

SUMMARY
Although the outcome of flavivirus infection can vary from asymptomatic to lethal, environmental factors modulating disease severity are poorly defined. Here, we observed increased susceptibility of mice to severe West Nile (WNV), Dengue, and Zika virus infections after treatment with oral antibiotics (Abx) that depleted the gut microbiota. Abx treatment impaired the development of optimal T cell responses, with decreased levels of WNV-specific CD8+ T cells associated with increased infection and immunopathology. Abx treatments that resulted in enhanced WNV susceptibility generated changes in the overall structure of the gut bacterial community and in the abundance of specific bacterial taxa. As little as 3 days of treatment with ampicillin was sufficient to alter host immunity and WNV outcome. Our results identify oral Abx therapy as a potential environmental determinant of systemic viral disease, and they raise the possibility that perturbation of the gut microbiota may have deleterious consequences for subsequent flavivirus infections.
In Brief
Thackray et al. observed increased susceptibility to West Nile, Zika, and Dengue virus infections following oral antibiotic treatment in mice. Antibiotics altered the bacterial abundance and community structure and the development of optimal T cell immunity. These data suggest that antibiotics may have deleterious consequences for subsequent flavivirus infections.
INTRODUCTION
Flaviviruses including West Nile (WNV), Dengue (DENV), and Zika (ZIKV) viruses account for approximately 400 million infections annually, with billions at risk and no specific therapy available. In humans, the majority of WNV and DENV infections are subclinical, with severe illness occurring in only a subset of individuals (Chancey et al., 2015; Halstead, 2007). Patient age, underlying immune or chronic disease status, and polymorphisms in immune genes (e.g., CCR5, OAS1, and TNF) are associated with increased susceptibility to WNV and DENV infections (Bigham et al., 2011; Lim et al., 2009, 2010; Lindsey et al., 2012; Loeb, 2013; Racsa et al., 2014). However, these factors do not account for most adverse outcomes. The contribution of environmental factors to flavivirus disease severity remains poorly defined.
Host defense mechanisms that control flavivirus replication and dissemination have been studied extensively (Diamond and Pierson, 2015; Suthar et al., 2013). Immune factors that modulate disease severity of WNV infection include type I interferon (IFN) responses, serum anti-WNV IgM titers, FoxP3+ CD4+ T regulatory cells (Tregs), and CD8+ T cell responses (Diamond et al., 2003; Lanteri et al., 2009; Samuel and Diamond, 2005; Shrestha and Diamond, 2004). Mechanisms that determine disease severity of DENV infection include Fc-γ receptor-mediated antibody-dependent enhancement of infection of myeloid cells and plasma levels of secreted NS1 protein (Diamond and Pierson, 2015; Katzelnick et al., 2017).
Antibiotic (Abx) consumption in humans continues to increase with greater than 800 courses of Abx prescribed annually per 1,000 people in outpatient populations in the United States alone (Hicks et al., 2015; Suda et al., 2014; Van Boeckel et al., 2014). Although anti-flavivirus activity has been demonstrated in vitro for some Abx (De Burghgraeve et al., 2012; Kaptein et al., 2010; Michaelis et al., 2007; Retallack et al., 2016; Zhang et al., 2009), their ability to modulate flavivirus pathogenesis in vivo has not been demonstrated. Oral Abx treatment that perturbs the gut microbiota (herein, microbiota) of mice inhibited poliovirus and reovirus replication, mouse mammary tumor virus transmission, and murine norovirus (MNoV) infection and persistence (Baldridge et al., 2015; Jones et al., 2014; Kane et al., 2011; Kuss et al., 2011). However, oral Abx treatment also delayed lymphocytic choriomeningitis virus (LCMV) clearance and increased influenza virus (IAV) pathogenesis in mice (Abt et al., 2012; Ichinohe et al., 2011; Wu et al., 2013), suggesting that differential interactions between specific viruses, microbial community members, and/or the host may modulate the outcome of infection.
Here we show that treatment of mice with oral Abx resulted in increased susceptibility to severe WNV, DENV, and ZIKV infections. Oral administration of an Abx cocktail comprised of vancomycin (V), neomycin (N), ampicillin (A), and metronidizole (M), herein referred to as VNAM, resulted in lethality, impaired host immunity, and increased WNV burden. VNAM treatment resulted in fewer WNV-specific CD8+ T cells and Tregs in the draining popliteal lymph node (DLN) and spleen. Adoptive transfer of both primed CD4+ and CD8+ T cells was required to reverse the the effect of VNAM treatment on WNV susceptibility. Treatment of mice with A or V alone was sufficient to alter WNV outcome. However, the addition of M was required to generate the fully vulnerable phenotype. A and AM treatment depleted the gut bacterial community biomass and differentially altered bacterial community structure and the abundance of specific bacterial taxa. Our results identify oral Abx treatment as a potential risk factor for disease severity during subsequent flavivirus infection, and they suggest that depletion and/or perturbation of the microbiota may adversely impact host antiviral immunity.
RESULTS
Oral Abx Treatment Alters Susceptibility to Flavivirus Infections
We treated C57BL/6J mice for 14 days before subcutanteous inoculation of WNV in the footpad, and then, throughout the course of infection (14 days continuous; Figure 1A), with a broad-spectrum Abx cocktail, VNAM, that depletes the microbiota (Baldridge et al., 2015). VNAM-treated mice were more susceptible to WNV infection than mice treated with vehicle alone (Figure 1B). Since reciprocal interactions between the host immune system and the microbiota can influence virus infection (Hand, 2016; Kuhn and Stappenbeck, 2013; Pfeiffer and Virgin, 2016), we evaluated the effect of Abx treatment on the outcome of WNV infection in several immunodeficient strains of mice. We observed increased susceptibility to WNV infection following VNAM treatment, even in the absence of signaling through the interferon lambda (IFN-λ) receptor (Ifnlr1, also called IL-28Rα) (Figure S1A). In contrast, previous studies showed that signaling through IFN-λ is required for the effects of oral Abx treatment on MNoV clearance (Baldridge et al., 2015). The effect of Abx on WNV outcome also occurred in the absence of Toll-like receptor (TLR)2- and TLR5-pathogen sensing, caspase 1/11-mediated inflammasome activation, cGAS- and STING-mediated DNA sensing, and STAT6-mediated induction of type 2 immunity (Figures S1B–S1G), all of which are pathways linked to host-microbiota interactions in prior studies (Burrows et al., 2015; Fricke et al., 2015; Ichinohe et al., 2011; Oh et al., 2014; Watson et al., 2015).
Figure 1. Oral Abx Treatment Increases Susceptibility to Flavivirus Infection.

(A) Outline of treatment regimens.
(B and D–G) Wild-type (WT) (B, D, and E), Ifnar1−/− (F), or Ifnar1f/fLysMCre+/+ (G) C57BL/6 mice were treated with vehicle (Kool-Aid) or vehicle supplemented with VNAM for 14 days before virus inoculation and during infection (14-day continuous) (B and F), for 3 days before virus inoculation and during infection (3-day continuous) (D), or for 3 days followed by drinking water for 3 days before virus inoculation (3-day pulse) (E and G).
(C) WT mice were treated with VNAM for 14 days, and then VNAM was replaced with vehicle for 3, 7, or 14 days before WNV inoculation (VNAM-stop). Other WT mice were treated for 17, 21, or 28 days before WNV inoculation and continuously during infection (vehicle, VNAM).
(B–G) Mice were inoculated subcutaneously with 102 focus-forming units (FFU) of WNV at 12 weeks of age (B–E), intravenously with 105 FFU of DENV at 6 weeks of age (F), or subcutaneously with 103 FFU of ZIKV at 4 weeks of age (G).
Survival curves were compared using the log-rank test with a Bonferroni correction (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Results were combined from two to five independent experiments: vehicle (n = 30), VNAM (n = 29) (B); 3 day-stop: vehicle (n = 10), VNAM (n = 10), VNAM-stop (n = 10); 7-day stop: vehicle (n = 15), VNAM (n = 15), VNAM-stop (n = 15); 14-day stop: vehicle (n = 10), VNAM (n = 10), VNAM-stop (n = 10) (C); vehicle (n = 15), VNAM (n = 15) (D); vehicle (n = 15), VNAM (n = 15) (E); vehicle (n = 16), VNAM (n = 15) (F); and vehicle (n = 21), VNAM (n = 22) (G). See also Figure S1.
We examined the durability of the effect of oral Abx on WNV outcome. Mice were treated with VNAM for 14 days, and then VNAM was replaced with vehicle for 3 (3-day stop), 7 (7-day stop), or 14 (14-day stop) days before WNV inoculation (Abx stop, blue lines, Figure 1A). As a control, mice were treated with vehicle or Abx for 17 (3-day stop), 21 (7-day stop), or 28 (14-day stop) days before WNV inoculation and continuously during infection (vehicle, black lines; Abx, red lines, Figure 1A). Increased lethality was observed for Abx-stop mice when VNAM was replaced with vehicle for 3 or 7 days before WNV inoculation; however, resistance to WNV infection was restored between 7 and 14 days after cessation of oral Abx (Figure 1C). Mice treated with VNAM starting 3 days before inoculation and throughout the course of infection (3-day continuous; Figure 1A) showed increased susceptibility to WNV infection compared to vehicle-treated controls (Figure 1D). In addition, mice given VNAM for 3 days followed by unsupplemented drinking water for 3 days before inoculation (3-day pulse; Figure 1A) still showed increased susceptibility to WNV compared to vehicle-treated controls (Figure 1E). Thus, the continued presence of oral Abx was not required to promote WNV infection, consistent with the idea that perturbation of the microbiota alters flavivirus pathogenesis.
To examine the generality of the effect of Abx treatment on flavivirus infection, we treated mice lacking the IFN-α/β receptor (Ifnar1−/−) for 14 days with VNAM before inoculation with DENV and then throughout the course of infection; Ifnar1−/− mice were used as DENV replicates poorly in immunocompetent mice (Zellweger and Shresta, 2014). In contrast to vehicle-treated Ifnar1−/− controls, VNAM-treated Ifnar1−/− mice succumbed to DENV infection (Figure 1F), demonstrating that oral Abx can enhance susceptibility to another flavirus infection and that this occurs even in the absence of type I IFN immunity. Indeed, we also administered a 3-day pulse treatment of VNAM to Ifnar1f/fLysMCre+/+ mice, which lack Ifnar1 expression only in macrophages, granulocytes, and some dendritic cell subsets; these mice were used because wild-type and Ifnar1−/− mice are fully resistant or vulnerable to subcutaneous inoculation of ZIKV, respectively (Lazear et al., 2016). We observed increased susceptibility to severe ZIKV infection in VNAM-treated Ifnar1f/fLysMCre+/+ mice compared to vehicle-treated controls (Figure 1G). Thus, even a short exposure to oral Abx increased the disease severity of distinct flaviviruses.
Oral Abx Treatment Increases Viral Burden
To define the basis for the enhanced virulence of WNV associated with Abx treatment, we evaluated viral burden in different tissues after 14 days of continuous VNAM treatment. We observed little alteration of virus levels in the DLN or spleen, but we detected more WNV in the brain and spinal cord of VNAM-treated mice compared to vehicle-treated controls at days 9 and 10 after infection (Figures 2A and 2B). More ZIKV also was detected in the brain of Ifnar1f/fLysMCre+/+ mice treated with a 3-day pulse of VNAM compared to vehicle-treated controls (Figure S2A).
Figure 2. Oral Abx Treatment Increases WNV Burden.

(A–D) Mice were treated with vehicle or VNAM (n = 9–15) and inoculated with WNV as in Figure 1B. Indicated tissues were harvested and WNV burden was assessed by qRT-PCR (A, C, and D) or plaque assay (B).
The dotted lines represent the limit of detection. Statistical significance was determined using the Mann-Whitney test (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Results were combined from two to three independent experiments. DLN, draining popliteal lymph node; MLN, mesenteric lymph node. See also Figure S2.
Since previous reports have documented WNV infection in the gut (Brown et al., 2007; Kimura et al., 2010; Williams et al., 2015), we examined viral burden along the length of the gastrointestinal tract. We observed more WNV at days 8–10 after infection in the duodenum, ileum, and colon of VNAM-treated mice compared to vehicle-treated controls (Figure 2C). In contrast to the DLN, we observed more WNV at days 2, 4, 8, 9, and 10 in the mesenteric lymph node (MLN) of VNAM-treated mice compared to vehicle-treated controls (Figure 2D).
Oral Abx Treatment Impairs Virus-Specific CD8+ T Cell Responses
Since we mainly observed enhanced WNV burden later during the course of infection, we examined components of adaptive immunity at day 8 in various tissues after a 14-day continuous VNAM treatment. We observed decreased numbers of leukocytes, B cells, and CD4+ and CD8+ T cells in the DLN and spleen, as well as diminished numbers and percentages of CD8+ T cells in the brain of VNAM-treated mice compared to vehicle-treated controls (Figures 3A and 3B; Figure S3A). In contrast, we observed increased numbers of leukocytes and CD4+ and CD8+ T cells, as well as increased numbers and percentages of B cells, in the MLN of VNAM-treated mice compared to vehicle-treated controls, suggesting that the effects of oral Abx treatment may differ between gut-proximal (MLN) and gut-distal (DLN and spleen) lymphoid tissues.
Figure 3. Oral Abx Treatment Decreases Treg and WNV-Specific CD8+ T Cell Responses.

(A–F) Mice were treated with vehicle or VNAM (n = 10–15) and inoculated with WNV as in Figure 1B. Tissues were harvested at day 8 after WNV inoculation. (A and B) The numbers (A) and percentages (B) of leukocytes, B cells, and CD4+ and CD8+ T cells.
(C) The numbers (left) and percentages (right) of Tregs.
(D) The numbers (left) and percentages (right) of WNV-specific CD8+ T cells.
(E and F) The numbers (E) and percentages (F) of CD8+ T cells expressing granzyme B (GzB), and IFN-γ and/or TNF-α after ex vivo stimulation with a Db-NS4B peptide. Statistical significance was determined using an unpaired t test (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Results were combined from two to three independent experiments. See also Figures S2–S4.
Consistent with an effect of Abx treatment on WNV burden later rather than earlier in infection, we did not observe differences in serum cytokine levels or WNV-specific neutralizing activity in mice after a 14-day continuous VNAM treatment compared to vehicle-treated controls (Figures S4A and S4B). Since decreased Treg frequencies are associated with increased disease severity following WNV infection (Lanteri et al., 2009), we profiled Tregs in the DLN, MLN, and spleen (Figure S3A). We observed fewer Tregs in the DLN and spleen of VNAM-treated mice compared to vehicle-treated controls, but no differences in the percentages of Tregs (Figure 3C). In contrast, we observed equivalent numbers of Tregs in the MLN of VNAM-treated mice compared to vehicle-treated controls (Figure 3C), suggesting that oral Abx treatment may alter the accumulation of Tregs in a tissue-specific manner. As equivalent numbers of Tregs were found in the spleen of ZIKV-infected vehicle- and VNAM-treated Ifnar1f/fLysMCre+/+ mice (Figure S2C), an impact of oral Abx treatment on Tregs may not be required to increase susceptibility to severe flavivirus infection.
Since virus-specific effector CD8+ T cells are required to clear WNV from the CNS (Brien et al., 2007; Shrestha and Diamond, 2007; Shrestha et al., 2006a, 2012), we examined the antigen specificity and polyfunctionality of the CD8+ T cells after 14 days of continuous VNAM treatment (Figures S3A and S3B). Decreased numbers and percentages of Db-NS4B-tetramer+ (WNV-specific) CD8+ T cells were detected in the spleen of VNAM-treated mice compared to vehicle-treated controls (Figure 3D). Consistent with these data, diminished ZIKV-specific CD8+ T cell responses were found in the spleen of VNAM-treated Ifnar1f/fLysMCre+/+ mice compared to vehicle-treated controls (Figure S2D). As decreased numbers of WNV-specific CD8+ T cells were observed in the DLN and brain of VNAM-treated mice whereas equivalent numbers were detected in the MLN (Figure 3D), we conclude that oral Abx treatment appears to impair antigen-specific CD8+ T cell responses in a tissue-specific manner.
Although we observed no statistically significant Abx-induced alterations in the numbers and percentages of polyfunctional CD8+ T cells in the DLN or MLN (Figures 3E and 3F), decreased numbers and percentages of CD8+ T cells expressing granzyme B (GzB), IFN-γ, and/or tumor necrosis factor alpha (TNF-α) were evident in the spleen and brain of VNAM-treated mice compared to vehicle-treated controls. As the levels of effector molecules per cell or the ratio of single-, double-, or triple-positive effector CD8+ T cells were similar between vehicle- and VNAM-treated mice (Figures S4C and S4D), oral Abx treatment appears to affect priming, proliferation, and/or trafficking rather than maturation of effector CD8+ T cells.
Oral Abx Treatment Impacts a Host Niche Important for Optimal Virus-Specific T Cell Responses
To explore further the role of CD8+ T cells in the effect of oral Abx treatment on WNV pathogenesis, we assessed viral infection in mice lacking CD8α+ T cells (Cd8−/−) after a 14-day continuous treatment with VNAM. We examined viral burden in lieu of survival as Cd8−/− mice uniformly succumb to WNV (Shrestha and Diamond, 2004). Similar levels of WNV were observed in all tissues examined at 8 days after inoculation in vehicle- and VNAM-treated Cd8−/− mice (Figure 4A), suggesting that Abx-induced changes in CD8+ T cells are important for increasing the virulence of flavivirus infection.
Figure 4. Oral Abx Treatment Impacts Optimal T Cell Responses during WNV Infection.

(A–F) Cd8−/− (A), NS4B+/− Tg (B), WT (D), and Tcrβδ−/− (F) mice were untreated or treated with vehicle or VNAM for 14 days continuously and inoculated with WNV as in Figure 1B or as illustrated in (C) and (E). Tissues (A) were harvested at day 8 after virus inoculation, and WNV burden was assessed as in Figure 2 (all comparisons were not statistically significant (p > 0.05, Mann-Whitney test). Survival curves (B, D, and F) were compared using the log-rank test with a Bonferroni correction (**p < 0.01 and ***p < 0.001). Results were combined from two to four independent experiments: vehicle (n = 8), VNAM (n = 9) (A); vehicle (n = 17), VNAM (n = 16) (B); VNAM (n = 15), VNAM + naive T cells (n = 10), VNAM + WNV-primed T cells (n = 10), VNAM + WNV-primed CD8+ T cells (n = 14) (D); and no T cells (n = 9), WNV-primed T cells (vehicle) (n = 8), WNV-primed T cells (VNAM) (n = 8) (F).
We evaluated the role of virus-specific CD8+ T cells following oral Abx treatment in transgenic (Tg) mice expressing a CD8+ T cell receptor recognizing the Db-restricted WNV NS4B peptide (Kim et al., 2014). Even though more than 90% of the CD8+ T cells of NS4B+/− Tg mice recognized the NS4B peptide (Figure S3C), we still observed increased susceptibility to WNV infection following a 14-day continuous VNAM treatment compared to vehicle-treated controls (p < 0.001; Figure 4B), consistent with the idea that more WNV-specific CD8+ T cell precursors cannot reverse the effect of Abx treatment on immunity when primed in an Abx-treated host.
We next evaluated whether adoptive transfer of T cells from mice that had not received Abx was sufficient to reverse the susceptibility phenotype to WNV infection observed after 14 days of VNAM treatment. Increased survival was observed in recipient VNAM-treated mice when WNV-primed, but not naive, donor T cells were transferred 1 day after WNV inoculation (Figures 4C and 4D). Transfer of WNV-primed CD8+ T cells, however, was not sufficient to fully overcome the effect of Abx treatment (Figure 4D).
To determine whether Abx treatment intrinsically impairs the function of WNV-specific T cells, we transferred WNV-primed T cells from wild-type mice treated for 14 days with vehicle or VNAM into untreated mice lacking T cells (Tcrβδ−/−) (Figure 4E). Resistance to WNV infection was observed in Tcrβδ−/− mice reconstituted with WNV-primed T cells from either vehicle- or VNAM-treated mice at 1 day after WNV inoculation (Figure 4F). These data suggest that oral Abx treatment impairs optimal T cell responses in specific contexts, i.e., T cells that control WNV pathogenesis can develop in an Abx-treated environment but function optimally in an untreated environment, and, conversely, T cells can control WNV pathogenesis in an Abx-treated environment when they are primed in an untreated environment.
Oral Abx Treatment Impairs Host Immunity prior to Virus Infection
Since hematopoiesis is modulated by the microbiota (Balmer et al., 2014; Khosravi et al., 2014), we examined whether oral Abx treatment affects progenitor populations in the bone marrow in the absence of infection (Figure S5A). After a 14-day treatment with VNAM, we observed decreased numbers of lineage− ckithi Sca1+ Flt3− hematopoietic stem cells (HSCs), lineage− CD27+ Flt3+ IL7rα− B220− CD11c− ckithi Sca1+ Ly6D− multi-potent progenitors (MPPs), lineage− CD27+ Flt3+ IL7Rα+ B220− CD11c− common lymphoid progenitors (CLPs), lineage− CD27+ Flt3+ IL7Ra+ B220− CD11c− ckitint/hi Ly6D− all-lymphoid progenitors (ALPs), lineage− CD27+ Flt3+ IL7Ra+ B220− CD11c− ckitlo/int Ly6D+ B cell-biased lymphoid progenitors (BLPs), lineage− CD27+ Flt3+ IL7Ra+ B220+ CD11c− Ly6D+ pre-pro B cell progenitors (pre-pro-Bs), but increased numbers of lineage− ckithi Sca1− CD16/32int common myeloid progenitors (CMPs) in the bone marrow (Figure 5A). In comparison, differences in the numbers of lineage− ckithi Sca1− CD16/32hi granulocyte-macrophage progenitors (GMPs), lineage− ckithi Sca1− CD16/32− megakaryocyte-erythrocyte progenitors (MEPs), lineage− Flt3+ ckitint CD115+ MHCII− CD11c− common DC progenitors (CDPs), lineage− Flt3+ ckithi CD115+ monocyte-macrophage DC progenitors (MDPs), lineage− Flt3+ ckitint MHCIIlow-int CD11c+ SiglecH− pre-DC1 progenitors (pre-DC1s), and lineage− Flt3+ ckitlo CD115+ MHCII− CD11c+ pre-DC2 progenitors (pre-DC2s) were not observed in the bone marrow (Figure 5A). These data show that oral Abx treatment can impact hematopoiesis in the bone marrow, similar to results from a previous study (Josefsdottir et al., 2017).
Figure 5. Oral Abx Treatment Impairs Immunity prior to Virus Infection.

(A–C) Mice were treated with vehicle or VNAM (n = 10) for 14 days and tissues were harvested.
(A) The numbers of total cells, hematopoietic stem cells (HSCs), multi-potent progenitors (MPPs), common lymphoid progenitors (CLPs), all-lymphoid progenitors (ALPs), B cell-biased lymphoid progenitors (BLP), pre-pro B cell progenitors (pre-pro-Bs), common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), megakaryocyte-erythrocyte progenitors (MEPs), common DC progenitors (CDPs), monocyte-macrophage DC progenitors (MDPs), pre-DC1 progenitors (pre-DC1s), and pre-DC2 progenitors (pre-DC2s) were measured.
(B) The numbers (left) and percentages (right) of leukocytes, B cells, CD4+ and CD8+ T cells, and Tregs.
(C) The numbers (left) and percentages (middle and right) of myeloid dendritic cells (mDCs), plasmacytoid dendritic cells (pDCs), and CD24+ (DC1) and CD172+ (DC2) dendritic cells. Statistical significance was determined using an unpaired t test (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Results were combined from two independent experiments.
(D) Sorted splenic DCs from vehicle- or VNAM-treated mice (n = 5) were cultured in duplicate for 3 days with 5-(and 6)-Carboxyfluorescein diacetate succinimidyl ester (CFDA SE or CFSE)-labeled OT-I T cells and different doses of heat-killed Listeria monocytogenes expressing ovalbumin (HKLM-OVA), and they were assayed for OT-I T cell proliferation and activation (CFSE−CD44+). Statistical significance was assessed using 2-way ANOVA with Tukey’s multiple-comparison test. Comparisons between vehicle- and VNAM-treated mice were not significant (p > 0.05). Results were combined from two independent experiments. See also Figure S5.
Cross-presentation by dendritic cells (DCs) contributes to the priming of WNV-specific CD8+ T cell responses (Hildner et al., 2008). We examined the number and percentage of DCs, as well as other immune cell subsets, in the spleen of naive vehicle- and VNAM-treated mice (Figure S5B). Fewer total leukocytes, B cells, CD4+ and CD8+ T cells, Tregs, as well as CD11c+ B220− MHCII+ myeloid and CD11cINT Bst2+ plasmacytoid DCs, were present in the spleens of VNAM-treated naive mice compared to vehicle-treated controls (Figures 5B and 5C). Although we observed lower numbers of CD11c+ B220− MHCII+ CD24+ CD172− (DC1) and CD11c+ B220− MHCII+ CD24− CD172+ (DC2) splenic DC subsets in VNAM-treated mice compared to vehicle-treated naive controls (Figure 5C), the ability of DC1 cells to cross-present cell-associated ovalbumin to MHC class I-restricted, ovalbumin-specific CD8+ T cells (OT-1) was not impaired (Figure 5D). These findings suggest that decreased numbers of DCs, rather than their antigen-presenting potential, may contribute to impaired virus-specific CD8+ T responses after Abx treatment.
The Impact of the Microbiota on Susceptibility to WNV Infection
To address whether the effects of Abx treatment on flavivirus infection were related to changes in the configuration of host microbial communities, we examined whether transfering microbiota after cessation of Abx, either by co-housing coprophagic animals or by gavage of cecal contents, could reverse the susceptibility phenotype. Female mice treated with vehicle or VNAM for 14 days and then co-housed for 7 days before WNV inoculation in the absence of treatment (Figure S6A) retained disparate susceptibility to WNV infection (Figure S6B). VNAM-stop mice gavaged with control PBS or cecal microbiota from vehicle- or VNAM-treated mice (Figure S6C) all exhibited similar susceptility to WNV infection compared to vehicle-treated controls (Figure S6D), suggesting that the effect of oral Abx on WNV infection was refractory to these microbial transfer approaches. However, VNAM-stop mice exposed to cecal micro-biota from other VNAM-treated mice had increased susceptibility to WNV compared to VNAM-stop mice that received cecal microbiota from vehicle-treated mice. In comparison, transfer of cecal microbiota from VNAM-treated mice did not increase the susceptibility of vehicle-treated mice to WNV (Figure 6D). These data indicate that the microbiota in VNAM-treated mice may contribute to increased susceptibility to WNV pathogenesis, but only in Abx-treated mice.
Figure 6. Treatment with A Alone or in Combination with M Depletes and Perturbs Gut Bacteria.

(A–F) Mice were treated with vehicle and single or combinations of Abx for 14 days before and during infection. Mice were inoculated with WNV as in Figure 1B. (A, B, and E) Survival curves were compared using the log-rank test (B) with a Bonferroni correction (A and E) (*p < 0.05 and ****p < 0.0001, comparisons in E were not significant).
(C and F) Fecal samples were harvested at 14 days after treatment (C) or 0–20 days after treatment (F). Bacterial 16S rRNA copy number was determined by qPCR. The dotted line represents the limit of detection. Statistical significance was determined using ANOVA with Dunnett’s multiple-comparison test (*p < 0.05 and ****p < 0.0001).
(D) The numbers (left) and percentages (right) of WNV-specific CD8+ T cells. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple-comparison test (*p < 0.05 and **p < 0.01).
(G) The effect of treatment on β-diversity (defined using the UniFrac phylogenetic dissimilarity metric). Statistical significance of six pairwise comparisons (V versus M, V versus A, V versus AM, M versus A, M versus AM, and A versus AM) for each time point from 0 to 20 days was determined using ADONIS with p < 0.05 considered significant (day 0, all comparisons were not significant, except for V versus AM and M versus AM; days 3–20, all comparisons were significant, except for day 16 V versus M).
(H) The effect of treatment on the abundance of bacterial taxa. Statistical significance was determined using the likelihood-ratio test of pairwise comparisons between all treatments and vehicle from 0 to 20 days. Unique sequences determined to be significant (p < 0.05) with a mean abundance of more than 100 reads were identified for each pairwise comparison. Plots display the abundances of a set of these taxa over time using the general additive model smoothed line for each treatment. Results were combined from two to five independent experiments using male and female mice, except for two experiments in which a single group of individually housed female mice was used (B and E–H): vehicle (n = 24), A (n = 20), V (n = 25), M (n = 20), N (n = 20), AM (n = 20), VM (n = 20), VNA (n = 25), VAM, (n = 15) VNAM (n = 15) (A); vehicle (n = 15), A (n = 30) (B); vehicle (n = 9–10), A (n = 9–10), M (n = 10), N (n = 9–10), AM (n = 10) (C and D); and vehicle (n = 10), A (n = 15), M (n = 10), AM (n = 10) (E–H). See also Figures S6 and S7 and Table S1.
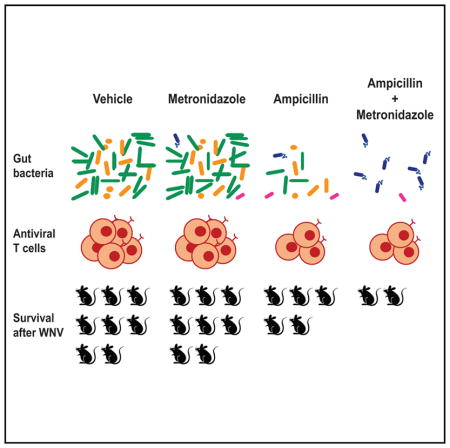
We next assessed which Abx in the VNAM cocktail was required for the effect on WNV outcome and whether this effect was associated with alterations in the microbiota. We treated mice for 14 days before and during WNV infection with single or combinations of Abx. Mice treated with either A or V had an intermediate susceptibility to severe WNV infection compared to vehicle-treated controls (Figures 6A and 6B). N was not required for increased susceptibility to WNV infection (Figure 6A). Although treatment with M did not confer increased susceptibility, it was required for optimal effects, since mice treated with VNA had an intermediate susceptibility and mice treated with AM or VM had increased susceptibility to WNV infection (Figure 6A). The increased susceptibility of mice treated with A, V, AM, VM, or VNAM, but not M or N alone, allowed us to identify a correlation between decreased microbiota biomass, as defined by the reduced number of bacterial 16S rRNA copies per fecal pellet, and viral virulence (Figure 6C). Vulnerability to WNV infection also was associated with decreased WNV-specific CD8+ T cell responses in the spleen of A- or AM-treated, but not M- or N-treated, mice compared to vehicle-treated controls (Figure 6D).
To assess further the effects on the microbiota of Abx treatments that modulated WNV outcome, we re-examined the impact of treatment with A, M, or AM on WNV outcome in an experiment using a single batch of 45 age-matched female mice obtained from a single source and that were individually housed. Mice treated for 2 weeks before and during WNV infection with A or AM had intermediate (8 of 15) or substantial (8 of 10) susceptibility to WNV infection, respectively, compared to vehicle-treated controls (3 of 10) (Figure 6E). A- and AM-treated mice, but not M-treated mice, had a substantial reduction in total bacterial biomass 3 days after treatment compared to vehicle-treated controls (Figure 6F). Sequencing of 16S rRNA amplicons revealed a decrease in the proportional representation of Bacteroidetes and an increase in the Firmicutes and Proteobacteria over time in A-treated mice. A-treated mice also had an expansion of Tenericutes at 3 days after treatment (Figure S7A). In contrast, Bacteroidetes and Firmicutes decreased in AM-treated mice over time to below the limits of detection, with a concomitant expansion of Protobacteria (Figure S7A). Comparisons of Abx treatments showed differences in measures of α-diversity (Figure S7B) and β-diversity between vehicle-treated controls and A-, M-, and AM-treated mice, as well as among A-, M-, and AM-treated mice (Figure 6G). These analyses identified differences in overall bacterial community structure that are associated with increased WNV virulence.
To determine whether the Abx treatments that differentially altered WNV outcome could be distinguished by the abundance of specific bacterial taxa, we performed likelihood ratio tests at different time points. We identified 72 bacterial 16S rRNA amplicon sequence variants (ASVs) across the six pairwise comparisons (vehicle versus A, vehicle versus M, vehicle versus AM, M versus A, M versus AM, A versus AM) (Table S1). Taxonomic assignment revealed an ASV with similarity to Alistipes onderdonkii that was decreased in A-, M-, and AM-treated mice compared to vehicle-treated controls (Figure 6H). Other ASVs were altered in M-treated mice compared to vehicle-treated controls (Figure 6H; Table S1). An ASV with 100% identity to Stenotrophomonas hibiscicola (known clinically as Stenotrophomonas maltophilia), an emerging multi-drug-resistant opportunistic pathogen in humans (Chang et al., 2015; Sánchez, 2015), was increased in AM-treated compared to A-treated mice (Figure 6H). These data show that changes in specific bacterial taxa were associated with increased virulence of WNV in A-compared to AM-treated mice.
3 Days of Ampicillin Alone Reduces T Cell Responses
We examined further the effect of a treatment with A, alone or in combination with M, on WNV outcome. Even a short 3-day pulse treatment with A, alone or in combination with M, resulted in increased susceptibility to WNV infection compared to vehicle-treated controls (Figure 7A). While we did not observe differences in the numbers or percentages of total B cells and CD4+ or CD8+ T cells in the spleen at day 8 after WNV inoculation (Figure 7B), we did observe decreased numbers and percentages of Tregs and WNV-specific CD8+ T cells in the spleens of A- and AM-treated mice compared to vehicle-treated controls (Figures 7C and 7D). Also, in contrast to what was observed for a 14-day continuous VNAM treatment (Figure 5A), equivalent numbers of all hematopoietic progenitor cell subsets were detected in the bone marrow of vehicle- and AM-pulsed mice (Figure 7E), suggesting that alterations in hematopoiesis are not required for oral Abx to impair Treg and virus-specific CD8+ T cell responses. Thus, even a short 3-day exposure to A was sufficient to impair T cell responses that likely are important for resolving the pathogenic outcome of WNV infection.
Figure 7. A Short Treatment of A Is Sufficient to Impair T Cell Responses Important for Resolving WNV Pathogenesis.

(A–E) Mice were treated for 3 days followed by drinking water for 3 days before WNV inoculation. Naive bone marrow was harvested (E) or mice were inoculated with WNV (A–D), as in Figure 1E.
(A) Survival curves were compared using the log-rank test with a Bonferroni correction (**p < 0.01 and ****p < 0.0001). (B) The numbers (left) and percentages (right) of leukocytes, B cells, and CD4+ and CD8+ T cells.
(C) The numbers (left) and percentages (right) of Tregs.
(D) The numbers (left) and percentages (right) of WNV-specific CD8+ T cells. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple-comparison test (*p < 0.05 and **p < 0.01; comparisons in B were not significant, p > 0.05).
(E) The numbers of total cells, HSCs, MPPs, CLPs, ALPs, BLPs, pre-pro-Bs, CMPs, GMPs, MEPs, CDPs, MDPs, pre-DC1s, and pre-DC2s. Statistical significance was determined using an unpaired t test (all comparisons not significant, p > 0.05). Results were combined from two to five independent experiments: vehicle (n = 25), A (n = 20), AM (n = 25) (A); vehicle (n = 14), A (n = 14), AM (n = 14) (B and C); and vehicle (n = 10), AM (n = 10) (D).
DISCUSSION
Here we showed that oral treatment with a cocktail of Abx that alters the microbiota results in increased susceptibility of mice to severe disease during infection with three different flaviviruses. Treatment of mice with VNAM resulted in fewer virus-specific effector CD8+ T cells in the DLN, spleen, and brain; increased virus burden in the gastrointestinal tract and brain; and increased susceptibility. Treatment of mice with a 3-day pulse of A or AM also resulted in decreased WNV-specific CD8+ T cell responses and increased susceptibility. The requirement for primed, but not unprimed, CD4+ and CD8+ T cells to reverse the VNAM effect upon adoptive transfer suggests that alterations in virus-specific T cells contribute to the effect of Abx treatment on WNV pathogenesis. Indeed, WNV-specific and effector CD4+ and CD8+ T cells control WNV burden, promote clearance of WNV from the CNS, and limit disease (Brien et al., 2007, 2008; Shrestha and Diamond, 2007; Shrestha et al., 2006a, 2006b, 2008).
VNAM treatment blunted virus-specific effector CD8+ T cell responses against IAV and LCMV infection, yet spared CD8+ T cell responses against intranasally administered herpes simplex virus 2 (Abt et al., 2012; Ichinohe et al., 2011). We observed that VNAM treatment altered a host environment important for the induction of optimal virus-specific T cell responses, as primed T cells transferred from VNAM-treated mice could reconstitute host immunity in Tcrβδ−/− mice. The relative preservation of WNV-specific CD8+ T cell responses in the MLN suggests that the effect of Abx treatment may be tissue specific.
Tregs are induced by WNV infection, however, their role is complex (Lanteri et al., 2009). Reduced Treg frequencies in the blood are associated with increased disease severity in patients and in mice, potentially due to the ability of Tregs to dampen WNV-induced immunopathology. Depletion studies in mice also suggest that Tregs function early in WNV infection to shape optimal immune control and, thus, limit disease. In our study, treatment of mice with A, AM, or VNAM resulted in fewer Tregs in the spleen during WNV infection. However, VNAM treatment did not decrease Treg numbers in the spleen of ZIKV-infected mice, suggesting that oral Abx can alter the outcome of flavivirus infection without necessarily affecting Treg numbers.
The effect of oral Abx treatment occurred after cessation of treatment, demonstrating that continuous Abx treatment was not required to increase susceptibility to flavivirus infection. Treatment with A or AM was sufficient to impair virus-specific CD8+ T cell responses and alter the outcome of WNV infection. While both types of Abx similarly reduced bacterial biomass, they could be distinguished by their effects on community diversity, phylogenetic configuration, and abundance of specific bacterial taxa. Although the relative contribution of each to the oral Abx phenotype remains to be established, the additional susceptibility in VNAM-stop-treated mice that received cecal microbiota from other VNAM-treated mice suggests that both the loss and gain of specific taxa might contribute to increased susceptibility.
The inability of co-housing or cecal microbiota transfer to reverse the severe WNV disease phenotype upon VNAM treatment is consistent with the idea that (1) the immune response deficit induced by depletion and/or perturbation of the micro-biota requires time for restoration, or (2) critically important bacterial effectors are inefficiently transmitted between mice by co-housing or cecal content inoculation. Our data do not formally exclude the possibility that oral Abx treatment itself may contribute to impaired antiviral immunity. Establishing that components of the microbiota per se are critical mediators of Abx effects will require studies where (1) microbial source tracking is performed that follows the immigration of members of donor cecal microbiota to recipient animals and/or the immigration and emigration of taxa from the microbiota of cagemates in co-housing experiments, and (2) microbiota transplants are performed from vehicle donors or donors treated with Abx to germ-free animals with subsequent WNV challenge.
In summary, we have shown that oral Abx treatment increases the risk of severe disease during flavivirus infection in mice, and we have illuminated a potential role for host microbial communities in promoting antiviral T cell responses during systemic virus infection. Our data in mice suggest the need for studies in humans to examine whether exposure to oral Abx, as well as other agents that perturb the microbiota, is associated with severe disease during infection by flaviviruses, as well as other pathogens requiring the rapid induction of T cell responses for protection.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-B220 AF488 | BD Biosciences | RA3-6B2 |
| Anti-B220 BV510 | BD Biosciences | RA3-6B2 |
| Anti-Bst2 APC | eBiosciences | CD317 |
| Anti-CD115 BV 711 | BioLegend | AFS98 |
| Anti-CD11c APC-Cy7 | Tonbo Biosciences | N418 |
| Anti-CD127 BV 421 | BD Biosciences | SB/199 |
| Anti-CD16/32 (Fc block) | BD Biosciences | 2.4G2 |
| Anti-CD172α APC | BD Biosciences | P84 |
| Anti-CD172α PerCP-ef710 | eBiosciences | P84 |
| Anti-CD19 A700 | Biolegend | 6D5 |
| Anti-CD19 PE | BD Biosciences | 1D3 |
| Anti-CD24 PE-Cy7 | BioLegend | M1/69 |
| Anti-CD27 APC | BD Biosciences | LG.3A10 |
| Anti-CD3 BUV 496 | BD Biosciences | 145-2c11 |
| Anti-CD3 BUV 395 | BD Horizon | 145-2C11 |
| Anti-CD4 FITC | BioLegend | RM4-5 |
| Anti-CD4 APC-Cy7 | Biolegend | GK1.5 |
| Anti-CD44 APC-Cy7 | BD Biosciences | IM7 |
| Anti-CD45 BUV 395 | BD Biosciences | 30-F11 |
| Anti-CD45 Alexa 488 | BD Biosciences | 30-F11 |
| Anti-CD45.1 PE-Cy7 | Tonbo Biosciences | A20 |
| Anti-CD45.1 PE-Cy7 | Biolegend | A20 |
| Anti-CD45R AF488 | BD Biosciences | RA3-6B2 |
| Anti-CD8α PerCP-Cy5.5 | BioLegend | 53-6.7 |
| Anti-CD8α V450 | BD Biosciences | 53-6.7 |
| Anti-CD8β PerCp-Cy5.5 | Biolegend | YT5156.7.7 |
| Anti-ckit BUV 395 | BD Biosciences | 2B8 |
| Anti-ckit PE-Cy7 | BD Biosciences | 2B8 |
| Anti-Flt3 PE Texas Red | BD Biosciences | A2F10.1 |
| Anti-FoxP3 BV421 | BioLegend | MF-14 |
| Anti-Granzyme B PE | Invitrogen | GB11 |
| Anti-IFN-γ BV421 | BioLegend | XMB1.2 |
| Anti-Ly6D PE | BioLegend | 49-H4 |
| Anti-MHCII I-A/I-E V450 | BioLegend | M5/114.15.2 |
| Anti-MHCII I-A/I-E V500 | BD Biosciences | M5/114.15.2 |
| Anti-Sca-1 PE | BD Biosciences | D7 |
| Anti-Sca-1 PE-Cy7 | eBiosciences | D7 |
| Anti-SiglecH PerCP-Cy5.5 | eBioscience | 440c |
| Anti-TCR-V α2 PE | Invitrogen | B20.1 |
| Anti-TNF-α BV605 | BioLegend | MP6-XT22 |
| Biotin-B220 | BioLegend | RA3-6B2 |
| Biotin-CD105 | eBiosciences | MJ7/18 |
| Biotin-CD11b | BioLegend | M1/70 |
| Biotin-CD19 | BD Biosciences | 1D3 |
| Biotin-CD3 | Tonbo Biosciences | 145-2C11 |
| Biotin-Ly6C | BD Biosciences | AL-21 |
| Biotin-Ly6G | BioLegend | 1A8 |
| Biotin-NK1.1 | BioLegend | PK136 |
| Biotin-Ter119 | BioLegend | Ter119 |
| Db-WNV NS4B tetramer Alexa647 | NIH Tetramer Core Facility | SSVWNATTA |
| Db-ZIKV envelope tetramer APC | Chiips Core, Washington University | IGVSNRDFV |
| Virus Strains | ||
| DENV2 strain D2S20 | Diamond lab | Makhluf et al., 2013 |
| WNV NY2000 strain 30000259 | Diamond lab | Ebel et al., 2001 |
| ZIKV Dakar strain 41671 | Diamond lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ampicillin sodium salt | Sigma-Aldrich | A9518 |
| CFSE | eBiosciences | 65-0850-84 |
| Db-NS4B peptide | NIH Tetramer Core Facility | Purtha et al., 2007 |
| Fixable Viability Dye eFluor 506 | eBiosciences | 65-0866-14 |
| Metronidazole | Sigma-Aldrich | M1547 |
| Neomycin trisulfate salt hydrate | Sigma-Aldrich | N1876 |
| Streptavidin conjugate | ThermoFisher Scientific | Qdot 605 |
| Vancomycin hydrochloride | Sigma-Aldrich | V2002 |
| Critical Commercial Assays | ||
| Bio-Plex Pro Mouse cytokine 23-plex assay | Bio-rad | M60009RDPD |
| FoxP3/transcription factor staining buffer set | eBiosciences | 00-5523-00 |
| Pan T cell isolation kit II, mouse | MACS Miltenyi Biotech | 130-095-130 |
| Cd8α+ T cell isolation kit, mouse | MACS Milteny Biotech | 130-104-075 |
| Deposited Data | ||
| Raw data | This paper | ENA: PRJEB15424 |
| Experimental Models: Cell Lines | ||
| African green monkey kidney (Vero) cells | ATCC; WHO Reference Cell Bank | Vero 76 clone E6; WHO Vero cells |
| Baby hamster kidney (BHK) cells | ATCC | BHK-21 clone 15 |
| Aedes albopictus cells | ATCC | C6/36 |
| Experimental Mouse Models: | ||
| C57BL6/J | The Jackson Laboratory | 000664 |
| Caspase1/11−/− | The Jackson Laboratory | 016621 |
| Cd8α−/− | The Jackson Laboratory | 002665 |
| cGas−/− | Virgin laboratory | Schoggins et al., 2014 |
| Ifnar1−/− | Michel Aguet | Müller et al., 1994 |
| Ifnarf/f | Ulrich Kalinke | Kamphuis et al., 2006; |
| LysMCre | The Jackson Laboratory | 004781 |
| Ifnlr1−/− | Zymogenetics | Ank et al., 2008 |
| WNV NS4B+/− | Diamond laboratory | Kim et al., 2014 |
| Stat6−/− | The Jackson Laboratory | 005977 |
| Sting−/− | The Jackson Laboratory | 017537 |
| Tcrβδ−/− | The Jackson Laboratory | 002122 |
| Tlr2−/− | The Jackson Laboratory | 004650 |
| Tlr5−/− | The Jackson Laboratory | 008377 |
| Oligonucleotides | ||
| WNV specific primers and probes | IDT | Brien et al., 2013 |
| ZIKV Dakar specific primers and probes | IDT | Govero et al., 2016 |
| V4 region specific 16S rRNA primers (515f/805r) | IDT | N/A |
| V4 region specific Golay-barcoded 16S rRNA primers (515f/806r) | IDT | N/A |
| Recombinant DNA | ||
| 16S rRNA plasmid standard in pCR2.1 TOPO | Diamond laboratory | This paper |
| Software and Algorithms | ||
| FlowJo | Treestar, Inc. | v10.4.1 |
| Prism 7 | GraphPad | v7.0a |
| dada2 R package | Callahan et al., 2016 | v.1.5.2 |
| QIIME | Caporaso et al., 2010 | v.1.8.0 |
| MSA | Bodenhofer et al., 2015 | v.1.8.0 |
| Phangorn | Schliep, 2011 | v.2.3.1 |
| PhyloSeq | McMurdie and Holmes, 2013 | v.1.23.1 |
| Other | ||
| 16S rRNA sequence data | This paper | https://www.ebi.ac.uk/ena/data/view/PRJEB15424 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael S. Diamond (diamond@wusm.wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
Vero and BHK21 cells were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). C6/36 Aedes albopictus cells were cultured at 28°C in L15 supplemented with 10% FBS and 25 mM HEPES pH 7.3. All cell lines were originally acquired from American Type Culture Collection and were tested and judged free of mycoplasma contamination.
Viruses
WNV strain 3000259, New York 2000 (Ebel et al., 2001) and DENV strain D2S20 (Makhluf et al., 2013)] were propagated in Aedes albopictus C6/36 cells as described (Brien et al., 2013; Pinto et al., 2015). ZIKV strain Dakar 41671 was propagated in Vero cells as described (Lazear et al., 2016). A full-length contig derived from next generation sequencing confirmed that our ZIKV stock was identical to the published sequence for ZIKV Dakar 41671 (GenBank: KU955595.1). The titers of WNV, DENV and ZIKV stocks were determined by focus-forming assay on Vero cells (Brien et al., 2013; Lazear et al., 2016; Pinto et al., 2015).
Mice
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381-01). Dissections and footpad injections were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize suffering.
C57BL/6J wild-type mice were purchased commercially (000664, Jackson Laboratories). The original Ifnar1tm1Agt (abbreviated Ifnar1−/−) and Ifnar1tm1Uka (abbreviated Ifnar1f/f) mice were provided by Michel Aguet (Swiss Institute for Experimental Cancer Research, Lausanne, Switzerland), and Ulrich Kalinke (Paul-Ehrlich-Institut, Langen, Germany), respectively (Kamphuis et al., 2006; Müller et al., 1994) Ifnar−/− mice were backcrossed for 10 generations onto the C57BL/6 background. Ifnarf/fLysMCre+/+ mice were backcrossed using speed congenic analysis to 99% C57BL/6 as judged by microsatellite analysis. Ifnlr1tm1Palu (abbreviated Ifnlr1−/−) mice lacking the entire IL28rα coding sequence (Ank et al., 2008) were obtained from Zymogenetics/Bristol-Myers Squibb (a gift of S. Doyle). WNV-specific CD8+ T cell receptor transgenic mice (abbreviated NS4B+/−) have been published (Kim et al., 2014). B6.129S2-Cd8atm1Mak/J (abbreviated Cd8−/−), B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J (abbreviated Tcrβδ−/−), Tlr2tm1Kir (abbreviated Tlr2−/−), Tlr5tm1flv (abbreviated Tlr5−/−), Stat6tm1Gru (abbreviated Stat6−/−), Caspase 1tm1Flv (abbreviated Caspase 1/11−/−), and Tmem173gt (abbreviated Sting−/−) all were purchased commercially (002665, 002122, 004650, 008377, 005977, 016621 and 017537, respectively) from Jackson Laboratories. Mb21d1tm1a mice (abbreviated cGas−/−) lacking the catalytic residues E211 and D213 were generated as described (Schoggins et al., 2014). Cd8−/−, cGas−/−, Ifnar1−/−, Ifnar1f/fLysMCre+/+, Ifnlr−/−, NS4B+/−, Tlr2−/−, Tlr5−/−, Stat6−/−, Sting−/−, Tcrβδ−/− and Caspase 1/11−/− mice were bred and housed at Washington University under specific-pathogen-free conditions. Mice were fed a 20% protein diet (PicoLab 5053, Purina) and maintained on a 12 h light/dark cycle (6 am to 6 pm). Male and female mice were housed in groups of up to 5 mice/cage in most studies, except in co-housing and individual-housing experiments where female mice were used.
Age- and sex-matched male and female mice were inoculated subcutaneously in the footpad with WNV at 12 weeks of age or ZIKV diluted in PBS at 4 weeks of age, or retroorbitally with DENV diluted in PBS at 6 weeks of age.
METHODS DETAILS
Abx Treatments
Cages of mice were treated ad lib at 4, 10 or 11 weeks of age with an Abx cocktail [1 g/L ampicillin (A), 0.35 g/L vancomycin (V), 1 g/L metronidazole (M), 1 g/L neomycin (N), Sigma] in 25 g/L grape Kool-Aid (Kraft Foods) or with Kool-Aid alone. In some experiments, mice were treated with a single or combinations of Abx. Water bottles were changed once per week. In experiments in which mice were treated with M either alone or in combination, 5 food pellets were soaked for 5 min in Kool-Aid supplemented with Abx. Soaked food pellets were placed on the cage bottom, and uneaten food pellets were removed three times per week.
Cecal Microbiota Transfer
Cecal contents from mice were collected, diluted in PBS (15 mL/mouse), homogenized by vortexing and minimally clarified by low-speed centrifugation for 5 min at 300 x g. Mice were then gavaged twice with 150 uL of fresh diluted cecal contents.
16S rDNA qPCR
Fecal samples were harvested into 2 mL tubes (Sarstedt, Germany) with 1-mm diameter zirconia/silica beads (Biospec). Fecal pellets were flash frozen in a bath of ethanol and dry ice, and stored at −80°C. Fecal nucleic acid was extracted from fecal pellets as described previously (Baldridge et al., 2015). SYBR green quantitative PCR for bacterial 16S rRNA genes was performed using primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GACTACCAGGGTATCTAATCC-3′), and the following cycling conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. Quantitation of 16S rRNA genes was performed by comparison to a plasmid standard of pCR2.1 TOPO (ThermoFisher Scientific) containing a 16S rRNA sequence from an uncultured intestinal bacterium from mice: GACTACCAGGGTATCTAATCCTGTTCGATCCCCACGCTTTCGTGCCTCAGCGTCAGTTGAGCGCCGGTATG CTGCCTTCGCAATCGGAGTTCTGCGTGATATCTATGCATTTCACCGCTACACCACGCATTCCGCATACTTCTCGCTCACTCAAGAC CCGCAGTTTCAACGGCGATACGGCGTTGAGCACCGCATTTTTACCGCTGACTTACTAATCCGCCTACGCACCCTTTAAACCCAAT AAATCCGGATAACGCTCGCATCCTCCGTATTACCGCGGCTGCTGGCAC.
16S rDNA Amplicon Sequencing and Analyses
Bar-coded PCR primers directed at the V4 region of bacterial 16S rRNA genes were used to generate amplicons from nucleic acid isolated from fecal pellets (Baldridge et al., 2015). Multiplex sequencing of amplicons with sample-specific barcodes was performed using an Illumina MiSeq instrument (paired end 2x250 nt reads).
Demultiplexing of 16S rRNA amplicon sequences was performed using QIIME (Quantitative Insights Into Microbial Ecology, v.1.8.0) (Caporaso et al., 2010). Read quality control and the resolution of sequence variants (ASVs) was completed using the dada2 R package (Callahan et al., 2016). Reads were quality-controlled by the removal of the 5′ 10 bases of the forward read and 3′ 10 bases of the reverse read. All sequences with Ns and reads mapping to phiX were removed. Error estimation was calculated on a random subset of 75 samples from the full dataset prior to application of the error model to the full data. Taxonomy was assigned independently to three 16S rRNA gene sequence databases: (1) GreenGenes 1997, (2) RDP training set 14 and (3) Silva training set v123 (Cole et al., 2014; McDonald et al., 2012; Quast et al., 2013).
Additional species level assignment to the RDP and Silva training sets was accomplished using the addSpecies function of dada2. ASVs were aligned using an R implementation of MSA (Bodenhofer et al., 2015) and arranged into a maximum likelihood phylogeny (GTR model with optimization of the proportion of invariable sites and the gamma rate parameter) using the phangorn package (Schliep, 2011). The resulting phylogenetic tree was combined with the table of ASVs and merged with sample data for loading into the PhyloSeq package (McMurdie and Holmes, 2013). ASVs not assigned to the kingdom bacteria or assigned to the mitochondrial family or chloroplast class were removed.
PhyloSeq was used to calculate alpha diversity measures of Shannon Diversity and observed richness and beta diversity using UniFrac (Lozupone et al., 2007). Differential abundance testing of taxa between groups was completed using the DeSeq2 algorithm using the likelihood-ratio test (Love et al., 2014). Full analysis workflows for dada2 ASV resolution, statistical analysis and plotting, and count tables are available at https://github.com/shandley/WNV-antibiotics.
Measurement of Viral Burden
At specific days after virus infection, serum was obtained by intracardiac heart puncture and followed by extensive perfusion. Tissues were harvested into 2 mL tubes (Sarstedt, Germany) with 1-mm diameter zirconia/silica beads (Biospec), flash frozen in a bath of ethanol and dry ice, weighted and stored at −80°C. Tissues were homogenized using a bead-beater apparatus and levels of virus were determined by plaque assay on BHK21 or Vero cells, or by quantitative reverse transcription-PCR using WNV- or ZIKV-specific primers and probes as described previously (Brien et al., 2013; Govero et al., 2016).
Measurement of Antibody, Cytokines, and Cellular Immune Responses
Focus reduction neutralization assays were performed after mixing serial dilutions of serum with a fixed amount (50 FFU) of WNV as previously described (Diamond et al., 2003).
Serum cytokine levels at day 8 after WNV inoculation were measured using the 23-plex Bio-Plex Pro assay according to the manufacturer’s protocol (Bio-Rad).
Cells from the DLN, MLN and spleen were dispersed into single cell suspensions with a cell strainer. Brains were minced and digested with 0.05% collagenase D, 0.1 μg/ml trypsin inhibitor TLCK, and 10 μg/ml DNase I in HBSS supplemented with 10 mM HEPES (pH 7.4; Life Technologies). Cells were dispersed into single cell suspensions with a cell strainer and pelleted through a 30% Percoll cushion for 30 min (1,200 × g at 4°C). Intracellular IFN-γ and TNF-α staining was performed after ex vivo restimulation with a Db-restricted NS4B immunodominant peptide SSVWNATTA using 1 μM peptide and 5 μg/ml brefeldin A (Sigma) as described previously (Purtha et al., 2007). Cells were stained with the following reagents and antibodies: Viability Dye (eBioscience, 506), Fc Block (BD Biosciences, clone 2.4G2), CD3 (BD Biosciences, clone 145-2c11), CD4 (Biolegend, clone RM4-5 or GK1.5), CD8α (Biolegend, Clone 53-6.7), CD8β (Biolegend, clone YT5156.7.7), CD45 (BD Biosciences, clone 30-F11), CD19 (Biolegend, clone 6D5; BD Biosciences, clone 1D3), FoxP3 (Biolegend, clone MF-14), Granzyme B (Invitrogen, clone GB11), IFN-γ (Biolegend, clone XMG1.2), and TNF-α (Biolegend, clone MP6-XT22). WNV-specific CD8+ T cells were stained using an Alexa-647 tetramer for the NS4B peptide as described previously (Purtha et al., 2007). ZIKV-specific CD8+ T cells were stained using an APC-labeled tetramer for the Db-restricted envelope protein peptide IGVSNRDFV (Elong Ngono et al., 2017). Data were collected on a BD LSRII flow cytometer and analyzed using FlowJo software (Treestar, Inc.).
Isolation of T cells and adoptive cell transfers were performed as previously described (Richner et al., 2015). Briefly, splenocytes were isolated from mice and erythrocytes were lysed. T cells were purified by negative selection to greater than 90% using the Pan T Cell or CD8α+ Isolation kits (MACS Miltenyi Biotech). Naive or WNV-primed T cells were counted and adjusted to equal number for adoptive transfer. Cells (5 × 106) were transferred via the retroorbital route into recipient mice that had been inoculated subcutaneously with WNV 24 h earlier.
DC analyses were performed as described previously (Satpathy et al., 2013). Briefly, spleens were minced and digested in 5 mL Iscove’s modified Dulbecco’s media + 10% FCS (cIMDM) with 250 μg/ml collagenase B (Roche) and 30 U/ml DNase I (Sigma-Al-drich). Cells were dispersed into single cell suspensions with a cell strainer before red blood cells were lysed with ACK lysis buffer. Cells were counted on a Vi-CELL analyzer (Beckman Coulter). Staining of 2 × 106 cells with the following antibodies was performed at 4°C in the presence of Fc Block in DPBS + 0.5% BSA + 2 mm EDTA): CD8 (BD Biosciences, 53-6.7), CD24 (BD Biosciences, M1/69), CD172a (BD Biosciences, P84), Bst2 (BD Biosciences, CD317), CD4(Biolegend, RM4-5), TCR-V α2(Biolegend, B20.1), CD11c (eBioscience, N418), MHC-II (eBioscience, I-A/I-E; M5/114.15.2), B220 (eBioscience, RA3-6B2), CD44 (eBioscience, IM7), CFSE (eBioscience), and CD45.1 (Tonbo Biosciences, A20). Cells were analyzed on a BD FACSCanto II or FACSAria II flow cytometer, and analyzed using FlowJo software (TreeStar, Inc.).
Cross-presentation analyses were performed as described (Kretzer et al., 2016). Briefly, splenic DCs were sorted as B220– MHCII+ CD11c+ CD24+ CD172– (DC1) and B220– MHCII+ CD11c+ CD24– CD172+ (DC2) cells. OT-1 T cells were sorted as B220– CD4–CD11c– CD45.1+ CD8+ Vα2+ cells. 10,000 DC from vehicle- or VNAM-treated mice were incubated with various doses of heat killed Listeria monocytogenes and 25,000 CFSE-labeled OT-1 T cells for 3 days in a 37°C 5% CO2 incubator and assayed for CFSE dilution and CD44 expression of OT-1 cells.
For BM analyses, cells were harvested from the pelvis, tibia, and femurs, ground with a mortar and pestle in PBS supplemented with 0.5% BSA and 2 mM EDTA, and passed through a 70-μm strainer. Red blood cells were lysed with ammonium chloride–potassium bicarbonate lysis buffer. Staining with the following antibodies was performed for 2 h at 4°C and 2 h at RT in DPBS + 0.5% BSA + 2 mm EDTA in the presence of Fc Block: SiglecH (eBioscience, 440c), c-kit (BD Biosciences, 2B8), Flt3 (BD Biosciences, A2F10.1), Sca-1 (BD Biosciences, D7), CD11c (Tonbo Biosciences, N418), CD34 (BD Biosciences, RAM34), CD115 (Biolegend, AFS98), biotin-B220 (Biolegend, RA3-6B2), biotin-CD3 (Tonbo Biosciences, 145-2C11), biotin-CD19 (BD Biosciences, 1D3), biotin-NK1.1 (Biolegend, PK136), biotin-Ter119 (Biolegend, Ter119), biotin-Ly6G (Biolegend, 1A8), biotin-CD105 (eBioscience, MJ7/18), Qdot605 Streptavidin Conjugate (ThermoFisher Scientific), MHCII (BD Biosciences, M5/114.15.2), and CD127 (BD Biosciences, SB/199). Staining with the following antibodies was performed for 1–2 h at 4°C in DPBS + 0.5% BSA + 2 mm EDTA + in the presence of Fc Block: CD45R (BD Biosciences, RA3-6B2), Sca-1 (eBioscience, D7), Flt3 (BD Biosciences, A2F10.1), Ly6D (Biolegend, 49-H4), CD11c (Tonbo Biosciences, N418), CD27 (BD Biosciences, LG.3A10), c-kit (BD Biosciences, 2B8), biotin-CD3 (Tonbo Biosciences, 145-2C11), biotin-CD19 (BD Biosciences, 1D3), biotin-Ly6G (Biolegend, 1A8), biotin-CD11b (Biolegend, M1/70), biotin-Ly6C (BD Biosciences, AL-21), Qdot605 Streptavidin Conjugate (ThermoFisher Scientific), and CD127 (BD Biosciences, SB/199). Data were collected on a FacsAria Fusion flow cytometer, and analyzed using FlowJo software.
Statistical Analyses
No statistical methods were used to predetermine sample size a priori. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment. All data were analyzed using Prism software (GraphPad10, San Diego, CA). Statistical details of experiments and tests used to analyze experiments are described in the Figure Legends. An unpaired t test or nonparametric Mann-Whitney test was used depending on data distribution for analyses of cell populations and virus burden.
DATA AVAILABILITY
The accession number for the bacterial 16S rRNA sequence datasets reported in this paper is ENA: PRJEB15424 (https://www.ebi.ac.uk/ena/data/view/PRJEB15424).
Supplementary Material
Highlights.
Oral antibiotics increase susceptibility of mice to severe flavivirus disease
Oral antibiotics impair development of optimal antiviral T cell responses
Oral antibiotics alter bacterial abundance and community structure
Just 3 days of oral ampicillin is sufficient to alter flavivirus outcome
Acknowledgments
NIH grants (U19AI106772 and AI07375 to M.S.D., RO1DK30292 to J.I.G., RO1DK101354 and U19AI109725 to H.W.V., and RFAAI12027 to L.B.T.) and a pilot grant from cWIDR at Washington University School of Medicine to L.B.T. supported this work. K.M.M. is a Howard Hughes Medical Institute Investigator. We thank J. White for preliminary data, G. Zhao for data analysis, and L. Droit, J. Govero, J. Hong, M. Karlsson, C. Liu, M. Noll, D. O’Donnell, and S. Venkatesh for technical assistance.
Footnotes
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.001.
AUTHOR CONTRIBUTIONS
L.B.T., Q.T., and H.L. performed the mouse studies and analyses of virus and bacteria burden. L.B.T., M.J.G., S.P., P.B., C.G.B., D.J.T., and T.M.L. performed analyses of the immune response. S.A.H., B.L.H., and C.D. performed bacteria 16S rRNA data analyses. L.B.T. and M.S.D. designed the experiments, with assistance from H.W.V., K.M.M., and J.I.G. L.B.T. and M.S.D. wrote the initial draft of the manuscript, with the other authors contributing to editing into the final form.
DECLARATION OF INTERESTS
M.S.D. is a consultant for Inbios, Aviana, and Sanofi and on the Scientific Advisory Board of Moderna.
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science. 2015;347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Schürch CM, Saito Y, Geuking MB, Li H, Cuenca M, Kovtonyuk LV, McCoy KD, Hapfelmeier S, Ochsenbein AF, et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol. 2014;193:5273–5283. doi: 10.4049/jimmunol.1400762. [DOI] [PubMed] [Google Scholar]
- Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, Rutherford A, Astakhova NM, Perelygin AA, Busch MP, et al. Host genetic risk factors for West Nile virus infection and disease progression. PLoS ONE. 2011;6:e24745. doi: 10.1371/journal.pone.0024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhofer U, Bonatesta E, Horejš-Kainrath C, Hochreiter S. msa: an R package for multiple sequence alignment. Bioinformatics. 2015;31:3997–3999. doi: 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Nikolich-Zugich J. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur J Immunol. 2007;37:1855–1863. doi: 10.1002/eji.200737196. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Nikolich-Zugich J. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J Immunol. 2008;181:8568–8575. doi: 10.4049/jimmunol.181.12.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD, Lazear HM, Diamond MS. Propagation, quantification, detection, and storage of West Nile virus. Curr Protoc Microbiol. 2013;31:15D.3.1–15D.3.18. doi: 10.1002/9780471729259.mc15d03s31. [DOI] [PubMed] [Google Scholar]
- Brown AN, Kent KA, Bennett CJ, Bernard KA. Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology. 2007;368:422–430. doi: 10.1016/j.virol.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci USA. 2015;112:9973–9977. doi: 10.1073/pnas.1508740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chancey C, Grinev A, Volkova E, Rios M. The global ecology and epidemiology of West Nile virus. BioMed Res Int. 2015;2015:376230. doi: 10.1155/2015/376230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YT, Lin CY, Chen YH, Hsueh PR. Update on infections caused by Stenotrophomonas maltophilia with particular attention to resistance mechanisms and therapeutic options. Front Microbiol. 2015;6:893. doi: 10.3389/fmicb.2015.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Burghgraeve T, Kaptein SJ, Ayala-Nunez NV, Mondotte JA, Pastorino B, Printsevskaya SS, de Lamballerie X, Jacobs M, Preobrazhenskaya M, Gamarnik AV, et al. An analogue of the antibiotic teicoplanin prevents flavivirus entry in vitro. PLoS ONE. 2012;7:e37244. doi: 10.1371/journal.pone.0037244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Pierson TC. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell. 2015;162:488–492. doi: 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med. 2003;198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel GD, Dupuis AP, 2nd, Ngo K, Nicholas D, Kauffman E, Jones SA, Young D, Maffei J, Shi PY, Bernard K, Kramer LD. Partial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Infect Dis. 2001;7:650–653. doi: 10.3201/eid0704.010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elong Ngono A, Vizcarra EA, Tang WW, Sheets N, Joo Y, Kim K, Gorman MJ, Diamond MS, Shresta S. Mapping and Role of the CD8+T Cell Response During Primary Zika Virus Infection in Mice. Cell Host Microbe. 2017;21:35–46. doi: 10.1016/j.chom.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WF, Song Y, Wang AJ, Smith A, Grinchuk V, Mongodin E, Pei C, Ma B, Lu N, Urban JF, Jr, et al. Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome. 2015;3:40. doi: 10.1186/s40168-015-0103-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govero J, Esakky P, Scheaffer SM, Fernandez E, Drury A, Platt DJ, Gorman MJ, Richner JM, Caine EA, Salazar V, et al. Zika virus infection damages the testes in mice. Nature. 2016;540:438–442. doi: 10.1038/nature20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB. Dengue. Lancet. 2007;370:1644–1652. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- Hand TW. The Role of the Microbiota in Shaping Infectious Immunity. Trends Immunol. 2016;37:647–658. doi: 10.1016/j.it.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks LA, Bartoces MG, Roberts RM, Suda KJ, Hunkler RJ, Taylor TH, Jr, Schrag SJ. US outpatient antibiotic prescribing variation according to geography, patient population, and provider specialty in 2011. Clin Infect Dis. 2015;60:1308–1316. doi: 10.1093/cid/civ076. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinjé J, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsdottir KS, Baldridge MT, Kadmon CS, King KY. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood. 2017;129:729–739. doi: 10.1182/blood-2016-03-708594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptein SJ, De Burghgraeve T, Froeyen M, Pastorino B, Alen MM, Mondotte JA, Herdewijn P, Jacobs M, de Lamballerie X, Schols D, et al. A derivate of the antibiotic doxorubicin is a selective inhibitor of dengue and yellow fever virus replication in vitro. Antimicrob Agents Chemother. 2010;54:5269–5280. doi: 10.1128/AAC.00686-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, Balmaseda A, Harris E. Antibody-dependent enhancement of severe dengue disease in humans. Science. 2017;358:929–932. doi: 10.1126/science.aan6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15:374–381. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Pinto AK, Myers NB, Hawkins O, Doll K, Kaabinejadian S, Netland J, Bevan MJ, Weidanz JA, Hildebrand WH, et al. A novel T-cell receptor mimic defines dendritic cells that present an immunodominant West Nile virus epitope in mice. Eur J Immunol. 2014;44:1936–1946. doi: 10.1002/eji.201444450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Sasaki M, Okumura M, Kim E, Sawa H. Flavivirus encephalitis: pathological aspects of mouse and other animal models. Vet Pathol. 2010;47:806–818. doi: 10.1177/0300985810372507. [DOI] [PubMed] [Google Scholar]
- Kretzer NM, Theisen DJ, Tussiwand R, Briseño CG, Grajales-Reyes GE, Wu X, Durai V, Albring J, Bagadia P, Murphy TL, Murphy KM. RAB43 facilitates cross-presentation of cell-associated antigens by CD8α+ dendritic cells. J Exp Med. 2016;213:2871–2883. doi: 10.1084/jem.20160597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn KA, Stappenbeck TS. Peripheral education of the immune system by the colonic microbiota. Semin Immunol. 2013;25:364–369. doi: 10.1016/j.smim.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanteri MC, O’Brien KM, Purtha WE, Cameron MJ, Lund JM, Owen RE, Heitman JW, Custer B, Hirschkorn DF, Tobler LH, et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J Clin Invest. 2009;119:3266–3277. doi: 10.1172/JCI39387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe. 2016;19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, Johnson H, Pape J, Foster GA, Krysztof D, et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009;5:e1000321. doi: 10.1371/journal.ppat.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, Stramer SL, Murphy PM. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis. 2010;201:178–185. doi: 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey NP, Staples JE, Lehman JA, Fischer M. Medical risk factors for severe West Nile Virus disease, United States, 2008–2010. Am J Trop Med Hyg. 2012;87:179–184. doi: 10.4269/ajtmh.2012.12-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb M. Genetic susceptibility to West Nile virus and dengue. Public Health Genomics. 2013;16:4–8. doi: 10.1159/000345934. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhluf H, Buck MD, King K, Perry ST, Henn MR, Shresta S. Tracking the evolution of dengue virus strains D2S10 and D2S20 by 454 pyrosequencing. PLoS ONE. 2013;8:e54220. doi: 10.1371/journal.pone.0054220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M, Kleinschmidt MC, Doerr HW, Cinatl J., Jr Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J Antimicrob Chemother. 2007;60:981–986. doi: 10.1093/jac/dkm307. [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, Hakimpour P, Gill KP, Nakaya HI, Yarovinsky F, et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity. 2014;41:478–492. doi: 10.1016/j.immuni.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Virgin HW. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science. 2016;351:aad5872. doi: 10.1126/science.aad5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AK, Brien JD, Lam CY, Johnson S, Chiang C, Hiscott J, Sarathy VV, Barrett AD, Shresta S, Diamond MS. Defining New Therapeutics Using a More Immunocompetent Mouse Model of Antibody-Enhanced Dengue Virus Infection. MBio. 2015;6:e01316–15. doi: 10.1128/mBio.01316-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Myers N, Mitaksov V, Sitati E, Connolly J, Fremont DH, Hansen TH, Diamond MS. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007;37:1845–1854. doi: 10.1002/eji.200737192. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racsa L, Gander R, Chung W, Southern P, Le J, Beal S, Lee F, Cavuoti D, Reisch J, Alatoom A. Clinical features of West Nile virus epidemic in Dallas, Texas, 2012. Diagn Microbiol Infect Dis. 2014;78:132–136. doi: 10.1016/j.diagmicrobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Retallack H, Di Lullo E, Arias C, Knopp KA, Laurie MT, Sandoval-Espinosa C, Mancia Leon WR, Krencik R, Ullian EM, Spatazza J, et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci USA. 2016;113:14408–14413. doi: 10.1073/pnas.1618029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner JM, Gmyrek GB, Govero J, Tu Y, van der Windt GJ, Metcalf TU, Haddad EK, Textor J, Miller MJ, Diamond MS. Age-Dependent Cell Trafficking Defects in Draining Lymph Nodes Impair Adaptive Immunity and Control of West Nile Virus Infection. PLoS Pathog. 2015;11:e1005027. doi: 10.1371/journal.ppat.1005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez MB. Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front Microbiol. 2015;6:658. doi: 10.3389/fmicb.2015.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. Panviral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J Virol. 2007;81:11749–11757. doi: 10.1128/JVI.01136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006a;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, Fikrig E, Diamond MS. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol. 2006b;80:5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Pinto AK, Green S, Bosch I, Diamond MS. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J Virol. 2012;86:8937–8948. doi: 10.1128/JVI.00673-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda KJ, Hicks LA, Roberts RM, Hunkler RJ, Taylor TH. Trends and seasonal variation in outpatient antibiotic prescription rates in the United States, 2006 to 2010. Antimicrob Agents Chemother. 2014;58:2763–2766. doi: 10.1128/AAC.02239-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Diamond MS, Gale M., Jr West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- Van Boeckel TP, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, Laxminarayan R. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis. 2014;14:742–750. doi: 10.1016/S1473-3099(14)70780-7. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JH, Mentoor JD, Van Wilpe E, Venter M. Comparative pathology of neurovirulent lineage 1 (NY99/385) and lineage 2 (SPU93/01) West Nile virus infections in BALBc mice. Vet Pathol. 2015;52:140–151. doi: 10.1177/0300985813520246. [DOI] [PubMed] [Google Scholar]
- Wu S, Jiang ZY, Sun YF, Yu B, Chen J, Dai CQ, Wu XL, Tang XL, Chen XY. Microbiota regulates the TLR7 signaling pathway against respiratory tract influenza A virus infection. Curr Microbiol. 2013;67:414–422. doi: 10.1007/s00284-013-0380-z. [DOI] [PubMed] [Google Scholar]
- Zellweger RM, Shresta S. Mouse models to study dengue virus immunology and pathogenesis. Front Immunol. 2014;5:151. doi: 10.3389/fimmu.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XG, Mason PW, Dubovi EJ, Xu X, Bourne N, Renshaw RW, Block TM, Birk AV. Antiviral activity of geneticin against dengue virus. Antiviral Res. 2009;83:21–27. doi: 10.1016/j.antiviral.2009.02.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the bacterial 16S rRNA sequence datasets reported in this paper is ENA: PRJEB15424 (https://www.ebi.ac.uk/ena/data/view/PRJEB15424).