 2J).
We also made attempts to produce an antibody against XBP1s (which is phosphorylated on the Ser148 residue) but we failed. Thus, to determine the role of IKKβ-mediated phosphorylation of the Thr48 and Ser148 residues of XBP1s, we replaced Thr48 and Ser148 of XBP1s with alanines (XBP1s(T48A) and XBP1s(S148A)). We also created a double mutant XBP1s (XBP1s(DM)) bearing both substitutions. HEK293 cells were transfected with XBP1s, XBP1sT48A, XBP1sS148A, or XBP1sDM constructs, with or without a ca-IKKβ-expressing vector. The T48A or S148A single mutations substantially blocked the IKKβ-mediated up-regulation of nuclear, cytoplasmic and total l XBP1s (Figure S2C), whereas double Thr48A and Ser148A mutation greatly reduced the effects of IKKβ on XBP1s (Figure S2C).
Next, to determine whether Thr48 and Ser148 are the main sites phosphorylated by IKKβ, we transfected HEK293 cells with flag-tagged XBP1s or flag–tagged XBP1s(DM), along with an IKKβ-HA, and subsequently immunoprecipitated XBP1s. Upon probing the XBP1s immunoprecipitates with an anti-phosphoserine/threonine antibody, we observed that IKKβ-mediated phosphorylation of XBP1s was greatly diminished when the Thr48 and Ser148 residues were mutated to alanine (Figure S2D).
To assess how IKKβ-mediated phosphorylation of XBP1s affects its expression, we transfected HEK293 cells with XBP1s or XBP1s(DM), along with one that expresses IKKβ; the cells were then treated with cycloheximide (20 μg ml-1). The rate of degradation of XBP1s(DM) protein was significantly higher than that of XBP1s (Figures S2E and S2H). Combined densitometric analysis of three independent experiments (Figure S2E-S2G) further demonstrated that the Thr48A/Ser148A double mutation reduced the half-life of XBP1s by more than 50% (Figure S2H).
The Interaction between IKKβ and XBP1s is Lost in Obesity
We next asked whether the interaction between IKKβ and XBP1s is altered in obesity. Wild-type (WT) and ob/ob mice were fasted for 24 hours, then given food ad libitum for 1 hour. Immunoprecipitation experiments showed that XBP1s was present in the IKKβ immunoprecipitates from the liver lysates of lean mice, in which XBP1s expression had been induced by re-feeding (Figure 3A); however, this interaction between XBP1s and IKKβ was not detected in ob/ob mice under the same re-feeding condition (Figure 3A). Meanwhile, as per our previous findings (Park et al., 2010), total XBP1s levels were lower in the liver of ob/ob mice than in those of lean mice (Figure 3A and 3B). The total levels of IKKβ in both mice (lean and ob/ob) were comparable (Figure 3A). These results indicate that the interaction between XBP1s and IKKβ is reduced in the liver of ob/ob mice and also raised the question whether IKKβ is activated during the re-feeding. Indeed, there was a significant up-regulation in IKKβ phosphorylation in response to re-feeding in lean mice (Figure 3C and 3D and S3A). The basal phosphorylation status of IKKβ in the obese mice was higher in fed state than the lean mice. However, re-feeding did not lead to any further increase (Figure 3C and 3D and S3A) In other words, obese mice were un-responsive to the stimuli created by re-feeding in increasing IKKβ phosphorylation. An IKKβ kinase assay also documented that re-feeding significantly up-regulated the kinase activity of IKKβ, but similar to the phosphorylation status, there was no up-regulation of IKKβ kinase activity in obese mice following the re-feeding (Figure 3E). Finally we investigated whether NF-κB activity is also regulated by re-feeding in the lean and obese mice. In parallel to the IKKβ activity, p65 nuclear levels were significantly upregulated after re-feeding in the lean mice, but this response was blunted in the obese mice (Figure 3F and 3G). Figure 3G was created by combination of three independent experiments (Figure 3F, S3B and S3C). Finally analysis of NF-κB target genes following the re-feeding has shown a similar pattern namely that various target gene expression such as Chemokine (C-X-C motif) ligand 10 (Cxcl10), Chemokine (C-C motif) ligand 2 (Ccl2), Chemokine (C-C motif) ligand 4 (Ccl4), Chemokine (C-C motif) ligand 5 (Ccl5), Hepatocyte growth factor (Hgf) and interferon gamma (Ifng) were significantly up-regulated in the lean mice but there were no response in the obese mice to refeeding (Figure 3H).
Figure 3. IKKβ Activation Response to Refeeding is Impaired in Ob/Ob Mice.
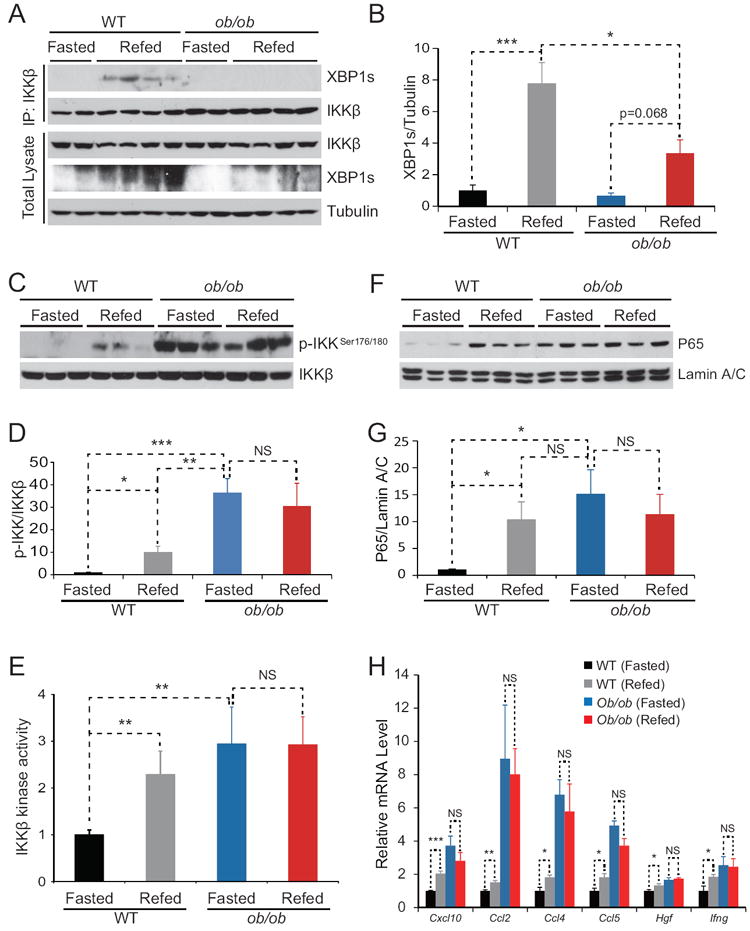
Lean and age-matched ob/ob mice were fasted for 24 hours and refed for 1 hour.
(A) XBP1s Immunoblotting in the IKKβ immunoprecipitates from the liver lysates (top). We specifically loaded two times more amounts of IP samples from the liver of ob/ob mice liver when compared to those of lean mice. XBP1s and tubulin protein levels in total liver lysates (bottom).
(B) Densitometric quantification of ratio of XBP1s protein level to tubulin protein in Figure 3A.
(C) Phospho-IKKβSer176/180 and IKKβ protein levels in total liver lysates.
(D) Densitometric quantification of ratio of Phospho-IKKβSer176/180 level to IKKβ protein level in Figure 3B and Figure S3A (n=6 for each group).
(E) IKKβ activity in mice livers (n=8 for each group). The average of the signals of IKKβ activities in the liver of fasted WT mice were used for normalization of IKKβ activities in the liver in other groups. The IKKβ activities in the four groups are expressed as fold difference.
(F) p65 and Lamin A/C protein levels in nuclear extracts of the liver of WT and ob/ob mice.
(G) Densitometric quantification of the ratio of p65 protein level to Lamin A/C protein in Figure 3E, Figure S3B and S3C (n=9 for each group).
(H) Gene expression levels of Cxcl10, Ccl2, Ccl4, Ccl5, Hgf and Ifng in the liver of WT and ob/ob mice (n=5 for each groups). 18S was used for normalization of gene expression. Error bars are represented as mean ± SEM. Significance was determined by student’s t test. *P < 0.05 **P < 0.01, ***P < 0.001, NS: Non-significance.
Contrary to previous reports (Cai et al., 2005), the above results collectively suggest a positive involvement of IKKβ in the regulation of glucose homeostasis and normal physiological responses. We thus examined what effect up-regulated IKKβ activity has in livers of obese mice by using different experimental approaches.
Adenovirus-Mediated Hepatic IKKβ Gain-Of-Function Improves Glucose Homeostasis
To increase IKKβ activity in liver, we injected Ad-ca-IKKβ (or Ad-LacZ, as a control) into ob/ob mice through tail vein. Ad-ca-IKKβ injection resulted in significant increase in total IKKβ and nuclear levels of p65 in the liver of ob/ob mice relative to the control group (Figures S3D and S3E).
Analysis of blood glucose of ob/ob mice showed that ca-IKKβ expression significantly reduced the blood glucose levels at fed and fasted states (Figures S3F and S3G). Glucose tolerance test (GTT) done on day 4 post-injection revealed a major improvement in glucose disposal rate in ca-IKKβ expressing mice (Figure S3H). Results of an insulin tolerance test (ITT), done on day 6 post injection, also showed a significant improvement in whole-body insulin sensitivity in IKKβ-expressing ob/ob mice, when absolute blood glucose levels were taken into account (Figure S3I). However, analysis of ITT results as percent values of the initial resting blood glucose values had shown no difference between the groups (Figure S3J), which lead us to conclude that whole body insulin sensitivity is not altered in the Ad-ca-IKKβ group. Furthermore, circulating insulin levels were significantly reduced in the Ad-ca-IKKβ-injected mice when compared to controls (Figure S3K); and plasma aspartate transaminase (AST) and alanine transaminase (ALT) levels were also significantly reduced in Ad-ca-IKKβ-injected mice (Figures S3L and S3M). Food intake (Figures S3N) and body weight (Figures S3O) were not altered.
We next investigated whether total and nuclear levels of XBP1s are up-regulated, and consequently whether ER stress was reduced, when IKKβ activity was increased. As shown in Figure S3P, total as well as nuclear levels of XBP1s were increased in the ca-IKKβ-expressing group, along with a marked reduction in PERK phosphorylation and expression of DNA damage inducible transcript 3 (DDIT3) (Figure S3Q). Collectively, these observations indicate that ER stress was reduced. Phosphorylation levels of c-Jun amino terminal kinase (JNK), p38 mitogen activated protein kinase (p38) and its downstream target activating transcription factor-2 (ATF2) were not altered by ca-IKKβ expression (Figure S3R).
AAV-Mediated Expression of IKKβ in Livers of Ob/ob Mice Improves Glucose Metabolism
We next used adeno-associated virus (AAV), which cannot induce immune responses by itself in vivo (Daya and Berns, 2008; Kotterman and Schaffer, 2014; Rehman et al., 2008), to increase activation of IKKβ in the liver. AAV-ca-IKKβ injection increased both total IKKβ and nuclear p65 levels in liver of ob/ob mice (Figure 4A). In parallel, expression levels of IKKβ target genes (e.g., chemokine (C-X-C motif) receptor 2 (Cxcr2); CD14 antigen (Cd14); superoxide dismutase 2 (Sod2); CD40 antigen (Cd40); and nitric oxide synthase 2 (Nos2)) were increased when mice were injected with AAV-ca-IKKβ (Figure S4A). Expression of ca-IKKβ also up-regulated the mRNA amounts of cytokines such as interleukin 1 (Il-1), interleukin 4 (Il-4), and interleukin 6 (Il-6) (Figure S4B), but not TNFα mRNA levels in the liver (Figure S4B). IL-6 plasma levels were also significantly up-regulated (Figure S4C). The AAV-ca-IKKβ used in this study employed a liver-specific thyroxine-binding globulin (TBG) promoter. Analysis of IKKβ levels in the epididymal white adipose tissue (WAT), in brown adipose tissue (BAT), and in muscle and kidney tissues documented that the increase in IKKβ levels was confined to the liver (Figure S4D).
Figure 4. IKKβ Gain of Function in the Liver of Ob/ob Mice is Beneficial.
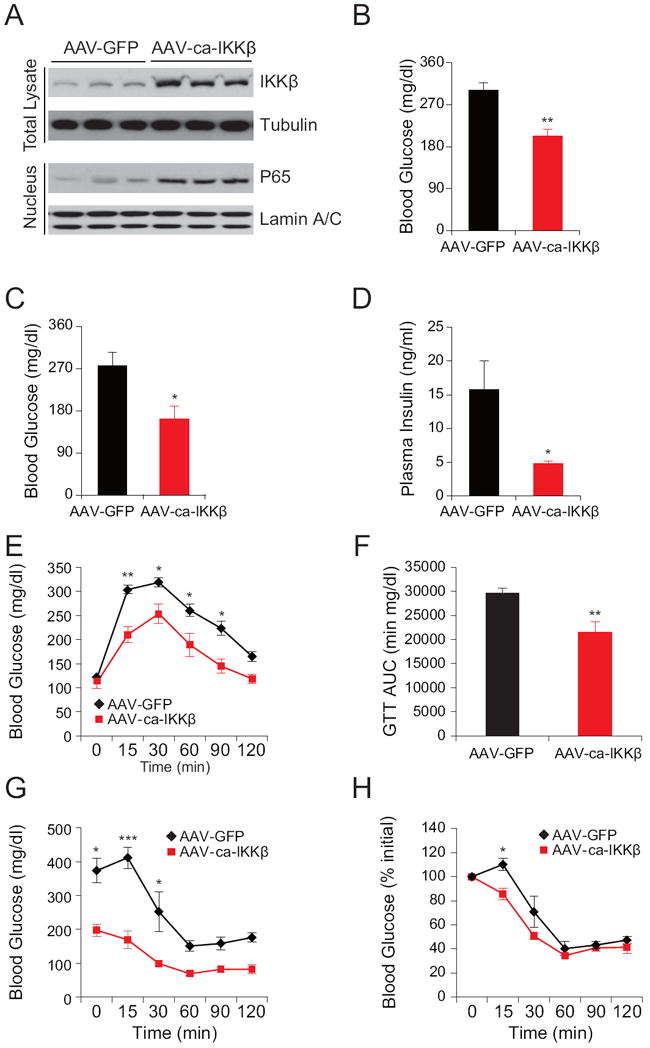
Male ob/ob mice (8 weeks old) were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 genome copies (GC) g-1 (n=5 in each group) through the tail vein.
(A) IKKβ and tubulin protein levels in total lysates and p65 and Lamin A/C protein levels in nuclear extracts of the liver on post-injection day 30.
(B-C) Blood glucose levels (mg dl-1) (B) at fed state and (C) after 6 hours of fasting on day 5 post injection.
(D) Plasma insulin levels (ng ml-1) on post injection day 24. Each experiment was repeated in two independent groups.
(E) GTT on post injection day 11. (F) Area under curve (AUC) analysis of GTT from Figure 4E.
(G-H) ITT performed 13 day after the AAV injection. (G) The curve depicts the absolute blood glucose (mg dl-1) levels, (H) Percent values of initial blood glucose concentration during ITT.
Each experiment was reproduced in four independent groups. Error bars are represented as mean ± SEM. Significance was determined by student’s t test (B-D and F) or two-way ANOVA with Bonferroni multiple-comparison analysis (E, G and H). *P < 0.05, **P < 0.01, ***P < 0.001.
Once liver-specific pattern of expression of IKKβ was successfully established for AAV-ca-IKKβ, we injected the vector (or AAV-GFP as a control) into the tail vein of ob/ob mice. On day 5 post injection, measurement of blood glucose at fed and fasted state showed that the ca-IKKβ-expressing mice had significantly lower blood glucose levels compared to control mice (Figures 4B and 4C); in addition, circulating insulin levels in the ca-IKKβ-expressing mice were also markedly reduced (Figure 4D). GTT, performed on day 8 after injection, revealed a significant improvement in the AAV-ca-IKKβ-injected group (Figures 4E and 4F). ITT done on day 13 post injection also showed a significantly enhanced whole-body insulin sensitivity in the ca-IKKβ-expressing group (Figures 4G and 4H).
Next, we investigated whether hepatic insulin receptor signaling is altered in ob/ob mice as a result of ca-IKKβ expression. Insulin-stimulated IR tyrosine phosphorylation, AKTSer473, AKTThr308, and GSK3β phosphorylations were significantly increased in AAV-ca-IKKβ-injected ob/ob mice when compared with the GFP-expressing control mice (Figure 5A-E). These results indicate that increased hepatic IKKβ activity improves IR signaling.
Figure 5. IKKβ Reduces ER Stress in the Liver of Ob/ob Mice.
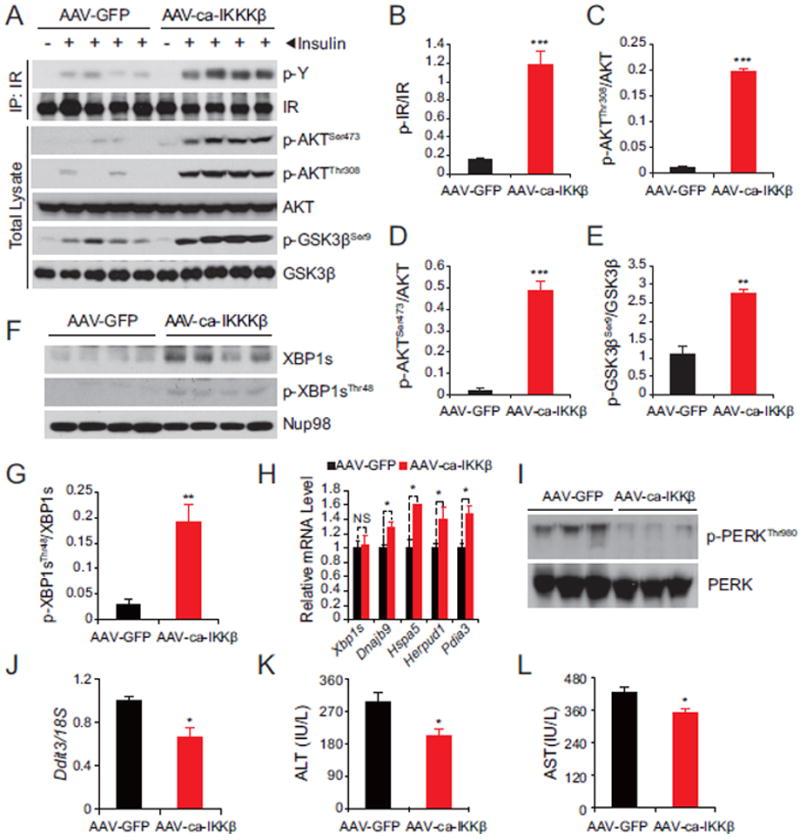
Male ob/ob mice (8 weeks old) were injected with AAV-ca-IKKβ and AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) through the tail vein.
(A) Protein lysates were subjected to IR-IP and subsequently phospho-tyrosine and IR immunoblotting. Direct lysates were used to analyze phospho AKTSer473, AKTThr308 and GSK3βSer9 and the total protein levels of AKT and GSK3β.
(B-E) Densitometric quantifications of the western blots bands in (A). Graphs depict the ratio of the autoradiographical signals of (B) phospho-IR/total IR, (C) phospho-AKTThr308/total AKT, (D) phospho-AKTSer473/total AKT and (E) phospho-GSK3βSer9/total GSK3β.
(F) Nuclear phospho-XBP1sThr48 and total XBP1s protein levels in the liver of ob/ob mice. Nup98 was used as a loading control.
(G) The graph depicts the ratio of the signals of phospho-XBP1sThr48 protein to XBP1s protein shown in (F).
(H) Gene expression levels of Xbp1s, Dnajb9, Hspa5, Herpud1 and Pdia3 in livers of ob/ob mice.
(I) PERKThr980 phosphorylation and total PERK protein levels in livers of ob/ob mice.
(J) Ddit3 mRNA levels in livers of ob/ob mice. 18S was used for normalization of gene expression.
(K-L), (K) Plasma alanine transaminase (ALT; IU L-1) and (L) Aspartate transaminase (AST; IU L-1) of ob/ob mice 12 days after injection.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
Increased activity of IKKβ also led to substantial upregulation of XBP1s protein amounts, and increased XBP1sThr48 phosphorylation (Figures 5F and 5G). Expression of XBP1s target genes - such as DnaJ homolog, subfamily B, member 9 (Dnajb9); heat shock protein 5 (Hspa5); homocysteine-inducible, ER stress-inducible, ubiquitin-like domain member 1 (Herpud1); protein disulfide isomerase associated 3 (Pdia3) - were also significantly increased in the IKKβ-expressing group, pointing to increased XBP1s activity (Figure 5H). Finally, increased IKKβ activity reduced PERKThr980 phosphorylation (Figure 5I), expression of Ddit3 (Figure 5J) and activating transcription factor-4 (ATF4) protein levels (Figure S4E). Phosphorylation of JNK, P38 and ATF2 were not altered (Figure S4E). Additionally, ALT and AST levels were significantly reduced (Figures 5K and 5L). No differences in food intake, bodyweight and body temperature were detected (Figure S4F-S4H)
Five weeks post injection, no necrosis or liver damage was detected in AAV-GFP- or AAV-ca-IKKβ-injected mice (Figure S4I). Immunostaining for active caspase-3 also did not reveal increased apoptosis in the liver of AAV-ca-IKKβ-injected mice (Figure S4I). And Trichrome staining (to evaluate the presence of any fibrosis caused by IKKβ activity) revealed no difference between the groups (Figure S4I).
AAV-Mediated Hepatic IKKβ Expression is Beneficial in DIO Mice
We next examined the effects of hepatic overexpression of IKKβ in DIO mice. As with results obtained from the ob/ob models, DIO mice injected with AAV-ca-IKKβ displayed significantly lower blood glucose and insulin levels compared to the control group (Figure 6A and 6B). GTT, performed on post-injection day 19, revealed a significant improvement in the IKKβ-expressing group (Figures 6C and 6D). ITT, performed on post-injection day 21, showed that whole-body insulin sensitivity was also significantly enhanced in the IKKβ group (Figures 6E and 6F). As shown in Figure 6G, analysis of blood glucose levels 10 weeks after the injections still showed a significant decline in the IKKβ-expressing group. GTT was performed again eight weeks after the injection and it showedimproved glucose tolerance in the IKKβ-expressing group (Figures 6H and 6I).
Figure 6. IKKβ Improves Glucose Homeostasis in DIO Mice.
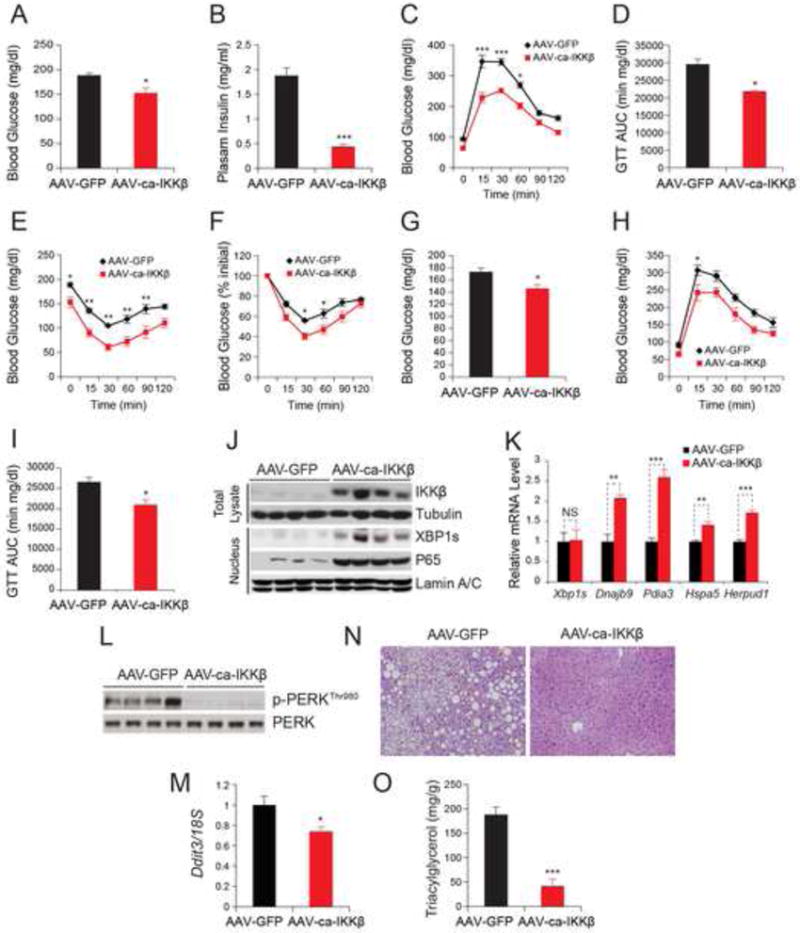
The diet induced obese (DIO) mice were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) via the tail vein.
(A) Blood glucose levels (mg dl-1) after 6 hours fasting on post injection day 21.
(B) Plasma insulin concentrations (ng ml-1) on day 22 post injection.
(C) GTT on day 19 post injection.
(D) Area under the curve of GTT in Figure 6C.
(E-F) ITT on day 21 post injection. The curves depict (E) the absolute blood glucose concentration and (F) the percentage of initial glucose concentration. Each experiment was reproduced in two independent groups.
(G) Blood glucose levels (mg dl-1) at 6-hour fasted state 10 weeks after injection.
(H) GTT 11 weeks after injection.
(I) AUC analysis of GTT in Figure 6H.
(J) IKKβ and tubulin protein levels in total lysates and XBP1s, p65 and Lamin A/C protein levels in nuclear extracts of the liver 14 weeks after injection.
(K) Gene expression levels of Xbp1s, Dnajb9, Hspa5, Herpud1 and Pdia3 in livers of DIO mice 24 days after injection.
(L) PERKThr980 phosphorylation and total PERK protein levels in livers of DIO mice 4 weeks after injection.
(M) Ddit3 mRNA levels in the liver of DIO mice 14 weeks after injection. 18S was used for normalization of gene expression.
(N) H&E staining of liver sections from DIO mice 14 weeks after injection.
(O) Triglyceride contents (mg g-1) in the liver 14 weeks after injection.
All Error bars are represented as mean ± SEM. Significance was determined by student’s t test (A, B, D, G, I, K, M and O) or two-way ANOVA with Bonferroni multiple-comparison analysis (C, E, F and H). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
At the time of sacrifice, IKKβ and XBP1s levels were analyzed. Expression of IKKβ was maintained (Figure 6J) and its target genes were significantly elevated (Figure S5A). XBP1s protein levels were also markedly upregulated, in parallel with the high expression levels of IKKβ (Figure 6J), without any changes in the corresponding mRNA levels (Figure 6K); and in addition, expression of XBP1s target genes was significantly increased (Figure 6K). AAV-mediated expression of IKKβ led to a marked decrease in PERK phosphorylation, ATF4 protein levels (Figure 6L and S5B) and a significant reduction in Ddit3 expression levels (Figure 6M) even 14 weeks post injection.
Histological analysis showed that expression of IKKβ in the liver for 14 weeks almost completely abolished hepatosteatosis (Figure 6N); and consistent with this, liver triglyceride levels were also significantly reduced (Figure 6O). q-PCR analysis of lipogenic genes (such as acetyl-Coenzyme A carboxylase alpha (Acaca), acetyl-Coenzyme A carboxylase beta (Acacb), diacylglycerol O-acyltransferase 2 (Dgat2) and fatty acid synthase (Fasn)) revealed a significantly decreased expression in the IKKβ-expressing mice when compared to controls (Figure S5C). Furthermore, IKKβ-expression in the liver did not alter in body temperature (Figure S5D), body weight (Figure S5E) or food intake (Figure S5F). Finally, ALT and AST levels were significantly reduced in the IKKβ group, confirming a beneficial role of IKKβ in the liver (Figure S5G and S5H).
To investigate the role of IKKβ over expression on glucose homeostasis in more detail, we performed hyperinsulinemic euglycemic clamp (HEC) studies in the DIO mice. As shown in supplementary Figure S5I, hepatic glucose production was significantly reduced during the HEC procedure in the ca-IKKβ expressing group, whereas, glucose infusion rate was significantly increased (Figure S5J and S5K). Glucose uptake rate was not different between the groups (Figure S5L). Finally analysis of blood glucose levels during the whole HEC experiment documented that euglycemic state were reached in both groups (Figure S5M).
Glucose homeostasis is improved in Liver-specific IKKβ transgenic mice
Earlier studies report that glucose homeostasis is disturbed in liver-specific constitutively active IKKβ transgenic mice (Cai et al., 2005). Based on our findings from experiments on Adenovirus and AAV-mediated constitutively active IKKβ expression, we created a transgenic mouse model in which the expression of constitutively active human IKKβS177E/S181E was driven under the albumin promoter (LIKKβ+/+) to investigate the phenotype of liver-specific IKKβ transgenic mice. Expression of constitutively active human IKKβ was increased about 3-fold, compared to endogenous IKKβ (Figure 7A and 7B). In parallel, amounts of nuclear p65 were also markedly increased (Figure 7A) and NF-κB target genes, such as Cd14, Cd40, Ccl2, Il-10, Il-1r1 and Il-6 were up-regulated (Figure S6A).
Figure 7. LIKKβTg(+/+) Mice Have Improved Metabolic Homeostasis.
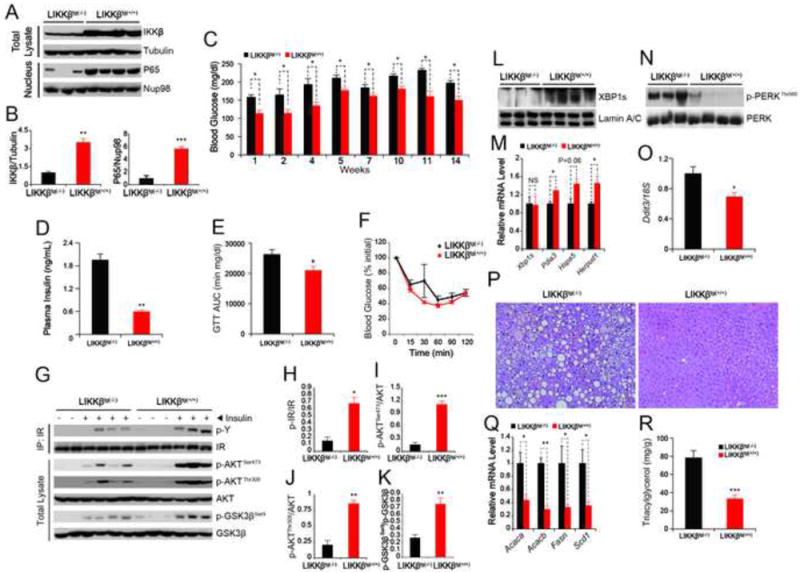
LIKKβTg(+/+) mice and control mice LIKKβTg(-/-)were fed with a HFD (n=4 in LIKKβTg(+/+) group and n=5 in LIKKβTg(-/-) group) for 14 weeks.
(A) IKKβ and tubulin protein levels in total liver lysates, and p65 and Lamin A/C protein levels in nuclear extracts of the liver.
(B) Densitometric quantifications of the western blot showing the ratio of IKKβ to tubulin and p65 to Nup98 in Figure 7A.
(C) Blood glucose levels (mg dl-1) at 6-hour fasting after at indicated weeks of HFD-feeding.
(D) Plasma insulin concentrations (ng ml-1) after 12 weeks of HFD-feeding.
(E) The graph depicts AUC analysis of the GTTs from three independent groups of LIKKβTg(+/+) and their corresponding controls after 8 weeks of HFD-feeding (n=14 in LIKKβTg(+/+) group and n=18 in LIKKβTg(-/-) group). For each GTT data and analysis of the three independent groups, see Figure S6B-S6D.
(F) The graph depicts ITT (percent values) results from three independent groups of LIKKβTg(+/+) and their corresponding controls after 6 weeks of HFD-feeding (n=14 in LIKKβTg(+/+) group and n=18 in LIKKβTg(-/-) group). For each ITT data and analysis of the absolute blood glucose levels of the three independent groups, see Figure S6E-S6G.
(G) Phospho-tyrosine and IR levels in IR-IP samples. Phospho AKTSer473, AKTThr308 and GSK3βSer9 and the total protein levels of AKT and GSK3β were determined from the direct lysates of the liver of mice, which was infused with insulin through portal vein.
(H-K) Densitometric quantifications of the western blot bands in Figure 7G. Graphs depict the ratios of the autoradiographic signals of (H) phospho-IR/total IR, (I) phospho-AKTThr308/total AKT, (J) phospho-AKTSer473/total AKT and (K) phospho-GSK3βSer9/total GSK3β.
(L) XBP1s and Lamin A/C protein levels in the nuclear extracts of the liver of LIKKβTg(+/+) and LIKKβTg(-/-) mice.
(M) Gene expression levels of Xbp1s, Pdia3, Hspa5 and Herpud1 in the liver of LIKKβTg(+/+) and LIKKβTg(-/-) mice.
(N) PERKThr980 phosphorylation and total PERK protein levels in the liver of LIKKβTg(+/+) and LIKKβTg(-/-) mice.
(O) Ddit3 mRNA levels in livers of LIKKβTg(+/+) and LIKKβTg(-/-) mice after 14 weeks of HFD-feeding. 18S was used for normalization of gene expression.
(P) H&E staining of liver sections from LIKKβTg(+/+) and LIKKβTg(-/-) mice.
(Q) Gene expression levels of Acaca, Acacb, Fasn and Scd1 in the liver of LIKKβTg(+/+) and LIKKβTg(-/-) mice.
(R) Triglyceride level in the liver of LIKKβTg(+/+) and LIKKβTg(-/-) mice.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (B, C, D, E, F, H, I, J, K, M, O, Q and R) or two-way ANOVA with Bonferroni multiple-comparison analysis (E). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
We placed LIKKβTg(+/+) and control mice (LIKKβTg(-/-)) on a HFD and investigated the phenotype of this model for a period of 14 weeks in three independent cohorts. Analysis of blood glucose levels throughout the experimental period revealed that LIKKβTg(+/+) mice have significantly reduced blood glucose levels (Figure 7C); consistent with this, plasma insulin levels in the LIKKβTg(+/+) mice were also significantly lower than in WT mice (Figure 7D). GTTs were performed in all three independent cohorts (Figures S6B-S6D): the combined AUC analysis (Figure 7E) documented a significant improvement in the glucose tolerance of LIKKβTg(+/+) mice. With ITTs, when the data points were analyzed as absolute blood glucose levels measured during the experiments in each cohort (Figures S6E-S6G), two of them (Figures S6E and S6F) displayed significant differences, and one did not (Figure S6G). However, when we analyzed percent changes as a function of the starting blood glucose levels of these three independent ITTs (Figure 7F), no differences were detected, leading us to conclude that general insulin sensitivity in the whole body of LIKKβTg(+/+) mice is not altered.
HFD feeding led to a blunted insulin-stimulated IR tyrosine phosphorylation in the liver of LIKKβTg(-/-) mice, while IKKβ overexpression significantly enhanced it (Figures 7G and 7H). Analysis of downstream IR signaling, reflected by AKTSer473, AKTThr308 and GSK3βSer9 phosphorylations (Figures 7G, and 7I-K), also documented significant up-regulation of insulin signaling in the LIKKβ+/+ liver, when compared to the LIKKβ-/- group.
XBP1s levels in these mice were consistent with the results of adenovirus- and AAV-mediated IKKβ gain-of-function experiments, with a marked increase in the levels of XBP1s protein (Figure 7L), but no change in the corresponding mRNA levels (Figure 7M). XBP1s target genes were also significantly upregulated (Figure 7M). Analysis of PERKThr980 phosphorylation (Figure 7N) and Ddit3 expression levels (Figure 7O) revealed significant downregulation in both, indicating a reduction in ER stress.
After 14 weeks of HFD feeding, mice were sacrificed and liver pathology was examined histologically by H&E staining. Hepatosteatosis induced by 14 weeks of HFD feeding in the LIKKβTg(-/-) group was completely blocked in LIKKβTg(+/+) mice (Figure 7P), indicating that IKKβ has strong anti-lipogenic properties in the liver. Indeed, the levels of expression of lipogenic genes such as, Acetyl-coenzyme A carboxylase alpha (Acaca), Acetyl-coenzyme A carboxylase beta (Acacb), Fatty acid synthase (Fasn), stearoyl-coenzyme A desaturase (Scd1) were dramatically reduced in the LIKKβTg(+/+) mice (Figure 7Q) and hepatic triglyceride levels were also significantly depressed (Figure 7R). In parallel, the ALT and AST levels were significantly lower in LIKKβTg(+/+) mice (Figures S6H and S6I). There were no increases in JNK, p38 MAPK and ATF2 activity (Figures S6J). Food intake (Figure S6K), body weight (Figure S6L) and body temperature (Figure S6M) were unaltered.
Depletion of XBP1s diminishes IKKβ’s beneficial effect in the liver
To address whether IKKβ’s effect on improvement of glucose homeostasis is mainly mediated by increased XBP1s activity, we depleted XBP1s while simultaneously overexpressing ca-IKKβ in the liver. For this reason, we first generated an adenovirus that expresses XBP1 shRNA (Ad-shXBP1). To test whether Ad-shXBP1 can effectively deplete XBP1s protein and mRNA, we first infected the MEFs with Ad-shXBP1 or with an adenovirus that expresses LacZ shRNA (Ad-shLacZ) as a control. Subsequently, cells were either incubated with vehicle or tunicamycin. As shown in Figure S7A, stimulation of cells with tunicamycin in Ad-shLacZ-infected cells led to robust production of XBP1s. However, this increase was completely abolished in the Ad-shXBP1-infected cells. Next, we infected the cells with Ad-shLacZ and Ad-shXBP1 and subsequently divided each group into two subgroups, one of which was stimulated with vehicle (DMSO) or tunicamycin. In both vehicle- and tunicamycin-treated cells, Ad-shXBP1 infection led to a highly significant reduction in XBP1s mRNA levels (Figure S7B). These results indicate that the Ad-shXBP1 that we produced efficiently silences XBP1s expression.
Next we injected AAV-GFP or AAV-ca-IKKβ together with Ad-shLacZ or Ad-shXBP1 into the tail vein of obese and diabetic db/db mice. Three days after the injections, AAV-ca-IKKβ + Ad-shLacZ-injected group had significantly lower blood glucose levels when compared with AAV-GFP- + Ad-shLacZ-injected group. However, the mice injected with AAV-ca-IKKβ + Ad-shXBP1 had significantly higher levels of blood glucose starting from the third day of injections (Figure S7C). Subsequently we have performed a GTT on post injection day six. AAV-ca-IKKβ + Ad-shLacZ- injected group had a significantly higher rate of glucose clearance from circulation when compared to the AAV-GFP+ Ad-shLacZ-injected group (Figure S7D). However, depletion of XBP1s in ca-IKKβ overexpressing conditions by injection of AAV-ca-IKKβ + Ad-shXBP1 significantly lowered the glucose clearance rate from circulation when compared to the AAV-ca-IKKβ+Ad-shLacZ-injected group, indicating that improved glucose tolerance with ca-IKKβ overexpression significantly diminishes when the XBP1s’ expression is silenced (Figure S7D and S7E). Analysis of blood glucose levels on post injection day 6 showed significant difference between the AAV-ca-IKKβ + Ad-shXBP1 and the AAV-ca-IKKβ + Ad-shLacZ groups (Figure S7F). In parallel, insulin levels were significantly reduced in the AAV-ca-IKKβ + Ad-shLacZ group and returned back to control levels in the AAV-ca-IKKβ + Ad-shXBP1 group (Figure S7G). All of these alterations happened in the absence of any change in the bodyweight or food intake of the groups (Figure S7H and S7I).
On day 10 post injections, mice were sacrificed and liver was collected for further analysis. Analysis of total XBP1s protein levels from the liver of three groups showed that ca-IKKβ overexpression increased XBP1s protein levels, and use of Ad-shXBP1 have blocked this up-regulation (Figure S7J and S7K), which confirms that XBP1shRNA has successfully depleted XBP1s protein. Ca-IKKβ overexpression mediated increase in XBP1s nuclear levels was also reduced with the expression of XBP1 shRNA (Figure S7L and S7M). Next, we analyzed PERKThr980 phosphorylation. In parallel to the results obtained from previous experiments ca-IKKβ overexpression reduced PERKThr980 phosphorylation (Figure S7N and S7O). However, depleting XBP1s in this setting significantly increased PERK phosphorylation, indicating that reduced ER stress after ca-IKKβ overexpression is mainly mediated by up-regulation of XBP1s protein levels (Figure S7N and S7O).
DISCUSSION
IKKβ is one of the most highly studied kinases in the intracellular inflammatory pathways (Hacker and Karin, 2006; Karin, 2005a; O’Dea and Hoffmann, 2009; Senftleben and Karin, 2002), and is regarded as a major intracellular mediator of inflammation-related hepatic insulin resistance.
In this report, we have uncovered an unexpected signaling network between IKKβ and XBP1s, and documented that IKKβ increases the stability and activity of XBP1s both in vitro and in vivo. Further analysis has shown that hepatic IKKβ is phosphorylated and activated at postprandial states and re-feeding induced increase in IKKβ activity leads to its binding to XBP1s. It is interesting to note that basal phosphorylation levels of hepatic IKKβ at fasted states are high, but it does not respond to the stimuli created by re-feeding in obesity. The majority of signaling systems work by ligand-dependent increase in the activity of the elements of those signaling systems. For example, in obese and insulin resistant conditions, basal phosphorylation of IRS proteins or down stream signaling molecules such as AKT are high compared to the lean states. However, insulin-stimulated up-regulation of IRS activity at postprandial conditions is severely suppressed in obese insulin resistant states and does not respond to stimuli, and therefore cannot activate downstream signaling. What we see in the case of IKKβ is very similar to the phenomenon observed in insulin resistant conditions for the insulin receptor signaling molecules.
Based on the initial biochemical findings, we used three gain-of-function approaches to explore whether IKKβ could have beneficial effects on hepatic glucose homeostasis. These three approaches yielded similar results, and established with certainty that IKKβ increases XBP1s activity, reduces ER stress, enhances hepatic IR signaling, and improves glucose homeostasis, blocks the development of hepatosteatosis or reduces already existing fatty liver disease.
An earlier study used an animal model similar to the LIKKβTg(+/+) mice reported that glucose homeostasis was disturbed in these mice. It is not uncommon to see different phenotypes from the same genetic animal models in different animal facilities of different institutes, or even on different backgrounds within the same animal facility. However, the findings we present here are based on three different experimental approaches, and the results from all three are consistent showing that hepatic IKKβ activity is beneficial for maintaining glucose homeostasis.
One question remains to be answered is that why both deletion (Arkan et al., 2005) and re-activation of IKKβ in the liver improves glucose homeostasis. One possible explanation is that embryonic deletion of IKKβ in the liver could elicit a compensatory response through hyper-activation of IKKα and creating the outcome observed in liver specific IKKβ knockout mice. Indeed, we have shown that IKKα has also a strong activity towards XBP1s.
For the last two decades, research on obesity and type 2 diabetes has viewed increased local or systemic inflammation as a major contributor to the development of type 2 diabetes in obesity (Gregor and Hotamisligil, 2011; Hotamisligil, 2006; Olefsky and Glass, 2010; Shoelson et al., 2006). The proposed role for inflammation in type 2 diabetes has shifted from involvement of systemic inflammation to local inflammation, sub-acute inflammation to chronic inflammation, and finally to a condition called metaflammation (Hotamisligil, 2006). Emerging evidence points to a contrasting, or at least much more complicated role, for inflammation in type 2 diabetes. Increased inflammatory stimuli are believed to block insulin receptor signaling through inhibition of insulin receptor substrates (IRS), by increasing their serine phosphorylation (Hotamisligil et al., 1996). In contrast, Copps et al. have shown that the main phosphorylation site (serine 307) in the IRS1 protein (that is responsive to inflammatory stimuli) (Copps et al., 2010) is a beneficial phosphorylation site in vivo; also, that phosphorylation of this site enhances insulin action, rather than blocking it (Copps et al., 2010). Several groups also report that interleukin 6 (IL-6), a cytokine that mediates inflammation, is beneficial for glucose homeostasis in obesity (Awazawa et al., 2011; Sadagurski et al., 2010), and that depletion of IL1R1 in these mice leads to a more severe obesity phenotype and glucose intolerance (Garcia et al., 2006; McGillicuddy et al., 2013). Consistent with these results, local inactivation of the main inflammatory cytokine TNF in WAT, reportedly worsens glucose homeostasis (Wernstedt Asterholm et al., 2014); this landmark study directly contradicts and challenges the central hypothesis (Hirosumi et al., 2002; Hotamisligil et al., 1993) that increased local TNF-mediated inflammation in WAT creates glucose intolerance and insulin resistance. Activation of IKKβ in adipose tissue also improves glucose homeostasis (Jiao et al., 2012). In addition, a recent study(Lancaster et al., 2016) has shown that double-stranded RNA-dependent protein kinase (PKR), which was claimed as a critical mediator and critical component of an inflammatory complex leading to insulin resistance (Nakamura et al., 2010) does not cause insulin resistance. Moreover, a number of anti-inflammatory approaches in human clinical trials have failed to improve insulin sensitivity: for example, statins, which have anti-inflammatory activity, reduce the activity of AP-1 and NF-κB. A recent report analyzed five trials involving 32,752 patients, and found that 12 percent of these patients were more likely to have diabetes (Preiss et al., 2011). Another study employed TNFα antagonism (e.g., a TNFα-specific antibody, or Fc-fusion TNFα receptors) to treat type 2 diabetes: these trials also failed to improve patients’ insulin sensitivity (Gao and Ye, 2012).
The convergence of the p38 MAPK (Lee et al., 2011) and IKKβ signaling pathways on XBP1s suggests that XBP1s is an important inflammatory signaling node, which has a reduced activity in obesity (Lee et al., 2011; Park et al., 2010). Given that p38 MAPK and IKKβ are mainly activated by cytokines (Hacker and Karin, 2006; Karin, 2005b; Senftleben and Karin, 2002), and that they are unresponsive to the stimuli in obesity conditions, we propose that obesity could be a condition of generalized cytokine resistance, wherein cytokines are unable to act on target tissues to activate relevant pathways to keep glucose homeostasis under control. Cytokine resistance may be the cause of up-regulated circulating cytokines in obesity.
In conclusion, our results elucidate the relationship between obesity and inflammation, and question the concept that hepatic inflammation or hepatic activation of IKKβ is detrimental for glucose homeostasis. Recent advances in our understanding of the obesity-inflammation-type 2 diabetes axis underscore the beneficial role of inflammation, and suggest that therapeutic developments for obesity and type 2 diabetes may, in fact, arise from a shift in our thinking about the mechanisms that underlie these disorders.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Umut Ozcan (umut.ozcan@childrens.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
To generate the Diet-induced obese (DIO) mice, wild-type C57BL/6J male mice were feed with a high-fat diet (HFD, 45 kcal% from fat) for 16-20 weeks. Liver-specific IKKβ transgenic hemizygous mice (LIKKβtg(+/-)) were kindly provided by Dr. Michael Karin. Homozygous (LIKKβtg(+/+)) and control (LIKKβtg(-/-)) mice were obtained by interbreeding IKKβ transgenic hemizygous (LIKKβtg(+/-)) mice. BKS.Cg-Dock7m(+/+)Leprdb//J mice and BKS.Cg -Leprob//J mice were obtained from the Jackson Laboratory. All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) at Boston Children’s Hospital.
To increase the protein levels and nuclear translocation of XBP1s in the livers of lean mice, C57BL/6J were fasted for 24 hours, then re-fed for one hour, at which point the mice were sacrificed.
Cell Lines
Human embryonic kidney 293 (HEK293), Chinese hamster ovary (CHO), Hepa-1c1c7 (murine hepatoma cell), HepG2 (human hepatoma cell) and MDA-MB-231 (breast adenocarcinoma cell) cells were maintained in DMEM with 10% fetal bovine serum (FBS), 10 U ml-1 penicillin, and 1 mg ml-1 streptomycin at 37°C in a 5% CO2 humidified atmosphere.
METHOD DETAILS
Biochemical Reagents
Anti-XBP1s, anti-insulin receptor (IR), anti-phosphotyrosine (PY99), anti-myc, anti-HA antibodies, anti-ATF4, anti-ATF6, and HRP-conjugated goat anti-mouse and goat anti-rabbit antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-IKKβ (Rabbit), anti-IKKα, anti-IKKγ, anti-p-JNK, anti-JNK2, anti-p-P38, anti-P38, anti-p-c-Jun, anti-c-Jun, anti-ATF2, anti-p-ATF2, anti-ubiquitin, anti-phospho-AKT (Thr308), anti-phospho-AKT (Ser473), anti-AKT, anti-phospho-GSK3α/β(Ser21/9), anti-GSK3β, anti-phospho-PERK (Thr980), PERK antibodies, anti-p65, anti-Nup98, anti-Lamin A/C and anti-α-tubulin antibodies and mouse anti-ubiquitin antibody were purchased from Cell Signaling Technology (Beverley, MA). Anti-Flag and anti-His antibody and chemical compound [(aminocarbony)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) and cycloheximide (CHX) were purchased from Sigma Aldrich (St. Louis, MO). Anti-phosphoserine/threonine antibody was purchased from ECM Bioscience (Versailles, KY). Rabbit polyclonal anti-phospho-XBP1sThr48 was raised against CRAAGSEASGT(p)PQARKRQR by Covance (Denver, PA) as described previously (Lee et al., 2011). Mouse anti-XBP1s was purchased from Biolegend (San Diego, CA). Mouse anti-IKKβ was purchased from Novus (Littleton, CO). Anti-Phosphoserine/threonine antibody was from Abcam (Cambridge, MA). Recombinant Human TNF-α protein was purchased from R&D systems (Minneapolis, MN).
Plasmids
Mouse IKKβ plasmid, pCR-Flag-IKKβ was purchased from Addgene (Cambridge, MA). Plasmids expressing constitutively active IKKβ (pcDNA3.1-IKKβ) with amino acid substitution mutations (S177E/S181E) were generated by PCR-based mutagenesis using pCR-Flag-IKKβ as a template and QuikChange II site-directed mutagenesis kit (Agilent Technologies), following manufacturer’s instruction. Constitutively active IKKβ (S177E/S181E) was subsequently cloned into pcDNA3.1 (Life Technologies).
Primer sequences for amino acid substitution of IKKβ (S177E/S181E) were as follows:
(forward),5’-GGAGCTGGATCAGGGCGAGCTGTGCACGGAATTTGTGGGGACTCTGC-3’; (reverse),
5’-GCAGAGTCCCCACAAATTCCGTGCACAGCTCGCCCTGATCCAGCTCC-3’.
Human XBP1s plasmid and mouse plasmid expressing kinase death IKKβ (pCR-Flag-IKKβ-KM) were purchased from Addgene. Vectors expressing mouse XBP1s (no tagged or flag-tagged XBP1s) and NH2-terminal (1–373 aa) of activating transcription factor 6a (ATF6n) were constructed previously (Ozcan et al. 2009). The XBP1s plasmids with amino acid substitution mutations (T48A, S148A or double mutations T48A/S148A) were generated by PCR-based mutagenesis using pcDNA3.1-Flag-XBP1s as a template and QuikChange II site-directed mutagenesis kit.
Primer sequences for T48A were as following:
(forward), 5’-GGGTCGGAGGCGAGCGGGGCACCGCAGGCTCGCAAGCGG-3’;
(reverse), 5’-CCGCTTGCGAGCCTGCGGTGCCCCGCTCGCCTCCGACCC-3’.
Primer sequences for S148A were as following:
(forward), 5’-CACGCTTGGGAATGGACGCGCTGGATCCTGACGAGG-3’;
(reverse), 5’-CCTCGTCAGGATCCAGCGCGTCCATTCCCAAGCGTG-3’.
Total Protein Extraction from Cells
Cells were lysed in lysis buffer (25 mM Tris-HCl (pH 7.4), 10 mM NaF, 10 mM Na4P2O7, 2 mM Na3VO4, 1 mM EGTA, 1 mM EDTA, 1% NP-40, protease inhibitor (Roche) and phosphatase inhibitor cocktails (Roche)). After rotation at 4°C for 20 minutes, cell lysates were centrifuged at 16,000 × g for 20 minutes at 4°C and supernatants were collected. Protein concentrations were measured by using Lowry protein assay (Bio-Rad) and normalized with lysis buffer for each sample to have an equal concentration. Proteins were denatured by boiling at 100°C for 5 minutes in 1X Laemmli buffer. The lysates were cooled to room temperature before loading to western blot gel.
Total Protein Extraction from Tissue
Liver, adipose, muscle and kidney tissues (ranging 50-100 mg) were homogenized with TissueLyser II (QIAGEN) in 1 mL of ice-cold tissue lysis buffer (25 mM TrisHCl, pH 7.4; 10 mM Na3VO4, 100 mM NaF, 50 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, 1% NP-40, protease inhibitor (Roche) and phosphatase inhibitor cocktails (Roche)). After homogenization, lysates were rotated at 4°C for 20 minutes and centrifuged at 16,000 × g for 20 minutes. The top lipid layer was carefully removed and the remaining supernatant was transferred to 1.5 mL Eppendorf tubes for further centrifugation. This step was repeated tiwce to completely remove the lipid. Protein concentrations were determined by using Lowry protein assay (Bio-Rad) and were normalized with lysis buffer for each sample. Proteins were denatured in 1X Laemmli buffer by boiling at 100°C for 5 minutes. The lysates were cooled to room temperature before loading for western blot analysis.
Western Blotting
Cell or tissue lysate samples, prepared as described above, were resolved on SDS-PAGE. Proteins were then transferred to polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in Tris-buffered saline solution with 0.05 % Tween 20 (TBST; pH 7.4) with 10% blocking reagent provided with BM Chemiluminescence Western blotting substrate (POD) assay system for 1 h, and then incubated with primary antibody in TBST with 10% blocking reagent at 4°C overnight. After the incubation, the membrane was washed three times in TBST and incubated with secondary antibody in TBST with 10% blocking reagent for 1 h at room temperature. After subsequent three-time washing in TBST, membrane was developed using a chemiluminescence assay system (Roche) and exposed to X-ray films (Denville Scientific). Relative protein levels were quantified by Image J program (NIH, Bethesda, MD).
For stripping, membrane was vigorously shaken in stripping buffer (62.5 mM Tris-HCl, pH 6.7; 2% SDS; 100 mM 2-mecaptomethanol) at 55°C for 15 minutes. After stripping, membrane was washed three times in TBST (3 times for 20 minutes).
Preparation anti-IKKβ-coated Dynabeads
Mouse anti-IKKβ antibody (8 μg per sample) (Novus, Littleton, CO) was conjugated to Dynabeads Protein G (50 μL per sample) (Life Technologies) with disuccininmidyl suberate (DSS) and bis(sulfosuccinimidyl) suberate (BS3) corsslinker (Thermo Scientific #21585) following its instructions with modifications, which is as following: For each sample, 50 μL of dynabeads were spinned down and the supernatant was discarded, and then dynabeads were incubated with 8 μg of antibody and 160 μL of conjugation buffer (PBS + 0.05% Tween-20) for 10 minutes at room temperature. The beads-antibody complex was washed with conjugation buffer (20 mM sodium phosphate, 0.15 M NaCl, 0.05% Tween-20, pH 8.5) twice by gentle pipetting. 2 mg of Bis (sulfosuccininidyl) suberate (BS3) was dissolved in 0.69 of mL conjugation buffer and 250 μL of BS3 solutions was immediately added to the beads-antibody complex.
The mixture was rotated for 30 minutes at room temperature, and then the reaction was stopped by adding 12.5 μL of quenching buffer (1 M Tris HCl, pH 7.5). After four times washing with immunoprecipitation buffer (PBST: 0.02% Tween, 0.1% sodium azide), the beads were stored in immunoprecipitation buffer at 4°C, until used for immunoprecipitation later.
Immunoprecipitation
Cell lysates (1-4 mg per sample) or tissue lysates (2-8 mg per sample) were incubated with antibody (1-8 μg per sample) overnight at 4°C with a gentle rotati on. Protein A Sepharose CL-4B beads (for rabbit immunoglobulin G [IgG], GE Healthcare Life Sciences) or protein G Sepharose CL-4B beads (for mouse IgG, GE Healthcare Life Sciences) (10-30 μLper sample) were added to the tubes and rotated at 4°C for 1 h. Beads were precipitated by centrifugation at 800 × g for 30 seconds and washed three times with cold lysis buffer. The pellet was resuspended in 2X Laemmli buffer and incubated at 100°C for 5 minutes. The supernata nts were collected and used for western blot.
For the immunoprecipation experiments in figure 2E, we immunoprecipitated the total lysates with anti-XBP1 antibody, then loaded three times more immunoprecipitated samples from XBP1s-transfected cells than the samples from the co-transfected cells with XBP1s and IKKβ.
For the immunoprecipation experiments in figure 3B, we used three times more immunoprecipitation samples from XBP1s-transfected cells, XBP1s(DM)-transfected cells and XBP1s(DM)-together with- IKKβ-transfected cells than that of XBP1s-together with-IKKβ-transfected cells when loading to SDS-PAGE gels to perform western blot.
For the immunoprecipation experiments in figure 4A, after immunoprecipitation of IKKβ from the liver lysates, we loaded two times more immunoprecipitates (IP) samples from ob/ob mice liver than that of lean mice liver to SDS-PAGE gels, and XBP1s and IKKβ protein levels were detected by immunoblotting.
Nuclear Protein Extraction
Cells were lysed with 400 μL of cytoplasmic lysis buffer (10 mM HEPES (pH 7.5), 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 10 mM KCl, 10 mM NaF, 0.1 mM Na3VO4, 1 mM dithiothreitol [DTT], protease inhibitor (Roche) and phosphatase inhibitor cocktails (Roche)). After 15 minutes of incubation on ice, 25 μL of 10% NP-40 was added to the cell lysates. The lysates were then incubated for 5 minutes on ice and vortexed 2 times for 10 sec. The lysates were centrifuged for 30 seconds at 16,000 × g, and supernatants were collected as cytoplasmic fractions. The pellets were re-suspended in 50 μL of nuclear extraction buffer (25 mM HEPES (pH 7.5], 500 mM NaCl, 10 mM NaF, 10% glycerol, 0.2% NP-40, 5 mM MgCl2, and 1 mM DTT, protease inhibitor (Roche) and phosphatase inhibitor cocktails (Roche)) and sonicated 3 times on ice. The nuclear fractions were centrifuged for 5 minutes at 16,000 × g and the supernatant was collected to obtain nuclear proteins. Both cytoplasmic and nuclear proteins were quantified using Bio Rad protein assay and normalized with lysis buffer to have an equal concentration. The proteins were denatured by boiling at 100°C following mixing with 5X Laemmli buffer.
For nuclear extraction from liver tissues, 80 mg of liver tissue was cut into small pieces and washed with PBS. Nuclear proteins were isolated using a commercially available kit from Thermo according to their instructions.
100 mg liver tissues was cut into small pieces and then homogenized using a Dounce homogenizer or a tissue grinder in 800 μl CER I. And then the tissue mixture was vortexed vigorously on the highest setting for 15 seconds to fully suspend the tissue samples, followed by 10minutes incubation on ice. 44 μL ice-cold CER II was added to the mixture and the tubes were vortexed for 5 seconds on the highest setting. The samples were incubated on ice for one minute, vortexed for another 5 seconds, and centrifuged for 5 minutes at the speed of 16,000 × g. The supernatant which contains cytoplasmic extract, was immediately transferred to a new pre-chilled tube and further lysed with Laemmli buffer. The pellet (nuclear fraction) was suspended in 200 μL ice-cold NER buffer by vortexing, followed by ultrasound for 20 sec. The lysate was centrifuged at maximum speed (~16,000 × g) for 10 minutes. The supernatant (nuclear extract) was immediately transferred to a new pre-chilled tube, and further lysed by adding Laemmli buffer.
Glucose Tolerance Test
Mice were fasted overnight (6 p.m.–9 a.m.), and D-glucose (0.3 g kg-1 for ob/ob, 1.25 g kg-1 for DIO and 1.5 g kg-1 for LIKKβtg(+/+) mice) was administrated intraperitoneally. Blood glucose levels were measured from the tail using a glucose meter (Contour, Bayer) at 0, 15, 30, 60, 90, and 120 minutes after D-glucose administration.
Insulin Tolerance Test
Mice were fasted for 6 h (8 a.m.–2 p.m.), and recombinant human insulin from Eli Lilly was administered intraperitoneally (2 IU kg-1 for ob/ob, 1.5 IU kg-1 for DIO and 1.4 IU kg-1 for LIKKβtg(+/+) mice. Blood glucose levels were measured from the tail using a glucose meter (Contour, Bayer) at 0, 15, 30, 60, 90, and 120 minutes after insulin administration.
Blood Glucose, Plasma Insulin and Plasma IL-6 Measurements
Mice were fasted for 6 h and their blood glucose levels were measured with a glucose meter (Bayer) after the blood was collected from tail vein. For enzyme-linked immunosorbent assay (ELISA), blood was collected from the tail vein and plasma was separated by centrifugation at 2,000 × g, 4°C f or 30 minutes. Plasma insulin levels were determined by an Ultra-Sensitive Mouse Insulin ELISA kit from Crystal Chem (Downers Grove, IL). Plasma IL-6 levels were measured with a Mouse IL-6 ELISA kit from Boston Biochem, Inc (Cambridge, MA).
Analysis of in vivo insulin signaling
For in vivo insulin signaling analysis, mice were anaesthetized with xylazine (100 mg per kg) / ketamine (20mg per kg) (Pfizer) after 6 h of fasting. Insulin (1 IU kg–1) or saline was infused into the liver via the portal vein. Five minutes after infusion, livers were harvested and frozen in liquid nitrogen immediately and stored at -80°C until use.
Blood Alanine Transaminase (ALT) and Aspartate Transaminase (AST) Measurements
Serum sampl preparation
The fresh blood were collected with heparin sodium coated capillary and the sample were centrifugated at 3,000 rpm at 4°C for 30 minutes. The supernatant blood plasma were collected and the ALT and AST levels in the plasma were measured with ALT Color Endpoint Assay and AST Color Endpoint Assay kits (Bio Scientific, Austin, TX), respectively, according to manufacturer’s instructions with no modifications.
AST Assay
Firstly, 6 serial dilutions of the Oxaloacetate Control were prepared by using the Pyruvate Dilution Buffer and the concentration of these Control Dilutions were as follows: 800 IU/L, 400 IU/L, 200 IU/L, 100 IU/L, 50 IU/L and 0 IU/L. 5 μL of each sample or standard (in duplicate) was added to the bottom of the microplate (96 well-plate) wells and 50 μL of AST Reagent Solution was added to the wells. The plate were covered with adhesive film and incubated the plate at 37°C for 10 minutes. The adhesive film were carefully removed and 50 μL AST Color Reagent was added to the wells. The plate was covered with adhesive film again and incubated at 37°C for 10 minutes. Then, 200 μL 0.1 M HCl was added to each well and the absorbance of each wells were measured at 510 nm. The AST concentration in each sample was calculated by the equation:
Use the mean absorbance values for each serum sample to determine the corresponding concentration of AST from the standard curve.
ALT Assay
Firstly, we made 6 serial dilutions of the Pyruvate Control using the Pyruvate Dilution Buffer and the concentration of these Pyruvate Control Dilutions are as follows:
150 IU/L, 70 IU/L, 32 IU/L, 15 IU/L, 7 IU/L and 0 IU/L.
10 μL of each sample or standard (in duplicate) was added to the bottom of the microplate (96 well-plate) wells and 50 μL of ALT Reagent Mix was added to the wells. The plate were covered with adhesive film and incubated the plate at 37°C for 30 minutes. The adhesive film were carefully removed and 50 μL DNPH Color Solution was added to the wells. The plate was covered with adhesive film again and incubated at 37°C for 10 minutes. Then, 200 μL 0.5 M NaOH was added to each well and the plate was incubated at 37°C for 5 minutes. The absorbance of each wells were measured at 510 nm.
RNA Preparation and Real-Time Quantitative PCR
Total RNA was extracted from cells or the liver of mice using Trizol reagent (Life Technologies) following its instruction. Purified total RNA was used to obtain cDNA using a cDNA synthesis kit (Bio-Rad) following these conditions: 25°C for 5 minutes, 42°C for 30 minutes, and 85°C for 5 minutes. The ge ne expression was analyzed with iQ5 Multicolor Real-Time PCR Detection System (BioRad, Hercules, California) with SYBR Green Supermix (BioRad, Hercules, California). The mRNA levels were normalized to 18S as a house keeping gene.
Genomic DNA Purification for Genotyping
Mouse tails were lysed in 100 μL 1X lysis buffer (4.5 mL of 1.5M Tris pH 8.8; 1.7 mL of 1M (NH4)2SO4; 0.3 mL of 0.5M MgCl2; 0.5 mL of Triton X100; 92 mL autoclaved H2O and 1 mL betamercaptoethanol) with 2.5 μL of Proteinase K and 0.5 μL of betamercaptoethanol, at 55°C for 15 hours. Samples were then boiled at 100°C for 20 minutes and cooled to room temperature, followed by a 20-sec centrifugation at 800 × g. The supernatant was removed to new tubes, and equal volume of phenol chloroform solution was added. Samples were vortexed for 20 seconds and centrifuged at 16,000 × g at 4°C for 20 minutes. The top layer was transferred to new tubes and two volumes of isopropanol were added to each tube. Samples were quickly vortexed and then centrifuged at 16,000 × g at 4°C for 10 minutes. The supernatant was discarded and the DNA pellet was washed with 500 μL of 70% ethanol by centrifugation at 16,000 × g at 4°C for 10 minutes. Same step was repeated with 100% ethanol. Then, the supernatant was discarded and the DNA pellet was let dry at room temperature. Finally, 100 μL of distilled H2O was added to the purified DNA, and the tubes were heated at 55°C for 10 minutes. After vortexing, the samples were centrifugated at 16,000 × g at 4°C for 10 minutes and the purified genomic DNA was used for genotyping.
Adenovirus Production
Adenovirus-producing plasmids containing a gene of constitutively active IKKβ or XBP1s were constructed using adenoviral expression kit from Life Technologies. The adenovirus vectors were digested with Pac I, and then transfected into 293A producer cells in 6-well-plates. The media was replaced with DMEM containing 10% FBS and 1% penicillin/streptomycin the next day. The cells were transferred to 10 cm tissue culture dishes 48 h after the transfection. The culture media was replaced with fresh media every 2-3 days until cytopathic effect (CPE) was observed. The cells were collected when 80% CPE was observed and adenovirus was harvested by freezing at -80°C and thawing at 37°C and repeating it for four times. Finally, cell lysates were removed by centrifugation at 2,000 × g for 30 minutes at 25°C and the supernatant containing adenovirus particles was stored at -80°C.
Adenovirus Transduction
To transduce cells in 10 cm dishes with adenovirus, cells were washed with culture medium containing 1% FBS and incubated with 2.5 mL of media containing 1% FBS and adenovirus. The dishes were gently rocked every 15 minutes for 1 h and 7.5 mL of media containing 1% FBS was added to each dish. The cells were incubated for additional 16 h.
Adenovirus or Adeno-Associated Virus (AAV) Injection to Mice
Virus (either adenovirus or AAV) was introduced to mice through the tail vein injection. Virus was thawed at 25°C before injection and the desired amount of virus was diluted with saline to a final volume of 100 μL per mouse. Mice were restrained and their tails were heated mildly with a heating lamp to achieve vasodilatation. Virus was injected through the tail vein. To prevent the back flow of virus solution, mild pressure was applied at the spot of injection immediately after injection until no bleeding was achieved.
Production of AAV
The constitutively active IKKβ coding sequence was generated by PCR-based method using pcDNA3.1-IKKβ(S177E/S181E) as a template. The following primer pair was used in this PCR:
(forward), 5’-TACTTACGCGTACCATGGACTACAAGGACGACGAT-3’;
(reverse), 5’-ACAAGTCGACCTACTAATCGCAGGCCTGCTCC-3’.
The resulting constitutively active IKKβ coding sequence was cloned into an AAV plasmid (TBG-AAV2.1) to create TBG-IKKβ vector, in which thyroid hormone-binding globulin (TBG) promoter controls hepatocyte-specific expression of IKKβ. The subsequent AAV virus production and purification were finished by Gene Therapy Center and Vector Core at University of Massachusetts Medical School.
Recombinant XBP1s Protein
Recombinant XBP1s proteins were produced and purified as described previously (Lee et al., 2011). Briefly, XBP1s coding sequence with 6× His-TF tag (N terminus) sequence were cloned into a pGSC1 plasmid. The final protein sequence of the construction (6× His +TF tag + 3C protease cleavage site) was as follows:
MNHKVHHHHHHMQVSVETTQGLGRRVTITIAADSIETAVKSELVNVAKKVRIDGFRKGKVPMNIVAQRYGASVRQDVLGDLMSRNFIDAIIKEKINPAGAPTYVPGEYKLGEDFTYSVEFEVYPEVELQGLEAIEVEKPIVEVTDADVDGMLDTLRKQQATWKEKDGAVEAEDRVTIDFTGSVDGEEFEGGKASDFVLAMGQGRMIPGFEDGIKGHKAGEEFTIDVTFPEEYHAENLKGKAAKFAINLKKVEERELPELTAEFIKRFGVEDGSVEGLRAEVRK NMERELKSAIRNRVKSQAIEGLVKANDIDVPAALIDSEIDVLRRQAAQRFGGNEKQALELPRELFEEQAKRRVVVGLLLGEVIRTNELKADEERVKGLIEEMASAYEDPKEVIEFYSKNKELMDNMRNVALEEQAVEAVLAKAKVTEKETTFNELMNQQASAGLEVLFQGP.
To produce recombinant XBP1s protein, 5 ng of pGSC1 plasmid containing XBP1s with His-TF tag was transformed into ArcticExpress TM (DE3) RP host stain E. coli (Agilent Technologies) to produce recombinant XBP1s protein. The total protein was extracted and purified by Ni-affinity resin (Life Technologies).
Protein Degradation Assay
HEK293 cells were transfected with XBP1s vector alone or together with IKKβ vector. After 24 h of transfection, the cells were treated with cycloheximide (CHX 20 μg ml-1, Sigma), a translation inhibitor. Cells without treatment and at 5, 10, 15, 25 and 45 minutes following CHX incubation were flash frozen in liquid nitrogen. Protein levels were determined by western blotting and the subsequent quantification was performed from scanned immunoblotting results with ImageJ software.
Genotyping of Liver-Specific IKKβ Transgenic Mice Using TaqMan-based Real-Time PCR
Litters from breedings of IKKβ transgenic hemizygous parents were genotyped using TaqMan-based real-time PCR in order to distinguish between the homozygous and hemizygous transgenic mice. Real-time PCR was conducted in triplicate in total 10 μL of reaction solution with 5 μL of 2X Taqman Genotyping Master Mix (Applied Biosystems, Life Technologies), 0.5 μL of 20X IKKβ gene expression assay, 0.5 μL of 20X GAPDH gene expression assay, and 10 ng genomic DNA (purified as described above) in each well of a 384-well plate. Real-time PCR was conducted on the ABI 6 (Applied Biosystems) using the following program: hold stage (50°C for 2 minutes, 95°C for 10 minutes) and PCR stage (40 cycles of 95°C for 15 sec, 60°C for 1 min). SDS Analysis (version 2.3, a software from ABI) was used to determine the Ct values of each reaction. Primer probe combinations were purchased as proprietary reagents from ABI-Life Technologies Inc:
Gapdh, Life Technologies, Cat. # 4400291; Assay ID: Mm00186822_cn;
IKKβ, Life Technologies, Cat. # 4351372; Assay ID: Hs01559467_m1.
Histological Analysis
The liver was collected immediately from AAV-injected ob/ob mice, AAV-injected DIO and LIKKβtg(+/+) and LIKKβtg(-/-) mice and stored in 10% formalin in phosphate buffer. After two weeks of incubation, the samples were embedded in paraffin and sectioned. The tissue sections were used for Hematoxylin and Eosin (H&E) Staining, and immunohistochemical staining for cleaved caspase-3 (active caspase-3) and Trichrome staining (Rodent Histopathology core, Harvard Medical School; Specialized Histopathology Services-Longwood, Brigham and Women’s Hospital).
IKKβ Kinase Activity Assays
IKK complexes were immunoppreciated from mouse liver lysates using anti-IKKγ antibodies (BD PharMingen, San Jose, CA) and protein A-Sepharose beads (GE Healthcare Life Sciences, Pittsburgh, PA) as described above. In vitro IKKβ Kinase Activity assay was conducted with the IKK complexes harvested, using Cyclex IKK Assay kit from MBL International (Woburn, MA), following its instructions, with no modifications.
To assay partially purfied recombinant IKKβ, add 10 μL of each sample or standard to the wells of the assay plate on ice. Duplicate wells containing 10 m units/10 μL IKKβ positive control (Cat# CY-E1176-2) has been included in each assay as a positive control for phosphorylation. Begin kinase reaction by addition of 90 μL Kinase Reaction Buffer (Kinase Buffer plus 20X ATP) and incubate for 30 minutes at 30°C. Wash the wells four times with Wash Buffer (containing 2% Tween-20) making sure each well is filled completely. Remove residual Wash Buffer by gentle tapping or aspiration. Pipette 100 μL of Anti-phospho-IκBα serine32 monoclonal antibody (AS-2E8) into each well and incubate for 30 minutes at RT. Wash the wells five times. Pipette 100 μL of HRP conjugated anti-mouse IgG into each well and incubate for 30 minutes at RT. Wash the wells five times. Add 100 μL of Substrate Reagent (containing chromogenic substrate, tetra-methylbenzidine (TMD)) to each well and incubate for 5-15 minutes at RT. Add 100 μL of Stop Solution (containing 1 N H2SO4) to each well. The absorbant signal in each well is measured with a spectrophotometric plate reader at dual wavelenghts of 450/540 nm. All samples and standards have been assayed in duplicate.
IKKβ Kinase Assay with XBP1s as Substrates
The in vitro IKKβ kinase assay was conducted as described previously (Hu et al., 2005; Shinohara et al., 2005). HEK293 cells were transfected with flag-IKKβ (S177E/S181E) vector, and the constitutively active IKKβ protein was purified through immunoprecipitation with an anti-flag antibody. Equal amount of purified IKKβ protein was incubated with His-TF-XBP1s fusion protein produced as described above or with commercial IKKβ substrate (Millipore, Darmstadt, Germany), in a kinase assay buffer (20 mM Hepes, pH 7.6; 25 mM β-glycerophosphate; 0.1 mM Na3VO4; 4mM NaF; 2 mM MnCl2; 10 mM MgCl2; 10 μM ATP) at 37°C for 30 minutes. For a negative control reaction, TPCA-1 (20 μg mL-1), an IKKβ inhibitor, was added to the reaction solution of IKKβ together with His-TF-XBP1s or IKKβ substrate at 37°C for 30 minutes. The kinase reaction was stopped by adding 1X Laemmli buffer and boiling at 100°C for 10 minutes. IKKβ-mediated phosphorylation of XBP1s or IKKβ substrate was analyzed by western blotting with anti-serine/threonine antibody.
Mass Spectrometric Analysis by LC-MS/MS
His-TF-XBP1s and purified constitutively active IKKβ protein (described above) were incubated in a kinase assay buffer (20 mM Hepes, pH 7.4; 25 mM β-glycerophosphate; 0.1 mM Na3VO4; 4 mM NaF; 2 mM MnCl2; 10 mM MgCl2; 10 μM ATP) at 37°C for 30 minutes. His-TF-XBP1s was pulled down with anti-His antibody, and resolved in SDS-PAGE and stained with Coomassie blue (Bio-Rad). Excised His-TF-XBP1s from Coomassie-stained gel was extracted and digested with trypsin and digested XBP1s peptide mixtures were subjected to microcapillary liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis (Taplin Biological Mass Spectrometry Facility, Harvard Medical School). Resulting MS/MS spectra of XBP1s fragments were analyzed by the SEQUEST (Thermo Scientific) algorithm.
Tissue Lipid Extraction
Around 75 mg of liver tissue (exact weight was recorded and used to normalize lipid amount) was homogenized with Homogenizer (Thermo Fisher Scientific) in 1 mL 50 mM NaCl on ice, following the addition of 5 mL of chloroform/methanol mixture (chloroform : methanol = 2:1). The homogenized tissue solution was vortexed for 10 seconds and centrifuged at 16,000 × g for 10 minutes. The aqueous phase was carefully removed, and the left oleic phase was mixed with 1.5 mL of methanol. The resulting mixture was vortexed for 10 seconds and centrifuged at 16,000 X g for 10 minutes. The lipid extract (top phase) was carefully moved to a new tube and mixed with 75 μL of 10% Triton X100 in acetone, and dried in fume hood. The resulting pellet was used for cholesterol or triglyceride determination with cholesterol assay kit (Wako, Richmond, VA) or Triglyceride assay kit (Wako, Richmond, VA) according to manufacture instructions.
Hyperinsulinemic euglycemic clamp
Prior to the clamp experiment, one catheter was inserted into the jugular vein for infusions. After 5–7 days of recovery, mice that had lost less than 10% of their preoperative weight were subjected to the clamp. On the day of experimentation, each mice was deprived of food for 3.5 h in the morning and then infused continuously with D-[3-3H]-glucose (PerkinElmer) (0.05 μCi/min) at a rate of 1μL/min for 1.5 h. After basal sampling, a primed continuous infusion of human regular insulin (Humulin, Eli Lilly) is infused at a rate of 4 mU/kg/min. The insulin solutions were prepared with 3% BSA. 20% glucose was infused at variable rates as needed to maintain plasma glucose at ~120 mg/dl. All infusions were done using micro infusion pumps (KD Scientific). Blood glucose concentrations were monitored regularly according to a fixed scheme from tail vein. Steady state was considered achieved during 75-120 minutes, when a fixed glucose-infusion rate maintained the glucose concentration in blood constantly for 45 minutes. The [3-3H] glucose content in serum during basal conditions and at steady state was measured in a liquid scintillation counter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars are represented as mean ± SEM. Statistical significance was calculated by Student’s t test, one-way analysis of variance (ANOVA) or two-way analysis of variance (ANOVA). When ANOVA indicated a significant difference among the groups, the statistical difference between two groups were compared using a stricter criterion for statistical significance according to the Bonferroni rule (corrected P value = pairwise P value × no. of comparisons). Significance was accepted at *P < 0.05, **P < 0.01 or ***P < 0.001.
DATA AND SOFTWARE AVAILABILITY
Image J software was downloaded from the website: https://imagej.nih.gov/ij/. GraphPad Prism 5 software was used for two-way analysis of variance (ANOVA) with Bonferroni multiple-comparison analysis
Supplementary Material
(A) p65 and Lamin A/C protein levels in nuclear extracts of HEK293 cells infected with XBP1s-expressing adenovirus (Ad-XBP1s)
(B) mRNA levels of IKKβ, CD14 antigen (CD14), CD40 antigen (CD40), chemokine (C-X-C motif) ligand 10 (CXCL10), chemokine (C-X-C motif) receptor 2 (CXCR2), superoxide dismutase 2, mitochondrial (SOD2) and nitric oxide synthase 2 (NOS2) in HEK293 cells infected with Ad-ca-IKKβ. HEK293 cells infected with Ad-LacZ were used as control. 18S was used for normalization of gene expression.
(C-F) XBP1s and tubulin levels in (C) CHO, (D) Hepa-1c1c7, (E) HepG2 and (F) MDA-MB-231 cells infected with Ad-XBP1s and Ad-ca-IKKβ.
(G) Human XBP1s (hXBP1s) and tubulin levels in HEK293 cells transfected with the vector expressing hXBP1s alone or together with ca-IKKβ expressing vector. These experiments were reproduced in two independent experiments.
(H) XBP1s mRNA levels in HEK293 cells infected with Ad-XBP1s alone or together with Ad-ca-IKKβ. 18S was used for normalization of gene expression.
(I) XBP1s mRNA levels in HepG2 cells infected with Ad-LacZ or Ad-ca-IKKβ. 18S was used for normalization of gene expression.
(J) The protein levels of cleaved forms of ATF6 (ATF6n), IKKβ and tubulin levels in HEK293 cells transfected with ATF6n expressing vector alone or together with ca-IKKβ expressing vector.
(K) XBP1s, IKKα and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with IKKα expressing vector.
(L) XBP1s, IKKγ and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with IKKγ expressing vector.
(M-N) XBP1s and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with ca-IKKβ expressing vector and further treated with cycloheximide (CHX, 20 μg ml-1) at indicated time points. The blots of the cells only transfected with XBP1s expressing vector were specifically exposed for a longer time (left bands) and the blots from cells co-transfected with XBP1s and ca-IKKβ expressing vectors together were exposed for a shorter time (right bands) to achieve similar levels of total XBP1s signal at the zero time point.
(O) XBP1s, phospho-IKKβSer176/180 and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector and further treated with vehicle or IL-1β (100 ng ml-1) for one hour. These results were reproduced in three independent experiments.
(P) 293T cells were transfected with XBP1s expressing vector alone or together with ca-IKKβ expressing vector. Sixteen hours after transfection, cells were treated with MG132 (10 mM) or DMSO. XBP1s and tubulin protein levels were analyzed with immunoblotting. These results were reproduced in three independent experiments.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
(A-B) Tandem mass spectroscopy (MS/MS) analysis of XBP1s that was immunoprecipitated from IKKβ kinase assay. (A) Phosphorylated thr48 residue of XBP1s in the protein fragments. (B) Phosphorylated ser148 residue of XBP1s in the protein fragments.
(C) Cytoplasmic, nuclear and total protein levels of XBP1s in HEK293 cells that were transfected with indicated vectors. These results were reproduced in three independent experiments.
(D) Phospho-XBP1s levels in HEK293 transfected with the indicated vectors. We specifically loaded three times more amount of IP samples from XBP1s-transfected cells (line 2), XBP1s(DM)-transfected cells (line 4) and XBP1s(DM)/ca-IKKβ-co-transfected cells (line 5) when compared to those of XBP1s/ca-IKKβ-co-transfected cells (line3) to achieve comparable XBP1s signal from each sample.
(E-G) XBP1s and tubulin protein levels in HEK293 cells transfected with the indicated vectors and further treated with CHX (20 μg ml-1) at indicated time points. Western blotting results in (E), (F) and (G) are from three independent experiments. The blots of the cells co-transfected with XBP1s and ca-IKKβ expressing vectors were exposed for a shorter time (left bands), and the blots from cells co-transfected with XBP1s(DM) and ca-IKKβ expressing vectors were exposed for a longer time (right bands) to achieve similar levels of total XBP1s signal at the zero time point.
(H) The graph depicts the averaged ratio of the signals of XBP1s to tubulin levels from three independent protein degradation experiments in Figure S2E-S2G. The dotted lines show the half-life of XBP1s protein.
Error bars are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni multiple-comparison analysis. *P < 0.05 **P < 0.01, ***P < 0.001.
(A-C) Eight-week-old lean C57BL/6J (WT) and age-matched ob/ob mice were fasted for 24 hours and refed for 1 hour.
(A) Phospho-IKKβSer176/180 and IKKβ protein levels in total liver lysates.
(B-C) p65 and Lamin A/C protein levels in nuclear extracts of the livers of WT and ob/ob mice that were refed for 1 hour following a 24-hour fasting.
(D-R) Male ob/ob mice (8 weeks old) were injected with Ad-ca-IKKβ or Ad-LacZ at a dose of 4 × 107 plaque-forming unit (PFU) g-1 (n=5 in each group) via the tail vein.
(D) IKKβ and tubulin protein levels in total lysates and p65 and Lamin A/C in nuclear extracts of the liver of ob/ob mice 12 days after the injection.
(E) Densitometric quantification of the western blot bands in (A) depicts the ratio of p65 protein to Lamin A/C protein.
(F-G), (F) Blood glucose levels (mg dl-1) before and (G) after 6 hours of fasting on day 11 post injection. Each experiment was reproduced in two independent groups.
(H) GTT on day 4 post injection.
(I-J) ITT on day 6 post injection. The curves depict (I) the absolute blood glucose concentration and (J) the percentage of initial glucose concentration. Each experiment was reproduced in two independent groups.
(K-M), (K) Plasma insulin concentrations (ng ml-1), (L) plasma ALT (IU L-1) and (M) AST (IU L-1) of ob/ob mice on day 12 post injection. Each experiment was reproduced in two independent groups.
(N) Daily food intake (g) of the ob/ob mice during 12 days after injection. Each experiment was reproduced in two independent groups.
(O) Body weight (g) of the ob/ob mice on 12 day post injection. Each experiment was reproduced in two independent groups.
(P) PERKThr980 phosphorylation, total PERK, XBP1s and Tubulin protein levels in the total lysate, and XBP1s and Lamin A/C protein levels in the nuclear extracts of the liver of ob/ob mice 12 days after injection.
(Q) Ddit3 mRNA levels in livers of ob/ob mice 12 days after injection. 18S was used for normalization of gene expression.
(R) Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71) and ATF2 protein amounts in livers of ob/ob mice 12 days after injection.
All Error bars are represented as mean ± SEM. Significance was determined by student’s t test (E-G, K-O, and Q) or two-way ANOVA with Bonferroni multiple-comparison analysis (H-J). *P < 0.05, **P < 0.01, ***P < 0.001.
Male ob/ob mice (8 weeks old) were injected with AAV-ca-IKKβ and AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) through the tail vein. The mice were sacrificed on post injection day 30.
(A) Gene expression levels of Cxcr2, Cd14, Sod2, Cd40 and Nos2 in the liver.
(B) Gene expression levels of Il-1, Il-4, Il-6 and Tnfα in the liver.
(C) Plasma IL-6 levels of ob/ob mice 30 days after AAV injection.
(D) IKKβ and tubulin protein levels in white adipose tissue (WAT), brown adipose tissue (BAT), muscle and kidney tissues from ob/ob mice.
(E) ATF4, Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71), ATF2 and tubulin protein amounts in livers of ob/ob mice 30 days after AAV injection.
(F) Averaged daily food intake (g) during 4 weeks after the AAV injection.
(G) Body weight (g) of the ob/ob mice during 4 weeks after injection.
(H) Body temperature (°C) of the ob/ob mice measured on day 8, 15 and 25 post injection.
(I) Hematoxylin and eosin (H&E) staining (left row), active caspase-3 imunostaining (middle row) and trichrome staining (right row) of liver sections from the ob/ob mice 30 days after injection.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (A, B, C, and F) or two-way ANOVA with Bonferroni multiple-comparison analysis (G and H). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
(A-H) Wild-type lean C57BL/6J male mice (3 weeks old) were placed on 45 kcal % High-Fat Diet (HFD) for 16-20 weeks. The diet induced obese (DIO) mice were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) via the tail vein.
(A) Gene expression levels of Cxcr2, Cd14, Sod2, Cd40, Nos2, interleukin 1 complex(Il-1), interleukin 4(Il-4), and interleukin 6 (Il-6) in the livers of DIO mice 4 weeks after injection.
(B) ATF4 and tubulin protein levels in the livers of DIO mice 4 weeks after injection.
(C) Gene expression levels of Acaca, Acacb, Dgat2 and Fasn in the liver of DIO mice 4 weeks after injection.
(D) Body temperature (B.T.) (°C) of the DIO mice measu red on day 20 post injection.
(E-F), (E) Body weight (g) and (F) average daily food intake (g) of the DIO mice during 14 weeks after the injection.
(G-H), (G) Plasma AST (IU L-1) and (H) ALT (IU L-1) of DIO mice 24 days after injection.(I-M) Wild-type lean C57BL/6J male mice (3 weeks old) were placed on 45 kcal % High-Fat Diet (HFD) for 16-20 weeks. The diet induced obese (DIO) mice were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=6 in each group) via the tail vein. Hyperinsulinemic-euglycemic clamp (HEC) was performed on these DIO mice 21 days after the AAV injection.
(I) Hepatic Glucose production (HGP) (mg/kg/min) measured under basal and steady-state conditions (clamp) during the clamp.
(J) Glucose infusion rates (GIR) (mg/kg/min) during HEC in the DIO mice.
(K) The graph depicts the average glucose infusion rate (mg/kg/min) of the DIO mice under steady-state conditions shown in (B).
(L) Glucose disappearance rates (mg/kg/min) during HEC.
(M) Blood glucose levels (mg/dl) during HEC.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (A, C, D, F, G, H, I, K and L) or two-way ANOVA with Bonferroni multiple-comparison analysis (E, J and M). *P < 0.05 NS: Nonsignificant.
LIKKβtg(+/+) and LIKKβtg(-/-) mice (4 weeks old) were challenged with a HFD.
(A) Gene expression levels of Cd14, Cd40, Ccl2, Il-10, interleukin 1 receptor, type I (Il-1r1), and Il-6 in the livers of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 14 weeks of HFD-feeding.
(B) GTT of the first group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(C) GTT of the second group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=4 in each group).
(D) GTT of the third group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=9 in LIKKβtg(+/+); n=6 in LIKKβtg(-/-) group).
(E) ITT of the first group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(F) ITT of the third group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=9 in LIKKβtg(+/+); n=6 in LIKKβtg(-/-) group).
(G) ITT of the second group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=4 in each group).
(H-I), (H) Plasma ALT (IU L-1) and (I) AST (IU L-1) of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 11 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(J) Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71) and ATF2 protein amounts in livers of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 14 weeks of HFD-feeding.
(K) Food intake (g) during 14 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(L) Body weight (g) of the LIKKβtg(+/+) and LIKKβtg(-/-) mice during 14 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(M) Body temperature (°C) of the hIKK β+/+ and LIKKβtg(-/-) mice measured after 9 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
Error bars are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni multiple-comparison analysis (A-F, I and J) or student’s t test (G, H and K). *P < 0.05, **P < 0.01, ***P < 0.001.
(A) MEF cells were infected with Ad-shLacZ and Ad-shXBP1s. Sixteen hours after infection, cells were treated with Tunicamycin (3 ug/mL) for 2 hr. XBP1s and Lamin A/C levels in the nucleus of MEFs cells were detected by western blotting.
(B) MEF cells were infected with Ad-shLacZ and Ad-shXBP1s viruses. Sixteen hours after infection, cells were treated with Tunicamycin (3 μg/mL) or DMSO for 2 hr. Xbp1s mRNA level was determined by QPCR.
(C-O) Male db/db mice (8 weeks old) were injected with AAV-GFP+Ad-shLacZ (n=6), AAV-ca-IKKβ+Ad-shLacZ (n=7), or AAV-ca-IKKβ+Ad-shXBP1s (n=6) through the tail vein; AAV-GFP and AAV-ca-IKKβ viruses were injected at a dose of 2.4 × 109 GC g-1 and Ad-shLacz and Ad-shXBP1 viruses were injected at a dose of 6 × 107 PFU g-1. 10 days after the injection, the mice were sacrificed. (C) Blood glucose levels (mg dl-1) after 6 hours of fasting on day 3 after the injections.
(D) GTT on post injection day 6.
(E) Area under curve (AUC) analysis of GTT from Figure S7D.
(F) Blood glucose levels (mg dl-1) after 12 hours of fasting on day 6 post injection.
(G) Plasma insulin levels (ng ml-1) on post injection day 10.
(H) Body weight (g) of the ob/ob mice during 9 days after injection.
(I) Average food intake (g) of the mice during 10 days after the AAV injection.
(J) XBP1s, IKKβ and tubulin protein levels in total lysates of the liver of db/db mice 10 days after injection.
(K) The graph depicts the ratio of the signals of XBP1s protein to Tubulin protein shown in (C).
(L) XBP1s and Lamin A/C protein levels in nuclear extracts of the liver of db/db mice 10 days after injection.
(M) The graph depicts the ratio of the signals of XBP1s protein to Lamin A/C protein shown in (L).
(N) PERKThr980 phosphorylation and total PERK protein levels in the livers of db/db mice 10 days after injection.
(O) The graph depicts the ratio of the signals of p-PERKThr980 protein to total PERK protein shown in (N).
Error bars are represented as mean ± SEM. Significance was determined by one-way ANOVA with Bonferroni multiple-comparison analysis (B, C, E, F, G, I, K, M and O) or two-way ANOVA with Bonferroni multiple-comparison analysis (D and H). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-XBP1s | Santa Cruz Biotechnology | Cat#sc-7160 |
| Rabbit polyclonal anti-insulin receptor (IR) | Santa Cruz Biotechnology | Cat#sc-711 |
| Mouse monoclonal anti-phosphotyrosine (PY99) | Santa Cruz Biotechnology | Cat#sc-7020 |
| Mouse monoclonal anti-myc | Santa Cruz Biotechnology | Cat#sc-40 |
| Mouse monoclonal anti-HA antibodies | Santa Cruz Biotechnology | Cat#sc-7392 |
| Rabbit polyclonal anti-ATF4 | Santa Cruz Biotechnology | Cat#sc-200 |
| Rabbit polyclonal anti-ATF6 | Santa Cruz Biotechnology | Cat#sc-22799 |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Cat#sc-2005 |
| Goat anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat#sc-2030 |
| Rabbit polyclonal anti-IKKβ | Cell Signaling Technology | Cat#2684 |
| Rabbit polyclonal anti-IKKα | Cell Signaling Technology | Cat#2682 |
| Rabbit polyclonal anti-IKKγ | Cell Signaling Technology | Cat#2685 |
| Rabbit polyclonal anti-p-JNK | Cell Signaling Technology | Cat#9251 |
| Rabbit polyclonal anti-JNK2 | Cell Signaling Technology | Cat#4672 |
| Rabbit monoclonal anti-p-P38 | Cell Signaling Technology | Cat#4631 |
| Rabbit polyclonal anti-P38 | Cell Signaling Technology | Cat#9212 |
| Rabbit polyclonal anti-p-c-Jun | Cell Signaling Technology | Cat#9164 |
| Rabbit monoclonal anti-c-Jun | Cell Signaling Technology | Cat#9165 |
| Rabbit monoclonal anti-ATF2 | Cell Signaling Technology | Cat#9226 |
| Rabbit polyclonal anti-p-ATF2 | Cell Signaling Technology | Cat#9225 |
| Mouse monoclonal anti-ubiquitin | Cell Signaling Technology | Cat#3936 |
| Rabbit monoclonal anti-phospho-AKT (Thr308) | Cell Signaling Technology | Cat#4056 |
| Rabbit monoclonal anti-phospho-AKT (Ser473) | Cell Signaling Technology | Cat#4058 |
| Rabbit polyclonal anti-AKT | Cell Signaling Technology | Cat#9272 |
| Rabbit polyclonal anti-phospho-GSK3α/β(Ser21/9) | Cell Signaling Technology | Cat#9331 |
| Rabbit monoclonal anti-GSK3β | Cell Signaling Technology | Cat#9315 |
| Rabbit monoclonal anti-phospho-PERK (Thr980) | Cell Signaling Technology | Cat#3179 |
| Rabbit monoclonal PERK antibodies | Cell Signaling Technology | Cat#3192 |
| Rabbit monoclonal anti-p65 | Cell Signaling Technology | Cat#4764 |
| Rabbit monoclonal anti-Nup98 | Cell Signaling Technology | Cat#4283 |
| Rabbit polyclonal anti-Lamin A/C | Cell Signaling Technology | Cat#2032 |
| Rabbit monoclonal anti-α-tubulin | Cell Signaling Technology | Cat#2125 |
| Mouse monoclonal Anti-Flag | Sigma Aldrich | Cat#F3165 |
| anti-His | Sigma Aldrich | SAB1305538 |
| Rabbit polyclonal anti-phosphoserine/threonine | ECM Biosciences | Cat#PP2551 |
| Rabbit polyclonal anti-phospho-XBP1sThr48 | Covance | Raised against CRAAGSEASGT(p)PQARKRQR |
| Mouse monoclonal anti-XBP1s | Biolegend | Cat#647502 |
| Mouse monoclonal anti-IKKβ | Novus | Cat#10AG2 |
| Rabbit polyclonal anti-Phosphoserine/threonine | Abcam | Cat#Ab17464 |
| Mouse monoclonal anti-IKKγ | BD PharMingen | Cat#559675 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (aminocarbony)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) | Sigma Aldrich | T1452 |
| Cycloheximide (CHX) | Sigma Aldrich | C1988 |
| Recombinant Human TNF-α protein | R&D systems | 210-TA |
| Recombinant human insulin | Eli lilly | HI-210 |
| Recombinant XBP1s | This paper | N/A |
| D-[3-3H]-glucose | PerkinElmer | NET331C250UC |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: Human embryonic kidney 293 (HEK293) | ATCC | CRL-1573 |
| Hamster: CHO cells | ATCC | CRL-2092 |
| Mouse: Hepa-1c1c7 (hepatoma cell) | ATCC | CRL-2026 |
| Human: HepG2 (hepatoma cell) | ATCC | HB-8065 |
| Human : MDA-MB-231 (breast adenocarcinoma cell) | ATCC | HTB-26 |
| Experimental Models: Organisms/Strains | ||
| Mouse: | ||
| B6.Cg-Lepob/J | The Jackson Laboratory | Cat# JAX:000632 |
| LIKKβtg(+/-) | Michael Karin | N/A |
| B6.BKS(D)-Leprdb/J | The Jackson Laboratory | Cat# JAX:000697 |
| Recombinant DNA | ||
| Plasmid: pCR-Flag-IKKβ(mouse) | Addgene | Cat#15465 |
| Plasmid: pcDNA3.1-IKKβ(S177E/S181E)-pcw107 | Addgene | Cat#64609 |
| Plasmid: Human XBP1s plasmid | Addgene | Cat#63680 |
| Plasmid: pcDNA3.1-Flag-XBP1s(mouse) | Jaemin L. et al., 2011 | N/A |
| Plasmid: pCR-Flag-IKKβ-KM(mouse) | Michael Karin | N/A |
| Plasmid: Vector expressing NH2-terminal (1–373 aa) of activating transcription factor 6n (ATF6n) | Ozcan L. et al., 2009 | N/A |
| Plasmid: pcDNA3.1-Flag-XBP1s(T48A) | This paper | N/A |
| Plasmid: pcDNA3.1-Flag-XBP1s(S148A) | This paper | N/A |
| Plasmid: pcDNA3.1-Flag-XBP1s(T48A/S148A) | This paper | N/A |
| Ad-ca-IKKβ | Michael Karin | N/A |
| Ad-XBP1s | This paper | N/A |
| AAV-GFP | Gene Therapy Center and Vector Core, University of Massachusetts Medical School | N/A |
| AAV-ca-IKKβ | This paper | N/A |
| Sequence-Based Reagents | ||
| Primers for Realtime analysis, see Table S1 | This paper | N/A |
Primer: sequences for amino acid substitution of IKKβ (S177E/S181E):
|
This paper | N/A |
Primer: sequences for amino acid substitution of XBP1(T48A):
|
This paper | N/A |
Primer: sequences for amino acid substitution of XBP1 (S148A):
|
This paper | N/A |
| Software and Algorithms | ||
| Image J | https://imagej.nih.gov/ij/ | N/A |
| GraphPad Prism 6 | Purchased by Boston Children Hospital | N/A |
| Other | ||
HIGLIGHTS.
IKKβ phosphorylates XBP1s on at least on two residues.
Phosphorylation of XBP1s increases its stability.
Increased hepatic activity of IKKβ does not cause insulin resistance.
IKKβ is beneficial for glucose homeostasis.
Acknowledgments
We thank Morris F. White and Rongya Tao for helping us to perform the hyperinsulinemic-euglycemic clamp experiments. This work was supported by funds provided to UO from Department of Medicine, Boston Children’s Hospital, by NIH grants R01DK081009 and R01DK098496 to UO, by the American Diabetes Association’s Career Development grant #7-09-CD-10, and by NIH grant AI043477 to MK. UO is a scientific founder, shareholder, and member of the scientific advisory board and board of directors of ERX Pharmaceuticals, Inc.
Footnotes
AUTHOR CONTRIBUTION
UO came up with the idea to investigate role of IKKβ in regulation of ER stress and XBP1s activity. UO and JL designed the experiments. JL has performed most of the experiments with help from DI, JL, HH, BA, PM, MASH, MFU and SWP. The LIKKβTg(+/+) mouse model was created by KT and MK. Data were analyzed by UO, MK and JL. The manuscript was written by UO, JL, SWP and MK.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-[beta] links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K, Kobayashi M, Iwane A, Sasako T, Okazaki Y, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab. 2011;13:401–412. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nature medicine. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 Serine 307 Promotes Insulin Sensitivity in Mice. Cell Metabolism. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clinical microbiology reviews. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang ZV, Tao C, Gao N, Holland WL, Ferdous A, Repa JJ, Liang G, Ye J, Lehrman MA, et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. The Journal of clinical investigation. 2013;123:455–468. doi: 10.1172/JCI62819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Ye JP. Why do anti-inflammatory therapies fail to improve insulin sensitivity? Acta pharmacologica Sinica. 2012;33:182–188. doi: 10.1038/aps.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MC, Wernstedt I, Berndtsson A, Enge M, Bell M, Hultgren O, Horn M, Ahren B, Enerback S, Ohlsson C, et al. Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes. 2006;55:1205–1213. doi: 10.2337/db05-1304. [DOI] [PubMed] [Google Scholar]
- Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor perspectives in biology. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory Mechanisms in Obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Science’s STKE : signal transduction knowledge environment. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hu FB. Globalization of Diabetes: The role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, Xu H. Constitutive activation of IKKbeta in adipose tissue prevents diet-induced obesity in mice. Endocrinology. 2012;153:154–165. doi: 10.1210/en.2011-1346. [DOI] [PubMed] [Google Scholar]
- Karin M. Inflammation-activated Protein Kinases as Targets for Drug Development. Proceedings of the American Thoracic Society. 2005a;2:386–390. doi: 10.1513/pats.200504-034SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Inflammation-activated protein kinases as targets for drug development. Proceedings of the American Thoracic Society. 2005b;2:386–390. doi: 10.1513/pats.200504-034SR. discussion 394-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nature reviews Genetics. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster GI, Kammoun HL, Kraakman MJ, Kowalski GM, Bruce CR, Febbraio MA. PKR is not obligatory for high-fat diet-induced obesity and its associated metabolic and inflammatory complications. Nature communications. 2016;7:10626. doi: 10.1038/ncomms10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. The Journal of biological chemistry. 2014;289:1203–1211. doi: 10.1074/jbc.R113.534743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sun C, Zhou Y, Lee J, Gokalp D, Herrema H, Park SW, Davis RJ, Ozcan U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat Med. 2011;17:1251–1260. doi: 10.1038/nm.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillicuddy FC, Reynolds CM, Finucane O, Coleman E, Harford KA, Grant C, Sergi D, Williams LM, Mills KH, Roche HM. Long-term exposure to a high-fat diet results in the development of glucose intolerance and insulin resistance in interleukin-1 receptor I-deficient mice. American journal of physiology Endocrinology and metabolism. 2013;305:E834–844. doi: 10.1152/ajpendo.00297.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140:338–348. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dea E, Hoffmann A. NF-κB signaling. Wiley interdisciplinary reviews Systems biology and medicine. 2009;1:107–107. doi: 10.1002/wsbm.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Park SW, Ozcan U. Potential for therapeutic manipulation of the UPR in disease. Seminars in immunopathology. 2013;35:351–373. doi: 10.1007/s00281-013-0370-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nature medicine. 2010;16:429–437. doi: 10.1038/nm.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. Jama. 2011;305:2556–2564. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- Rehman KK, Trucco M, Wang Z, Xiao X, Robbins PD. AAV8-mediated gene transfer of interleukin-4 to endogenous beta-cells prevents the onset of diabetes in NOD mice. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:1409–1416. doi: 10.1038/mt.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Norquay L, Farhang J, D’Aquino K, Copps K, White MF. Human IL6 enhances leptin action in mice. Diabetologia. 2010;53:525–535. doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U, Karin M. The IKK/NF-kappa B pathway. Critical care medicine. 2002;30:S18–26. [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of clinical investigation. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, et al. Prevalence and control of diabetes in Chinese adults. Jama. 2013;310:948–959. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu CM, Cao XC, Zang Y, Zhou YB, Li J. Involvement of transcription factor XBP1s in the resistance of HDAC6 inhibitor Tubastatin A to superoxidation via acetylation-mediated proteasomal degradation. Biochemical and biophysical research communications. 2014;450:433–439. doi: 10.1016/j.bbrc.2014.05.134. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Lee J, Fisher SJ, White MF, Biddinger SB, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nature medicine. 2011;17:356–365. doi: 10.1038/nm.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) p65 and Lamin A/C protein levels in nuclear extracts of HEK293 cells infected with XBP1s-expressing adenovirus (Ad-XBP1s)
(B) mRNA levels of IKKβ, CD14 antigen (CD14), CD40 antigen (CD40), chemokine (C-X-C motif) ligand 10 (CXCL10), chemokine (C-X-C motif) receptor 2 (CXCR2), superoxide dismutase 2, mitochondrial (SOD2) and nitric oxide synthase 2 (NOS2) in HEK293 cells infected with Ad-ca-IKKβ. HEK293 cells infected with Ad-LacZ were used as control. 18S was used for normalization of gene expression.
(C-F) XBP1s and tubulin levels in (C) CHO, (D) Hepa-1c1c7, (E) HepG2 and (F) MDA-MB-231 cells infected with Ad-XBP1s and Ad-ca-IKKβ.
(G) Human XBP1s (hXBP1s) and tubulin levels in HEK293 cells transfected with the vector expressing hXBP1s alone or together with ca-IKKβ expressing vector. These experiments were reproduced in two independent experiments.
(H) XBP1s mRNA levels in HEK293 cells infected with Ad-XBP1s alone or together with Ad-ca-IKKβ. 18S was used for normalization of gene expression.
(I) XBP1s mRNA levels in HepG2 cells infected with Ad-LacZ or Ad-ca-IKKβ. 18S was used for normalization of gene expression.
(J) The protein levels of cleaved forms of ATF6 (ATF6n), IKKβ and tubulin levels in HEK293 cells transfected with ATF6n expressing vector alone or together with ca-IKKβ expressing vector.
(K) XBP1s, IKKα and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with IKKα expressing vector.
(L) XBP1s, IKKγ and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with IKKγ expressing vector.
(M-N) XBP1s and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector alone or together with ca-IKKβ expressing vector and further treated with cycloheximide (CHX, 20 μg ml-1) at indicated time points. The blots of the cells only transfected with XBP1s expressing vector were specifically exposed for a longer time (left bands) and the blots from cells co-transfected with XBP1s and ca-IKKβ expressing vectors together were exposed for a shorter time (right bands) to achieve similar levels of total XBP1s signal at the zero time point.
(O) XBP1s, phospho-IKKβSer176/180 and tubulin protein levels in HEK293 cells transfected with XBP1s expressing vector and further treated with vehicle or IL-1β (100 ng ml-1) for one hour. These results were reproduced in three independent experiments.
(P) 293T cells were transfected with XBP1s expressing vector alone or together with ca-IKKβ expressing vector. Sixteen hours after transfection, cells were treated with MG132 (10 mM) or DMSO. XBP1s and tubulin protein levels were analyzed with immunoblotting. These results were reproduced in three independent experiments.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
(A-B) Tandem mass spectroscopy (MS/MS) analysis of XBP1s that was immunoprecipitated from IKKβ kinase assay. (A) Phosphorylated thr48 residue of XBP1s in the protein fragments. (B) Phosphorylated ser148 residue of XBP1s in the protein fragments.
(C) Cytoplasmic, nuclear and total protein levels of XBP1s in HEK293 cells that were transfected with indicated vectors. These results were reproduced in three independent experiments.
(D) Phospho-XBP1s levels in HEK293 transfected with the indicated vectors. We specifically loaded three times more amount of IP samples from XBP1s-transfected cells (line 2), XBP1s(DM)-transfected cells (line 4) and XBP1s(DM)/ca-IKKβ-co-transfected cells (line 5) when compared to those of XBP1s/ca-IKKβ-co-transfected cells (line3) to achieve comparable XBP1s signal from each sample.
(E-G) XBP1s and tubulin protein levels in HEK293 cells transfected with the indicated vectors and further treated with CHX (20 μg ml-1) at indicated time points. Western blotting results in (E), (F) and (G) are from three independent experiments. The blots of the cells co-transfected with XBP1s and ca-IKKβ expressing vectors were exposed for a shorter time (left bands), and the blots from cells co-transfected with XBP1s(DM) and ca-IKKβ expressing vectors were exposed for a longer time (right bands) to achieve similar levels of total XBP1s signal at the zero time point.
(H) The graph depicts the averaged ratio of the signals of XBP1s to tubulin levels from three independent protein degradation experiments in Figure S2E-S2G. The dotted lines show the half-life of XBP1s protein.
Error bars are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni multiple-comparison analysis. *P < 0.05 **P < 0.01, ***P < 0.001.
(A-C) Eight-week-old lean C57BL/6J (WT) and age-matched ob/ob mice were fasted for 24 hours and refed for 1 hour.
(A) Phospho-IKKβSer176/180 and IKKβ protein levels in total liver lysates.
(B-C) p65 and Lamin A/C protein levels in nuclear extracts of the livers of WT and ob/ob mice that were refed for 1 hour following a 24-hour fasting.
(D-R) Male ob/ob mice (8 weeks old) were injected with Ad-ca-IKKβ or Ad-LacZ at a dose of 4 × 107 plaque-forming unit (PFU) g-1 (n=5 in each group) via the tail vein.
(D) IKKβ and tubulin protein levels in total lysates and p65 and Lamin A/C in nuclear extracts of the liver of ob/ob mice 12 days after the injection.
(E) Densitometric quantification of the western blot bands in (A) depicts the ratio of p65 protein to Lamin A/C protein.
(F-G), (F) Blood glucose levels (mg dl-1) before and (G) after 6 hours of fasting on day 11 post injection. Each experiment was reproduced in two independent groups.
(H) GTT on day 4 post injection.
(I-J) ITT on day 6 post injection. The curves depict (I) the absolute blood glucose concentration and (J) the percentage of initial glucose concentration. Each experiment was reproduced in two independent groups.
(K-M), (K) Plasma insulin concentrations (ng ml-1), (L) plasma ALT (IU L-1) and (M) AST (IU L-1) of ob/ob mice on day 12 post injection. Each experiment was reproduced in two independent groups.
(N) Daily food intake (g) of the ob/ob mice during 12 days after injection. Each experiment was reproduced in two independent groups.
(O) Body weight (g) of the ob/ob mice on 12 day post injection. Each experiment was reproduced in two independent groups.
(P) PERKThr980 phosphorylation, total PERK, XBP1s and Tubulin protein levels in the total lysate, and XBP1s and Lamin A/C protein levels in the nuclear extracts of the liver of ob/ob mice 12 days after injection.
(Q) Ddit3 mRNA levels in livers of ob/ob mice 12 days after injection. 18S was used for normalization of gene expression.
(R) Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71) and ATF2 protein amounts in livers of ob/ob mice 12 days after injection.
All Error bars are represented as mean ± SEM. Significance was determined by student’s t test (E-G, K-O, and Q) or two-way ANOVA with Bonferroni multiple-comparison analysis (H-J). *P < 0.05, **P < 0.01, ***P < 0.001.
Male ob/ob mice (8 weeks old) were injected with AAV-ca-IKKβ and AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) through the tail vein. The mice were sacrificed on post injection day 30.
(A) Gene expression levels of Cxcr2, Cd14, Sod2, Cd40 and Nos2 in the liver.
(B) Gene expression levels of Il-1, Il-4, Il-6 and Tnfα in the liver.
(C) Plasma IL-6 levels of ob/ob mice 30 days after AAV injection.
(D) IKKβ and tubulin protein levels in white adipose tissue (WAT), brown adipose tissue (BAT), muscle and kidney tissues from ob/ob mice.
(E) ATF4, Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71), ATF2 and tubulin protein amounts in livers of ob/ob mice 30 days after AAV injection.
(F) Averaged daily food intake (g) during 4 weeks after the AAV injection.
(G) Body weight (g) of the ob/ob mice during 4 weeks after injection.
(H) Body temperature (°C) of the ob/ob mice measured on day 8, 15 and 25 post injection.
(I) Hematoxylin and eosin (H&E) staining (left row), active caspase-3 imunostaining (middle row) and trichrome staining (right row) of liver sections from the ob/ob mice 30 days after injection.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (A, B, C, and F) or two-way ANOVA with Bonferroni multiple-comparison analysis (G and H). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.
(A-H) Wild-type lean C57BL/6J male mice (3 weeks old) were placed on 45 kcal % High-Fat Diet (HFD) for 16-20 weeks. The diet induced obese (DIO) mice were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=5 in each group) via the tail vein.
(A) Gene expression levels of Cxcr2, Cd14, Sod2, Cd40, Nos2, interleukin 1 complex(Il-1), interleukin 4(Il-4), and interleukin 6 (Il-6) in the livers of DIO mice 4 weeks after injection.
(B) ATF4 and tubulin protein levels in the livers of DIO mice 4 weeks after injection.
(C) Gene expression levels of Acaca, Acacb, Dgat2 and Fasn in the liver of DIO mice 4 weeks after injection.
(D) Body temperature (B.T.) (°C) of the DIO mice measu red on day 20 post injection.
(E-F), (E) Body weight (g) and (F) average daily food intake (g) of the DIO mice during 14 weeks after the injection.
(G-H), (G) Plasma AST (IU L-1) and (H) ALT (IU L-1) of DIO mice 24 days after injection.(I-M) Wild-type lean C57BL/6J male mice (3 weeks old) were placed on 45 kcal % High-Fat Diet (HFD) for 16-20 weeks. The diet induced obese (DIO) mice were injected with AAV-ca-IKKβ or AAV-GFP at a dose of 2.4 × 109 GC g-1 (n=6 in each group) via the tail vein. Hyperinsulinemic-euglycemic clamp (HEC) was performed on these DIO mice 21 days after the AAV injection.
(I) Hepatic Glucose production (HGP) (mg/kg/min) measured under basal and steady-state conditions (clamp) during the clamp.
(J) Glucose infusion rates (GIR) (mg/kg/min) during HEC in the DIO mice.
(K) The graph depicts the average glucose infusion rate (mg/kg/min) of the DIO mice under steady-state conditions shown in (B).
(L) Glucose disappearance rates (mg/kg/min) during HEC.
(M) Blood glucose levels (mg/dl) during HEC.
Error bars are represented as mean ± SEM. Significance was determined by student’s t test (A, C, D, F, G, H, I, K and L) or two-way ANOVA with Bonferroni multiple-comparison analysis (E, J and M). *P < 0.05 NS: Nonsignificant.
LIKKβtg(+/+) and LIKKβtg(-/-) mice (4 weeks old) were challenged with a HFD.
(A) Gene expression levels of Cd14, Cd40, Ccl2, Il-10, interleukin 1 receptor, type I (Il-1r1), and Il-6 in the livers of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 14 weeks of HFD-feeding.
(B) GTT of the first group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(C) GTT of the second group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=4 in each group).
(D) GTT of the third group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 8 weeks of HFD-feeding (n=9 in LIKKβtg(+/+); n=6 in LIKKβtg(-/-) group).
(E) ITT of the first group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(F) ITT of the third group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=9 in LIKKβtg(+/+); n=6 in LIKKβtg(-/-) group).
(G) ITT of the second group of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 6 weeks of HFD-feeding (n=4 in each group).
(H-I), (H) Plasma ALT (IU L-1) and (I) AST (IU L-1) of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 11 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(J) Phospho-JNK(Thr183/Tyr185), JNK, Phospho-P38(Thr180/Tyr182), P38, Phospho-ATF2(Thr69/71) and ATF2 protein amounts in livers of LIKKβtg(+/+) and LIKKβtg(-/-) mice after 14 weeks of HFD-feeding.
(K) Food intake (g) during 14 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(L) Body weight (g) of the LIKKβtg(+/+) and LIKKβtg(-/-) mice during 14 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
(M) Body temperature (°C) of the hIKK β+/+ and LIKKβtg(-/-) mice measured after 9 weeks of HFD-feeding (n=5 in LIKKβtg(+/+) group; n=4 in LIKKβtg(-/-) group).
Error bars are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni multiple-comparison analysis (A-F, I and J) or student’s t test (G, H and K). *P < 0.05, **P < 0.01, ***P < 0.001.
(A) MEF cells were infected with Ad-shLacZ and Ad-shXBP1s. Sixteen hours after infection, cells were treated with Tunicamycin (3 ug/mL) for 2 hr. XBP1s and Lamin A/C levels in the nucleus of MEFs cells were detected by western blotting.
(B) MEF cells were infected with Ad-shLacZ and Ad-shXBP1s viruses. Sixteen hours after infection, cells were treated with Tunicamycin (3 μg/mL) or DMSO for 2 hr. Xbp1s mRNA level was determined by QPCR.
(C-O) Male db/db mice (8 weeks old) were injected with AAV-GFP+Ad-shLacZ (n=6), AAV-ca-IKKβ+Ad-shLacZ (n=7), or AAV-ca-IKKβ+Ad-shXBP1s (n=6) through the tail vein; AAV-GFP and AAV-ca-IKKβ viruses were injected at a dose of 2.4 × 109 GC g-1 and Ad-shLacz and Ad-shXBP1 viruses were injected at a dose of 6 × 107 PFU g-1. 10 days after the injection, the mice were sacrificed. (C) Blood glucose levels (mg dl-1) after 6 hours of fasting on day 3 after the injections.
(D) GTT on post injection day 6.
(E) Area under curve (AUC) analysis of GTT from Figure S7D.
(F) Blood glucose levels (mg dl-1) after 12 hours of fasting on day 6 post injection.
(G) Plasma insulin levels (ng ml-1) on post injection day 10.
(H) Body weight (g) of the ob/ob mice during 9 days after injection.
(I) Average food intake (g) of the mice during 10 days after the AAV injection.
(J) XBP1s, IKKβ and tubulin protein levels in total lysates of the liver of db/db mice 10 days after injection.
(K) The graph depicts the ratio of the signals of XBP1s protein to Tubulin protein shown in (C).
(L) XBP1s and Lamin A/C protein levels in nuclear extracts of the liver of db/db mice 10 days after injection.
(M) The graph depicts the ratio of the signals of XBP1s protein to Lamin A/C protein shown in (L).
(N) PERKThr980 phosphorylation and total PERK protein levels in the livers of db/db mice 10 days after injection.
(O) The graph depicts the ratio of the signals of p-PERKThr980 protein to total PERK protein shown in (N).
Error bars are represented as mean ± SEM. Significance was determined by one-way ANOVA with Bonferroni multiple-comparison analysis (B, C, E, F, G, I, K, M and O) or two-way ANOVA with Bonferroni multiple-comparison analysis (D and H). *P < 0.05, **P < 0.01, ***P < 0.001, NS: Non-significance.