

Abstract
Studying the cell biological processes during converting the identities of specific cell types provides important insights into mechanism that maintain and protect cellular identities. The conversion of germ cells into specific neurons in the nematode Caenorhabditis elegans (C. elegans) is a powerful tool for performing genetic screens in order to dissect regulatory pathways that safeguard established cell identities. Reprogramming of germ cells to a specific type of neurons termed ASE requires transgenic animals that allow broad over-expression of the Zn-finger transcription factor (TF) CHE-1. Endogenous CHE-1 is expressed exclusively in two head neurons and is required to specify the glutamatergic ASE neurons fate, which can easily be visualized by the gcy-5prom::gfp reporter. A trans gene containing the heat-shock promoter-driven che-1 gene expression construct allows broad mis-expression of CHE-1 in the entire animal upon heat-shock treatment. The combination of RNAi against the chromatin-regulating factor LIN-53 and heat-shock-induced che-1 over-expression leads to reprogramming of germ cell into ASE neuron-like cells. We describe here the specific RNAi procedure and appropriate conditions for heat-shock treatment of transgenic animals in order to successfully induce germ cell to neuron conversion.
Keywords: Developmental Biology, Issue 131, Germ Cell, Transcription Factor-Induced Reprogramming, Caenorhabditis elegans, Epigenetic Reprogramming Barrier, RNA Interference, Heat-Shock Induction, Neuronal Reprogramming
Introduction
Cell fate reprogramming by ectopic TF expression such as the myogenic TF MyoD, which, when overexpressed, converts fibroblasts directly into muscle-like cells1, has been a research focus for decades. However, differentiated cells are often refractory to this process, due to mechanisms that safeguard their cellular identity. Germ cells show a similar degree of protection and there are underlying mechanisms that often act as a barrier to prevent germ cell conversion into other cell types upon over-expression of a fate-inducing TF. Typically, epigenetic regulation such as histone modifications and chromatin structure imposes an impediment for the conversion of cell fates2,3. We have previously applied reverse genetics using RNAi knock-downs in C. elegans and identified factors that safeguard cellular identities and thereby counteract reprogramming of germ cells into neurons4. Specifically, the polycomb repressive complex 2 (PRC2) and the highly conserved histone chaperone LIN-53 (CAF-1/RBBP/4/7 in humans) prevent the direct conversion of germ cells into specific somatic cell types such as glutamatergic taste neurons in C. elegans4,5. To identify these germ cell reprogramming barrier factors, we used transgenic animals carrying the heat-shock inducible Zn-finger TF CHE-1. CHE-1 is normally expressed in two head neurons of C. elegans, where it specifies the fate of glutamatergic taste neurons termed ASE6. The ASE neuron fate can be visualized by expression of the ASE-specific fluorescent reporter gcy-5prom::gfp. GCY-5 is a chemoreceptor only expressed in ASE right neuron7. Upon lin-53 depletion by RNAi and induction of broad ectopic expression of CHE-1 by heat-shock, germ cells can be converted into neurons in vivo4,5. Importantly, timing of RNAi treatment is crucial as only adult worms (P0 generation) exposed to lin-53 RNAi give rise to F1 progeny animals with a germline that is permissive for reprogramming into neurons4.
The above-described germ cell to neuron conversion procedure in C. elegans can be used to study a variety of biological questions such as the implication of signaling pathways in cell fate reprogramming. For instance, we used the CHE-1-induced germ cell reprogramming into ASE neurons upon RNAi against lin-53 in order to study the implication of the Notch signaling pathway in the process of germ cell reprogramming8. By performing double RNAi to co-deplete lin-53 and the histone demethylase-encoding gene utx-1 we showed that the Notch signaling pathway counteracts PRC2-mediated gene silencing by activating expression of UTX-1 in the germline8. This finding demonstrates that the germ cell conversion to neurons described in this protocol could be used to study a complex biological process such as cellular reprogramming in an intact organism and in the context of different developmental and environmental conditions. Furthermore, it provides the perspective to challenge germ cells by over-expressing different fate-inducing TFs to study their role in restriction of cellular plasticity9, or to assess their potential for inducing reprogramming of germ cells to other cell types in vivo.
While mammalian tissue cultures also allow studying different aspects of cell fate reprogramming, cells grown in culture have limited capacity to mimic signaling pathway conditions compared to an intact organism. Therefore, C. elegans is a versatile model for examination of the roles of various biological processes such as Notch signaling during reprogramming in vivo. Additionally, it is a powerful model for genetic screens that is relatively easy to maintain and genetically modify for low costs.
Protocol
All methods described here with regards to animal care have been approved by the LaGeSo Berlin, Germany
1. Solution Preparation
- NGM-Agar plates (1 L)
- Add 3 g of NaCl, 2.5 g of peptone media (e.g., Bacto-Peptone), and 20 g of agar. After autoclaving, add 1 mL of cholesterol (5 mg/mL in 95% EtOH stock), 1 mL of 1 M MgSO4, 1 mL of 1 M CaCl2, 25 mL of 1 M K2PO4, and 1 mL of amphotericin B (2.5 mg/mL stock).
- NGM-Agar RNAi plates (1 L)
- Add 3 g of NaCl, 2.5 g of peptone media, and 20 g of agar. After autoclaving add, 1 mL of cholesterol (5 mg/mL in 95% EtOH stock), 1 mL of 1 M MgSO4, 1 mL of 1 M CaCl2, 25 mL of 1 M K2PO4, 1 mL of amphotericin B (2.5 mg/mL stock), 1 mL of 1 M IPTG, and 1 mL carbenicillin (50 mg/mL).
- LB-Agar (1 L)
- Add 10 g of peptone media, 5 g of yeast extract, 5 g of NaCl, 20 g of agar, 10 mL of 1 M Tris pH 8.0. Add H2O to 1 L. After autoclaving, add 1 mL of 50 mg/mL carbenicillin and 2.5 mL of 5 mg/mL tetracycline.
- LB Medium (1 L)
- Add 10 g of peptone media, 5 g of yeast extract, 5 g of NaCl, and 10 mL of 1 M Tris pH 8.0. Add H2O to 1 L. After autoclaving, add 1 mL of 50 mg/mL carbenicillin.
- M9-buffer (1 L)
- Add 6.0 g of Na2HPO4, 3 g of KH2PO4, 5 g of NaCl, and 50 mg of gelatin. Before usage, add 1 mL of 1 M MgSO4.
- Bleaching solution (10 mL)
- Add 1 mL of NaClO and 2 mL of 5 N NaOH. Add H2O to 10 mL.
2. Preparation of RNAi Plates
Note: C. elegans is typically cultured in the laboratory on 6 cm Petri plates containing 7.5 mL of Nematode Growth Medium Agar (NGM-Agar)10,11. This protocol is optimized for worms kept at 15 °C. To prepare plates with NGM, use standard sterile techniques to prevent fungal and bacterial contamination. The following is the protocol to prepare NGM plates supplemented with reagents for RNAi.
- Prepare 6-cm NGM-Agar RNAi plates containing 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 50 µg/mL carbenicillin. Let them dry for 24 – 48 h at room temperature in the dark12.
- Keep RNAi plates at 4 °C in the dark. Do not use if older than 14 days.
- Select the RNAi bacteria (Escherichia coli HT115) clone containing the L4440 plasmid with the lin-53 gene DNA sequence from the frozen glycerol stock from the available RNAi library13 on LB-agar plates containing 50 µg/mL carbenicillin and 12.5 µg/mL tetracycline by using the three-phase streaking pattern. Grow the bacteria at 37 °C overnight12. This clone allows IPTG-dependent production of lin-53 dsRNA in the bacteria.
- Additionally, grow RNAi bacteria that contain the L4440 plasmid without any gene sequence as the empty vector control14.
The next day, pick a single colony from lin-53 or empty vector LB-agar plates using a 200 µL pipette tip and inoculate each of them into a separate culture tube containing 2 mL of liquid LB medium14 supplemented with 50 µg/mL carbenicillin. Grow cultures overnight at 37 °C until they reach an optical density (OD) at 600 nm of 0.6 – 0.8. Measure the OD using a spectrophotometer15. CAUTION: The liquid LB media should not contain tetracycline in contrast to the LB-agar plate used for streaking the bacteria from the glycerol stock, since inclusion of tetracycline during feeding leads to a decreased RNAi efficiency 12
- Add 500 µL of each bacterial culture (lin-53 or empty vector) to 6 cm NGM-Agar RNAi plates using a multipipette. Incubate plates with a closed lid overnight at room temperature in the dark to dry. During this time, the IPTG in the NGM plates will induce production of dsRNA in the bacteria.
- Use at least 3 plates per bacterial culture per experiment. This will provide 3 technical replicates for each experiment.
Store dried NGM-agar RNAi plates with bacteria at 4 °C in the dark for up to two weeks.
3. Preparation of C. elegans Strain BAT28
- Maintain the worm strain BAT288 containing the transgenes otIs305 [hsp-16.2prom::che-1, rol-6(su1006)] and ntIs1[gcy-5prom::gfp] on OP50 bacteria using standard NGM-Agar plates at 15 °C. Note: For detailed protocol on Nematode culture, see10. The rol-6(su1006) is a dominant allele and commonly used phenotypic injection marker16 that causes the typical rolling movement. It is used in the otIs305 transgene to track hsp-16.2prom::che-1.
- Keep the BAT28 strain at 15 °C at all times in order to prevent precautious activation of the hsp-16.2prom::che-1 transgene. Reduce exposure to temperatures higher than 15 °C while handling the strain as much as possible.
- To age-synchronize worms, use the bleaching technique17.
- Wash off 6 cm NGM-Agar plates containing adults and eggs of BAT28 using 900 µL M9 buffer. Pellet worms by centrifuging at 900 x g for 1 min. Remove the supernatant.
- Add 0.5 – 1 mL of bleaching solution and shake the tube for approximately 1 min until adult worms start to burst open. Monitor using a standard stereomicroscope.
- Pellet worms by centrifuging at 900 x g for 1 min. Remove the supernatant.
- Wash the worm pellet 3 times by adding 800 µL of M9 buffer and centrifuging at 900 x g.
- Place the cleaned eggs on fresh NGM-plates seeded with OP50 bacteria. Grow a bleached population on OP50 bacteria at 15 °C until they reach L4 stage (approximately 4 days). L4 larvae can be recognized by a white patch approximately halfway along the ventral side of the worm18 using a standard stereomicroscope. Note: To achieve the germ cell to neuron conversion phenotype upon depletion of lin-53, it needs to be depleted in the parental generation (P0). The scoring for the conversion phenotype is undertaken in the following generation (filial generation F1). To achieve the deplete lin-53 already in the P0, L4 animals are subjected to RNAi.
- Manually transfer 50 L4 stage worms per replicate using a platinum wire to an NGM plate that does not contain any bacteria and let worms move away from any transferred OP50 bacteria. Let worms move on the plate for around 5 minutes. Use bacteria from the NGM-Agar RNAi plates previously seeded with lin-53 RNAi bacteria (or empty vector control) to transfer to the respective RNAi plates.
- Avoid transferring OP50 bacteria to the actual RNAi plates. Work fast to minimize exposure time of the strain to temperatures higher than 15 °C.
- Incubate worms on NGM-Agar RNAi plates at 15 °C for approximately 7 days until F1 progeny of the worms reaches L3 – L4 stage. They can be clearly separated from the P0 animals since they are bigger and thicker than the F1 progeny.
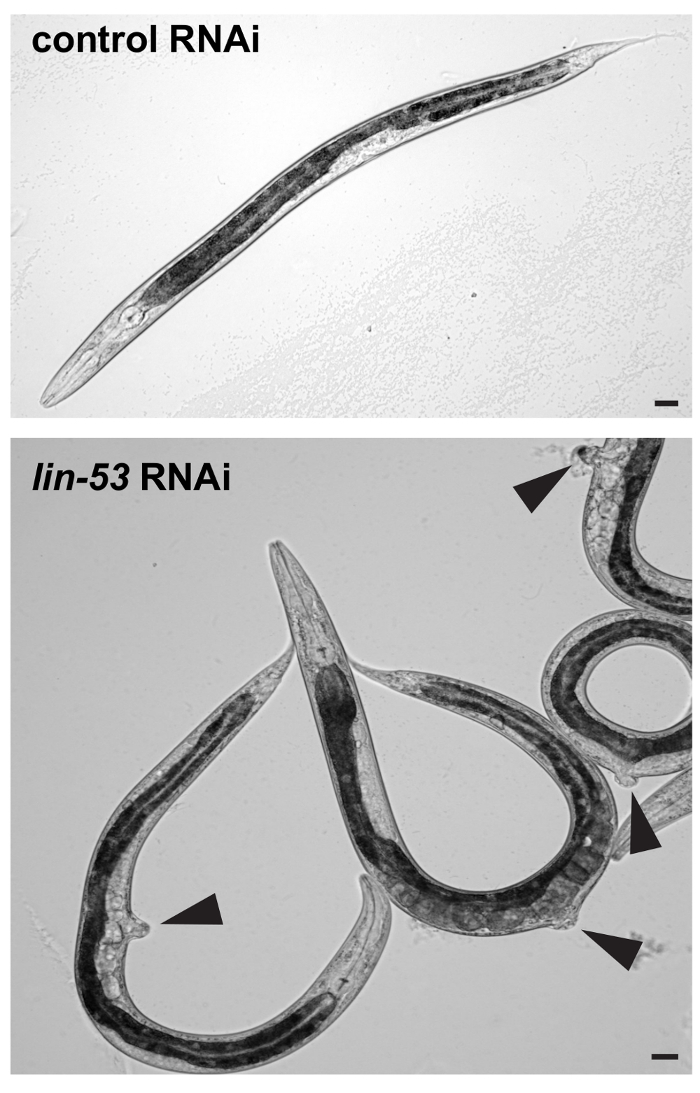
- Visually check under a standard stereomicroscope whether F1 progeny worms show the protruding vulva (pvul) phenotype as shown in Figure 1. RNAi against lin-53 has pleiotropic effects and causes the pvul phenotype which can be used to assess whether RNAi against lin-53 has been successful. Note: RNAi against lin-53 can also cause lethality of F1 embryos. This effect increases if plates are exposed to higher degrees than 15 °C before animals reached the L3 – L4 stage. Dead embryos can be recognized by arrested development and lack of hatching.
- Incubate plates in the dark since IPTG is light sensitive.
4. Induction of Germ Cell Reprogramming
Incubate examined RNAi plates containing lin-53 and empty vector RNAi-treated F1 progeny for 30 min at 37 °C in the dark to activate CHE-1. Use a vented incubator since it allows for a more efficient heat-shock.
Allow heat-shocked animals to recover for 30 min at room temperature in the dark and then incubate plates at 25 °C overnight in the dark.
Using a standard stereomicroscope coupled to a fluorescence light source, examine animals under the GFP filter for gcy-5prom::gfp-derived signals in the mid-body area of the worms as shown in Figure 2. Set microscope for GFP signals: excitation maximum at 488 nm with emission maximum at 509 nm.
- Evaluate the degree of germ cell to neuron conversion by mounting animals with GFP signals in the mid-body area on agar pad-containing microscopy slides19.
- Count the number of animals showing the germ cell to neuron conversion vs. the dark animals to assess the phenotype penetrance. Typically, around 30% of the animals show discrete GFP signals in the germline upon lin-53 depletion, whereas not more than 5% of the animals show diffuse GFP signals in the germline upon empty vector RNAi.
Representative Results
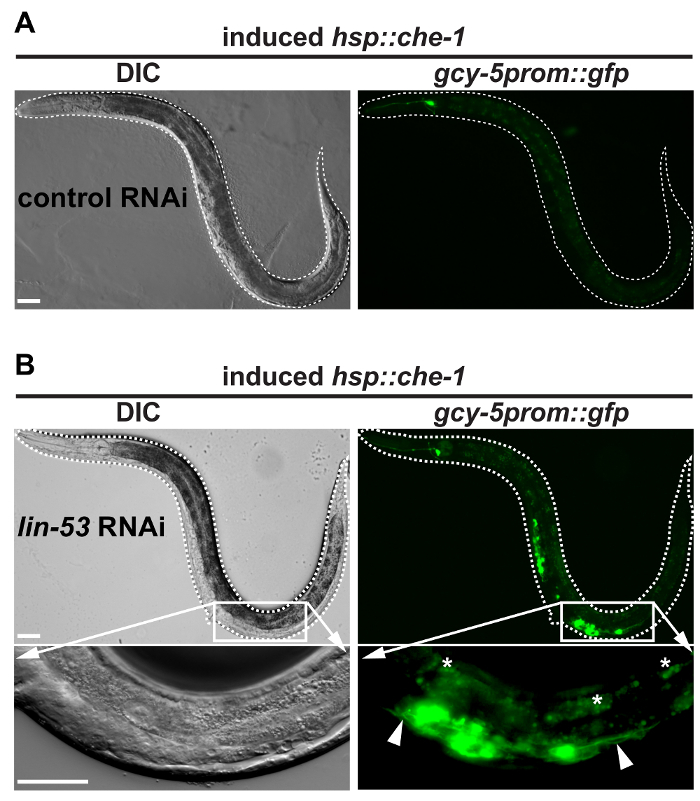
As shown in Figure 1, F1 animals that exhibit the pvul phenotype illustrate successful application of lin-53 RNAi. These animals are prone to allow conversion of their germ cells into ASE neuron-like cells upon heat-shock induction of che-1 over-expression as illustrated in Figure 2. In contrast to worms that grew on the control empty vector RNAi lin-53 RNAi treatment will yield animals that display ectopic GFP-signals in the mid-body area after heat-shock and 25 °C incubation overnight as described earlier4,8. Closer examination under an epifluorescence microscope of these worms reveals that the cells showing GFP signals also display neuron-like projections (Figure 2B, white arrows) that are typical for neuronal cells. If RNAi against lin-53 was not successful or heat-shock conditions were inappropriate GFP signals will resemble to those in control RNAi treated animals (Figure 2). Importantly, avoid inducing over-expression of che-1 by heat induction before animals reach mid L3 stage. Animals that were too young at the time of che-1 overexpression induction can display ectopic gcy-5prom::gfp expression in other regions of the body such as the developing vulva as shown in Figure 313.

Figure 1: Pvul phenotype caused by RNAi against lin-53. (Top) DIC picture of L4/young adult stage F1 progeny worms derived from control or lin-53 RNAi treated mothers. Scale bars = 20 µm. (Bottom) Animals treated with lin-53 RNAi display the protruding vulva (pvul) phenotype (black arrow-heads). The pvul phenotype confirms that RNAi against lin-53 was successful. Scale bars = 20 µm. Please click here to view a larger version of this figure.
Figure 2: Worms with successfully reprogrammed germ cell into neurons. (A) BAT28 strain animals grown on the empty vector do not show GFP signals in the germline after heat-shock induction of che-1 overexpression. Dashed white line outlines worm. Scale bars = 20 µm. (B) In contrast to BAT28 strain animals grown on the empty vector control RNAi animals grown on lin-53 RNAi display gcy-5prom::gfp signals in the mid-body area. Pictures of the mid-body section with GFP signals taken at 63X magnification reveal GFP-positive cells showing protrusions (white arrow-heads). This morphological feature indicates that germ cells underwent conversion into ASE neuron-like cells. White asterisks mark intestine-derived auto-fluorescence. All pictures were acquired using an epifluorescence microscope with 20X magnification and 63X magnification. Dashed white line outlines worms. Scale bars = 20 µm. Please click here to view a larger version of this figure.
Figure 3: Early stage worm with ectopic GFP. BAT28 strain animals grown on the empty vector RNAi bacteria that were heat-shock induced for che-1 overexpression at early larval stages such as L2 show GFP signals in the mid-body area (white asterisks). These signals are not due to germ cell reprogramming. Dashed white line outlines worm. Scale bars = 20 µm. Please click here to view a larger version of this figure.
Discussion
While the described protocol using the transgenic C. elegans strain BAT28 and RNAi against lin-53 is straightforward, a number of steps are critical in order to ensure the expected result of germ cell to neuron reprogramming. It is important that the strain BAT28 is kept at 15 °C at all times prior to the experiments. During handling and maintenance of the strain, the time at temperatures above 15 °C needs to be minimized as much as possible. Proper heat-shock conditions are important since insufficient induction of che-1 overexpression will not result in germ cell to ASE neuron conversion. This can be detected by measuring the phenotype penetrance as described in the protocol. Extensive heat-shock can lead to lethality of the animals. For instance, plates that have been placed near the inner walls or on the bottom ground of a static air incubator often experience extensive heat treatment. Therefore, it is recommended to use a vented heat incubator. Additionally, timing of the heat-shock induction is critical since over-expression of che-1 during larval stages earlier then L3 can lead to ectopic gcy-5prom::gfp induction in different body regions such as the vulva (Figure 3)9. Furthermore, extensive heat-shock can be detected by the phenotype penetrance in empty vector RNAi animals which should not extend 5% and increased auto-fluorescence of other tissues, namely the intestine.
Sporadically, RNAi bacteria can lose their activity to efficiently produce dsRNA of the target gene. Unfortunately, this can not be detected prior to the RNAi experiment. In such cases a fresh streak from the glycerol should be grown as described in Section 2 of the protocol. If there is still no RNAi-caused pleiotropic phenotype visible such as the pvul phenotype caused by successful RNAi against lin-53, then a plasmid DNA isolation from the respective bacteria should be performed and fresh HT115 bacteria need to be transformed with the L4400 plasmid containing the lin-53 sequence.
Importantly, the BAT28 strain has a the roller phenotype caused by the injection marker pRF4, which was used during transgenesis of the animals with the hsp-16.2prom::che-1 DNA construct. Occasionally, animals lose the rolling phenotype, which can indicate silencing of the hsp-16.2prom::che-1 transgene. To properly maintain the BAT28 strain, pick 10 roller animals to a fresh plate and propagate selecting for rolling animals.
The application of the protocol is limited to the F1 RNAi procedure, which means that animals whose parents (P0 generation) have not been exposed to lin-53 RNAi will not show germ cell to neuron conversion due to maternal rescue4. An alternative procedure to convert germ cells to neurons has been described by Ciosk and colleagues15 using RNAi knock-down of gld-1 and mex-3 without over-expression of a transcription factor. However, the obtained neurons do not belong to a specific type of neurons and germ cells also convert to muscle-like cells upon RNAi-mediated depletion of gld-1 and mex-315. Previously, it has been demonstrated the RNAi against lin-53 allowed germ cell conversion into GABAergic neuron-like cells upon over-expression of the Pitx-type homoedomain transcription factor UNC-3016 instead of CHE-14. For future directions, different fate-inducing transcription factors can be tested whether lin-53 depleted germ cells can be converted to other cell types than specific neurons. In this context, overexpression of the myogenic bHLH transcription factor HLH-1, homolog of the mammalian MyoD17, induces conversion of germ cell to muscles upon lin-53 RNAi5. The established reprogramming of germ cells to a specific somatic cell type upon lin-53 RNAi and over-expression of an appropriate fate-inducing transcription factor can be used for a number of different genetic screens. Such suppressor or enhancer screens can help dissecting regulatory pathways that play a role during cell fate conversion.
Disclosures
The authors have declared that no competing interests exist.
Acknowledgments
We thank Alina El-Khalili, and Martina Hajduskova for technical assistance. We thank members of the Tursun group for comments on the manuscript. This work was sponsored by the ERC-StG-2014-637530 and ERC CIG PCIG12-GA-2012-333922 and is supported by the Max Delbrueck Center for Molecular Medicine in the Helmholtz Association.
References
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51(6) doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Ebrahimi B. Reprogramming barriers and enhancers: strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. 2015. pp. 1–12. [DOI] [PMC free article] [PubMed]
- Becker JS, Nicetto D, Zaret KS. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Gent. 2016;32(1):29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tursun B, Patel T, Kratsios P, Hobert O. Direct Conversion of C. elegans Germ Cells into Specific Neuron Types. Science. 2011;331(6015):304–308. doi: 10.1126/science.1199082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T, Tursun B, Rahe DP, Hobert O. Removal of Polycomb Repressive Complex 2 Makes C.elegans Germ Cells Susceptible to Direct Conversion into Specific Somatic Cell Types. Cell Rep. 2012. pp. 1–9. [DOI] [PMC free article] [PubMed]
- Etchberger JF, et al. The molecular signature and cis-regulatory architecture of a C. elegans gustatory neuron. Genes Dev. 2007;21(13):1653, 1674. doi: 10.1101/gad.1560107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Avery L, Baude E, Garbers DL. Guanylyl cyclase expression in specific sensory neurons: a new family of chemosensory receptors. Proc Nat Acad Sci. USA. 1997;94(7):3384–3387. doi: 10.1073/pnas.94.7.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelk S, et al. Increasing Notch signaling antagonizes PRC2-mediated silencing to promote reprograming of germ cells into neurons. eLife. 2016;5 doi: 10.7554/eLife.15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T, Hobert O. Coordinated control of terminal differentiation and restriction of cellular plasticity. eLife. 2017;6:e24100. doi: 10.7554/eLife.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The Genetics of Caenorhabditis Elegans. Genetics. 1974;77(1):71. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Parihar M, Pires-daSilva A. An Introduction to Worm Lab: from Culturing Worms to Mutagenesis. J Vis Exp. 2011. p. e2293. [DOI] [PMC free article] [PubMed]
- Ahringer J. Reverse genetics. WormBook. 2006.
- Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421(6920):231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2000;18(1):419357. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosk R, DePalma M, Priess JR. Translational regulators maintain totipotency in the Caenorhabditis elegans germline. Science. 2006;311(5762):851–853. doi: 10.1126/science.1122491. [DOI] [PubMed] [Google Scholar]
- Jin Y, Hoskins R, Horvitz HR. Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature. 1994;372(6508):780–783. doi: 10.1038/372780a0. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Branda CS, Krause M, Stern MJ, Fire A. MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development. 1998;125(13):2479–2488. doi: 10.1242/dev.125.13.2479. [DOI] [PubMed] [Google Scholar]
- Ullrich M, Liang V, et al. Bio-orthogonal labeling as a tool to visualize and identify newly synthesized proteins in Caenorhabditis elegans. Nat Protoc. 2014;9(9):2237–2255. doi: 10.1038/nprot.2014.150. [DOI] [PubMed] [Google Scholar]
- Shaham S. Methods in cell biology. WormBook. 2006. pp. 1–75.