

Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR) is an adaptive immunity system in prokaryotes that has been repurposed by scientists to generate RNA-guided nucleases, such as CRISPR-associated (Cas) 9 for site-specific eukaryotic genome editing. Genome engineering by Cas9 is used to efficiently, easily and robustly modify endogenous genes in many biomedically-relevant mammalian cell lines and organisms. Here we show an example of how to utilize the CRISPR/Cas9 methodology to understand the biological function of specific genetic mutations. We model calreticulin (CALR) mutations in murine interleukin-3 (mIL-3) dependent pro-B (Ba/F3) cells by delivery of single guide RNAs (sgRNAs) targeting the endogenous Calr locus in the specific region where insertion and/or deletion (indel) CALR mutations occur in patients with myeloproliferative neoplasms (MPN), a type of blood cancer. The sgRNAs create double strand breaks (DSBs) in the targeted region that are repaired by non-homologous end joining (NHEJ) to give indels of various sizes. We then employ the standard Ba/F3 cellular transformation assay to understand the effect of physiological level expression of Calr mutations on hematopoietic cellular transformation. This approach can be applied to other genes to study their biological function in various mammalian cell lines.
Keywords: Cancer Research, Issue 131, CRISPR, Cas9, gene editing, sgRNA, calreticulin, sequencing, reporter
Introduction
CRISPR/Cas9 technology has recently revolutionized targeted genome editing in living cells and organisms. It has become an extremely powerful tool for biomedical research and is currently being utilized as a potential avenue for therapy of genetic diseases1. The basis for all genome editing tools relies on the creation of a nuclease-induced DNA double stranded break (DSB) at the genomic locus to be modified. The DSBs can be repaired by non-homologous end-joining (NHEJ) or homology-directed repair (HDR)2,3. The advantage of the Cas9 nuclease over other genome engineering nucleases, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) is its dependence on RNA for targeting the nuclease to a desired DNA sequence, compared to the protein-DNA interactions found in ZFNs and TALENs2,3.
After the discovery of the CRISPR/Cas9 nuclease pathway as an adaptive immune system in prokaryotic cells4,5, much effort has gone into adapting the pathway for use in mammalian cell lines and model organisms2,3. As a tool for gene editing, the CRISPR/Cas9 pathway utilizes two main components: the Streptococcus pyogenes (Sp) derived Cas9 nuclease and sgRNAs targeting the gene of interest2,3. The sgRNA consists of 20 nucleotides that direct Cas9 to a specific site on the genome through RNA-DNA base pair complementarity2,3. The target site of the sgRNA must lie adjacent to a protospacer adjacent motif (PAM) site in the form of 5' NGG, which is recognized by the SpCas9 nuclease. With these tools, Cas9 can be directed to any DNA sequence by designing sgRNAs that target the region of interest. In addition to Sp derived Cas9, there are additional variants for Cas9 with different features depending on the specific application. For example, there are Cas9 variants with higher specificity for on-target editing or single-strand cleavage capacity for DNA nicking6,7. Moreover, catalytically inactive Cas9 has recently been developed for transcriptional regulation8. Scientists have now used the CRISPR/Cas9 system for a variety of applications, such as gene knockin and knockout to study the biological functions of genes9, loss-of-function and gain-of-function library screens10 and genetic engineering of model organisms11.
In this protocol, we combine the CRISPR/Cas9 methodology with the Ba/F3 cellular transformation assay to understand the biological function of CALR mutations. Ba/F3 cells are a murine IL-3 dependent hematopoietic cell line that can be rendered IL-3 independent upon expression of certain oncogenes such as BCR-ABL12. In order to understand whether mutant calreticulin can transform Ba/F3 cells to cytokine independent growth, we targeted exon 9 of the endogenous Calr locus using CRISPR/Cas9 to introduce indel mutations and then withdrew IL-3 from the cells to apply a positive selection pressure, with the goal of recapitulating gain-of-function CALR mutations found in MPN patients. The protocol includes the design, cloning and delivery of sgRNAs, the development of stable Cas9 expressing cells and screening for CRISPR on-target gene editing. This protocol can be applied to different genes and various cytokine-dependent cell lines of interest and is especially valuable in modelling and studying the biological function of genes involved in cancer.
Protocol
1. sgRNA Design Using Online Tools13
- Design sgRNAs targeting the gene of interest using freely available online tools.
- Copy and paste the NCBI reference sequence of the gene of interest into the Broad Institute sgRNA designer web tool: (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). NOTE: This tool identifies sgRNA sequences with cleavage sites within exons and those that span the intron/exon boundary but still cleave within the exon.
- Download and open the text output file in excel.
- Focus on the columns populated with the sgRNA sequences and the sgRNA context sequences. Note that the sgRNA sequences do not contain the protospacer adjacent motif (PAM) but the context sequences do.
- Note that the on-target efficacy score lists the predicted cleavage efficiency score on a scale of 0 to 1 where a score of 1 denotes a higher cleavage efficiency.
- Use the 'sort' function of excel to either order the targets by the efficacy score or by location within the gene of the target through the 'Target Cut %' column. NOTE: Sorting by location within the gene is useful for identifying sgRNAs that target a specific domain or exon of interest.
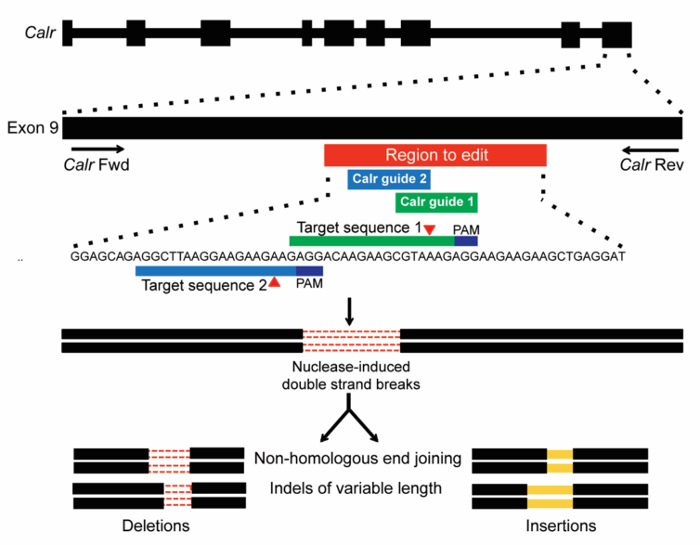
- Select 3-6 sgRNAs that target the area of interest with high (>0.6) on-target efficacy scores. It can be useful to know what kinds of mutations may be responsible for the phenotype that is desired (Please see Figure 1 explaining the targeting strategy used for calreticulin). NOTE: sgRNAs below the suggested 0.6 efficacy score threshold should be considered when there is a lack of other good candidates.
- Use the MIT gRNA analysis web tool to screen for potential off-target effects (http://crispr.mit.edu/). For each sgRNA selected, run the target sequence (including the PAM). NOTE: The Broad Institute sgRNA designer web tool could also be used to screen for off-target effects (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design).
- Note that the MIT sgRNA analysis tool scores each sgRNA on a scale of 1-100 where a score of 100 denotes higher specificity. A score greater than 70 is ideal and represents a sgRNA with minimum off-target effects. This website also generates a list of off-target hits with their location within the genome and how many mismatches they each have with the candidate guide. NOTE: Off-target hits with four mismatches or greater are considered safe14. Off-target hits with less than four mismatches may be problematic if they fall within exons14.
- Choose 2-3 sgRNAs which target distinct locations within the region of interest for the target gene, and which have the highest paired cleavage efficiency and off-target scores (See Table 1 for sgRNA sequences targeting exon 9 of Calr). NOTE: Depending on the experimental goals, greater emphasis may be placed upon the different values obtained from the mentioned tools.
2. Cloning sgRNA Oligos13
- Generate the forward oligo
- Create a list of sgRNA sequences without the PAM site.
- Add a G to the 5' end of the sequence if the 5'end of the sgRNA sequence is not a G. NOTE: This G is necessary for maximizing U6-driven sgRNA transcription level. Addition of a G is necessary for m1 sgRNA (Table 1).
- Add the sequence "CACC" to the 5' end of the G-optimized guide sequence (no PAM) (Table 2) in order to generate the overhangs required for ligation of annealed oligos into BsmBI digested lentiGuide vector ( see step 3).
- Generate the reverse oligo
- Reverse complement the G-optimized guide sequence (no PAM).
- Add the sequence "AAAC" to the 5' end of the reverse complemented G-optimized guide sequence (Table 2).
- Order the designed oligos from a primer synthesis company.
- Anneal and phosphorylate oligos
- Add 1 µL of forward oligo (100 µM), 1 µL of reverse oligo (100 µM), 1 µL of 10x T4 ligation buffer, 0.5 µL of T4 polynucleotide kinase (PNK) and 6.5 µL of H2O to a PCR tube (Total = 10 µL).
- Run the following program in the thermocycler: 37 ˚C for 30 min, 95 ˚C for 5 min , ramp from 95 to 25 at 0.1 °C/s.
- Dilute the reaction product at 1:250 in H2O.
3. Digestion of lentiGuide-Puro Vector13
- Digest lentiGuide-Puro vector
- Assemble a 50 µL digestion reaction with the following components: 5 µg of circular plasmid, 2 µL (30 units) of BsmBI restriction enzyme, 5 µL of restriction enzyme buffer and up to 50 µL of H2O.
- Run the reaction for 2 h at 55 °C. After the first hour, add 1 µL more of BsmBI restriction enzyme.
- Dephosphorylate the cut backbone
- Add 7 µL of phosphatase reaction buffer and 2 µL of phosphatase enzyme to the digested vector.
- Incubate for 30 min at 37 °C.
PCR purify the cut and dephosphorylated backbone using a commercial kit. NOTE: Substitute the provided pink colored columns with the miniprep kit blue colored columns since these can better accommodate large plasmid sizes. The yield can be increased by heating the elution buffer in a 90 ˚C heat block prior to elution and by eluting the DNA from the column twice at the end (run the eluted DNA from the first elution back through a second time).
4. Ligation ofAnnealed Oligos into Digested Backbone13
Add 50 ng of digested backbone, 1 µL of annealed oligo dilution, 1 µL of 10x T4 ligation buffer, 1 µL of T4 ligase and up to 10 µL of H2O to a PCR tube. Incubate for 1 h at room temperature
Transform Stbl3 cells with 2 µL of the ligation reaction. Plate onto a lysogeny broth (LB) agar plate with 100 µg/mL ampicillin and incubate overnight at 37 °C.
Pick 3 colonies and inoculate into a mini-prep culture.
- Validate correct oligo insertion
- Perform a miniprep for each culture and sequence through the oligo insertion site starting from a primer in the U6 promoter using a commercial kit.
- Perform a midi-prep or a maxi-prep of the sequence-verified culture.
5. Generation of Cell lLines Stably Expressing SpCas9
NOTE: This protocol involves the delivery of pLX_TRC311-Cas9 plasmid by lentiviral infection. This protocol is described in detail for murine interleukin-3 (mIL-3) dependent pro-B (Ba/F3) cells, a suspension cell line and could be adapted to other cell types using the preferred culture conditions for each cell type. The culture medium for Ba/F3 cells consists of RPMI supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin/ L-glutamine and 10 ng/mL of murine interleukin 3.
- Transfect HEK-293T cells
- Seed 3 x 106 HEK-293T cells per 10 cm tissue culture dish. Keep seeded cells overnight before transfection. NOTE: HEK-293T cells are an adherent cell line and are routinely used for virus production. The cells are maintained in DMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin/L-glutamine. Cells should be 80% confluent at time of transfection.
- Pre-warm reduced serum media (Opti-MEM) and growth medium.
- Add 500 µL of reduced serum media, 7 µg of pLX_TRC311-Cas9 construct, 4 µg of pCMV-VSV-G lentiviral packaging plasmid and 4 µg of psPAX2 lentiviral packaging plasmid into a tube NOTE: Total DNA per 10 cm dish of 293T cells is 15 µg.
- Mix the DNA well and add 45 µL of transfection reagent. Mix gently and let it stand for 20 min. NOTE: It is important to use reduced serum media because serum will inhibit the complex formation between the transfection reagent and plasmid DNA.
- Aspirate the old medium in the 293T plate and add 5 mL of new growth medium.
- Add the DNA and transfection reagent mix in a drop wise manner to the 293T plate. Swirl gently and incubate at 37 °C and 5% CO2 for 24 h.
- Harvest viral supernatants at 24 and 48 h post transfection. Pass through a 0.22 µm filter and aliquot into a cryovial tube (1.6 mL/tube and 1.5 mL will be used for infection).
- Store viral supernatant at -80 °C. NOTE: lentiviral supernatant can be used within 6 months. If possible, spinfection transductions should be performed with fresh virus).
- Lentiviral infection of Ba/F3 cells
- Thaw the viral supernatant on ice, if frozen.
- Centrifuge cells at 300 x g for 4 min.
- Aspirate supernatant and resuspend the cells at a concentration of 3 x 106 cells/mL.
- Add 500 µL of resuspended cells, 1.5 mL of viral supernatant, 4 µL of polybrene (stock: 2 mg/mL) and 10 ng/mL of m-IL3 in a 6-well tissue culture plate. Make sure to include an uninfected well with 1.5 mL of media instead of viral supernatant as a control. NOTE: The amount of viral supernatant added should correspond to a multiplicity of infection (MOI) <1.
- Centrifuge the plate at 440 x g for 120 min at 37 °C.
- Take the plate out of the centrifuge and place in 37 °C and 5% CO2 incubator and allow to grow overnight.
- Spin down the cells after 24 h, and resuspend with 5 mL of fresh warm media in a 6-well plate.
- Selection of Cas9 infected cells
- Spin down the cells and resuspend with fresh warm media supplemented with 5 µg/mL of blasticidin, 48 h post spinfection.
- Select the cells for 9 days with blasticidin or until uninfected (negative) control cells are dead.
- Once the selection is complete, transfer the resistant cells to medium with lower concentration of blasticidin.
6. Reporter Assay for Cas9 Activity15
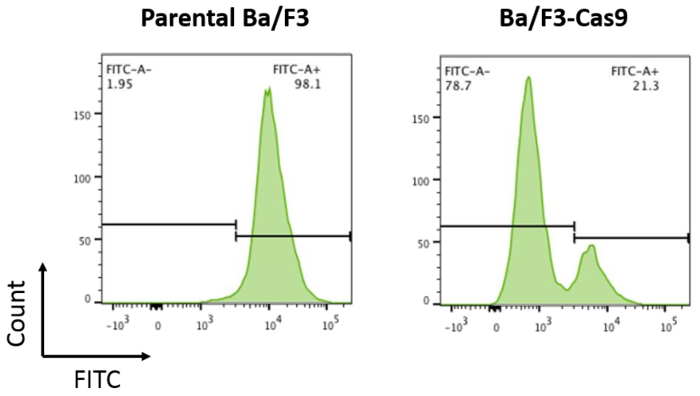
Transduce parental cells and cells stably expressing Cas9 with pXPR-011 by lentiviral infection (see steps 5.1-5.2). NOTE: This vector contains both GFP and a guide targeting GFP. Cells containing active Cas9 will result in a reduction of GFP (Figure 2).
Select the cells for 3 days with 2 µg/mL of puromycin, 48 h post spinfection .
Analyze the samples for GFP expression by flow cytometry. NOTE: A GFP reduction of 50% or more represents optimal Cas9 activity. A higher concentration of blasticidin selection could be used to increase Cas9 activity if less than a 50% reduction is observed. Not all cell lines can tolerate blasticidin selection. In this case, use of Cas9 vectors with other selection cassettes may be necessary. In addition, not all cell lines can tolerate stable expression of Cas9. In this case, transient transfection of Cas9 may be used.
7. Spinfection of sgRNAs into Cas9 Expressing Cells
Follow steps 5.1-5.2 for lentiviral infection of sgRNAs into cells of interest. NOTE: In step 5.1.3, replace pLX_TRC311-Cas9 construct with the lentiGuide-Puro construct validated from Section 4.
48 h post spinfection, select the cells for 3 days with 2 µg/mL of puromycin.
Transfer the resistant cells to medium with lower concentration of antibiotics and continue selection for another 4 days to allow for sufficient editing.
8. Ba/F3 Cellular Transformation and Positive Selection Using m-IL3 Withdrawal
NOTE: This assay is described for mIL-3 dependent Ba/F3 cells but could be applied to any cytokine dependent cell line.
Spin down exponentially growing Ba/F3 cells ectopically expressing Cas9 and the sgRNA of interest.
Aspirate supernatant and wash the cells with 5 mL of Phosphate Buffered Saline (PBS).
Spin down the cells and repeat the wash step in 8.2 four times (this step ensures that the media is free of IL-3).
Aspirate the PBS and resuspend the cells in 5 mL of fresh media without IL-3.
Count the cells using a hemocytometer or a cell viability analyzer.
Seed the cells in triplicate in a 6-well tissue culture plate at a concentration of 1 x 105 cells/mL in a total volume of 2 mL of fresh media without IL-3.
Monitor and count the cells every 2 days for a total of 8 days. NOTE: Ba/F3 cells lacking IL-3 typically die 2 days post IL-3 starvation. It is important to include a negative control in the assay (typically a non-targeting guide is used as a control).
9. Screening for CRISPR On-target Editing
Design CRISPR screening primers upstream and downstream of the sgRNA cleavage site NOTE: use primers at least 100 bp from the predicted cleavage site to ensure detection would not be impacted by a large insertion and/or deletion (indel) at the sgRNA target site.
Isolate genomic DNA (gDNA) from transformed cells (at the end of the growth curve) and from cells pre-cytokine withdrawal. NOTE: Always include cells overexpressing the non-targeting guide as a control for gene editing. Isolating gDNA from cells before and after transformation will identify clones that were positively selected for and have a proliferative advantage.
Assemble a 50 µL PCR with the following components: 25 µL of 2x PCR mix, 1 µL of forward primer (10 µM), 1 µL of reverse primer (10 µM), 50-100 ng of gDNA, and H2O up to 50 µL. NOTE: Numerous high-fidelity polymerases may be used in step 9.3.
Run samples in a thermocycler using the following parameters: 95°C for 1 min, 30 cycles of (94 °C for 1 min, 52 °C for 30 s, 72 °C for 30 s), and 72 °C for 10 min. This PCR is optimized for the Calr primers listed in Table 3. Optimize PCR conditions for the primer pair designed in step 9.1 based on testing it on gDNA.
Run 5 µL of the samples on a 2% agarose gel at 10 V/cm using 1x Tris-acetate-EDTA (TAE) buffer. Examine samples for the presence/ absence of an amplified band corresponding in size to the gene of interest.
Use the rest of the PCR reaction (45 µL) to perform a PCR purification.
Clone the amplicons with a PCR cloning kit into a plasmid vector. For example, pGEM T-easy vectors are typically used.
Transform the plasmid into Stbl3 cells or other compatible cells and plate onto LB agar plates with the relevant antibiotic. Select 10-20 colonies, mini-prep each one, and subject each clone to Sanger sequencing to characterize the indels created from CRISPR/Cas9 editing. NOTE: If desired, single cell fluorescence activated cell sorting (FACS) could be performed on the bulk edited population to isolate a clone with a specific indel. Moreover, next generation sequencing (NGS) methods can be used to more robustly quantify on-target editing using deep sequencing of PCR amplicons spanning the sgRNA target region. For example, Tracking of Indels by Decomposition or TIDE can be used instead of subcloning to precisely determine the spectrum and frequency of targeted mutations generated in a pool of cells (https://tide.nki.nl/#about)16.
Representative Results
Using the method outlined here, the goal of this experiment is to study the functional effects of introducing indel mutations to the endogenous Calr locus on hematopoietic cell transformation. The CRISPR/Cas9 system is used as a tool to create endogenous Calr mutations in Ba/F3 cells. Two sgRNAs were chosen to target exon 9 of Calr (Figure 1), in the region where insertions and/or deletion (indel) mutations typically occur in CALR-mutant MPN patients17,18. The first sgRNA (m1) was chosen based on its high cleavage efficiency and favorable off-target scores (Table 1). The second sgRNA (m2) was chosen primarily for its location within exon 9 and for lack of additional sgRNAs in the region to edit with high cleavage efficiency and favorable off-target scores (Table 1). Two distinct sgRNAs (m1 or m2) were used in separate infections to ensure that the observed effects were due to on-target gene editing. Off-target effects are unlikely to be shared by multiple independent sgRNAs. The non-targeting control (scramble) was also used as a negative control. Recruitment of the Cas9 endonuclease to Calr exon 9 is predicted to create DSBs at this locus. The DSBs would then be repaired by NHEJ, which can generate indels of variable sizes (Figure 1).
To develop the in vitro CRISPR/Cas9 system in Ba/F3 cells, cells stably expressing the Cas9 protein were made by lentiviral-mediated transduction as per the protocol described above. Stable Cas9 expression results in robust Cas9 activity. Cas9 activity in Ba/F3-Cas9 cells was measured by pXPR-011, a reporter construct that contains both GFP and a guide targeting GFP15. Cells containing active Cas9 will result in a reduction of GFP15. GFP was measured in the cells using flow cytometry. Ba/F3-Cas9 cells displayed a reduction of approximately 76% in GFP, corresponding to robust Cas9 activity (Figure 2).
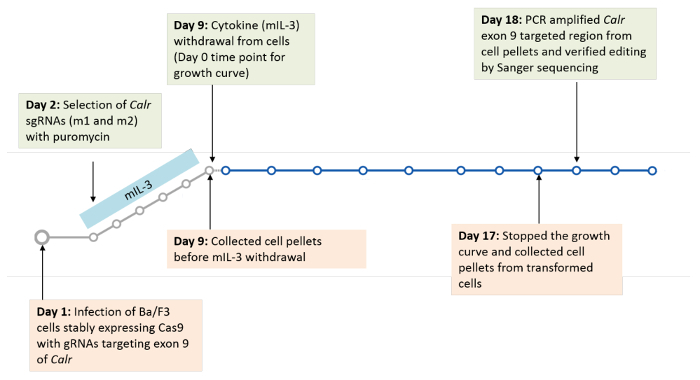
Type 1 cytokine receptors, such as the thrombopoietin receptor (MPL), the erythropoietin receptor (EPOR) and the granulocyte colony-stimulating factor receptor (G-CSFR) were each individually, stably expressed in Ba/F3-Cas9 cells to determine their cooperativity with mutant calreticulin in inducing transformation of Ba/F3 cells. Transduction of the sgRNA constructs (m1, m2 or a scramble (non-targeting guide)) was then carried out in each of the Ba/F3 cell lines stably expressing Cas9 and the receptor of interest. Cells were then selected for 7 days with puromycin to allow for sufficient time for CRISPR/Cas9 gene editing. A positive selection pressure was then applied by starving the cells of cytokine (mIL-3). The goal of this starvation pressure is to identify if indels in Calr exon 9, similar to those observed in MPN patients, are selected for, resulting in cytokine-independent growth and transformation of Ba/F3 cells. The growth curve was carried out for a total of 8 days and the cells were counted every 2 days to measure their transformation (Figure 3)19. Cell pellets for genomic DNA extraction were collected before the start of the growth curve and at the end of the growth curve to check for on-target editing and to monitor the indels that expanded post cytokine starvation (Figure 3)19.
Cytokine independent growth was observed in Calr-targeted Ba/F3-MPL-Cas9 cells (Figure 4B), but not Calr-targeted parental Ba/F3-Cas9 cells (Figure 4A), or Ba/F3-Cas9 cells ectopically expressing EPOR (Figure 4C) or G-CSFR (Figure 4D)19. To confirm that mIL-3 independent growth in Calr-targeted Ba/F3-MPL-Cas9 cells was a result of on-target gene editing, cells were harvested 8 days post mIL-3 withdrawal and genomic DNA was extracted19. A 422 bp region spanning the target site was amplified using the primers listed in Table 3. Sub-cloning of PCR amplicons was performed and 30 individual clones were sent for Sanger sequencing. For m1 or m2 Calr-targeted Ba/F3-MPL-Cas9 cells, all sub-clones contained indels of varying sizes at the intended cut site (Figure 5)19. 11 out of 13 m1 sub-clones were found to contain indels that led to +1 bp frameshift mutations (Figure 5), similar to those found in patients. For m2 Calr-targeted cells, 10 out of 17 sub-clones were found to contain indels that led to +1 bp frameshifts (Figure 5)19. These data confirm that CRISPR/Cas9- mediated introduction of +1bp frameshift mutations to the endogenous Calr exon 9 locus is sufficient to confer oncogenic activity to Calr19. The sequencing data in Figure 5 also suggests that the introduction of heterozygous +1bp frameshift mutations to the endogenous Calr locus is sufficient to transform Ba/F3-MPL cells, consistent with the observation that CALR mutations are typically heterozygous in MPN patients17,18. Since NHEJ repair results in high heterogeneity between the indels, single cell sorting to isolate a clone of interest could be performed. This would allow the researcher to study the effects of specific mutations created by CRISPR/Cas9.
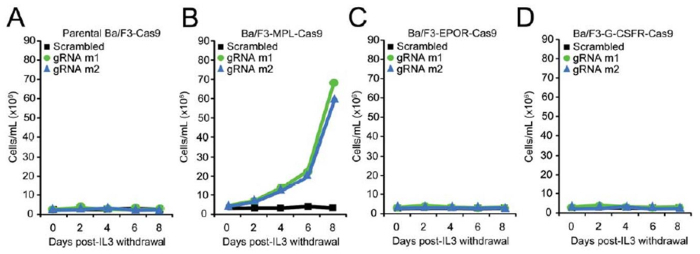

Figure 1: Schema of CRISPR/Cas9 gene-editing targeting of exon 9 of Calr. Two sgRNAs were designed to target a site within the mouse Calr exon 9 region corresponding to that targeted by the +1bp frameshift mutations in human MPN (red bar). Target sequences with PAMs (dark blue) are shown, and expected sites of cleavage by Cas9 are indicated by red triangles. DSBs generated by CRISPR/Cas9 are then repaired by NHEJ, which is expected to generate indels of variable length19. Please click here to view a larger version of this figure.
Figure 2: Cas9 activity assay. Parental Ba/F3 cells and Ba/F3 cells overexpressing Cas9 were transduced with pXPR-011 to measure Cas9 activity. A reduction in GFP correlates with increased Cas9 activity. Parental Ba/F3 cells are ~100% FITC+ compared to Ba/F3-Cas9 cells where the FITC is reduced by ~76%. Please click here to view a larger version of this figure.
Figure 3: Timeline for infection and selection of sgRNAs in Ba/F3 cells. Please click here to view a larger version of this figure.
Figure 4: Introduction of +1 bp frameshift mutations into the endogenous Calr locus is sufficient to confer oncogenic activity to Calr. (A-D) Growth curves in parental Ba/F3-Cas9 cells (A) Ba/F3-MPL-Cas9 cells (B), Ba/F3-EPOR-Cas9 cells (C), and Ba/F3-G-CSFR-Cas9 cells (D) demonstrates IL-3 independent growth in Calr-targeted Ba/F3-MPL-Cas9 cells only19. Please click here to view a larger version of this figure.
Figure 5: Sanger sequencing verification. Confirmation of on-target editing of endogenous Calr (exon 9) in Ba/F3-MPL cells. +1 bp frameshift mutations are indicated in black19. Please click here to view a larger version of this figure.
| Target gene | sgRNA ID | Target sequence (5' to 3') | Strand | Cleavage Efficiency | Off-target score |
| Calr | m1 | AGAGGACAAGAAGCGTAAAGAGG | + | 0.7 | 70 |
| Calr | m2 | GAGGCTTAAGGAAGAAGAAGAGG | + | 0.3 | 28 |
Table 1: 20-mer protospacer sequences including the PAM site (in bold) for two sgRNAs targeting exon 9 of Calr19.
| Primer ID | Sequence (5' to 3') |
| m1-F | CACCGAGAGGACAAGAAGCGTAAAG |
| m1-R | AAACCTTTACGCTTCTTGTCCTCTC |
| m2-F | CACCGAGGCTTAAGGAAGAAGAAG |
| m2-R | AAACCTTCTTCTTCCTTAACCTC |
Table 2: Protospacer sequences and their reverse complements with "CACC" and "AAAC" added for cloning into pLenti-Guide vector using BsmBI restriction enzyme19.
| Primer ID | Sequence (5' to 3') |
| Calr_Fwd | ACCACCTGTCTTTCCGTTCT |
| Calr_Rev | GGCCTCTACAGCTCATCCTT |
Table 3: CRISPR screening PCR primers upstream and downstream of the sgRNA cleavage site.
Discussion
Here we demonstrate the use of CRISPR/Cas9 gene editing to study the biological function of CALR mutations in hematopoietic cells. The success of this protocol is highly dependent on multiple factors. First, it is important to know what kinds of mutations may be responsible for the phenotype that is desired. In this protocol, the readout is the transformation of Ba/F3 cells to mIL-3 independence and the types of mutations are indels in exon 9 of CALR. However, if the desired mutation is a single base pair substitution, then HDR-mediated repair is the preferred method because it can introduce precise point mutations or insertions from a single-strand or double-stranded DNA donor template2,3. If it is desired to knockout the gene, then the creation of large-scale genomic deletions targeting early coding exons is preferred20.
Second, the cell line must be cytokine-dependent in order to allow for the positive selection pressure post cytokine withdrawal. It is important to note that not all cell lines can tolerate stable expression of Cas9 (see step 6.3). This could be due to the blasticidin selection, which is not tolerated by all cell lines. In this case, choice of other Cas9 constructs with different selection cassettes may be necessary. A caveat of using cytokine-dependent cell lines, such as Ba/F3 cells is that they are susceptible to spontaneous cytokine independent growth, sometimes as a result of the acquisition of mutations in the ectopically expressed gene of interest following cytokine withdrawal21. Accordingly, the use of appropriate controls is required in order to be confident that the observed cellular transformation is due to on-target CRISPR gene editing. In this protocol, we did not observe cellular transformation in Ba/F3-MPL cells transduced with a non-targeting sgRNA control or in Ba/F3-EPOR and Ba/F3-GCSFR cells transduced with Calr-targeted sgRNAs. This provides further confidence that cellular transformation in Ba/F3-MPL cells is a result of on-target editing of the endogenous Calr locus.
In this protocol, DNA repair by NHEJ introduces indels of variable sizes as seen in Figure 5. Analysis by Sanger sequencing was performed on a sub-sample of the bulk population of edited cells in order to look for the expansion of indels that lead to +1 bp frameshift mutations post cytokine withdrawal. Next generation sequencing mentioned in step 9.9 is a more robust way to quantify on-target editing for bulk cell populations. If analysis on a specific clone is desired, then single cell sorting of the bulk population is necessary. Single cell sorting is advantageous in understanding the biological effect resulting from a specific type of mutation and is useful when the goal is to understand the differences between two different types of mutations.
One concern with the CRISPR/Cas9 system is off-target effects, which are cleavage events outside the targeted region. To assess the specificity of the CRISPR/Cas9 system and to make sure that the transformed populations do not contain any off-target effects leading to the observed transformation, we would have to examine whether genome editing occurred outside the region of interest. This could be done by looking for evidence of Cas9/NHEJ-driven genome editing at potential off-target genomic loci with significant sequence similarity to the intended target. For this, next generation sequencing could also be used to quantitatively assess the frequency of off-target effects at specified locations along the genome. To reduce the potential for off-target effects, different factors could be incorporated into the protocol. As mentioned in the results, studying multiple sgRNAs targeting the region of interest ensures that the phenotype is a result of an on-target event. Moreover, recent studies have suggested that truncated sgRNAs with 17 nucleotides can reduce the frequency of off-target cleavage events22. Additionally, prolonged exposure to Cas9 from stable expression can result in a higher frequency of off-target effects. It is important to note that a dual vector system containing both Cas9 and the sgRNA could be used in the protocol instead of stable expression of Cas9; however, these vectors tend to be very large and result in a low lentiviral titer and lower infection efficiency. As a result, transient transfection of the Cas9 construct in the cells by nucleofection could be used. Recent reports have also developed Cas9 constructs with higher specificity for on-target editing, such as the eSpCas9 construct6.
In summary, CRISPR/Cas9 represents an efficient, inexpensive and reliable genome engineering tool, allowing for the study of genetic mutations at physiological expression levels. Here we employ CRISPR/Cas9 gene editing as a robust and efficient method to advance the understanding of the functional effects of CALR mutations in hematopoietic cells.
Disclosures
We have no conflicts of interest related to this report.
Acknowledgments
This work was supported by the NIH (R01HL131835), a Damon Runyon clinical investigator award and the Starr Cancer Consortium.
References
- DeWitt MA, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Trans Med. 2016;8:360. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;4:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gerbach CA, Barbas CF. ZFN, TALEN, CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Samuel SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archea. Nature. 2012;482(7385):331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8(11):2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, et al. CRISPR/Cas9-mediated Gene-knockout screens and target identification via Whole genome sequencing uncover host genes required for picornavirus infection. J Biol Chem. 2017;10:1074. doi: 10.1074/jbc.M117.782425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl D, et al. Generation of mouse models of myeloid malignancies with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32(9):941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific bcr/abl protein. Proc Natl Acad Sci USA. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, et al. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucl Acid Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampfl T, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- Nangalia J, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf S, et al. Mutant Calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discovery. 2016;6(4):368–381. doi: 10.1158/2159-8290.CD-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Canver MC, Orkin SH. Generation of Genomic Deletions in Mammalian Cell lines via CRISPR/Cas9. J. Vis. Exp. 2015. p. e52118. [DOI] [PMC free article] [PubMed]
- Watanabe-Smith K, et al. Analysis of acquired mutations in transgenes arising in Ba/F3 transformation assays: findings and recommendations. Oncotarget. 2017;8:12596–12606. doi: 10.18632/oncotarget.15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. NatBiotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]