

Abstract
Embedded in the extracellular matrix (ECM), normal and neoplastic epithelial cells intimately communicate with hematopoietic and non-hematopoietic cells, thus greatly influencing normal tissue homeostasis and disease outcome. In breast cancer, tumor-associated macrophages (TAMs) play a critical role in disease progression, metastasis, and recurrence; therefore, understanding the mechanisms of monocyte chemoattraction to the tumor microenvironment and their interactions with tumor cells is important to control the disease. Here, we provide a detailed description of a three-dimensional (3D) co-culture system of human breast cancer (BrC) cells and human monocytes. BrC cells produced high basal levels of regulated on-activation, normal T-cell expressed and secreted (RANTES), monocyte chemoattractant protein-1 (MCP-1), and granulocyte-macrophage colony-stimulating factor (G-CSF), while in co-culture with monocytes, pro-inflammatory cytokines Interleukin (IL)-1 beta (IL-1β) and IL-8 were enriched together with matrix metalloproteinases (MMP)-1, MMP-2, and MMP-10. This tumor stroma microenvironment promoted resistance to anoikis in MCF-10A 3D acini-like structures, chemoattraction of monocytes, and invasion of aggressive BrC cells. The protocols presented here provide an affordable alternative to study intra-tumor communication and are an example of the great potential that in vitro 3D cell systems provide to interrogate specific features of tumor biology related to tumor aggression.
Keywords: Cancer Biology, Issue 131, Three-dimensional (3D) culture, extracellular matrix extract (ECME), co-culture system, tumor microenvironment communication, breast cancer (BrC), monocytes, tumor malignancy
Introduction
Tumor biology is far more intricate than previously thought. Mounting evidence shows that tumor cells are more than a mere bulk of uncontrolled proliferating cells; rather, different neoplastic cells seem to perform different functions displaying high organization and hierarchy within the tumor1. Tumor cells are also in intimate communication with non-transformed cells: macrophages, fibroblasts, lymphocytes, adipocytes, endothelial cells, among other cells are all immersed in the scaffold proteins and polysaccharides that constitute the ECM. Numerous direct and indirect interactions are established between transformed and non-transformed cells and with the ECM, which exerts a powerful influence over the disease outcome2,3. In the specific case of BrC, it is particularly important to dissect the communication of BrC cells with TAMs, considering that TAMs have been found to play a critical role in the evolution of the tumor, increasing the risk of metastasis and of disease recurrence4,5.
To analyze intra-tumoral interactions and their possible outcomes, new 3D in vitro approaches have been developed based on the use of ECM extracts that provide a much more complex microenvironment, closer to the reality of tumor biology, in comparison to the conventional monolayer cell cultures in which the cells grow attached to plastic. Petersen and Bissell6 provided the first model of nonmalignant and malignant mammary epithelial cells cultured on a laminin-rich basement membrane and were the first to describe the 3D organotypic structures that discriminate nonmalignant human breast epithelial cells from their malignant counterparts. A decade later, the model developed by Debnath, Muthuswamy, and Brugge7,8,9 provided a valuable tool to elucidate the biological pathways compromised during malignant transformation of glandular acini, such as large acini formation due to uncontrolled proliferation, delocalization of tight junction proteins as evidence of impaired cell polarization, and loss of acini lumen as a result of cell resistance to anoikis, a type of programmed cell death that occurs in anchorage-dependent cells when they detach from the surrounding ECM. The models of Sameni, Jedeszko, and Sloane have focused on imaging proteolytic activity by cells, which is closely related to invasiveness, another crucial trait of tumor malignancy10,11,12. These models rely on protein matrices mixed with different fluorescence-quenched protein substrates (DQ-gelatin, DQ-collagen I, and DQ-collagen IV), in which fluorescent signals are indicative of the proteolytic degradation of collagen. 3D models are also used to study stem cell properties of both non-transformed and tumor cells, in which cell aggregates, also termed spheroids, can be cultured in suspension or in ECM-like proteins interrogating for mechanisms of cell differentiation, asymmetric cell division, cell-to-cell adherence, and cell motility13,14. Invasion assays allow testing of the intrinsic aggressiveness of the tumor and the identification of the molecules that serve as chemoattractants during the invasive process15. Overall, 3D models represent an affordable diversification of in vitro cell culture that more closely reflect normal and oncogenic tissue morphogenesis.
We have designed a 3D cell co-culture system based on the aforementioned models7,10,11, using both human commercial BrC cell lines of known aggressive potential (luminal and triple-negative types) and primary cells explanted from BrC patients. We first developed a model where either non-aggressive (MCF-7) or aggressive (MDA-MB-231) BrC cells were co-cultured with U937 monocytes in an extracellular matrix extract (ECME)-based 3D system that allowed direct cell-cell interactions. These co-cultures were used to determine how the communication between these two cell lineages influenced the transcription of a set of genes related to cancer aggressive behavior. A significant increase of cyclooxygenase-2 (COX-2) transcript was observed that coincided with an increased production of one of its products, prostaglandin E2 (PGE2), a finding that highlighted the role of inflammation in cancer progression. Increased transcription of MMP was also observed that correlated with greater collagen proteolysis when aggressive MDA-MB-231 cells were co-cultured with U937 monocytes in DQ-Collagen IV-containing cultures. Of note, our co-cultures did not support the assumption that cell-cell interaction mechanisms are needed for collagen degradation. It rather suggested that communication between the two cell lineages was mediated by secreted molecules. Furthermore, the supernatants harvested from these co-culture assays contained soluble factors that disorganized glandular acini formed by non-transformed MCF-10A cells13. It was found that aggressive and primary BrC cells secreted elevated levels of monocyte chemotactic molecules MCP-1, GM-CSF, and RANTES. Thus, we outlined a 3D culture in which cells were separated in cell culture inserts to prevent cell-cell interactions. These cultures were used to address the indirect communication between BrC cells and monocytes. For these assays, non-aggressive and aggressive commercial BrC cell lines and primary BrC cells, and three different types of human monocytes: commercial U937 and THP-1 cells, and primary monocytes (PMs) isolated from peripheral blood of healthy donors (all monocytes were used in a non-activated state) were used. Increased concentrations of inflammatory cytokines IL-1β and IL-8 were observed to be enriched in co-culture. Similarly, it was found that MMP-1, MMP-2, and MMP-10 were also increased in the BrC cells-monocyte co-cultures, and thus reinforcing previous findings14. In this manuscript, a point-by-point workflow of primary BrC cells isolation and testing in 3D cultures is presented along with representative results. This work constitutes a good example of the great potential that in vitro 3D cell systems provide to interrogate specific aspects of tumor biology.
Protocol
Samples from BrC patients were obtained from the tissue bank of the Unidad de Investigación en Virología y Cáncer, Hospital Infantil de México Federico Gómez. This study was approved by the scientific, ethical, and biosecurity review boards of the Hospital Infantil de México Federico Gómez: Comité de Investigación, Comité de Ética en Investigación and Comité de Bioseguridad. All patients were prospectively enrolled and were informed about the nature of the study: those willing to participate signed a written informed consent prior to specimen collection and were treated according to the ethical guidelines and best clinical practice of the institution. The identity of the participants was anonymized for the duration of the study. Patients included were diagnosed with invasive ductal carcinoma, histological grade 2 and clinical stage II, with no previous neoadjuvant therapy before tissue resection. Patients were all female aged on average 56.8 years old (range 42 to 75)14.
1. 3D Cell Cultures
Seed 0.1 x 106 MCF-10A cells or primary BrC cells in 25 cm2 culture flasks with DMEM/F12 culture medium supplemented with 5% horse serum, 100 U/mL penicillin, 100 µg/mL streptomycin, 100 ng/mL cholera toxin, 0.5 µg/mL hydrocortisone, 10 µg/mL insulin, and 20 ng/mL of Epidermal Growth Factor (EGF). Seed 0.1 x 106 commercial BrC cells, MCF-7 cells, and MDA-MB-231 cells in 75 cm2 culture flasks with DMEM/F12 culture medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. Incubate at 37 °C in a humidified 5% CO2 environment.
After 48 h, rinse the cell monolayer with 10 mL of sterile 1x phosphate buffered saline (PBS). Trypsinize the cell monolayer with 3 mL solution of 0.05% trypsin and 0.48 mM ethylenediaminetetraacetic acid (EDTA) for 5-10 min at 37 °C. Add 200 µL of the corresponding serum to stop trypsinization and 7 mL of medium without supplements. Resuspend the cells by pipetting and remove an aliquot of 10 µL to count cells in a Neubauer chamber.
In each well of an 8-well chamber slide system, spread a 40 µL base of pure ECME, followed by an incubation of 30 min at 37 °C.
In each well, place 800 cells resuspended in 400 µL of DMEM/F12 (without phenol red) supplemented as in step 1.1 but with the following changes: for primary BrC cells and MCF-10A cells, add 4 ng/mL of EGF and 2% ECME; for MCF-7 and MDA-MB-231, only add 2% ECME.
Incubate cultures at 37 °C in a humidified 5% CO2 environment. Replace culture media every 48 h.
Record morphological changes of cells every 24 h for 15 days with an optical microscope. To analyze the morphology of primary BrC cells, MCF-10A and MDA-MB-231 cells are good reference cell lines for examples of ordered acinar formation and disordered aggressive cancer-like structures, respectively.
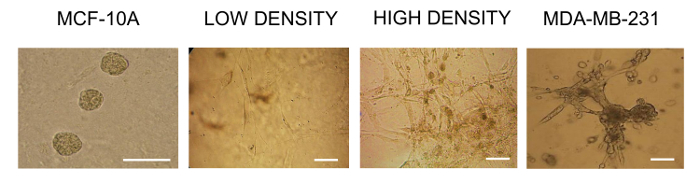
Obtain images of cells in bright field microscopy at 100X and 200X of magnification and compare cell morphology with reference MCF-10A and MDA-MB-231 cell lines (Figure 1).
2. Obtaining Primary Cancer Cells from Tumor Tissue
Note: To obtain tumor epithelial cells use tissue from resected primary tumors from BrC patients with no previous neoadjuvant therapy; avoid necrotic areas and work with a minimum of 0.5 cm3 of tumor tissue.
Rinse tumor tissue with sterile 1x PBS and mechanically disaggregate it with a scalpel into 1-2 mm fragments.
Digest the tissue in a sterile 20 mL glass vial with cap for 2 h at room temperature (RT) with 2 mL of the follow solution: mix 1 mg/mL collagenase type I and 100 U/mL hyaluronidase in DMEM/F12 medium containing 100 U/mL penicillin and 100 µg/mL streptomycin, with constant stirring.
Filter the resulting suspension through sterilized pieces of tulle to eliminate large pieces of undigested tissue. Then filter the cell suspension through a sterilized 100 µm-pore membrane.
Pellet the cells at 430 x g for 5 min at RT and wash them twice with sterile 1x PBS. Culture primary cells in DMEM/F12 medium supplemented with 5% horse serum, 100 U/mL penicillin, 100 µg/mL streptomycin, 100 ng/mL cholera toxin, 0.5 µg/mL hydrocortisone, 10 µg/mL insulin, and 20 ng/mL of EGF. Replace the medium every 48 h. Maintain cultures at 37 °C in a humidified 5% CO2 environment.
Ensure that the isolated cells are epithelial cells with a standard protocol of immunocytochemistry14. Test three epithelial markers: a panel of cytokeratins (PanCK), mucin-1 (Muc-1), and epithelial cell adhesion molecule (EpCAM).
3. Isolating PM cells from Peripheral Blood
Extract approximately 40 mL of peripheral blood from a healthy volunteer, dilute the blood in a 1:3 proportion with sterile endotoxin-free 1x PBS, and subject it to a density gradient separation.
In a conical tube, place 2 mL of a polysucrose and sodium diatrizoate medium with a density of 1.077 g/mL. Carefully and slowly overlay 8 mL of the diluted blood. Centrifuge for 30 min, 765 x g at RT. Use the slowest acceleration-deceleration speed of the centrifuge. Note: After the centrifugation, four layers will be formed from bottom to top: the red blood cells pellet, the density gradient medium layer, the mononuclear cells layer (it appears as a fine white ring), and the plasma layer.
Carefully retrieve the mononuclear cell layer from the gradient with a sterile Pasteur pipette and wash them three times with 1x PBS, each time followed by slower centrifugation (430, 275, and 191 x g) for 10 min at RT.
- Use negative selection protocols to avoid activation of cells. An example of such a protocol is the following:
- Wash the mononuclear cells once with a washing buffered solution (0.5% bovine serum albumin, 2 mM EDTA in 1x PBS). Count the cells in a Neubauer chamber and adjust them to a density of 1 x 107 cells/30 µL of a buffered solution.
- For every 1 x 107 cells, add 10 µL of a FcR blocking reagent and 10 µL of a monocyte biotin-antibody cocktail that contains anti-human CD3, CD7, CD16, CD19, CD56, CD123, and Glycophorin A (included in commercial kits).
- Mix the cells and incubate for 15 min at 4 °C. Add an additional 30 µL of the washing buffered solution plus 20 µL of anti-biotin microbeads (included in the kit); mix cells and incubate for 20 min at 4 °C.
- Wash the cells once with a buffered solution, centrifuge at 430 x g for 5 min at RT, and resuspend in 1.5 mL of buffered solution for magnetic separation.
- Load the cell suspension on a pre-rinsed magnetic separation column and add 7 mL of buffered solution. Collect the monocyte-enriched fraction in a 15-mL conical tube, count the cells, and if not cultured immediately, freeze monocytes at a density of 2 x 106 cells/1 mL of DMEM/F12 medium supplemented with 50% FBS and 10% DMSO at −80 °C. Note: Avoid using monocytes after more than 2 months of freezing, as their viability can be compromised.
Maintain cultures of PMs in DMEM/F12 medium supplemented with 6% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin, at 37 °C in a humidified 5% CO2 environment.
Perform each set of experiments utilizing PMs from at least two different donors independently, as pooling monocytes from different donors can result in monocyte activation.
4. Establishing 3D Co-cultures
- 3D co-cultures with indirect interaction Note: Perform these assays in 24-well flat-bottom culture plates using polycarbonate cell culture inserts with membranes of 0.4 µm pore size. In this assay, both supernatants and cells can be retrieved at the end of the experiment.
- Cultivate 2 x 106 U937 and THP-1 monocytes in 10 mL of RPMI 1640 medium supplemented with 10% FBS, 1% antibiotic/antimycotic, and cultivate fresh PMs in supplemented DMEM/F12 medium (as indicated earlier in step 3.5), at 37 °C in a humidified 5% CO2 environment.
- Plate 4 x 105 monocytes in 1 mL/well of their corresponding medium (supplemented with 2% ECME and 2% FBS for U937 and THP-1 monocytes, or 2% ECME and 6% FBS for PMs).
- Place an insert in each well and add 0.9 mL of the 4 x 105 BrC cell suspension in the corresponding medium (supplemented with 2% ECME and 2% FBS for commercial cell lines or 5% horse serum for primary BrC cells). Replace half of total media every 48 h.
- Incubate at 37 °C in a humidified 5% CO2 environment for 5 days and recover the supernatants. Retrieve cells by trypsinization if subsequent analysis is performed.
- Include controls of individual cell cultures with the respective media.
- 3D co-cultures with direct interaction
- Label BrC cells and monocytes with different fluorescent dyes before cell culture to allow independent sorting of each cell lineage after culture for analysis of mRNA and protein expression. Use commercially available derivatives of coumarin and rhodamine that freely pass through the cell membrane of living cells. A general protocol for labeling is the following:
- Prepare a monolayer of BrC cells of interest (2 × 106 cells in a 25 cm2 culture flask) and replace the standard medium with sufficient pre-warmed working solution of the fluorescent dye (1:2,000 of the fluorescent dye in standard base medium without any supplement). Incubate 30 min at 37 °C in a humidified 5% CO2 environment. Aspirate the dye solution and rinse gently with sufficient 1x PBS. Aspirate the PBS and add the standard medium.
- Trypsinize the cell monolayer with 2 mL of a solution of 0.05% trypsin and 0.48 mM EDTA for 5-10 min at 37 °C. Add 200 µL of FBS to stop trypsinization and 7 mL of the correspondent medium without supplements. Resuspend the cells by pipetting. Take an aliquot of 10 µL to count cells in a Neubauer chamber. Continue with the co-culture protocol after cell counting.
- Pellet the cells at 430 x g for 5 min at RT (perform this first since monocytes grow in suspension). Discard supernatant and gently resuspend cells with 3 mL of a pre-warmed working solution of the fluorescent dye (1:2,000 of the fluorescent dye in a standard base medium without supplements). Incubate for 30 min at 37 °C in a humidified 5% CO2 environment.
- Pellet the cells again, discard the dye working solution, and gently resuspend with 5-7 mL of 1x PBS. Pellet once more, discard the PBS, and resuspend cells in 5-7 mL of standard medium. Take an aliquot of 10 µL of cell suspension to count cells in a Neubauer chamber. Continue with the co-culture protocol after cell counting.
- Set the co-culture as follows: : spread enough ECME at the bottom of the well to form an even layer in each well of a 4-well chamber slide system. Plate 20 µL of a single cell suspension containing 5 × 105 labeled BrC cells per well.
- After 15-20 min of incubation at 37 °C, add a suspension of 2.5 x 105 labeled monocytes in 80 µL assay medium (supplemented with 60% ECME).
- Allow ECME to solidify for 15-20 min at 37 °C.
- Add 1 mL of a 1:1 mix of the BrC cells and monocyte culture media.
- Incubate co-cultures at 37 °C in a humidified 5% CO2 environment for 24 h, 48 h, or 5 days to track changes at different time points.
- Degrade ECM proteins to recover the cells from the cultures. Aspirate and discard the medium, add 0.5 mL of 1x PBS with 0.1% trypsin and 0.25% EDTA, and incubate for 3 h at 37 °C. After incubation, add 0.5 mL of 1x PBS with 10% FBS to neutralize the trypsin, and resuspend the cells by pipetting vigorously to obtain a single-cell suspension.
- Pellet cells at 765 x g for 5 min at RT, discard the supernatant, and resuspend the cells in 5 mL of 1x PBS with 10% FBS. Repeat this step once.
- Subject the cell suspension to fluorescence-activated cell sorting (FACS) with the appropriate instrument. Ensure that the final populations are at least 95% pure).
- After sorting, wash the cells with sterile 1x PBS, pellet (430 x g for 5 min at RT), and resuspend in sterile 1x PBS. Cells can be processed according to specific protocols for RNA isolation or protein analysis.
- Repeat each assay three times, including 3D cultures of each individual cell lineage as controls.
- Direct interaction of 3D co-cultures to assess collagen degradation
- Establish 3D cell co-cultures as described in step 4.2, with the additional step of adding 32.5 µg/mL of type IV collagen labeled with fluorescein isothiocyanate to the ECME. The final concentration of collagen IV in the ECME is 0.5%. Grow the cultures in a 35-mm coverslip placed in a Petri dishes.
- Spread a layer of 40 µL of ECME in the bottom of the Petri dish. Allow ECME to solidify at 37 °C.
- Add 2.5 x 105 BrC cells in 10 µL of the medium and allow cells to settle down.
- Add a suspension of 1.25 x 105 U937 monocytes in 40 µL assay medium (supplemented with 60% of DQ-Collagen IV labeled-ECME).
- Allow ECME to solidify for 15-20 min at 37 °C.
- Add 2 mL of a 1:1 mix of BrC cells and monocyte culture media.
- Incubate the co-cultures for 5 days at 37 °C in a humidified 5% CO2 environment. Replace culture media every 48 h.
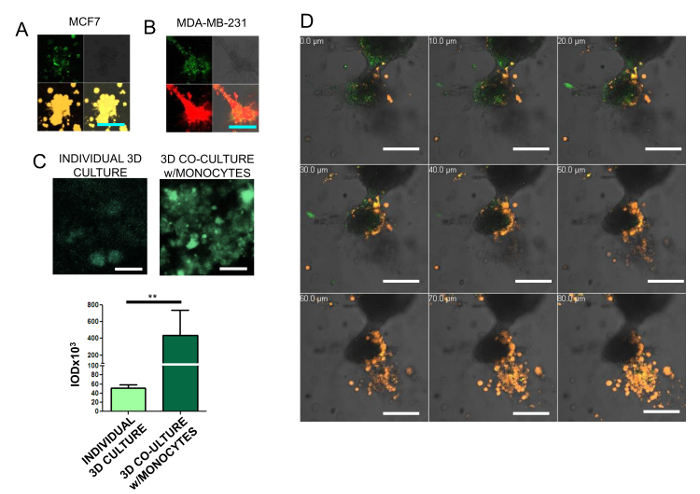
- Analyze the fluorescence emission in a confocal scanning microscope and measure the integrated optical density (IOD) per 50 µm2 of culture. Compare IODs against individual cultures and the culture at time zero (Figure 2).
5. Analysis of Supernatants from 3D Co-cultures with Indirect Interaction
After 5 days of co-culture, recover the supernatants from both the upper and the lower compartments of the 3D co-cultures, and from the individual 3D culture controls. Mix well each supernatant by pipetting. Aliquot, and store the supernatant at −20°C until use (up to one month). Note: Keep supernatants at −80°C if storage will be longer than one month.
Analyze the supernatants with multiplexing assay platforms, enzyme-linked immunosorbent assays (ELISA), or bead-based immunoassays following the manufacturer's recommended procedures. Note: Supernatants can also be analyzed by Western blot or concentrated through specific pore filters to obtain size enriched protein fractions to perform zymography analysis16,17. Alternatively, use the supernatants as conditioned media for other experiments, as described below. Conditioned media is usually obtained from a 5-day culture (Figure 3).
6. Characterization of the Effects of Supernatants from Primary BrC cells on Acini Formation and Acini Structure
In each well of an 8-well chamber slide system spread a 40 µL base of ECME and let it solidify for 30 min at 37 °C.
Seed 800 MCF-10A cells/well in 400 µL of supplemented DMEM/F12 culture medium, as described in step 1.2.
Add 400 µL of conditioned media or supplemented DMEM/F12 culture medium with 20 ng/mL of human recombinant IL-1β.
Place the chamber slide into a glass petri dish containing a 35 mm petri dish filled with 2 mL of PBS to create a humidity environment.
Replace the media/supernatant every 48 h.
Record glandular acini formation every 24 h for 14 days with an optical microscope.
After 14 days of culture, stain acini with 100 µL of 4',6-diamidino-2-phenylindole (DAPI) at a concentration of 100 nM in 1x PBS to observe cell nuclei. Incubate for 25 min at RT in constant stirring. Rinse three times with 1x PBS for 5 min, each time in constant stirring at RT. Mount the preparations with a mounting medium specific for fluorescent staining. Seal with transparent polish and maintain protected from light at 4 °C.
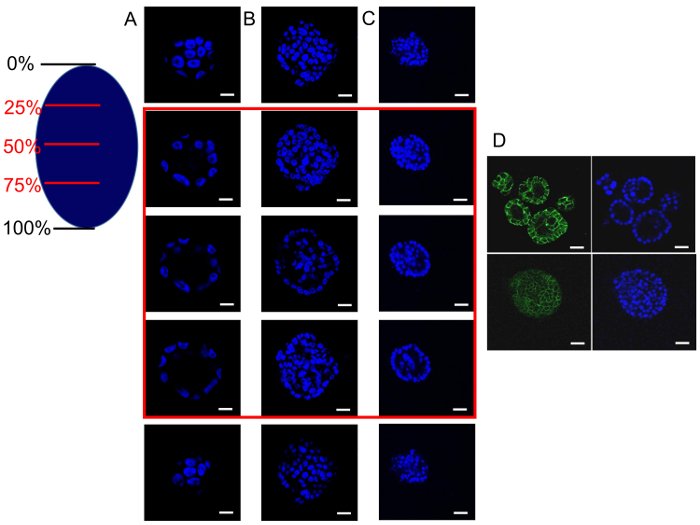
Analyze the acini with a confocal microscope taking images of transversal stacks at different depths of the acini (Figure 4A, B, C).
Visualize different cellular proteins in the acini with proper staining protocols. For instance, E-cadherin was stained to assess cell-to-cell adhesion, cell polarity, and/or lumen formation18 (Figure 4D).
7. Migration Assays
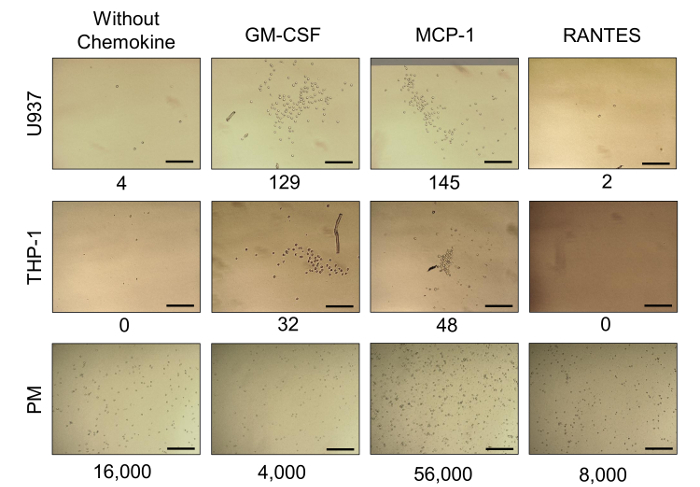
Note: Perform migration assays of U937, THP-1, and fresh PMs in 24-well flat-bottom culture plates using polycarbonate cell culture inserts with membranes of 8 µm pore size. It was previously demonstrated that the chemokines GM-CSF, MCP-1, and RANTES were found secreted at high concentrations in individual 3D cultures of the aggressive BrC cell lines. It was proposed that these cytokines were critical to attracting monocytes to the site of the primary tumor; this was tested in migration assays using the cytokines as chemoattractants.
Culture U937, THP-1, or fresh PMs as described in step 4.1.1.
Rinse once each insert with 200 µL of RPMI 1640 without sera to hydrate the membrane.
Fill it with 50 µL of cold ECME, place the inserts in a 24-well flat-bottom culture plate, and allow the ECME to polymerize for 1 h at 37 °C.
Add 180 µL of RPMI without FBS in the upper compartment of the plate. Add in the lower chamber without bubbling 800 µL of RPMI supplemented with either 100 ng/mL of GM-CSF, MCP-1, or RANTES as chemoattractant. Use medium without the cytokines as a negative control. Incubate for 30 min at 37 °C to establish a chemokine gradient.
Place 1.5 x 105 of each type of monocyte in a sterile tube, wash twice with 1 mL of 1x PBS, and centrifuge at 430 x g for 5 min at RT. Resuspend monocytes in 20 µL of RPMI without FBS, place them in the inserts, and very carefully homogenize the cell suspension (the final volume of the upper chamber is 200 µL).
Allow the cell migration to progress for 24 h at 37 °C in a humidified 5% CO2 environment. Note: As monocytes are non-adherent, migratory cells will usually be in the media of the lower compartment.
Remove the inserts, retrieve the media, and count cells at 2, 4, 6, and 24 h using a Neubauer chamber or a flow cytometer; alternatively, especially if no cells are found in the media, acquire images of the lower part of the inserts with a digital camera. The mean cell count from 3 random fields (at 100X magnification) is used for the analysis (Figure 5).
8. Invasion Assays
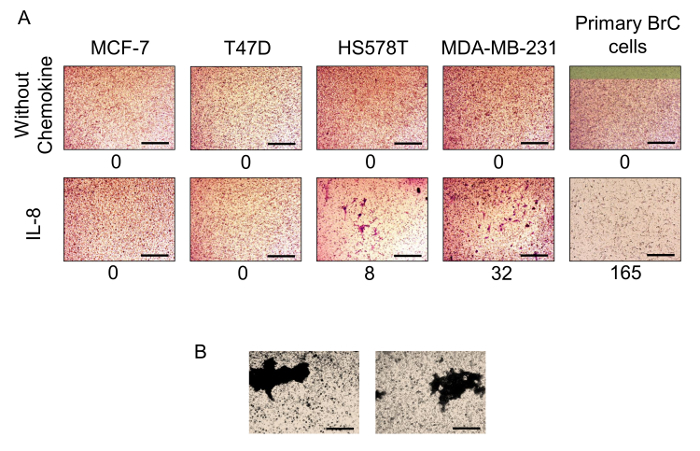
Note: Perform invasion assays of the BrC cells in 24-well flat-bottom culture plates using polycarbonate cell culture inserts with membranes of 8 µm pore size. In our original studies, IL-8 was one of the cytokines enriched in the supernatants of the BrC cell/monocyte 3D co-cultures. Whether this cytokine was participating in the invasion of the BrC cell lines was tested.
Expand BrC cells in 75 cm2 flasks. MCF-7, T47D, HS578T, and MDA-MB-231 as described in step 1.2 (but without ECME) and primary BrC cells as described in step 2.4.
Wash once each polycarbonate 8 µm-pore membrane cell culture insert with 200 µL of medium without sera (the medium used depends on the cell line tested) to hydrate the membrane.
Fill the insert with 50 µL of cold ECME (1:4 dilution ECME:cold medium without FBS), place the inserts in a 24-well flat-bottom culture plate, and allow the ECME to polymerize for 1 h at 37 °C.
Once the ECME has polymerized, add 180 µL of the respective cell media without FBS. Add 800 µL media without FBS through the slits of the insert. Incubate for 30 min at 37 °C to establish an IL-8 gradient. Note: Avoid creating bubbles in the media. Media used is without FBS but supplemented with 100 ng/mL of IL-8 as a chemoattractant, or pure media without FBS, as negative controls.
- Obtain a total of 6 x 105 cells of each cell line as follows:
- Discard supernatants, rinse once with 10 mL of 1x PBS, and add 2 mL of 0.05% trypsin/0.48 mM EDTA for 2 min at 37 °C to detach the cells. Add 4 mL of supplemented medium to stop the trypsin reaction and resuspend vigorously by pipetting.
Take a 10 µL aliquot of the cell suspension to count the cells in a Neubauer chamber. Take a volume of cell suspension containing 6 x 105 cells. Centrifuge at 430 x g for 5 min at RT, discard the supernatant, and rinse cells in 1 mL of 1x PBS. Repeat the rinse once more.
Resuspend the cells in 20 µL of the respective media without FBS and place in the inserts containing 180 µL of the respective media without FBS. Carefully homogenize the cell suspension by pipetting.
Allow the cell invasion to progress for 48 h at 37 °C in a humidified 5% CO2 environment.
After 48 h, remove the insert, discard the supernatant and rinse the insert once very carefully with 200 µL of 1x PBS. With a cotton-tipped applicator remove the excess of PBS. Note: The invasive cells usually are attached to the other side of the insert as in this case where cells are adherent.
Fix the cells by placing the insert in a well of a 24-well flat-bottom culture plate containing 1 mL/well of 4% paraformaldehyde for 15 min at RT. Afterwards, rinse the inserts once with 1x PBS.
Stain the cells by placing the insert in a well of a 24-well flat-bottom culture plate containing 1 mL/well of 1x PBS with 0.2% crystal violet for 45 min at RT. Carefully, with a cotton-tipped applicator remove all the ECME from the top of the membrane, which contains cells that did not migrate, and rinse very carefully with distilled water to avoid eliminating fixed invasive cells.
Count the invading cells by observing the lower side of the insert under the inverted microscope. The mean cell count from 3 random fields (at 100X magnification) is used. In cases where the cells have invaded as clusters and are difficult to count individually, acquire images from 3 random fields (at 100X magnification) with a digital camera. Analyze the images with software for image analysis and use the IOD to quantify cell invasion (Figure 6).
Representative Results
Morphological Analysis of BrC Cells in 3D Cultures:
The morphology of primary BrC cells growing in 3D cultures at low and high densities was studied over 5 days. During the first 48 h, cells adhere to the ECME and maintain a low density. At this time point, it can clearly be appreciated that cells present an elongated spindle-like shape, some with two or more long cytoplasmic projections and without showing apparent cell-cell contacts. After 5 days of culture, cells proliferated while maintaining their initial morphology. Moreover, they formed structures resembling nets without a specific structural organization, and appear piled up with no contact inhibition. A comparison with non-transformed MCF-10A cells was made, which form spherical structures resembling glandular acini. These cells display a characteristic morphology of rounded well-delimited and organized structures in homogenous sizes (of approximately 50 µm). On the contrary, primary BrC cells share a much closer resemblance with aggressive BrC cells MDA-MB-231 (Figure 1).
Analysis of Cell-to-cell Direct Interactions and Collagen Degradation:
We designed a 3D co-culture model to assess the following: cell morphology, collagen degradation, cell migration, and whether there is a direct interaction between BrC cells and monocytes in the co-culture. After 5 days, both MCF-7 and MDA-MB-231 commercial BrC cells form disorganized aggregates in 3D cultures, however, they differ in their morphology. Aggressive cells MDA-MB-231 form aggregates with elongated forms with some cells tending to separate from the rest, while non-aggressive MCF-7 cells form aggregates where cells maintain rounded shapes and they appear to be more densely compacted(Figure 2A, B). DQ collagen IV was incorporated to visualize proteolytic degradation (green), with both BrC cultures exhibiting fluorescent activity. To quantitatively assess proteolytic activity, green fluorescence was measured as IOD of six 50 µm2 fields. A comparison was made between individual 3D cultures of MDA-MB-231 cells and co-cultures with U937 monocytes. Statistically significant increase of proteolysis in the co-culture (Figure 2C) was observed. Also, a confocal microscopy analysis of serial transversal stacks every 10 µm of the MDA-MB-231 and U937 monocytes co-culture was performed (Figure 2D). This analysis revealed that U937 monocytes migrated towards aggressive cells MDA-MB-231 with a few monocytes reaching the layer of BrC cells. However, it was evident that cell-cell direct interaction sites were not necessarily coinciding with areas of higher proteolytic activity: instead, areas of collagen degradation were uniformly spread in the culture, leading to the inference that collagen degradation was the result of indirect communication mediated by secreted factors. Therefore, the 3D co-culture model was changed to place the cells in different compartments separated by a porous membrane to allow cell-cell communication based on secreted factors, and avoid cell labeling and sorting after culture.
Analysis of IL-1β in Supernatants from 3D Cultures:
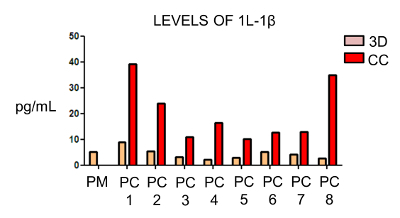
Working with the indirect interaction 3D cultures, the supernatants from individual primary BrC cultures and their 5 days co-cultures with PM were harvested. These supernatants were subjected to the simultaneous analysis of 18 cytokines and 5 MMPs using commercial kits and an instrument specifically designed for this analysis. Of all analytes tested, significantly increased levels of IL-1β and IL-8 were observed in the primary BrC cell-monocyte co-cultures, both crucial inflammatory cytokines that have been previously associated with malignant progression15,18,19,20 (Figure 3; results for IL-1β). This example is another type of analysis that 3D cultures allow to mimic the communication network that shapes the tumor microenvironment.
Characterization of Supernatants and Acini Structure:
On the basis of the 3D experimental system developed by Debnath, Muthuswamy, and Brugge7, where MCF-10A were established as a model of the mechanisms associated with oncogenesis, evaluation of whether the secreted factors from the primary BrC cell supernatants influenced the formation of the MCF-10A 3D acini-like structures, similar to the activity observed after transduction of viral and cellular oncogenes7,21 was performed. MCF-10A cells were cultivated with two different supernatants from two primary BrC cell lines to observe lumen formation (as a measure of resistance to anoikis). At day 14, the spheroids were evaluated by confocal microscopy transversal cuts at 0, 25, 50, 75, and 100% depth, and in both cases, it was found that MCF-10A cells evaded anoikis (measured by counting the number of central (luminal) cells in the acini (Figure 4B, C)). Thus, MCF-10A cell acini that are formed with the conditioned media from BrC cells, lack a well-formed hollow lumen, unlike MCF-10A that are grown with its standard culture medium (as described in step 1.2)(Figure 4A). In the 3D co-culture system, IL-1β was highly secreted; therefore, the MCF-10A cells were also cultivated with human recombinant IL-1β18. Figure 4D shows a confocal microscopy transversal cut at 50% depth of the acini in the presence of IL-1β (lower panels), and reveals that this cytokine also confers resistance to anoikis. Thus, soluble factors secreted by primary BrC cells, such as IL-1β, promote the survival of non-transformed MCF-10A cells that lose ECM interactions, a characteristic of invasive cancers.
3D Co-cultures Promote an Inflammatory Response Associated with Migration of Monocytes and Invasion of Cancer Cells:
We previously demonstrated that primary BrC cells in 3D culture secrete high basal levels of pro-inflammatory chemokines known to attract monocytes14. Thus, we analyzed the migratory capacities of monocytes in response to the chemokines found enriched in the conditioned media of the BrC cells. The migratory capacity of commercial monocytes U937 and THP-1 and fresh PMs was evaluated in response to three different chemokines: GM-CSF, MCP-1, and RANTES. U937 and THP-1 monocytes were capable of migrating in response to GM-CSF and MCP-1, with U937 as the most responsive, while both monocytes had a null response to RANTES. Importantly, PMs showed a high basal migratory activity and the most potent response to MCP-1 (Figure 5). IL-8 was also enriched after the 3D co-culture of primary BrC cells and monocytes. Thus, we analyzed whether IL-8 modifies the capacity of invasion of commercial and primary BrC cell lines. The commercial aggressive BrC cell lines (HS578T and MDA-MB-231) invaded in response to IL-8, while the non-aggressive (MCF-7 and T47D) did not invade. Surprisingly, primary BrC cells were more invasive than commercial aggressive BrC cell lines in response to IL-8 (Figure 6A, right panels). In some cases, cells invade as clusters and therefore IOD analysis was necessary (Figure 6B).
Figure 1: Representative images of BrC cells in 3D cultures. From left to right: control of non-transformed MCF-10A cells forming characteristic glandular acini, with rounded morphology, well-delimited cell contacts, and organized structures of homogenous sizes (approximately 50 µm on average, optical magnification of 200X). Middle panels show primary BrC cells cultured at low density (2 days) and high density (5 days). Cells adhere to the ECME and maintain a low density at early time points. It is evident that cells present an elongated spindle-like shape, some with two or more long cytoplasmic projections, and with no apparent cell-cell adhesion. On the other hand, high-density cells at later points formed structures resembling nets without a specific structural organization. These cells appeared piled up showing no contact inhibition. A control of commercial MDA-MB-231 cells after 5 days in 3D culture is shown; these aggressive BrC cells share a much closer resemblance with primary BrC cultures (optical magnification of 100X). Scale bar represents 50 µm. 800 cells were seeded at the beginning of the experiment. Please click here to view a larger version of this figure.
Figure 2: Analysis of ECM degradation. Non-aggressive MCF-7 cells stained with a yellow fluorochrome (A) and aggressive MDA-MB-231 cells stained with a red fluorochrome (B) cultivated for 5 days in 3D conditions; 0.5% of green fluorescence-labeled collagen IV was incorporated to visualize ECM degradation. In A and B, the upper left pictures represent the levels of collagen degradation, the upper right panels represent the optical pictures, the bottom left represent the BrC cells, and the bottom right panels show the merge of fluorescent and optical images. For A and B, the optical magnification of 200X and scale bar of 100 µm are presented. (C) Representative images of ECM proteolysis (green) in 3D culture of individual MDA-MB-231 cells as control, compared with ECM proteolysis of 3D co-culture of MDA-MB-231 with U937 monocytes. Scale bars represent 10 µm. The IOD (integrated optical density) of six 50 µm2 fields from each condition was measured. The mean and the standard error of the mean (SEM) from the quantifications are presented; two asterisks represent a statistically significant difference with p < 0.01 (Mann-Whitney test). (D) Direct interaction of MDA-MB-231 cells (gray shade mass) with fluorescently stained U937 monocytes (orange); green fluorescence corresponds to proteolysis. Serial slices were generated every 10 µm by confocal microscopy to address the localization and direct interactions between both cell lines. Optical magnification of 200X; scale bars represent 100 µm. Please click here to view a larger version of this figure.
Figure 3: IL-1β concentration is increased in 3D co-cultures. One representative result of IL-1β levels (pg/mL) in supernatants from eight (PC1-8) primary BrC cells, cultured individually in 3D (3D), and compared against their 3D co-cultures with PM (CC). A control of PM in individual 3D culture was included for comparison (PM). Results were extracted from a larger multiplexing analysis, where several cytokines were simultaneously detected in each sample by means of an immunodetection-based technique available as a commercial kit. Please click here to view a larger version of this figure.
Figure 4: Secreted factors from primary BrC cells induce resistance to anoikis in non-transformed MCF-10A cells. MCF-10A cells were grown in 3D conditions with standard culture media (column A and upper panels of D), in which it can be observed that the acini present typical hollow lumens (25-75% depth highlighted with a red square); when MCF-10A cells were grown with two different supernatants from two primary BrC cultures (columns B and C), lumens are not hollow but filled with cells denoting resistance to anoikis. Similarly, when MCF-10A cells were cultured in 3D conditions adding human recombinant IL-1β (20 ng/mL), no hollow lumen can be appreciated at 50% depth confocal microscopy sections of the acini (lower panels of D). The right oval caricature represents the confocal microscopy sections that were made at 0, 25, 50, 75 and 100% depth of the acini. Images show nuclei stained with DAPI (blue) and E-cadherin in green. Optical magnification of 200X, scale bars represent 10 µm. Please click here to view a larger version of this figure.
Figure 5: Migratory capacity of monocytes. Representative images of the migratory capacity of U937, THP-1 and PM in response to GM-CSF, MCP-1 and RANTES. Controls without chemokine are included (first column from left to right). The numbers below the images indicate the number of migratory cells in each condition; U937 and THP-1 monocytes were mainly responsive to GM-CSF and MCP-1, while PM showed a high migratory capacity per se, and were the most responsive cells to MCP-1. Optical magnification of 100X; scale bars represents 100 µm. Please click here to view a larger version of this figure.
Figure 6: Aggressive and primary BrC cells invade in response to IL-8. A. Invasion assays with two non-aggressive (MCF-7 and T47D), two highly-aggressive (HS578T and MDA-MB-231) BrC cell lines and one primary BrC culture in response to IL-8 used as chemoattractant. Upper panels correspond to controls without chemokine showing no invasion, lower panels show the response to IL-8, with highly-aggressive BrC cells and primary BrC cells invading through the membrane of the cell culture insert. B. Example of cells that invade in clusters; in these cases, measurement of invasion is done with an image analysis program to determine invasion in terms of IOD (integrated optical density) per area. The numbers below the images represent the number of invading cells in each condition. Optical magnification of 100X; scale bars represent 100 µm. Please click here to view a larger version of this figure.
Discussion
Epithelial cells grow in a 3D spatial conformation and their interaction with the ECM proteins is pivotal for tissue homeostasis. Many cancer studies have been based on cells grown in monolayers (2D) and although they have been critical to understanding many aspects of tumor formation and progression, monolayers do not recapitulate the characteristics that the ECM imposes on cells, for instance: limiting proliferation, adhesion-dependent cell survival, apical-basolateral polarity, ECM remodeling, cell differentiation, etc. Importantly, not only the interaction between tumor cells and ECM are essential for the progression of cancer, but also the communication established between tumor and non-tumor cells, such as immune cells. The protocols provided here facilitate the understanding of the interactions between immune cells and cancer cells within the environment provided by the ECM, the inflammatory response promoted by these interactions, and how this inflammatory condition create a positive loop fostering chemoattraction of monocytes and more invasion of cancer cells.
A great advantage of using ECME is that it provides a biological scaffold suitable to perform a variety of experimental 3D models22. It is enriched with ECM proteins such as laminin, collagen IV, entactin, heparan sulfate proteoglycan, and growth factors, which in vivo would be interacting with epithelial cells. A disadvantage is that because it comes from lysates of murine sarcomas, the concentration of their components varies between different lots. So, it is often necessary to characterize several lots to obtain more homogeneous data. In the laboratory, each new lot is tested in two ways: 1) by evaluating the correct acini formation of MCF-10A cells, and 2) by assessing the invasiveness of BrC cell lines of well-established invasive capacity. Another option is to work with other scaffolding products, such as Hydrogel, which is a synthetic nanofiber peptide scaffold. The stiffness of the 3D culture can be controlled by adjusting the concentration of Hydrogel23,24. Importantly, this system could be applied to study the interaction between any type of neoplastic cell and hematopoietic or non-hematopoietic cells. It could even be possible to design more complex systems using more than two different cell lineages. One important advantage of the 3D models is that they allow study of the cell morphology and cell organization shaped by ECM interactions, which are altered during oncogenic transformation. Likewise, one can study oncogenic features such as proteolytic degradation, and whether cells initially placed in different layers of the culture signal to each other to promote direct interaction/communication. Moreover, different cell lineages can be labeled with different fluorochromes so that they can be isolated after the experiment to address specific questions to each individual cell type, using, for example RNA and/or protein expression analysis13. Here, IL-1β secretion to the medium in primary BrC cells/PM co-cultures is shown, supporting a possible pivotal role of this cytokine in the indirect communication between tumor cells and monocytes that triggers malignant progression.
Another essential experimental protocol in the laboratory is the analysis of acini formation in 3D cultures of non-transformed MCF-10A cells. This assay was used to test for viral and cellular oncogene activity, and to test how the tumor microenvironment influences feature related to aggressive cancers. For instance, during the normal development of the acini, luminal central cells lose contact with the basal membrane proteins (provided by ECME) triggering mechanisms of cell death, known as anoikis. It was observed that both the supernatants from primary BrC cells or recombinant IL-1β promote MCF-10A cells resistance to anoikis. Because a high concentration of IL-1β is promoted upon BrC cell-monocyte interaction, this observation supports that the tumor stroma is an integral component of tumor progression to highly aggressive stages.
The migration and invasion protocols described here constitute useful 3D complementary assays in which we can support the capability of cells to migrate or invade in response to stimuli derived from the tumor stroma. It was shown that chemokines (GM-CSF and MCP-1) found in elevated concentrations in 3D cultures of primary BrC cells can promote migration of U937, THP-1, and PMs, supporting the ability of aggressive tumor cells to promote a pro-inflammatory microenvironment. Furthermore, IL-8 that is also found in increased concentration in the supernatants from co-cultures of BrC cells/monocytes, promoted increased invasion of both commercial aggressive BrC cells (HS578T and MDA-MB-231) and primary BrC cells.
Regarding the condition of cells, it is important to work with primary BrC cells before they reach ten passages to take advantage of their proliferation peak and avoid potential post-isolation genetic and epigenetic changes resulting from in vitro culture. Another essential aspect when working with primary cells is to confirm their identity after isolation. We used a panel of biomarkers to determine their epithelial lineage identity, which includes PanCK, Muc-1, and EpCAM (specified in step 2.5). Similarly, it is preferable to use fresh PMs, of which each batch is tested assuring that they are at the same stage of differentiation. Each new batch used presented the phenotype CD34neg CD11bpos CD14pos CD64pos CD68neg CD16neg, which corresponds to non-activated immature monocytes. Importantly, we do not mix monocytes from different donors, as mixing results in monocyte activation. Instead, we independently tested BrC co-cultures using monocytes from at least two different donors.
3D culture systems still have the limitation that only a few elements of tumor biology can be reliably modeled. A new alternative is the more complex 3D models of organoids, which are based on the expansion and differentiation of stem cells in vitro. Organoids also develop highly organized epithelial structures. Examples include the gastric 25,26, liver 27, and kidney organoids28. However, organoid models for every human organ remain to be achieved; furthermore, they require much more complex and expensive experimental conditions.
In conclusion, here we describe in detail a workflow of assays: starting from isolation of tumor cells from BrC patients, testing their inflammatory capacity in an ECME-based 3D culture model, testing how that inflammatory microenvironment serves them to recruit additional inflammatory cells, such as monocytes, to finally analyzing the communication between both types of cells, and how that communication further fosters an inflammatory microenvironment that promotes progression to more aggressive cancer stages. Additionally, we describe the morphology and phenotype of primary BrC cells. In future studies, it will be interesting to test other inflammatory cells or even test the communication between BrC cells and multiple cells often found in the tumor stroma. These multiple-cell 3D cultures will provide a larger picture of what happens biologically in vivo during cancer progression. Comparisons between in vitro data and the patient clinical outcome will identify potential mechanisms that have a higher impact on cancer aggressiveness.
Disclosures
The authors declare that they do not have any conflict of interest.
Acknowledgments
This work was supported by CONACyT FONSEC SSA/IMSS/ISSSTE Project No. 233061 to Ezequiel M. Fuentes-Pananá and by Fondo de Apoyo a la Investigación, Hospital Infantil de México Federico Gómez (project number HIM-2014-053). Espinoza-Sánchez NA is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship 231663 from CONACYT. E-S NA, also acknowledge the financial support provided by the Mexican Institute of Social Security (IMSS).
References
- Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore) 2016;95(1):2–7. doi: 10.1097/MD.0000000000004764. Suppl 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–773. doi: 10.7150/jca.17648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017. [DOI] [PMC free article] [PubMed]
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A. 1992;89(19):9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Debnath J, et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111(1):29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3(9):785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameni M, Dosescu J, Moin K, Sloane BF. Functional imaging of proteolysis: stromal and inflammatory cells increase tumor proteolysis. Mol Imaging. 2003;2(3):159–175. doi: 10.1162/15353500200303136. [DOI] [PubMed] [Google Scholar]
- Sameni M, et al. MAME models for 4D live-cell imaging of tumor: microenvironment interactions that impact malignant progression. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Jedeszko C, Sameni M, Olive MB, Moin K, Sloane BF. Visualizing protease activity in living cells: from two dimensions to four dimensions. Curr Protoc Cell Biol. 2008;4:20. doi: 10.1002/0471143030.cb0420s39. Chapter 4 Unit 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimal-Ramirez GK, et al. MMP1, MMP9, and COX2 expressions in promonocytes are induced by breast cancer cells and correlate with collagen degradation, transformation-like morphological changes in MCF-10A acini, and tumor aggressiveness. Biomed Res Int. 2013;2013:279505. doi: 10.1155/2013/279505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza-Sanchez NA, Chimal-Ramirez GK, Mantilla A, Fuentes-Panana EM. IL-1beta, IL-8, and Matrix Metalloproteinases-1, -2, and -10 Are Enriched upon Monocyte-Breast Cancer Cell Cocultivation in a Matrigel-Based Three-Dimensional System. Front Immunol. 2017;8:205. doi: 10.3389/fimmu.2017.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer. 2012;11:87. doi: 10.1186/1476-4598-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher S, Winston SE, Fuller SA, Hurrell JG. Immunoblotting and immunodetection. Curr Protoc Mol Biol. 2008;10:18. doi: 10.1002/0471142727.mb1008s83. Chapter 10 Unit 10. [DOI] [PubMed] [Google Scholar]
- Troeberg L, Nagase H. Zymography of metalloproteinases. Curr Protoc Protein Sci. 2004;21:15. doi: 10.1002/0471140864.ps2115s33. Chapter 21 Unit 21. [DOI] [PubMed] [Google Scholar]
- Arevalo-Romero H, Meza I, Vallejo-Flores G, Fuentes-Panana EM. Helicobacter pylori CagA and IL-1beta Promote the Epithelial-to-Mesenchymal Transition in a Nontransformed Epithelial Cell Model. Gastroenterol Res Pract. 2016;2016:4969163. doi: 10.1155/2016/4969163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Leon RP, Hall MK, Schwertfeger KL. Interleukin-1beta and fibroblast growth factor receptor 1 cooperate to induce cyclooxygenase-2 during early mammary tumourigenesis. Breast Cancer Res. 2009;11(2):21. doi: 10.1186/bcr2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, et al. Epidermal growth factor (EGF) and interleukin (IL)-1beta synergistically promote ERK1/2-mediated invasive breast ductal cancer cell migration and invasion. Mol Cancer. 2012;11:79. doi: 10.1186/1476-4598-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande SM, Bannish G, Fuentes-Panana EM, Katz E, Monroe JG. Tonic B-cell and viral ITAM signaling: context is everything. Immunol Rev. 2007;218:214–234. doi: 10.1111/j.1600-065X.2007.00535.x. [DOI] [PubMed] [Google Scholar]
- Benton G, Arnaoutova I, George J, Kleinman HK, Koblinski J. Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv Drug Deliv Rev. 2014;79-80:3–18. doi: 10.1016/j.addr.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Worthington P, Pochan DJ, Langhans SA. Peptide Hydrogels - Versatile Matrices for 3D Cell Culture in Cancer Medicine. Front Oncol. 2015;5:92. doi: 10.3389/fonc.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng. 2009;103(4):655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaermann P, et al. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut. 2016;65(2):202–213. doi: 10.1136/gutjnl-2014-307949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompaiah M, Bartfeld S. Gastric Organoids: An Emerging Model System to Study Helicobacter pylori Pathogenesis. Curr Top Microbiol Immunol. 2017;400:149–168. doi: 10.1007/978-3-319-50520-6_7. [DOI] [PubMed] [Google Scholar]
- Takebe T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499(7459):481–484. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- Takasato M, et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16(1):118–126. doi: 10.1038/ncb2894. [DOI] [PubMed] [Google Scholar]