

Abstract
Quiescence is a temporary, reversible state in which cells have ceased cell division, but retain the capacity to proliferate. Multiple studies, including ours, have demonstrated that quiescence is associated with widespread changes in gene expression. Some of these changes occur through changes in the level or activity of proliferation-associated transcription factors, such as E2F and MYC. We have demonstrated that mRNA decay can also contribute to changes in gene expression between proliferating and quiescent cells. In this protocol, we describe the procedure for establishing proliferating and quiescent cultures of human dermal foreskin fibroblasts. We then describe the procedures for inhibiting new transcription in proliferating and quiescent cells with Actinomycin D (ActD). ActD treatment represents a straightforward and reproducible approach to dissociating new transcription from transcript decay. A disadvantage of ActD treatment is that the time course must be limited to a short time frame because ActD affects cell viability. Transcript levels are monitored over time to determine transcript decay rates. This procedure allows for the identification of genes and isoforms that exhibit differential decay in proliferating versus quiescent fibroblasts.
Keywords: Genetics, Issue 131, Transcript decay, quiescence, fibroblasts, contact inhibition, half-life, actinomycin D, miR-29
Introduction
Steady state levels of transcripts reflect the contribution of both transcript synthesis and transcript decay. Regulated and coordinated transcript decay is an important mechanism for controlling biological processes1,2,3,4. For example, transcript decay rates have been shown to contribute to the temporal series of events following activation by the inflammatory cytokine tumor necrosis factor5.
We have previously shown that the transition between proliferation and quiescence in primary human fibroblasts is associated with changes in the transcript levels of many genes6. Some of these changes reflect differences in the activity of transcription factors between these two states.
Changes in a transcript's decay rate can also contribute to changes in the expression level of a transcript in two different states7,8. Based on our earlier findings that the transition between proliferation and quiescence is associated with changes in the levels of multiple microRNAs9, we asked whether there is also a contribution from differential transcript decay in the changes in gene expression in proliferating versus quiescent fibroblasts.
In order to monitor transcript stability, we determined the rate of decay of transcripts genome-wide in proliferating versus quiescent cells. To achieve this, we monitored decay rates in proliferating versus quiescent fibroblasts by introducing an inhibitor of new transcription and monitoring the rate at which individual transcripts disappeared over time. The advantage of this approach is that, as compared to methods that simply monitor overall gene expression, by inhibiting new transcript synthesis, we will be able to determine the decay rate for these transcripts separately from the rate at which they are transcribed.
ActD treatment to inhibit new transcription and determine transcript decay rates has been successfully applied in multiple previous studies. ActD has been used to dissect the importance of RNA stability in the changes in transcript abundance that result from treatment with pro-inflammatory cytokines5. A similar approach has also been used to dissect the differences in transcript decay rate in proliferating versus differentiated C2C12 cells as they adopt a differentiated muscle phenotype10. As another example, global mRNA half-lives have also been detected in pluripotent and differentiated mouse embryonic stem cells11. In these examples, mRNA decay has been shown to be important for regulating transcript abundance and for the transition of cells to different cell states.
Applying the methods described below, we discovered changes in transcript decay rates in approximately 500 genes when comparing fibroblasts in proliferating and quiescent states12. In particular, we discovered that the targets of the microRNA miR-29, which is downregulated in quiescent cells, are stabilized when cells transition to quiescence. We describe here the methodology we used to determine decay rates in proliferating and quiescent cells. This methodology is useful for comparing global mRNA decay rates in two distinct but similar conditions when information about rapidly decaying genes is sought. It could also be used to address other questions such as the effect of cell culture conditions on transcript decay, for instance, in two dimensional versus three dimensional cultures. Decay rates can be determined genome-wide with methods such as microarrays or RNA-Seq. Alternatively, real-time qPCR or Northern blotting can be used to determine decay rates on a gene-by-gene or isoform-by-isoform basis. These rates can then be used to calculate the half-life of each monitored gene and to identify genes with decay rates that are different in two conditions.
Protocol
All experiments described were approved by Institutional Review Boards at Princeton University and the University of California, Los Angeles.
1. Prepare Proliferating and Contact-inhibited Fibroblasts for ActD Time Course
NOTE: This protocol uses a timecourse with four timepoints. Three biologically independent samples can be collected per timepoint by collecting different tissue culture plates in one experiment, or the experiment can be repeated multiple times with different cultures of cells. In our experience, one 10 cm (diameter) tissue culture dish (one plate) provides sufficient RNA for analysis. If needed, multiple tissue culture dishes can be pooled for each sample to increase the number of cells at each timepoint. In addition, the same experiment can be performed with fibroblasts isolated from different individuals as truly independent biological replicates.
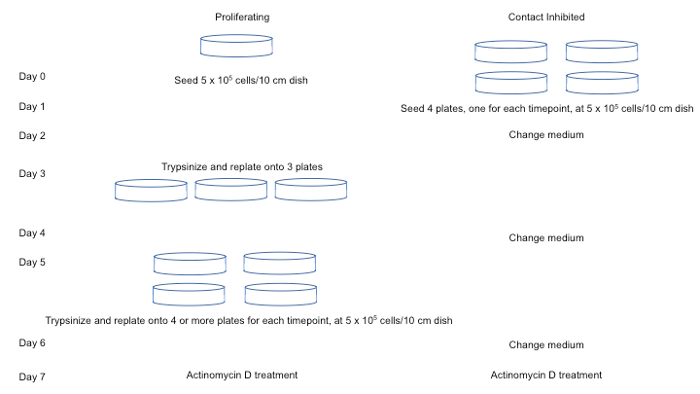
- Establish proliferating and quiescent cultures of cells for the transcript decay analysis. For proliferating cells, split the cells every three days while the contact-inhibited cells are becoming contact-inhibited.
- For the contact-inhibited cells, change the medium every 2 days for 7 days. Split the proliferating cells 2 days before the contact-inhibited fibroblasts are ready for collection. An example of a cell culture scheme is shown in Figure 1. The remaining steps in this section provide a detailed protocol for establishing and splitting the cells.
Isolate primary dermal fibroblasts from neonatal skin according to established protocols13. Passage fibroblasts to ensure a homogeneous population. NOTE: Fibroblasts can be frozen and thawed, if needed.
Thaw or replate fibroblasts, if needed. Grow fibroblasts in DMEM with 10% serum under sterile conditions in a tissue culture incubator at 37 °C and 5% CO2. Use fibroblasts that have been grown for fewer than 10 passages. Fibroblasts should be <80% confluent on the day of splitting cells to make proliferating and quiescent populations.
To split a tissue culture plate into multiple plates, prewarm PBS, trypsin, and medium with serum in a 37 °C water bath for 20 min.
Aspirate or use a pipet to remove the medium from each tissue culture plate with cells to be replated.
Pipet warm PBS onto each plate (10 mL of solution for a 150 mm plate, 5 mL of solution for a 100 mm plate, 2 mL for a well of a 6 well plate, and 1 mL for a well of a 12 well plate).
Aspirate or pipet to remove PBS from each tissue culture plate.
Gently pipet 1x Trypsin-EDTA onto each plate (8 mL of solution for a 150 mm plate, 3 mL of solution for a 100 mm plate, 2 mL for a well of a 6 well plate, and 1 mL for a well of a 12 well plate). NOTE: Make 1x Trypsin-EDTA by diluting sterile 10x Trypsin-EDTA in sterile PBS. The final 1x solution should be 0.05% trypsin and 0.02% EDTA.
Incubate trypsin with cells for 5 min.
Gently tap the plate to dislodge cells from the plate.
Pipet to resuspend cells into a single cell suspension.
Transfer cells in trypsin solution to a conical tube containing the same volume of DMEM with 10% serum (8 mL of solution for a 150 mm plate, 3 mL of solution for a 100 mm plate, 2 mL for a well of a 6 well plate, and 1 mL for a well of a 12 well plate).
Spin down cells at 4 °C for 5 minutes at 1,000 x g to remove trypsin.
Resuspend cells in an appropriate volume of medium and count with a hemacytometer to determine cell concentration.
Seed 5 x 105 cells per 100 mm tissue culture plate for proliferating and contact-inhibited cells.
Using this method, establish the proliferating and contact-inhibited samples required for analysis (Figure 1).
2. ActD Time Course
Resuspend ActD at a stock concentration of 1 mg/mL (For 1 mg of ActD, use 950 µL of sterile PBS and 50 µL of sterile DMSO). NOTE: Frozen stock solutions of ActD should be stable for a month at -20 °C. Dilute solutions should be discarded and not stored for further use.
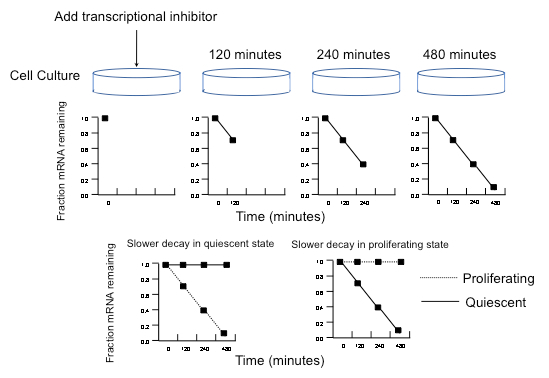
- Add ActD to the appropriate volume of prewarmed medium for all biological replicate cell culture plates that were set up for the 0, 120, 240, and 480 min timepoints (Figure 2). Add ActD at a concentration of 15 µg per mL of medium. Mix well, aspirate off the old medium, and add ActD-containing medium to the cells.
- For a 1 mg/mL stock, add 15 µL ActD for each mL of medium, e.g., for 10 mL of medium for a 100 mm plate, add 150 µL ActD.
To collect the zero timepoint sample, gently aspirate off the ActD medium, and pipet prewarmed PBS onto the cells. Pipet 10 mL of solution for a 150 mm plate, 5 mL of solution for a 100 mm plate, 2 mL for a well of a 6 well plate, and 1 mL for a well of a 12 well plate.
Gently aspirate or pipet off the PBS. NOTE: All steps involving RNA isolation should be performed with RNase-free water, RNase-free microcentrifuge tubes, and RNase-free pipettes. Gloves should be worn and changed frequently when working with RNA.
Add phenol-guanidine isothiocyanate solution (1 mL/10-cm plate with 4 x 105 to 4 x 107 cells) directly to the tissue culture plate. NOTE: Phenol-guanidine isothiocyanate solution is a monophasic solution that maintains RNA quality while lysing cells and separating RNA, DNA, and protein from each other. CAUTION: Phenol and guanidine isothiocyanate are corrosive. Use appropriate protection.
Pipet the phenol-guanidine isothiocyanate solution-cell mixture up and down several times to make a homogeneous mixture. NOTE: Phenol-guanidine isothiocyanate solution lysate can be stored at 4 °C overnight or at -20 °C for up to a year.
Collect each time point after the appropriate amount of time has passed using the method described in steps 2.3-2.6.
3. Isolation of Total RNA from Phenol-guanidine Isothiocyanate Solution Lysate
Incubate samples at room temperature for 5 min to ensure the complete dissolution of RNA-protein complexes. NOTE: The steps of the phenol-guanidine isothiocyanate solution protocol can be performed at room temperature unless otherwise noted.
Introduce spike-in control transcripts that can be detected with the methodology used. Introduce these controls into each sample at a known quantity and concentration. NOTE: The external RNA control consortium (ERCC) spike-in control mix14 has been specifically designed as a spike-in control for RNA-Seq. It can also be used for Northern blots or real-time qPCR. Spike-in controls specifically designed for microarray technologies are also available (see Table of Materials).
Transfer the phenol-guanidine isothiocyanate solution mixture to a 2.0 mL microcentrifuge tube with a pipette. Add 0.2 mL chloroform to the Phenol-guanidine isothiocyanate solution mixture for every 1 mL of phenol-guanidine isothiocyanate solution used (e.g., add 0.3 mL chloroform to 1.5 mL phenol-guanidine isothiocyanate solution mixture).
Shake the microcentrifuge tube vigorously for 15 s and then incubate chloroform-phenol-guanidine isothiocyanate mixture at room temperature for 3 min.
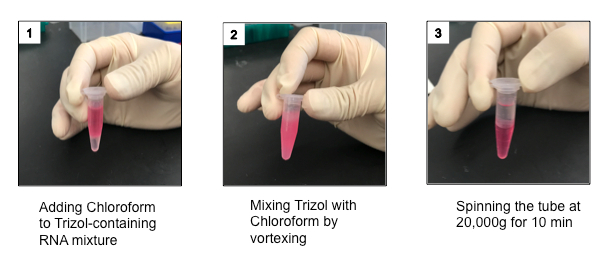
Spin samples at 12,000 x g for 20 minutes at 4 °C. NOTE: After the initial spin, the sample should separate into 3 layers - a red organic layer at the bottom, a white/pink interphase in the middle, and a clear aqueous layer on the top (Figure 3).
Transfer the aqueous layer to a new 2.0 mL microcentrifuge tube using a micropipette. NOTE: Be careful not to disturb the interphase during this process. It is okay to leave some of the aqueous fraction. It is better to leave some of the aqueous fraction than to also include some of the interphase layers, as including the interphase layer would contaminate the RNA with DNA. The aqueous layer will be approximately half of the initial volume, so about 0.9 mL if the phenol-guanidine isothiocyanate solution/chloroform mixture was 1.8 mL.
Discard the interphase and organic fractions.
Add an equal volume of 2-propanol to the aqueous fraction and invert the tube 10 times. Add up to 1 µL of 20 mg/mL aqueous solution of glycogen per 20 µL of sample, especially if the yield is expected to be low because fewer than 106 cells were collected. NOTE: This will result in a dense, solid pellet that is easy to monitor after the spin steps that follow.
Incubate samples at room temperature for 10 min.
Spin samples at 12,000 x g for 20 minutes at 4 °C. Ensure that, at the end of this spin, the RNA has precipitated to form a white pellet at the bottom of the tube.
With a pipette, remove and discard the supernatant taking special care not to disturb the white RNA pellet. NOTE: A loose pipet tip held in hand can be used to remove excess ethanol. If the RNA pellet is disturbed, but not discarded, the investigator can repeat the spin and remove the supernatant again. Avoid discarding the pellet as this will require the investigator to repeat the entire procedure.
Prepare a solution of 75% ethanol and 25% nuclease-free water. Add 1 mL of 75% ethanol per 1 mL of phenol-guanidine isothiocyanate solution reagent to wash the pellet.
Spin samples at 12,000 x g for 10 min at 4 °C.
After removing most of the supernatant, use a pipet to remove as much ethanol as possible, while still taking care not to disturb the pellet. NOTE: The RNA pellet is less compact at this step and tends to move. Be extra careful when handling the pellet at this point to avoid losing the RNA.
Spin samples 12,000 x g for 2 min at room temperature to pellet all excess ethanol.
Remove all excess ethanol from the tube using a micropipette taking care to not disturb the pellet.
Let the RNA pellet air dry for 5 min.
Add 50 µL of nuclease-free water to the RNA pellet and resuspend the pellet in the nuclease-free water.
Treat samples with 1 µL DNase (2 units/µL) to remove DNA and then inactivate DNase and cations (see Table of Materials).
Store RNA samples in 2.0 mL microcentrifuge tubes at -80 °C.
4. Analysis of Transcript Abundance
Quantify RNA and check RNA for purity using a spectrophotometer. NOTE: The ratio of absorbance at 260 nm to 280 nm should be approximately 2.0 for RNA.
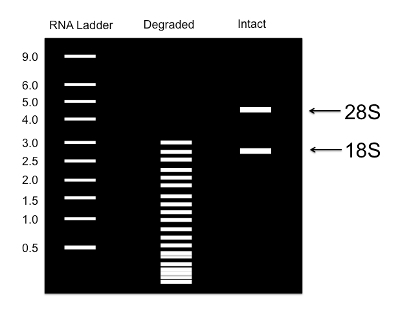
Analyze RNA with a 1% agarose gel or an equivalent technology to visualize the extent of degradation. NOTE: Two clear bands representing 18S and 28S rRNA should be clearly visible (Figure 4). If these bands are not visible, the RNA may be degraded. Trouble shooting tips for RNA degradation are provided in the Discussion.
- Monitor transcript abundance in the collected aliquots of RNA compared with the spiked-in internal standard using microarrays15, RNA-Seq16, real-time qPCR17, or Northern blots18.
- Determine the abundance for each transcript at each time point compared with a spiked-in control of known abundance. NOTE: For microarrays, the values will be fluorescence intensities. For Northern blots, bands on the X-ray film can be quantified. For real-time qPCR, transcript abundance can be determined by comparison with known standards with the 2-σσCT method19. For RNA-Seq, normalized values can be determined as FPKM or fragments per kilobase of exon per million reads mapped. For isoform-specific decay rates, RNA-Seq data can be analyzed using isoform-aware methodologies such as DEXSeq20. Isoform-specific decay rates are best determined with RNA-Seq. If they are to be determined with microarray data, then the data must be analyzed on a probe-by-probe basis rather than a gene-by-gene basis. The analyst must be careful when interpreting information from a limited number of probes. For instance, there may be 5 or fewer probes that are informative about a particular isoform. In this case, the analyst will need to determine whether the behavior of these probes is sufficiently consistent so that the results are reliable.
Perform real-time qPCR on the isolated RNA17 in the collected samples to analyze rapidly decaying genes, like c-Myc and a slowly decaying gene like GAPDH, to gain confidence that transcript decay rates were accurately detected. NOTE: For further confirmation, isoform-specific real-time qPCR probes can be developed and used to monitor the abundance of specific isoforms over time21.
5. Decay Rate Constant Determination
For each timepoint, log-transform the abundance values for each transcript (gene-specific or isoform-specific). NOTE: Log-transforming the values will convert exponential decay curves to lines that can be fit with a linear decay model22.
Fit the expression profile for each transcript (gene-specific or isoform-specific) to a linear decay model. The formula for such a model is f(t) = C - r*t. In this equation, C is a constant that represents the starting amount of a transcript, r is the rate of decline, and t is the time since the beginning of the experiment. From this equation, determine the decay rate as r*ln(10) (see reference11). NOTE: The maSigPro software package is a regression-based approach designed to detect significant gene expression profile differences in time course data23. Sample code and results for maSigPro are available at: https://www.bioconductor.org/packages/release/bioc/vignettes/maSigPro/inst/doc/maSigProUsersGuide.pdf.
Filter out genes that fail to fit a log-linear decay model. This can be accomplished with an ANOVA F-test. NOTE:The F test is based on the F-statistic, which is defined as the variation between sample means divided by the variation within samples. Correct the p-values for multiple comparisons by applying the linear step-up Benjamini and Hochberg false discovery procedure24. A q-value greater than 0.05 with the F-test can be used as a cutoff for genes that fail to fit a log-linear decay model.
Perform an ANOVA F-test to determine transcripts with a significant interaction term between a categorical cell cycle condition and time (false discovery rate, FDR <0.05).
Representative Results
We have previously reported the results of microarray analyses of transcript decay rates in proliferating and contact-inhibited primary human fibroblasts over an 8-hour time course12. A list of genes with a significant change in transcript stability comparing proliferating and contact-inhibited fibroblasts is provided in Supplementary Table 1. The fluorescence intensities at time zero and over a time course after ActD treatment are provided. Genes were included on the list by determining the genes that have significantly different slopes in proliferating and contact-inhibited conditions using an ANOVA F-test. Examples of genes with different decay rates in proliferating and quiescent fibroblasts are shown in Figure 5.
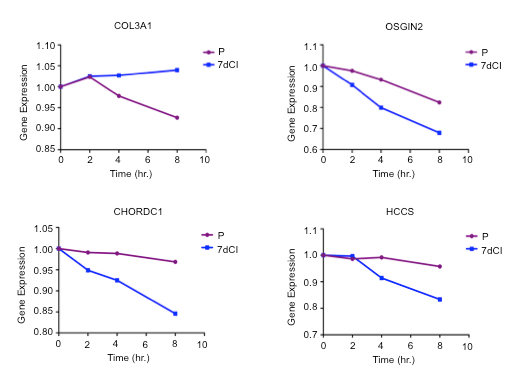
Figure 1: Schematic of protocol for establishing proliferating and contact-inhibited plates for ActD time course analysis. A day-by-day description of the necessary steps to establish cultures of cells for ActD treatment is provided, assuming four timepoints will be used for the time course. For simplicity, one plate is shown per timepoint, but additional plates can be added to generate biological replicates. The protocol can also be adjusted to add more plates of cells if more than four timepoints are desired. Please click here to view a larger version of this figure.
Figure 2: Schematic of ActD time course and decay rate calculations in quiescent compared to proliferating human fibroblasts. The expected fraction of mRNAs remaining over time for a hypothetical transcript after treatment with a transcriptional inhibitor is shown. Below, sample plots of the fraction of mRNA remaining over time are provided for genes that are more stable in quiescent than proliferating cells, and genes that are more stable in proliferating than quiescent cells. For each datapoint, only a single value is shown for clarity. In the actual protocol, there will be three datapoints for each timepoint. This figure has been modified from Elizabeth Pender Johnson's Ph.D. thesis with her consent. Please click here to view a larger version of this figure.
Figure 3: Photographs of mixing phenol-guanidine isothiocyanate reagent and chloroform before and after centrifugation. The mixture is shown when the phenol-guanidine isothiocyanate reagent and chloroform are initially combined, after mixing and after centrifugation. After the mixture is centrifuged, the bottom phase, which is red, is the organic phase. The interphase in the middle is white and cloudy. The top phase is the aqueous phase and it is clear. The RNA is in the aqueous phase, which should be collected with a pipette for precipitation. Please click here to view a larger version of this figure.
Figure 4: Sample RNA gel to monitor RNA quality. An illustration of an RNA gel showing samples with intact RNA. Intact RNA has prominent bands for the 28S and 18S rRNAs. Please click here to view a larger version of this figure.
Figure 5: Examples of genes with different decay rates in proliferating versus quiescent fibroblasts. Fluorescence intensities (gene expression) over time are shown for four genes that decay differently in proliferating versus quiescent fibroblasts. Please click here to view a larger version of this figure.
Supplementary Table 1: Gene-specific fluorescence intensities in proliferating and quiescent fibroblasts. Data are provided for time zero and over a time course after ActD treatment in proliferating and contact-inhibited fibroblasts. P values, q values, and false discovery rates are provided. Table is reprinted from its original publication in BMC Genomics12. Please click here to download this file.
Discussion
Quiescence can be induced by external signals including withdrawal of mitogens or serum, lack of cell adhesion, and contact inhibition. Contact inhibition, one of multiple possible methods for inducing quiescence, is a highly evolutionarily conserved process in which cells exit the proliferative cell cycle in response to cell-to-cell contact. We focus here on contact inhibition as an example of a method to induce quiescence. Previous studies have reported that cell-cell contact can affect microRNA biogenesis25, thus, the results provide information on one quiescence method and additional studies are required to determine which changes are associated with quiescence per se.
We used primary human diploid fibroblasts that we isolated from neonatal skin. We initiated these experiments with fibroblasts that were passaged fewer than 10 times. We discarded later-passage fibroblasts because they senesce. While this protocol focuses on proliferating and quiescent dermal fibroblasts, the procedures described can be used to monitor decay rates in different types of cells and under different conditions.
We monitored mRNA decay rates genome-wide using a transcription shut-off time course. Shut-off time courses are the most widely used method for decoupling mRNA decay from mRNA transcription in order to calculate mRNA half-lives1,26. In the method described here, ActD, a drug that complexes with the B-form of DNA to block elongation of RNA Polymerase II, is used to arrest transcription of mRNAs27. The rate that transcripts decrease in abundance over an 8-hour time period can be determined as the transcript's decay rate.
An important limitation of ActD-based approaches for monitoring transcript decay is that ActD prevents global transcription, which will affect the cells' viability. This should be kept in mind when interpreting ActD-based data, as decay rates are being determined in cells that are more stressed than their native state. Another important concern is that the effects of ActD may be different for cells in different states, including proliferating versus contact-inhibited fibroblasts. One approach for minimizing the side effects of ActD treatment is to keep time courses short in duration. For this reason, we selected an 8 h time course.
There are alternative approaches to ActD for monitoring transcript decay rates28,29. Yeast strains with temperature-sensitive RNA polymerase II alleles have been used to monitor transcript decay rates30. A variation on this approach called "anchor-away" has been used in yeast to achieve conditional depletion of RNA polymerase II in response to rapamycin treatment31,32. It is also possible to monitor decay in cells with active transcription. For instance, thio-substituted uracil nucleotides can be introduced into cells, incorporated into mRNA, and subsequently detected33,34,35. With this approach, the rate at which different transcripts are synthesized can be determined in cells that are continuing to synthesize mRNA. Such approaches have a significant advantage over ActD treatment because they are not toxic. In our experience, however, there can be variability in the recovery of transcripts with these methods. ActD treatment is a straightforward and reproducible approach for monitoring transcript decay.
A few steps in the protocol provided are critical for its success. One important issue is preventing RNA degradation during RNA isolation. At step 4.1, the RNA should be monitored for degradation. If the RNA does not contain two clear peaks for 18S and 28S rRNAs (Figure 4), this is a sign that transcript degradation has occurred. Some instruments provide RNA Integrity Number (RIN) scores ranging from 1 to 10. Samples with RIN scores in the 7-10 range are acceptable for further analysis. If the RNA is degraded, then it is important to take more precautions to prevent RNA degradation, such as ensuring that solutions and sterileware are RNase-free, and that gloves are worn when handling pipettes for RNA. Water can be pretreated with 0.1% diethyl pyrocarbonate (DEPC), incubated for at least 2 h at 37 °C, and autoclaved for at least 15 min. Note that DEPC cannot be used with Tris-based buffers. RNase decontaminating solutions can also be used to remove RNase from work surfaces, centrifuges, and pipettes, and reagents that inhibit RNases can be added to samples.
Another critical step is the separation of the phases of the phenol-guanidine isothiocyanate solution. It is important to ensure that only the top phase is collected for RNA processing. If the resulting RNA includes residual phenol, the ratio of absorbance at 230, 260, and 280 nm will not be in the ratio 1:2:1 as expected. Higher than expected absorbance at 230 nm can indicate phenol contamination. If this occurs, the RNA can be ethanol precipitated again and washed several additional times to remove any residual phenol.
Finally, when the RNA is pelleted and the supernatant is removed, the pellet can be sticky, thin, and easily disturbed. At this point, it is important to take extra care in taking off the liquid. It may be important to visualize the tube with good lighting to locate the pellet and make sure it is not disturbed. Added glycogen can help to ensure the pellet is visible at this point.
The selection of time points for the time course of decay is also an important issue to consider. While the four timepoints we selected allowed us to monitor decay for over 10,000 genes, some genes did not significantly decay during the 8 h time course. If short-lived transcripts are a high priority, the investigators may want to add additional sample collections prior to 120 min, for instance at 30 min. For better assessment of long-lived transcripts, the investigators may want to add additional sample collections at later timepoints.
Another potential issue could result if the decay rates do not follow the expected distribution. For instance, there may be too many or too few genes showing a log-linear decay rate over 8 h. If this happens, it is helpful to monitor the decay rates of genes that are known to be short or long-lived in order to determine whether they follow the expected pattern. If genes have a positive rather than negative slope over time, they can be excluded from further analysis as they do not demonstrate a log-linear decay rate. A final issue could arise if the cells in the two conditions being compared have very different responses to the ActD such that many genes decay faster in one condition. In this case, it may be helpful to refer not only to the difference in decay rate between the two states, but also to the spike-in controls that we suggest including prior to RNA isolation to provide information on absolute decay rates in the two states.
The ability to monitor transcript decay rates between proliferating and quiescent cells or any two similar states has the potential to provide further insight into the importance of transcript decay in regulating physiological changes. Applications include cells with and without oncogenic activation, before and after senescence or viral infection, with and without knockdown of a specific gene, or in two different states of differentiation. The findings can be coupled with RNA-Seq to provide information about isoform-specific changes in transcript decay rates, which may provide insight into the changes in isoform expression observed as cells undergo physiological transitions.
Disclosures
The authors have no competing interests to disclose.
Acknowledgments
HAC was the Milton E. Cassel scholar of the Rita Allen Foundation (http://www.ritaallenfoundation.org). This work was funded by grants to HAC from the National Institute of General Medical Sciences Center of Excellence grant P50 GM071508 (P.I. David Botstein), PhRMA Foundation grant 2007RSGl9572, National Science Foundation Grant OCI-1047879 to David August, National Institute of General Medical Sciences R01 GM081686, National Institute of General Medical Sciences R01 GM0866465, the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research, the Iris Cantor Women’s Health Center/UCLA CTSI NIH Grant UL1TR000124, and the Leukemia Lymphoma Society. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P50CA092131. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. HAC is a member of the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research, the UCLA Molecular Biology Institute, and the UCLA Bioinformatics Interdepartmental Program.
References
- Neff AT, Lee JY, Wilusz J, Tian B, Wilusz CJ. Global analysis reveals multiple pathways for unique regulation of mRNA decay in induced pluripotent stem cells. Genome Res. 2012;22(8):1457–1467. doi: 10.1101/gr.134312.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan A, Ogilvie RL, Reilly C, Abelson ML, Raghavan S, Vasdewani J, Krathwohl M, Bohjanen PR. Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res. 2002;30(24):5529–5538. doi: 10.1093/nar/gkf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle C, Fan J, Cho-Chung YS, Werner T, Ray J, Do L, Gorospe M, Becker KG. Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics. 2005;6:75. doi: 10.1186/1471-2164-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13(8):1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. 2009;10(3):281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4(3):83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001;2(4):237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8(2):113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Suh EJ, Remillard MY, Legesse-Miller A, Johnson EL, Lemons JM, Chapman TR, Forman JJ, Kojima M, Silberman ES, Coller HA. A microRNA network regulates proliferative timing and extracellular matrix synthesis during cellular quiescence in fibroblasts. Genome Biol. 2012;13(12):121. doi: 10.1186/gb-2012-13-12-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoen PA, Hirsch M, de Meijer EJ, de Menezes RX, van Ommen GJ, den Dunnen JT. mRNA degradation controls differentiation state-dependent differences in transcript and splice variant abundance. Nucleic Acids Res. 2011;39(2):556–566. doi: 10.1093/nar/gkq790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16(1):45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EL, Robinson DG, Coller HA. Widespread changes in mRNA stability contribute to quiescence-specific gene expression patterns in a fibroblast model of quiescence. BMC Genomics. 2017;18(1):123. doi: 10.1186/s12864-017-3521-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legesse-Miller A, Elemento O, Pfau SJ, Forman JJ, Tavazoie S, Coller HA. let-7 Overexpression leads to an increased fraction of cells in G2/M, direct down-regulation of Cdc34, and stabilization of Wee1 kinase in primary fibroblasts. J Biol Chem. 2009;284(11):6605–6609. doi: 10.1074/jbc.C900002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21(9):1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PK, Downey TJ, Spitznagel EL, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31(19):5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39(1):75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
- Streit S, Michalski CW, Erkan M, Kleeff J, Friess H. Northern blot analysis for detection and quantification of RNA in pancreatic cancer cells and tissues. Nat Protoc. 2009;4(1):37–43. doi: 10.1038/nprot.2008.216. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22(10):2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke II, Vandesompele J, Paepe AD, Messiaen L. Quantification of splice variants using real-time PCR. Nucleic Acids Res. 2001;29(13):68–68. doi: 10.1093/nar/29.13.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove JL, Hulsey MG, Beale EG. The kinetics of mammalian gene expression. Bioessays. 1991;13(12):667–674. doi: 10.1002/bies.950131209. [DOI] [PubMed] [Google Scholar]
- Conesa A, Nueda M. R package version 1.46.0. maSigPro: Significant Gene Expression Profile Differences in Time Course Gene Expression Data. 2016. Available from: http://bioinfo.cipf.es.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. Cell-cell contact globally activates microRNA biogenesis. Proc Natl Acad Sci U S A. 2009;106(17):7016–7021. doi: 10.1073/pnas.0811523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Dolken L, Ruzsics Z, Koszinowski UH, Zimmer R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 2009;37(17):115. doi: 10.1093/nar/gkp542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro R, Amos H. mRNA half-life measured by use of actinomycin D in animal cells - A caution. Biochimica et Biophysica Acta. 1966;129(2):406–409. [Google Scholar]
- Perez-Ortin JE, Alepuz P, Chavez S, Choder M. Eukaryotic mRNA decay: methodologies, pathways, and links to other stages of gene expression. J Mol Biol. 2013;425(20):3750–3775. doi: 10.1016/j.jmb.2013.02.029. [DOI] [PubMed] [Google Scholar]
- Chen CY, Ezzeddine N, Shyu AB. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008;448:335–357. doi: 10.1016/S0076-6879(08)02617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta I, Clauder-Munster S, Klaus B, Jarvelin AI, Aiyar RS, Benes V, Wilkening S, Huber W, Pelechano V, Steinmetz LM. Alternative polyadenylation diversifies post-transcriptional regulation by selective RNA-protein interactions. Mol Syst Biol. 2014;10:719. doi: 10.1002/msb.135068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisberg JV, Moqtaderi Z, Fan X, Ozsolak F, Struhl K. Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell. 2014;156(4):812–824. doi: 10.1016/j.cell.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31(6):925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Cleary MD, Meiering CD, Jan E, Guymon R, Boothroyd JC. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat Biotechnol. 2005;23(2):232–237. doi: 10.1038/nbt1061. [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29(5):436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi MJ, Ghini F, Cerruti B, de Pretis S, Bonetti P, Giacomelli C, Gorski MM, Kress T, Pelizzola M, Muller H, et al. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 2016;26(4):554–565. doi: 10.1101/gr.198788.115. [DOI] [PMC free article] [PubMed] [Google Scholar]