

Abstract
Hereditary leiomyomatosis renal cell cancer syndrome is an autosomal dominant disorder characterized by uterine and cutaneous leiomyomas and increased predisposition to renal cell carcinoma, papillary type II. The syndrome is caused by heterozygous mutations to the fumarate hydratase (FH) gene located on chromosome 1. Affected females generally present with early onset, atypical uterine leiomyomas and cutaneous findings, however, delays in diagnosis are very common in patients with isolated uterine findings. We present a case series of 2 sisters in their 20s who presented with isolated uterine leiomyomas and were found to carry a novel mutation for the fumarate hydratase gene. One patient was referred for treatment of infertility and recurrent miscarriages and the other was referred for acute symptomatic anemia due to myomas. Prompt diagnosis of hereditary leiomyomatosis renal cell cancer was made due to a high index of clinical suspicion based on early onset disease and familial clustering as well as characteristic pathologic findings on uterine leiomyoma surgical specimen. Timely diagnosis not only allowed for genetic counseling and renal cancer surveillance, but also for fertility counseling given the increased morbidity associated with uterine leiomyoma due to hereditary leiomyomatosis and renal cell cancer syndrome.
Key Words: Hereditary leiomyomatosis and renal cell cancer syndrome, Fumarate hydratase, Reed syndrome, Multiple cutaneous and uterine leiomyomatosis, Leiomyoma with bizarre nuclei
Uterine leiomyomas are the most prevalent tumors in reproductive aged woman and comprise the most common indication for hysterectomy worldwide. Indeed, 77% of histopathologic hysterectomy specimens demonstrate the presence of uterine leiomyomas 1,2. Cytogenetic studies have shown that 40% to 50% of leiomyoma specimens exhibit chromosomal rearrangements and up to 70% of specimens display somatic mutations 1,3. Despite the aforementioned genetic abnormalities, which may play a role in leiomyoma tumorigenesis, the only known genetic syndrome that leads to uterine leiomyomas is hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome.
First described as Reed syndrome in 1973 and later as HLRCC in 2001, this syndrome has been shown to arise from germline mutations in the fumarate hydratase (FH) gene 4. The FH gene, located on chromosome 1, encodes for the enzyme fumarate hydratase, which catalyzes the conversion of fumarate to malate in the Krebs cycle. Homozygotes for the FH gene, termed FH deficiency, present with neonatal encephalopathy and rarely survive beyond early childhood. Whereas heterozygotes present with a constellation of symptoms including multiple cutaneous leiomyomas, early onset uterine leiomyomas, and increased risk for renal cell carcinoma, papillary type II 5,6. The highest morbidity of the syndrome stems from its risk of renal cell carcinoma, which occurs in 20% to 25% of patients with an average mean age of onset of 46 yr 7,8. The syndrome is also named after its most prominent clinical features which occur in 70% to 80% of patients: multiple cutaneous and uterine leiomyomatosis 9.
Worldwide, the Leiden Open Variation Database reports more than 300 individuals to date have been diagnosed with clinical HLRCC and approximately 150 families exhibit unique DNA mutations of the FH gene 10. The current manuscript briefly reports on 2 sisters in their 20s who presented with isolated uterine leiomyomas and were found to carry a novel mutation for the FH gene. In addition, this report is the first documented case of familial HLRCC in patients of Arab descent.
CASE REPORT
Patient 1
A 27-yr-old woman, gravida 2, para 0, was referred to our center for recurrent pregnancy loss. Her obstetrical history was significant for 2 spontaneous miscarriages; the first at 6 wk after a natural conception and the second at 16 wk following an in vitro fertilization frozen embryo transfer. Her gynecologic history was significant for menorrhagia and uterine leiomyomas requiring an abdominal myomectomy (24 myomas removed) at age 25. She then underwent one unsuccessful in vitro fertilization cycle followed by a frozen embryo transfer, which resulted in the second trimester miscarriage. The patient also required a hysteroscopic resection of retained placenta after the 16-wk pregnancy loss. Baseline infertility assessment revealed normal thyroid stimulating hormone, prolactin, and ovarian reserve (anti mullerian hormone 3.66 pg/mL, antral follicle count 20). The partner had a normal semen analysis. Ultrasonography of the pelvis was notable for an enlarged uterus measuring 11×7.9×6.2 cm with multiple intramural myomas and poor visualization of the endometrial stripe. In addition, saline infusion sonogram confirmed persistent uterine adhesions with poor distension of the cavity.
Resolution of extensive Asherman syndrome and submucous myomas (Fig. 1A) was achieved following 2 hysteroscopic resection procedures. The latter procedure was performed under direct laparoscopic visualization and included lysis of pelvic adhesions and resection of peritoneal endometriosis in the posterior cul-de-sac and bladder mucosa (Fig. 1B). The couple was counseled that the primary component of their infertility was likely due to a uterine factor secondary to fibroids and intrauterine adhesions as well as endometriosis.
FIG. 1.
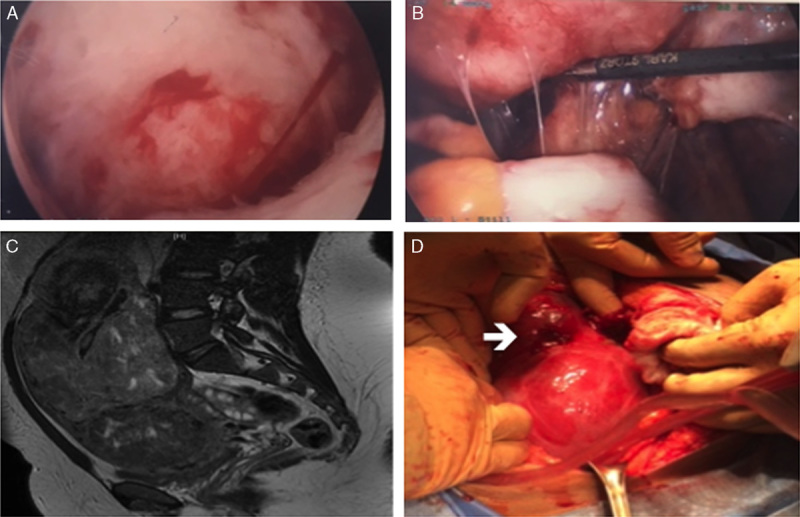
Preoperative and intraoperative photos of patients 1 and 2. (A) Intraoperative hysteroscopy for resection submucous fibroid showing extensive Asherman syndrome and submucous myoma (patient 1). (B) Intraoperative laparoscopy lysis of pelvic adhesions and resection of peritoneal endometriosis in the posterior cul-de-sac (patient 1). (C) Axial T2 image on pelvic magnetic resonance imaging of enlarged fibroid uterus (patient 2). (D) Intraoperative images of laparotomy myomectomy showing spontaneous rupture (arrow) from the fundal degenerated fibroid (patient 2).
Patient 2
This 21-yr-old, nulliparous woman was the younger sibling of patient 1 and was referred to our center for management of menorrhagia and a palpable abdominal mass. At presentation, the patient complained of heavy vaginal bleeding for the last month and increasing size of an abdominal mass over a 6-mo period. Physical examination was notable for a 25-wk size myomatous uterus. The patient was admitted to the hospital for treatment of symptomatic anemia with an initial hemoglobin of 6 g/dL on presentation. Pelvic magnetic resonance imaging demonstrated a 23.4×14.3×9.4 cm uterus with multiple confluent myomas (6–10 cm) replacing the myometrium and extending into the submucosa (Fig. 1C). She was administered 4 doses of IV conjugated estrogen (25 mg) to control vaginal bleeding and was transfused 5 units of packed RBCs. The patient was discharged on birth control pills with a hemoglobin of 10 g/dL.
Five days after discharge, the patient presented to the emergency room with increasing abdominal pain and shortness of breath. Computerized tomography scan of the abdomen and chest was diagnostic of a pulmonary embolus, degenerated leiomyoma, and hemoperitoneum. The clinical and radiologic findings were suggestive of spontaneous rupture of a leiomyoma. A diagnostic paracentesis was negative for malignant cells. Anticoagulation was initiated and an inferior vena cava filter was placed in preparation for surgical management. At laparotomy, she was found to have large volume serosanguineous ascites and a ruptured, degenerated leiomyoma (Fig. 1D). Inspection of the uterus revealed that the myometrium was comprised of multiple confluent myomas (7–10 cm) consistent with extensive degeneration and necrosis. She underwent an abdominal myomectomy after frozen section evaluation revealed benign leiomyoma with bizarre nuclei (atypical leiomyoma) with extensive inflammation. The patient was discharged on postoperative day #3 with anticoagulation for 6 mo.
Histologic Findings
The leiomyomas removed by hysteroscopy (patient 1) and by myomectomy (patient 2) showed similar pathologic features on permanent histologic specimens. The leiomyomata showed diffuse cytologic atypia characterized by enlarged nuclei, marked variation of nuclear sizes and shapes as well as multinucleated cells (Fig. 2A). Some nuclei had prominent purple or red nucleoli, with clearing of the chromatin resulting in characteristic perinucleolar halos (Fig. 2B). Occasional intranuclear and intracytoplasmic eosinophilic inclusions were present (Fig. 2A). The vessels were thin-walled with characteristic staghorn contours (Fig. 2C). There were no mitoses in tumor of patient 1 and 1 mitosis per 10 high-power field in patient 2, and in both cases Ki-67 immunostaining was positive in <5% of cells. The leiomyoma of patient 2 had benign infarct. No evidence of coagulative tumor cell necrosis or abnormal mitoses was identified. Immunostaining for fumarate hydratase showed lack of staining in the tumor cells, with preserved staining in vascular endothelial cells (Fig. 2D). The immunostaining for fumarate hydratase was performed using J-13 clone of antibody manufactured by Santa Cruz Biotechnology, at a dilution 1:5000.
FIG. 2.
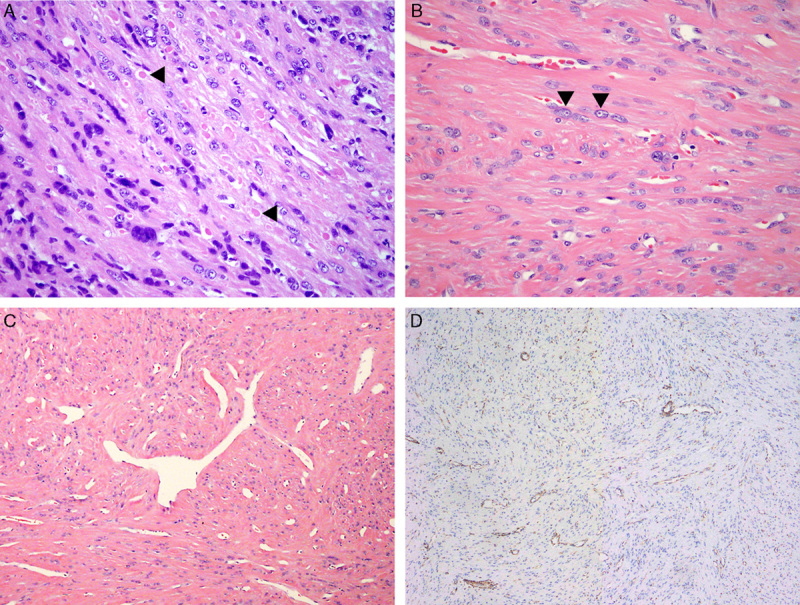
Histologic sections showing characteristic findings in hereditary leiomyomatosis renal cell cancer uterine leiomyoma. (A) Leiomyoma with atypical, hyperchromatic nuclei and multinucleation; intracytoplasmic eosinophilic inclusions are present (arrowheads) (patient 1). (B) Leiomyoma with atypical nuclei; prominent nucleoli and perinucleolar halos are present (arrowheads) (patient 2). (C) Leiomyoma with staghorn vasculature (patient 2). (D) Fumarate hydratase immunostain showing lack of staining in the tumor cells, but preserved staining in vascular endothelial cells (patient 1).
Genetic Testing
Genetic testing was performed by the Mount Sinai Genetic Testing Laboratory and analyzed a panel of 281 diseases using a combination of sequencing, targeted genotyping, and copy number analysis. Initial next-generation sequencing of known pathogenic variants for the fumarate hydratase gene was negative. Because of increased clinical suspicion for a novel mutation, next-generation sequencing with single gene full sequencing was subsequently performed and indicated that both siblings were heterozygous for a novel mutation in the FH gene (c.215C>A, p. T72N). This point mutation substitutes an asparagine for threonine at position 72 within the protein and in silico predictions suggest this variant to be deleterious. The DNA variant has not been published previously in the literature nor has its allele frequency been reported in any database.
Follow-up
The patients were re-examined and questioned regarding their personal and family history for manifestations of HLRCC. Both sisters did not report any history of painful cutaneous nodules and their dermatologic exams were unremarkable. The patients reported no symptoms of hematuria or flank pain and abdominal imaging was negative for renal abnormalities. The family pedigree was notable for the mother who underwent a hysterectomy in her 30s for symptomatic fibroids and a maternal grandmother who passed away from kidney problems of unknown etiology in her 40s (Fig. 3). The 2 siblings had 1 younger sister (19 yr), with no leiomyomas or other extrauterine manifestations of HLRCC. It should be noted that their parents were first cousins. Otherwise, the family history was unremarkable. Both patients were well at 1 and 2 mo follow-up.
FIG. 3.
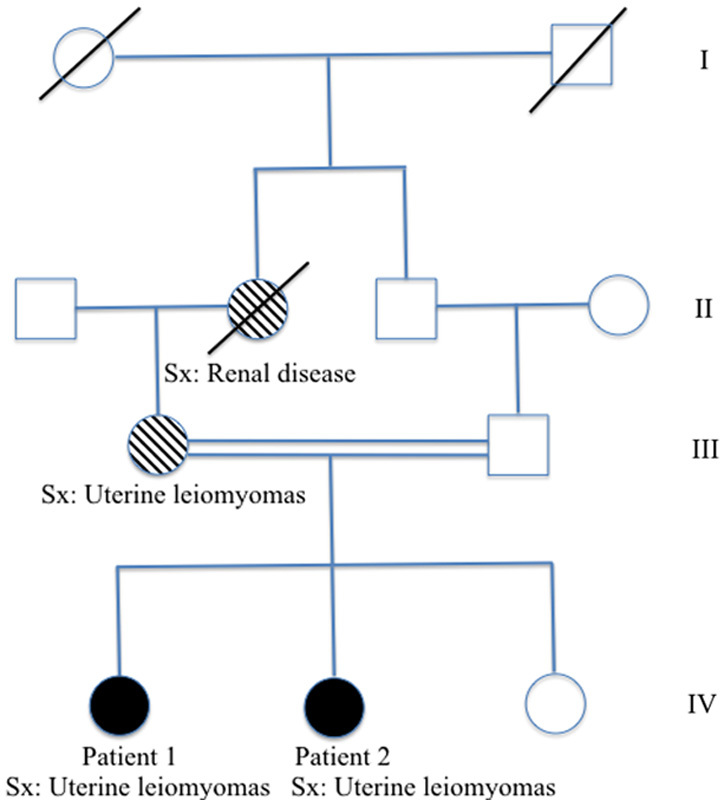
Pedigree of Arabic family with novel FH mutation (c.215C>A, p. T72N). Sx indicates symptom manifestation of hereditary leiomyomatosis renal cell cancer.
DISCUSSION
The current report depicts a familial case series of HLRCC with a novel mutation of the fumarate hydratase gene. Genetic testing was recommended based on the findings of familial clustering of early onset, isolated uterine leiomyomas that displayed characteristic histologic features of leiomyoma with bizarre nuclei. To date, there are approximately 150 novel mutations in the FH gene leading to HLRCC. In addition, patient 2 presented with a pulmonary embolus following hormonal treatment and uterine leiomyoma rupture, outcomes that have not previously been described in this specific subset of patients.
HLRCC exhibits a high clinical penetrance with 95% of patients developing characteristic findings by the age of 45; however, delays in diagnosis from initial symptoms still occur. The most prominent early clinical manifestations remain cutaneous and uterine leiomyomas, such that 80%–90% of patients with FH mutations will have myomas at the time of diagnosis 2,11. Although cutaneous symptoms may precede uterine findings by a mean of 6 yr (25 vs. 31 yr old), 40% of cutaneous leiomyomas can have subtle manifestations and therefore be missed 8. To that point, numerous case reports note a diagnosis of HLRCC by cutaneous biopsy more than 15 yrs after the patient had undergone surgical management for uterine myomas in their 20s to 30s 12–14. This delay further underscores the need for early renal carcinoma surveillance for these patients and their affected relatives. Given the strong penetrance of uterine leiomyomas with HLRCC, the current case report advises practicing physicians to consider genetic testing for patients with early onset, atypical, isolated uterine myomas in families with a history of heightened concern.
Uterine myomas associated with HLRCC present an earlier mean age (30 vs. 40 yr) and also cause increased morbidity when compared with benign leiomyoma with typical histologic features. It is of interest to note that prior studies report 28% of women with HLRCC had a coexisting diagnosis of secondary infertility or recurrent miscarriages in addition to symptomatic leiomyomas 8. Patients with HLRCC also sought surgical treatment at an earlier age compared with the general population (30–35 vs. 44 yr). The 3 largest HLRCC studies totaling 111 women reported a 50% hysterectomy rate in patients younger than 35 yr old, with a significant portion of those patients receiving definitive surgical management at younger than 30 yr of age 7,8,15. Thus, the early onset of symptomatic myomas significantly affects a woman’s ability to conceive as well as reduces her overall childbearing years. Prompt diagnosis will not only allow for proper counseling regarding childbearing—given the risk of hysterectomy, but also can also inform patients of options such as in vitro fertilization with prenatal genetic diagnosis to decrease the risk of transmission to future offspring.
In conclusion, the current report presents a novel mutation in the FH gene causing hereditary leiomyomatosis and renal cell cancer syndrome in 2 sisters. Both patients presented with early onset uterine leiomyomas in their 20s without any other manifestations of the syndrome. Prompt genetic diagnosis allowed for multidisciplinary management including genetic counseling for the entire family, urological assessment for renal cancer surveillance, and fertility counseling by a reproductive endocrinologist.
Acknowledgment
The authors thank Logan Stone for his editorial assistance.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Sparic R, Mirkovic L, Malvasi A, et al. Epidemiology of uterine myomas: a review. Int J Fertil Steril 2016;9:424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tolvanen J, Uimari O, Ryynanen M, et al. Strong family history of uterine leiomyomatosis warrants fumarate hydratase mutation screening. Human Reprod 2012;27:1865–9. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler KC, Warr DJ, Warsetsky SI, et al. Novel fumarate hydratase mutation in a family with atypical uterine leiomyomas and hereditary leiomyomatosis and renal cell cancer. Fertil Steril 2016;105:144–8. [DOI] [PubMed] [Google Scholar]
- 4.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 2001;98:3387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frey MK, Worley MJ, Jr, Heyman KP, et al. A case report of hereditary leiomyomatosis and renal cell cancer. Am J Obstet Gynecol 2010;202:e8–e9. [DOI] [PubMed] [Google Scholar]
- 6.Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 2010;79:49–59. [DOI] [PubMed] [Google Scholar]
- 7.Toro JR, Nickerson ML, Wei M-H, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 2003;73:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 2005;141:199–206. [DOI] [PubMed] [Google Scholar]
- 9.Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene 2016;589:127–32. [DOI] [PubMed] [Google Scholar]
- 10.Bayley J-P, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 2008;9:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubinova K, Tesarova M, Hansikova H, et al. Fumarate hydratase gene mutation in two young patients with sporadic uterine fibroids. J Obstet Gynaecol Res 2012;39:410–4. [DOI] [PubMed] [Google Scholar]
- 12.Mehrtens S, Veitch D, Kulakov E, et al. A case of hereditary leiomyomatosis and renal cell carcinoma. Case RepDermatol Med 2016;2016:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varol A, Stapleton K, Roscioli T. The syndrome of hereditary leiomyomatosis and renal cell cancer (HLRCC): the clinical features of an individual with a fumarate hydratase gene mutation. Australas J Dermatol 2006;47:274–6. [DOI] [PubMed] [Google Scholar]
- 14.Venables ZC, Ramaiya A, Holden S, et al. Hereditary leiomyomatosis associated with renal cell carcinoma. Clin Exp Dermatol 2014;40:99–100. [DOI] [PubMed] [Google Scholar]
- 15.Wei M-H. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2005;43:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]