

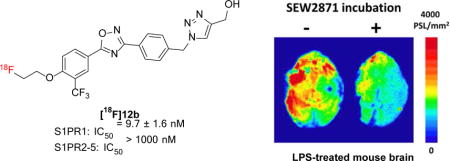
Abstract
Thirteen new S1PR1 ligands were designed and synthesized by replacing azetidine-3-carboxylic acid moiety of compound 4 with new polar groups. The in vitro binding potency of these new analogs toward S1PR1 was determined. Out of 13 new compounds, four compounds 9a, 10c, 12b, and 16b displayed high S1PR1 binding potency with IC50 values of 13.2 ± 3.2, 14.7 ± 1.7, 9.7 ± 1.6, and 6.3 ± 1.3 nM, respectively; further binding studies of these four ligands toward S1PR2-5 suggested they are highly selective for S1PR1 over other S1PRs. The radiosynthesis of the lead radiotracer [18F]12b was achieved with good radiochemical yield (~14.1%), high radiochemical purity (> 98%), and good specific activity (~54.1 GBq/μmol, decay corrected to the end of synthesis, EOS). Ex vivo autoradiography and initial biodistribution studies in rodents were performed, suggesting that [18F]12b was able to penetrate the blood-brain barrier (BBB) with high brain uptake (0.71% ID/g at 60 min post injection) and no defluorination was observed. In vitro autoradiography study in brain slices of lipopolysaccharides (LPS)-induced neuroinflammation mice indicated that SEW2871, a specific S1PR1 ligand was able to reduce the uptake of [18F]12b, suggesting [18F]12b has S1PR1 specific binding. These initial results suggested that [18F]12b has potential to be a F-18 labeled radiotracer for imaging S1PR1 in the brain of the animal in vivo.
Keywords: Sphingosine-1-phosphate receptor 1, F-18 radiotracer, PET, Multiple sclerosis, Autoradiography, Biodistribution
Graphical abstract
1. Introduction
Sphingosine-1-phosphate receptor 1 (S1PR1) is a G-protein-coupled receptor which is widely expressed in different cells such as immune cells, neural cells, endothelial cells, and smooth muscle cells. Acting with the naturally occurring lipid mediator molecule sphingosine 1-phosphate (S1P, Fig. 1), S1PR1 initiates signaling cascades that exert various biological functions, such as cell survival and differentiation, inflammation, cardiovascular function, and vascular permeability [1]. It was reported that S1P/S1PR1 signaling system plays a key role in immune-mediated diseases, such as multiple sclerosis (MS) [2, 3], colitis [4], influenza [5], rheumatoid arthritis [6], inflammatory bowel disease [7], and atherosclerosis [8, 9]. In MS, S1PR1 involves in immune-cell trafficking and activation, astrocyte proliferation, microglia activation, and interruption of the BBB function [10, 11]. Fingolimod (FTY720, Fig. 1), an S1PR modulator, is the first oral drug that US Food and Drug Administration (FDA) approved for treating remitting-relapsing MS [11]. In vivo, FTY720 is phosphorylated to form a bioactive metabolite, FTY720-P that exerts its disease-ameliorating effects by down-modulation of S1PR1 expression from the cell surface and altering lymphocyte trafficking [10]. Currently, targeting on S1PR1 represents a novel therapeutic strategy for MS and other inflammatory diseases.
Fig. 1.
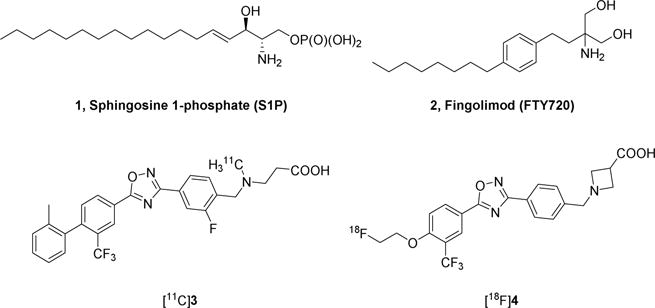
Structure of S1P, FTY720, and S1PR1 PET tracers.
Positron emission tomography (PET) provides a unique noninvasive imaging tool for quantitative assessment of target protein in vivo and it is used widely in preclinical and clinical investigations of assessing receptor occupancy, downstream functional changes, and diseases pathophysiology for guiding drug development [12]. Using a specific S1PR1 radiotracer, PET could advance our understanding of the S1PR1 pathophysiologic functions in diseases. Utilizing a specific C-11 labeled S1PR1 radiotracer [11C]3 (Fig. 1), we reported the feasibility of imaging measure S1PR1 expression for its inflammatory response in three animal models of diseases, the experimental autoimmune encephalomyelitis (EAE) rat model of MS [13], the femoral artery wire-injury mouse model of neointimal hyperplasia [14], and the carotid artery balloon injury rat model of vascular inflammation [15]. Because F-18 isotope has a longer half-life (t1/2 = 109.8 min) than C-11 (t1/2 = 20.4 min), F-18 tracers provide additional advantages of fewer constraints for production, delivery to satellite clinical PET sites with 3-4 hours’ drive distance, permission of longer scan sessions, and higher target-to-reference ratios. Therefore, identification of a F-18 radiotracer for imaging S1PR1 in vivo is imperative in PET community. To develop F-18 labeled S1PR1 radiotracers, Haufe’s group firstly reported three 18F-labeled FTY-720 analogs as S1PR radiotracers [16, 17]. These F-18 labeled radiotracers were evaluated in rodents with PET imaging modality for their inflammation responses by assessing S1PR1 expression at the binding sites. However, in vivo defluorination and low selectivity of these radiotracers prevented further transferring these radiotracers into clinical evaluation. Recently, we reported our in vivo investigation of a selective F-18 S1PR1 tracer [18F]4 for its response to acute liver inflammation of rat model that was induced by injection of LPS [18]. Although it showed positive response in liver inflammation, [18F]4 is unable to penetrate the BBB, preventing its implementation in the assessment of neuroinflammation in vivo. To overcome this limitation, herein, we reported our efforts on design, syntheses, and in vitro bioactivity evaluation of 13 new analogs; among these 13 new analogs, the F-18 labeled [18F]12b was further radiosynthesized and performed initial evaluation to check its specificity and capability of penetrating the BBB in rodents in vivo.
2. Results and discussion
2.1. Chemistry
Generally, the S1PR1 compound structure is composed of a lipophilic terminal group that interacts with the hydrophobic binding pocket of S1PR1, a polar head group, and a rigid aryl or heteroaryl linker. Generally, optimization of using a carboxylic acid as the polar head group lead to discovering high potent and highly selective S1PR1 agonists [19]. The promising in vitro binding data of compound 4 that has an aryl azetidine carboxylic acid group [18]encourages us to further optimize the aryl azetidine carboxylic acid group using other hydrophilic groups to identify new analogs having improved binding properties and physicochemical properties. In current work, we replaced the aryl azetidine carboxyl acid group with aryl sulfonamide, amide, N-hydroxy amide, hydrazide, alcohol, carboxyl acid, and amine groups to generate new analogs. The syntheses of these new analogs were illustrated in Scheme 1–4.
Scheme 1.
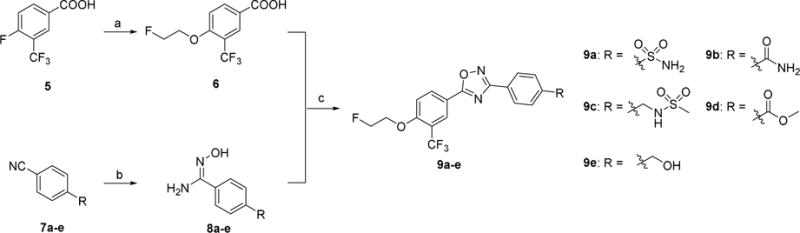
Syntheses of 9a–e. Reagents and conditions: (a) NaH, 2-fluoroethan-1-ol, DMSO, 1 M HCl, RT; (b) hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (c) TBTU, HOBt, DIPEA, DMF, RT-120 °C.
Scheme 4.

Syntheses of 21a–b. Reagents and conditions: (a) di-tert-butyl dicarbonate, triethylamine, CH2Cl2, RT; (b) hydroxylamine hydrochloride, NaHCO3, methanol, reflux; (c) TBTU, HOBt, DIPEA, 6, DMF, RT-120 °C; (d) 4 M HCl in dioxane, RT.
Compounds 9a–e were generated according to Scheme 1 by following reported procedure [18]. Alkylation of commercially available 4-fluoro-3-(trifluoromethyl)benzoic acid 5 and 2-fluoroethan-1-ol generated the intermediate 4-(2-fluoroethoxy)-3-(trifluoromethyl)benzoic acid 6 under the condition of sodium hydride in dimethyl sulfoxide (DMSO). Meanwhile, the 4-position substituted amidoximes 8a–e were prepared from corresponding benzonitriles 7a–e reacting with hydroxylamine hydrochloride and triethylamine. Coupling of 6 with 8a–e under the conventional peptide coupling conditions and subsequently thermally cyclization yielded the oxadiazoles 9a–e.
The syntheses of compounds 10a–c were depicted in Scheme 2. Starting from compound 9d, compounds 10a–c were synthesized by the following procedures. The hydrolysis of 9d using lithium hydroxide as base gave compound 10a, and the ammonolysis of 9d with hydroxylamine or hydrazine provided 10b or 10c, respectively.
Scheme 2.
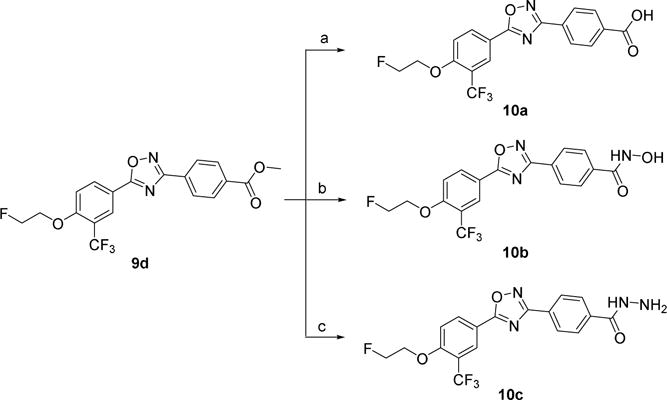
Syntheses of 10a–c. Reagents and conditions: (a) LiOH, THF, H2O, RT; (b) hydroxylamine, KOH, methanol, reflux; (c) hydrazine hydrate, ethanol, reflux.
The syntheses of analogs 12a, 12b, 15, and 16a–b were illustrated in Scheme 3. The alcohol intermediate 9e was easily transformed to azide 11a under the condition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and diphenyl phosphoryl azide (DPPA), which was used to proceed the copper(II)-catalyzed alkyne cycloaddition to afford compounds 12a–b. The alcohol intermediate 9e was converted to aldehyde 13 by Swern oxidation. After reductive amination from aldehyde 13 using corresponding amines afforded compounds 14 and 16a–b. Deprotection of tert-butyloxycarbonyl (Boc) group of 14 using 4 M HCl in dioxane afforded compound 15.
Scheme 3.
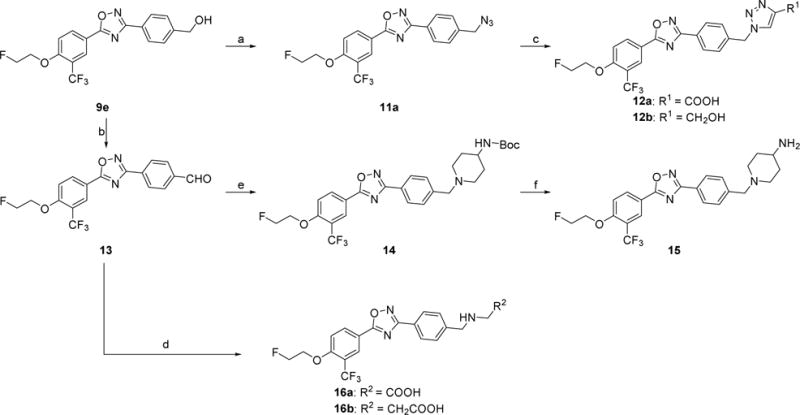
Syntheses of 12a–b, 15, and 16a–b. Reagents and conditions: (a) DPPA, DBU, toluene, 0 °C-RT; (b) oxalyl chloride, DMSO, CH2Cl2, triethylamine, −78 °C-RT; (c) propiolic acid or propargyl alcohol, CuSO4•5H2O, sodium ascorbate, THF, H2O, RT; (d) NaBH3CN, amines, acetic acid, methanol, RT; (e) 4-(N-Boc-amino)piperidine, NaBH(OAc)3, acetic acid, 1,2-dichloroethane, RT; (f) 4 M HCl in dioxane, RT.
The syntheses of the analogs 21a–b were shown in Scheme 4. Amines 17a–b were protected using Boc group to afford 18a–b. Oxadiazoles 20a–b were prepared via a similar procedure shown in Scheme 1, followed by deprotection of Boc group yielded compounds 21a–b.
2.2. In vitro binding assay
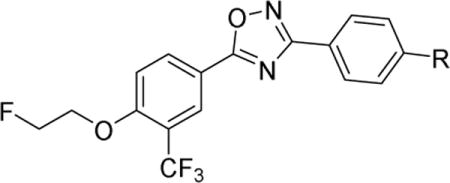
Newly synthesized compounds were firstly evaluated for binding potency toward S1PR1 by a [32P]S1P competitive binding assay following published procedure [20], S1P was used as a reference compound. The binding potency was expressed as the IC50 value. As shown in Table 1, most of the new analogs have good binding potency for S1PR1 with the IC50 values from 6.3 nM to 341 nM except compound 10a (> 1000 nM) that has a carboxyl group. Compounds 9a and 9c, bearing sulfonamides group exhibited good binding potency with the IC50 values of 13.2 and 40.0 nM, respectively. Compound 9b, having an amide group on the benzene ring showed a reduced potency with an IC50 value of 67.1 nM compared to sulfonamide compounds 9a and 9c. Introducing N-hydroxy amide or hydrazide group on the benzene ring displayed a different impact on S1PR1 binding potency. Compared to benzamide compound 9b, benzohydrazide compound 10c showed a 5-fold decreased IC50 value of 14.7 nM, but N-hydroxybenzamide compound 10b exhibited a 4-fold increased IC50 value of 272 nM. Replacing the azetidine carboxylic acid with 1,2,3-triazole-4-carboxylic acid, 2-amino acetic acid, and 3-amino propanoic acid resulted in different S1PR1 binding potency. Compared to the IC50 value of compound 4 (2.6 nM [18]), 1,2,3-triazole-4-carboxylic acid containing compound 12a (341 nM) has 130-fold increased IC50 value; the 2-amino acetic acid containing compound 16a (99.8 nM) has 38-fold increased IC50 value; 3-amino propanoic acid containing compound 16b (6.3 nM) has comparable binding potency. Compound 12b showed a 35-fold decreased IC50 value of 9.7 nM compared to the corresponding acid 12a (341 nM), which is also comparable to compound 4. Replacing the azetidine carboxylic acid of compound 4 with different amines were evaluated, and the piperidine-4-amine caused the most increased IC50 value, the IC50 value of compound 15 was 156 nM; both methylamine and O-ethylamine caused increased IC50 values too (34.2 nM for 21a, 44.2 nM for 21b), but less increase than compound 15. The representative competitive binding curves of compounds 12b, 16b, and S1P for S1PR1 were displayed in Fig. 2. Our in vitro binding data showed that compounds 9a, 10c, 12b, and 16b were potent for S1PR1 with the IC50 values less than 20 nM. Consequentially, their IC50 values of binding to other S1P receptor subtypes S1PR2, S1PR3, S1PR4, and S1PR5 were determined to check the binding selectivity. The IC50 values of these four compounds binding toward S1PR1-5 were displayed in table 2. Our data showed the IC50 values of compounds binding toward S1PR2-5 are > 1000 nM, indicating all four compounds 9a, 10c, 12b, and 16b are selective for S1PR1 over S1PR2-5. The physicochemical properties of compounds play crucial role in brain penetration, and studies showed marketed CNS drugs were found to generally have smaller molecular weight (MW), higher calculated partition coefficient (ClogP), fewer hydrogen bond donors (HBDs), and lower topological polar surface area (TPSA) compared with non-CNS drugs [21–23]. A useful guideline was concluded as follows to assess the molecules for CNS drugs: MW ≤ 470; 1 < ClogP ≤ 3; 40 < TPSA ≤ 90; and HBD ≤ 2 [24, 25]. Among these four compounds, both compounds 12b and 16b have good MW (463.4 and 453.4, respectively), ClogP (3.1 and 2.0, respectively), TPSA (91.4 and 92.5, respectively), and HBD (1 and 2, respectively) parameters for CNS drugs, suggesting the F-18 labeled version counterparts may have the capability of penetrating the BBB and entering the brain. Therefore, we further radiosynthesized [18F]12b and conducted preliminary in vivo and in vitro evaluation in rats via autoradiography and ex vivo biodistribution study.
Table 1.
| ||||||
|---|---|---|---|---|---|---|
| Compd. | R | S1PR1 IC50 (nM) | MW | ClogP | TPSA | HBD |
| S1P | 1.4 ± 0.3 | 379.5 | 4.1 | 113.0 | 4 | |
| 9a |
|
13.2 ± 3.2 | 431.4 | 3.4 | 103.3 | 1 |
| 9b |
|
67.1 ± 12.6 | 395.3 | 3.7 | 86.3 | 1 |
| 9c |
|
40.0 ± 17.8 | 459.4 | 3.7 | 89.4 | 1 |
| 10a |
|
> 1000 | 396.3 | 4.9 | 80.5 | 1 |
| 10b |
|
272 ± 65 | 411.3 | 3.3 | 92.5 | 2 |
| 10c |
|
14.7 ± 1.7 | 410.3 | 3.3 | 98.3 | 2 |
| 12a |
|
341 ± 126 | 477.4 | 4.2 | 108.4 | 1 |
| 12b |
|
9.7 ± 1.6 | 463.4 | 3.1 | 91.4 | 1 |
| 15 |
|
156 ± 47 | 464.5 | 4.1 | 72.4 | 1 |
| 16a |
|
99.8 ± 12.2 | 439.4 | 1.3 | 92.5 | 2 |
| 16b |
|
6.3 ± 1.3 | 453.4 | 2.0 | 92.5 | 2 |
| 21a |
|
34.2 ± 7.1 | 381.3 | 4.0 | 69.2 | 1 |
| 21b |
|
44.2 ± 6.8 | 425.4 | 4.1 | 78.4 | 1 |
IC50 values were determined at least 3 independent experiments, each run was performed in duplicate; Assays for compounds which showed no activity (IC50 > 1000 nM) were only repeated twice;
MW, ClogP, TPSA, and HBD were calculated by ChemDraw Professional 16.0.1.4 (77) (PerkinElmer Informatics, Inc.).
Fig. 2.
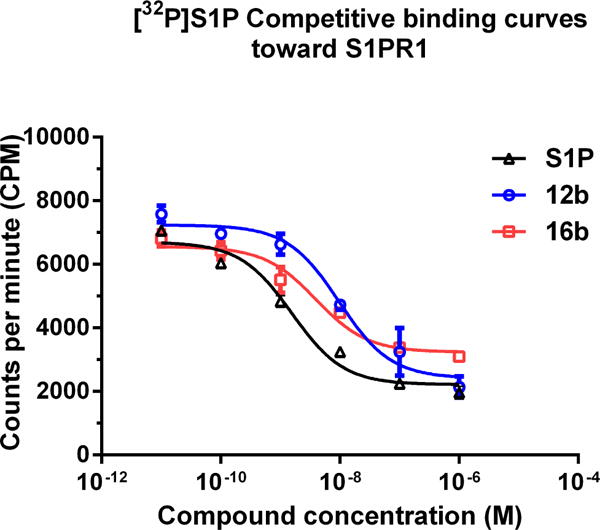
The presentative competitive binding curves of compounds 12b, 16b, and S1P for S1PR1. A CHO cell membrane containing recombinant human S1PR1 was used in a [32P]S1P competitive binding assay to measure the binding affinities. S1P (fitted IC50 = 1.4 ± 0.3 nM) 12b (fitted IC50 = 9.7 ± 1.6 nM); and 16b (fitted IC50 = 6.3 ± 1.3 nM).
Table 2.
The IC50 values (mean ± SD) of S1P, 9a, 10c, 12b, and 16b toward other S1PRsa
| Compd. | IC50 (nM)
|
||||
|---|---|---|---|---|---|
| S1PR1 | S1PR2 | S1PR3 | S1PR4 | S1PR5 | |
| S1P | 1.4 ± 0.3 | 3.6 ± 0.5 | 0.4 ± 0.2 | 151 ± 82 | 3.1 ± 1.1 |
| 9a | 13.2 ± 3.2 | > 1000 | > 1000 | > 1000 | > 1000 |
| 10c | 14.7 ± 1.7 | > 1000 | > 1000 | > 1000 | > 1000 |
| 12b | 9.7 ± 1.6 | > 1000 | > 1000 | > 1000 | > 1000 |
| 16b | 6.3 ± 1.3 | > 1000 | > 1000 | > 1000 | > 1000 |
IC50 values were determined at least 3 independent experiments, each run was performed in duplicate; Assays for compounds which showed no activity (IC50 > 1000 nM) were only repeated twice.
2.3. Radiosynthesis of [18F]12b
Compound 25, the precursor for radiolabeling [18F]12b, was prepared by following Scheme 5. The cyclization of compound 8e with 4-hydroxy-3-(trifluoromethyl)benzoic acid yielded compound 22, which was converted to azide 23 in the presence of DPPA and DBU. After alkylating with ethylene ditosylate, followed by azide-alkyne cycloaddition, tosylate precursor 25 was obtained. Radiosynthesis of [18F]12b was achieved by using the nucleophilic reaction between the tosylate precursor 25 and [18F]KF in acetonitrile with Kryptofix 222 followed by deprotection of methoxymethyl (MOM) group. After purification using semi-preparative high-performance liquid chromatography (HPLC) combined with solid-phase extraction (SPE), [18F]12b was formulated using 10% ethanol in 0.9% saline with high chemical and radiochemical purity (> 98%), good radiochemical yields (14.1 ± 2.9%), and high specific activity (54.1 ± 8.3 GBq/μmol, n = 6, decay corrected to EOS). The representative radio-UV analytical HPLC chromatograms (Fig. S1) and method were presented in the supporting material. It took approximately 90 min for the total radiosynthesis.
Scheme 5.
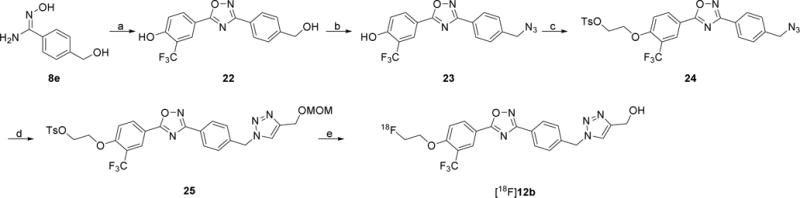
Syntheses of the precursor 25 and [18F]12b. Reagents and conditions: (a) 4-hydroxy-3-(trifluoromethyl)benzoic acid, TBTU, HOBt, DIPEA, DMF, RT-120 °C; (b) DPPA, DBU, toluene, 0 °C, RT; (c) ethylene ditosylate, K2CO3, CH3CN, 90 °C; (d) 3-(methoxymethoxy)prop-1-yne, CuSO4•5H2O, sodium ascorbate, THF, H2O, RT; (e) [18F]KF, Kryptofix 222, K2CO3, CH3CN, 110 °C, 15 min, 6 M HCl, 110 °C, 5 min, 6 M NaOH.
2.4. Ex vivo autoradiography study of [18F] 12b in normal rat
Ex vivo autoradiography study was performed to check if [18F]12b is able to cross the BBB in the rat. An adult male Sprague-Dawley (SD) rat was tail intravenously (i.v.) injected with [18F]12b and euthanized at 60 min post-injection. The brain was immediately removed and snap-frozen followed by sectioning and mounting on glass slides. The brain sections were exposed and the distribution of radioactivity was visualized by a Bio-Imaging analyzer. As shown in Fig. 3, [18F]12b was able to cross the BBB and showed high uptake in the rat brain, which is consistent to the expression of S1PR1 in the brain of rodents and human [26–28].
Fig. 3.
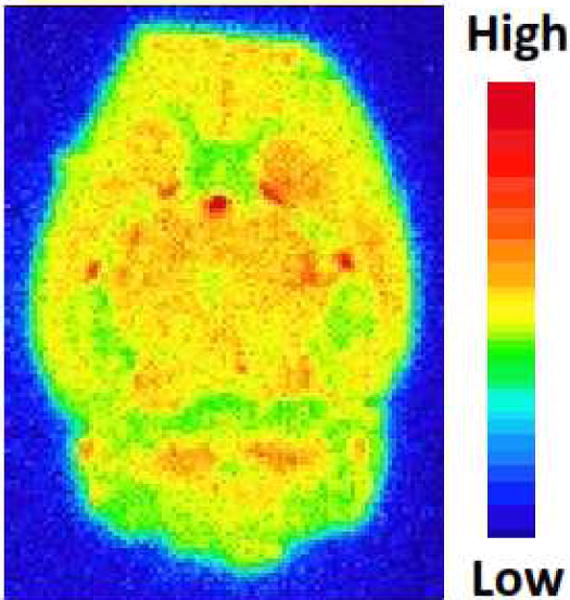
Ex vivo autoradiography of [18F]12b in rat brain.
2.5. In vitro autoradiography study of [18F]12b in LPS-treated mice
To confirm the binding specificity of [18F]12b toward S1PR1, in vitro autoradiography studies were performed using mouse brain slices that generated from LPS-induced murine neuroinflammation model, a widely used model for neuroinflammation, and the expression of S1PR1 was activated in the mouse brain following the LPS injection according to the report [29]. SEW2871, an S1PR1-specific ligand (IC50 = 37 nM) [30] was used as a blocking agent. Adult male C57BL/6 mice were euthanized at 24 h after receiving an intraperitoneal injection of LPS, brain tissues were collected and sectioned. For the control study, the slices were incubated with [18F]12b; for the blocking study, slices were incubated with [18F]12b in the presence of SEW2871 at 10 μM of concentration. In vitro autoradiography study showed that SEW2871 was able to reduce the uptake of [18F]12b by ~36%, suggesting the binding of [18F]12b toward S1PR1 is specific (Fig. 4).
Fig. 4.
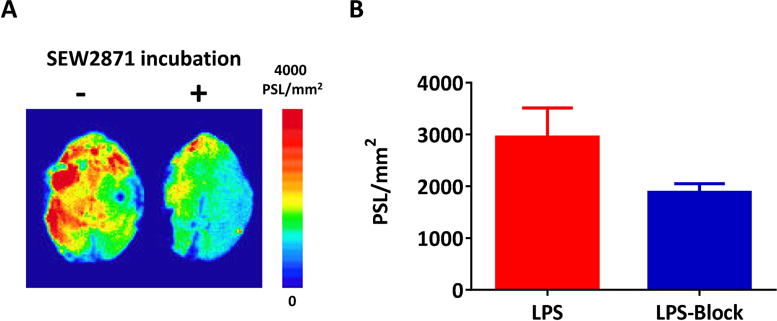
Representative in vitro autoradiographic images (A) and quantification of autoradiography (B) of brain sections from LPS-induced neuroinflammation mice under control or blocking condition. SEW2871, an S1PR1-specific ligand (IC50 = 37 nM), was used as the blocking agent.
2.6. Biodistribution study of [18F]12b in rats
To further investigate the tissue distribution of [18F]12b in rodents, the ex vivo biodistribution study was performed in adult female Lewis rats. Animals were euthanized at 5, 30, 60, and 120 min post-injection. As shown in the Table 3, the initial uptake (ID%/g) at 5 min post injection in blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, bone, thymus, and brain was 0.49, 1.65, 2.79, 0.39, 0.51, 1.96, 2.22, 3.43, 5.69, 0.55, 0.69, and 0.49, respectively. A rapid clearance of radioactivity was observed in tissues heart, lung, spleen, kidney, and liver; uptake (ID%/g) was decreased to 1.19, 1.96, 1.36, 2.11, and 4.30 at 30 min from 1.65, 2.79, 2.22, 3.43, and 5.69 at 5 min, respectively. The radioactivity accumulation in the bone has no change from 5 min to 120 min, suggesting [13F]12b had no defluorination in vivo. A high uptake (ID%/g) of [18F]12b in the brain was observed compared to radiotracer [18F]4 [18]. The brain uptake (ID%/g) of [18F]12b was 0.49, 0.57, 0.71, and 0.61 at 5, 30, 60, and 120 min, respectively. In brain regions including cerebellum, brain stem, cortex, striatum, thalamus, and hippocampus, [18F]12b had similar distribution, the uptake (ID%/g) in each region was 0.53, 0.50, 0.59, 0.47, 0.46, and 0.43, respectively at 5 min; the brain uptake at 60 min post injection reached the maximum, then gradually washed out as shown in Fig. 5. Our data suggested that [18F]12b has the potential to be a PET tracer for imaging S1PR1 expression in the brain in vivo.
Table 3.
Biodistribution of [18F]12b in female Lewis rats.(%ID/g values, mean ± SD, n = 4).
| Organs | Tissue uptake (%ID/g)
|
|||
|---|---|---|---|---|
| 5 min | 30 min | 60 min | 120 min | |
| Blood | 0.49 ± 0.07 | 0.42 ± 0.05 | 0.36 ± 0.02 | 0.35 ± 0.01 |
| Heart | 1.65 ± 0.27 | 1.19 ± 0.06 | 1.05 ± 0.08 | 0.94 ± 0.06 |
| Lung | 2.79 ± 0.57 | 1.96 ± 0.07 | 1.67 ± 0.09 | 1.54 ± 0.07 |
| Muscle | 0.39 ± 0.04 | 0.59 ± 0.05 | 0.51 ± 0.02 | 0.47 ± 0.02 |
| Fat | 0.51 ± 0.22 | 0.62 ± 0.10 | 0.69 ± 0.07 | 0.57 ± 0.07 |
| Pancreas | 1.96 ± 0.56 | 1.89 ± 0.16 | 1.52 ± 0.25 | 1.45 ± 0.15 |
| Spleen | 2.22 ± 0.23 | 1.36 ± 0.04 | 1.27 ± 0.08 | 1.14 ± 0.10 |
| Kidney | 3.43 ± 0.31 | 2.11 ± 0.08 | 1.91 ± 0.10 | 1.62 ± 0.09 |
| Liver | 5.69 ± 0.60 | 4.30 ± 0.37 | 4.21 ± 0.21 | 3.66 ± 0.14 |
| Bone | 0.55 ± 0.05 | 0.45 ± 0.03 | 0.45 ± 0.01 | 0.51 ± 0.05 |
| Thymus | 0.69 ± 0.08 | 0.83 ± 0.07 | 0.82 ± 0.04 | 0.78 ± 0.05 |
| Brain | 0.49 ± 0.04 | 0.57 ± 0.02 | 0.71 ± 0.03 | 0.61 ± 0.05 |
Fig. 5.
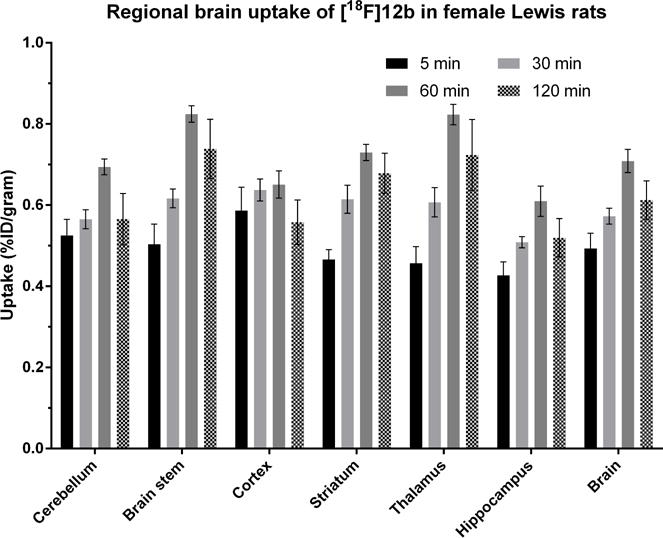
Regional brain uptake of [18F]12b in female Lewis rats. Animals were sacrificed at 5, 30, 60, and 120 min post-injection. %ID/gram values (mean ± SD) with 4 rats per group.
3. Conclusion
In summary, we explored the structure of lead compound 4 to afford new S1PR1 compounds by replacing the 3-N-azetedinyl carboxyl acid with different hydrophilic groups. Thirteen new analogs were synthesized and their in vitro binding affinities were determined. Four compounds 9a, 10c, 12b, and 16b had high S1PR1 binding potency with IC50 values ranging from 6.3 to 15 nM and also displayed highly selective for S1PR1 versus other S1P receptor subtypes (IC50 > 1000 nM for S1PR2-5). Radiosynthesis of [18F]12b was accomplished with good yield and high purity. Our initial evaluation of [18F]12b in rodent using autoradiography and biodistribution studies indicated [18F]12b was able to penetrate the BBB and had good brain retention; no defluorination was observed in vivo. In vitro autoradiography study in brain slices of LPS-induced neuroinflammation mice indicated that SEW2871, a specific S1PR1 ligand was able to reduce the uptake of [18F]12b, suggesting that [18F]12b had specific binding toward S1PR1. Further in vivo evaluation of [18F]12b in animal models of neuroinflammatory diseases warrants the suitability of [18F]12b to be an S1PR1 specific PET radiotracer for assessing inflammation response in CNS.
4. Experimental section
4.1. General
Commercially available starting materials, reagents, and solvents were used as purchased without further purification unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on pre-coated glass plates of silica gel 60 F254 from EMD Chemicals Inc. Visualization was accomplished with ultraviolet light (UV 254 nm). Compounds were purified by recrystallization or flash column chromatography using 230–400 mesh silica gel purchased from Silicycle. Melting points were determined on a MEL-TEMP 3.0 apparatus without corrected. 1H NMR and 13C NMR spectra were received from Varian 400 MHz instrument. Chemical shifts were reported in parts per million (ppm) and were calibrated using a residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; DMSO-d6: δ 2.50 ppm; Acetone-d6: δ 2.05 ppm). High-resolution positive ion mass was acquired by a Bruker MaXis 4G Q-TOF mass spectrometer with electrospray ionization source.
All animal experiments were conducted under Washington University’s Institutional Animal Care and Use Committee (IACUC)-approved protocols in accordance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals.
4.2. Chemistry
4.2.1. Synthesis of 9a–e
4-(2-Fluoroethoxy)-3-(trifluoromethyl)benzoic acid (6)
To a round-bottom flask equipped with a stir bar was added 4-fluoro-3-(trifluoromethyl)benzoic acid (8.32 g, 40.0 mmol), 2-fluoroethanol (3.84 g, 60 mmol), and DMSO (100 mL) under N2 atmosphere. NaH (2.07 g, 90.0 mmol) was added portion wise with stirring. The reaction was stirred at room temperature overnight and monitored by TLC. The mixture was poured into ice-water (1 L) and acidified with 1 M HCl to give a precipitate. The resulting precipitate was filtered off, washed with water and hexanes to give a tan solid (9.6 g, 95%). MP: 131–133 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.18 (br, 1H, -COOH), 8.37 − 8.04 (m, 2H, -ArH), 7.45 − 7.32 (m, 1H, -ArH), 4.89 − 4.74 (m, 1H, -CH2F), 4.72 − 4.58 (m, 1H, -CH2F), 4.52 − 4.39 (m, 1H, -CH2O-), 4.38 − 4.29 (m, 1H, -CH2O-).
General procedure for the synthesis of 8a–e
To a round-bottom flask equipped with a stir bar was added 7a–e (1.0 eq), hydroxylamine hydrochloride (2.0 eq), NaHCO3 (4.0 eq), and methanol (5.0 mL/mmol). The reaction was refluxed and stirred in a pre-heated 75 °C oil-bath for 6 h. The reaction mixture was cooled to room temperature, and the precipitate was filtered off and washed with methanol. The filtrate was concentrated in vacuo without further purification.
(E)-N′-hydroxy-4-sulfamoylbenzimidamide (8a)
Yield: 70%. MP: 217–219 °C. 1H NMR (400 MHz, DMSO-d6) δ = 7.88−7.78 (m, 4H, -ArH), 5.93 (s, 2H, -SO2NH2).
(E)-4-(N′-hydroxycarbamimidoyl)benzamide (8b)
Yield: 39%. MP: 221–224 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H, -OH), 7.98 (s, 1H,-NH2), 7.86 (d, J = 8.4 Hz, 2H, -ArH), 7.74 (d, J = 8.4 Hz, 2H, - ArH), 7.38 (s, 1H, -NH2), 5.88 (s, 2H, -CONH2).
(E)-N′-hydroxy-4-(methylsulfonamidomethyl)benzimidamide (8c)
Yield: 95%. MP: 178–180 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.62 (s, 1H, -OH), 7.63 (d, J = 8.3 Hz, 2H, -ArH), 7.32 (d, J = 8.4 Hz, 2H, -ArH), 5.78 (s, 2H, -NH2), 4.14 (s, 2H, -CH2-), 2.84 (s, 3H, -CH3).
Methyl (E)-4-(N′-hydroxycarbamimidoyl)benzoate (8d)
Yield: 86%. MP: 168–170 °C. 1H NMR (400 MHz, DMSO-d6) δ = 9.91 (s, 1H, -OH), 7.94 (d, J = 8.0 Hz, 2H, -ArH), 7.82 (d, J = 8.4 Hz, 2H, -ArH), 5.93 (s, 2H, - NH2), 3.85 (s, 3H, -OCH3)
(E)-N′-hydroxy-4-(hydroxymethyl)benzimidamide (8e)
Yield: 76%. MP: 152–155 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H, −OH), 7.71 − 7.57 (m, 2H, -ArH), 7.37 − 7.24 (m, 2H, -ArH), 5.77 (s, 2H, -NH2), 5.24 (s, 1H, -CH2OH), 4.51 (s, 2H, -CH2-).
General procedure for the synthesis of 9a–e
To a round-bottom flask equipped with a stir bar was added acid 6 (1.0 eq), HOBt (0.2 eq), TBTU (1.0 eq), DIPEA (3.0 eq), and DMF (5.0 mL/mmol). The reaction mixture was stirred for 0.5 h followed by adding amidoxime 8a–e (1.0 eq). The reaction mixture was stirred for 1 h at room temperature, then refluxed in a pre-heated 120 °C oil-bath for 4 h and monitored by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column.
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzenesulfonamide (9a)
Yield: 51%. MP: 198–199 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 − 8.38 (m, 1H, -ArH), 8.33 − 8.24 (m, 3H, -ArH), 8.04 (d, J = 8.4 Hz, 2H, -ArH), 7.62 − 7.52 (m, 3H, -ArH&-NH2), 4.89 − 4.81 (m, 1H, -CH2F), 4.76 − 4.70 (m, 1H, -CH2F), 4.61 − 4.55 (m, 1H, -CH2O-), 4.54 − 4.44 (m, 1H, -CH2O-). 13C NMR (101 MHz, DMSO-d6) δ 174.92, 167.79, 160.02, 147.12, 134.58, 129.34, 128.21, 127.08 (q, J = 5.1 Hz), 127.04, 123.31 (d, J = 272.7 Hz), 118.61 (q, J = 31.3 Hz), 116.10, 115.44, 82.10 (d, J = 168.7 Hz), 69.24 (d, J = 18.2 Hz). HRMS (ESI) calcd for C17H13F4N3O4S [M + H+] 432.0636. Found [M + H+] 432.0640.
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzamide (9b)
Yield: 29%. MP: 244–245 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J = 8.1 Hz, 1H, -ArH), 8.30 (s, 1H, -ArH), 8.19 − 8.10 (m, 3H, -ArH&-NH2), 8.10 − 8.03 (m, 2H, -ArH), 7.59 − 7.51 (m, 2H, -ArH), 4.89 − 4.82 (m, 1H, -CH2F), 4.76 − 4.71 (m, 1H, -CH2F), 4.62 − 4.55 (m, 1H, -CH2O-), 4.53 − 4.47 (m, 1H, -CH2O-). 13C NMR (101 MHz, DMSO-d6) δ 174.72, 168.14, 167.49, 159.96, 137.42, 134.54, 128.77, 127.42, 127.08 (q, J = 5.1 Hz), 123.31 (d, J = 273.7 Hz), 118.58 (q, J = 31.3 Hz), 116.17, 115.41, 82.10 (d, J = 168.7 Hz), 69.23 (d, J = 19.2 Hz). HRMS (ESI) calcd for C18H13F4N3O3 [M + H+] 396. 0966. Found [M + H+] 396.0959.
N-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)methanesulfonamide (9c)
Yield: 9%. MP: 208–300 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.41 (d, J = 8.9 Hz, 1H, -ArH), 8.31 (s, 1H, -ArH), 8.07 (d, J = 8.1 Hz, 2H, -ArH), 7.73 − 7.67 (m, 1H, -ArH), 7.61 − 7.52 (m, 3H, -ArH&-NHSO2-), 4.89 − 4.81 (m, 1H, -CH2F), 4.78 − 4.70 (m, 1H, -CH2F), 4.63 − 4.56 (m, 1H, -CH2O-), 4.55 − 4.48 (m, 1H, -CH2O-), 4.26 (d, J = 6.3 Hz, 2H, -ArCH2-), 2.91 (s, 3H, -SO2CH3). 13C NMR (101 MHz, DMSO-d6) δ 174.54, 168.46, 159.91, 142.71, 134.54, 128.77, 127.64, 127.07 (q, J = 5.1 Hz), 125.29, 123.34 (d, J = 273.7 Hz), 118.56 (q, J = 31.3 Hz), 116.31, 115.43, 82.12 (d, J = 168.7 Hz), 69.23 (d, J = 18.2 Hz), 46.13, 40.35. HRMS (ESI) calcd for C19H17F4N3O4S [M + H+] 460.0949. Found [M + H+] 460.0936.
Methyl 4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzoate (9d)
Yield: 62%. MP: 166–169 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.45 (d, J = 8.9 Hz, 1H, -ArH), 8.42 (s, 1H, -ArH), 8.26 (d, J = 8.1 Hz, 2H, -ArH), 8.18 (d, J = 8.2 Hz, 2H, -ArH), 7.57 (d, J = 8.7 Hz, 1H, -ArH), 4.96 − 4.92 (m, 1H, -CH2F), 4.85 − 4.80 (m, 1H, -CH2F), 4.68 − 4.64 (m, 1H, -CH2O-), 4.62 − 4.56 (m, 1H, -CH2O-), 3.94 (s, 3H, -OCH3). 13C NMR (101 MHz, Acetone-d6) δ 174.80, 168.03, 165.62, 160.06, 133.86, 132.69, 130.76, 129.94, 127.34, 126.97 (q, J = 5.1 Hz), 123.12 (d, J = 273.7 Hz), 119.22 (d, J = 31.3 Hz), 116.41, 114.50, 81.54 (d, J = 169.7 Hz), 68.95 (d, J = 19.2 Hz), 51.75.
(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanol (9e) [18]
Yield: 65%. MP: 190–192 °C. 1H NMR (400 MHz, CDCl3) δ = 8.45 (d, J = 2.0 Hz, 1H, -ArH), 8.33 (dd, J = 8.8 Hz, 2.0 Hz, 1H, -ArH), 8.13 (d, J = 8.0 Hz, 2H, -ArH), 7.50 (d, J = 8.0 Hz, 2H, -ArH), 7.16 (d, J = 8.4 Hz, 1H, -ArH), 4.90-4.74 (m, 4H, -CH2F &-CH2OH), 4.48-4.35 (m, 2H, -CH2O-).
4.2.2. Synthesis of 10a–c
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzoic acid (10a)
To a round-bottom flask equipped with a stir bar was added ester 9d (2.0 g, 4.87 mmol), lithium hydroxide (230 mg, 9.75 mmol), THF (20 mL), and water (4 mL). The reaction mixture was stirred overnight and acidified with 1 M HCl to pH = 1. The reaction mixture was extracted with ethyl acetate (20 mL), and the ethyl acetate layer was washed with saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, 10a was obtained as an off-white solid. (1.7 g, 87%). MP: 220-222 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H, -COOH), 8.39 (d, J = 8.8 Hz, 1H, -ArH), 8.28 (s, 1H, -ArH), 8.19 − 8.08 (m, 4H, -ArH), 7.54 (d, J = 8.8 Hz, 1H, -ArH), 4.91 − 4.81 (m, 1H, -CH2F), 4.79 − 4.69 (m, 1H, -CH2F), 4.63 − 4.47 (m, 2H, -CH2O-). 13C NMR (101 MHz, DMSO-d6) δ 174.80, 168.02, 167.02, 159.98, 134.55, 133.84, 130.13, 129.85, 127.70, 127.07 (q, J = 5.1 Hz), 123.30 (d, J = 273.7 Hz), 118.57 (q, J = 31.3 Hz), 116.11, 115.41, 82.09 (d, J = 168.7 Hz), 69.22 (d, J = 19.2 Hz).
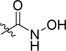
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)-N-hydroxybenzamide (10b)
To a round-bottom flask equipped with a stir bar was added ester 9d (150 mg, 0.37 mmol), hydroxylamine hydrochloride (76 mg, 1.1 mmol) and methanol (1.9 mL), followed by adding potassium hydroxide (12.5 mg, 2.22 mmol). The reaction mixture was refluxed overnight. After cooling to room temperature, the mixture was diluted with water (20 mL) to precipitate the product. The precipitate was filtered off and washed with diethyl ether to give an off-white solid (111 mg, 73%). MP: 186–189 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H, -OH), 9.20 (s, 1H, -NH-), 8.42 (d, J = 8.1 Hz, 1H, -ArH), 8.31 (s, 1H, -ArH), 8.18 − 8.11 (m, 2H, -ArH), 8.01 − 7.91 (m, 2H, -ArH), 7.61 − 7.51 (m, 1H, -ArH), 4.92 − 4.68 (m, 2H, -CH2F), 4.64 − 4.47 (m, 2H, -CH2O-). 13C NMR (101 MHz, DMSO-d6) δ 174.77, 168.12, 167.05, 159.99, 135.95, 134.59, 130.55, 128.23, 127.59, 127.12 (d, J = 5.1 Hz), 123.31 (d, J = 274.7 Hz), 118.57 (J = 30.3 Hz), 116.19, 115.45, 82.11 (d, J = 168.7 Hz), 69.24 (d, J = 19.2 Hz). HRMS (ESI) calcd for C18H13F4N3O4 [M + H+] 412.0919. Found [M + H+] 412.0915.
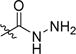
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzohydrazide (10c)
To a round-bottom flask equipped with a stir bar was added ester 9d (205 mg, 0.5 mmol), ethanol (1.3 mL), and hydrazine monohydrate (0.07 mL, 1.5 mmol). The reaction mixture was refluxed overnight. After cooling to room temperature, the precipitate was filtered off and washed with diethyl ether to give an off-white solid. (100 mg, 49%). MP: 186–188 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, 1H, -CONH-), 8.42 (d, J = 8.7 Hz, 1H, -ArH), 8.32 (s, 1H, -ArH), 8.14 (d, J = 8.2 Hz, 2H, -ArH), 8.01 (d, J = 8.2 Hz, 2H, -ArH), 7.56 (d, J = 8.8 Hz, 1H, -ArH), 4.89 − 4.81 (m, 1H, -CH2F), 4.77 − 4.70 (m, 1H, -CH2F), 4.68 − 4.55 (m, 3H, -CH2O-&-NH2), 4.55 − 4.48 (m, 1H, -CH2O-). 13C NMR (101 MHz, DMSO-d6) δ 174.75, 168.14, 165.35, 159.98, 136.46, 134.57, 128.62, 128.30, 127.52, 127.11 (q, J = 5.1 Hz), 123.32 (d, J = 273.7 Hz), 118.60 (q, J = 31.3 Hz), 116.20, 115.46, 82.11 (d, J = 167.7 Hz), 69.25 (d, J = 19.2 Hz). HRMS (ESI) calcd for C18H14F4N4O3 [M + H+] 411. 1075. Found [M + H+] 411.1067.
4.2.3. Synthesis of 12a–b, 15, and 16a–b
3-(4-(Azidomethyl)phenyl)-5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazole (11a)
To a round-bottom flask equipped with a stir bar was added 9e (1.0 g, 2.62 mmol), diphenyl phosphoryl azide (0.86 g, 3.14 mmol), and toluene (5.0 mL). The mixture was cooled to 0 °C before adding 1,8-diazabicyclo[5.4.0]undec-7-ene (0.48, 3.14 mmol) dropwise. The reaction was warmed to room temperature slowly and stirred overnight. Then, the reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (30 mL). The ethyl acetate layer was washed with 1 M HCl, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column, eluted with ethyl acetate/hexanes (V/V, 3/7) to give a white solid. (330 mg, 62%). MP: 117–119 °C. 1H NMR (400 MHz, CDCl3) δ = 8.47 (d, J = 2.0 Hz, 1H, -ArH), 8.35 (dd, J = 8.8 Hz, 2.0 Hz, 1H, -ArH), 8.19 (d, J = 8.4 Hz, 2H, -ArH), 7.47 (d, J = 8.0 Hz, 2H, -ArH), 7.18 (d, J = 8.8 Hz, 1H, -ArH), 4.94 − 4.73 (m, 2H, -CH2F), 4.50 − 4.35 (m, 4H, -CH2O-&-CH2N3). 13C NMR (101 MHz, Acetone-d6) δ 174.54, 168.40, 159.98, 139.55, 133.81, 129.35, 128.89, 127.60, 126.92 (q, J = 5.1 Hz), 123.15 (d, J = 273.7 Hz), 119.21 (d, J = 31.3 Hz), 116.60, 114.50, 81.55 (d, J = 169.7 Hz), 68.94 (d, J = 20.2 Hz), 53.69.
General procedure for the synthesis of 12a–b
To a round-bottom flask equipped with a stir bar was added azide 11a (1.0 eq) and THF. Once the azide was dissolved, water (0.6 mL/mmol) was added followed by propiolic acid or propargyl alcohol (1.05 eq), sodium ascorbate (0.10 eq), and copper sulfate pentahydrate (0.06 eq). The reaction mixture was stirred for 4 h and monitored by TLC. Then, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, a yellow solid was obtained.
1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)-1H-1,2,3-triazole-4-carbox-ylic acid (12a)
MP: 157–158 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.09 (s, 1H, -COOH), 8.86 (s, 1H, -ArH), 8.41 (d, J = 8.2 Hz, 1H, -ArH), 8.30 (s, 1H, -ArH), 8.10 (d, J = 7.5 Hz, 2H, -ArH), 7.53 (d, J = 7.7 Hz, 2H, -ArH), 7.17 − 7.11 (m, 1H, -ArH), 5.78 (s, 2H, -ArCH2-), 4.88 − 4.46 (m, 4H, -OCH2CH2F). 13C NMR (101 MHz, DMSO-d6) δ 174.64, 168.26, 162.01, 159.93, 140.38, 139.55, 134.57, 129.31, 129.20, 128.04, 127.08 (d, J = 5.1 Hz), 123.32 (d, J = 274.7 Hz), 120.33, 118.56 (q, J = 31.3 Hz), 116.24, 115.43, 82.11 (d, J = 167.7 Hz), 69.23 (d, J = 19.2 Hz), 53.04. HRMS (ESI) calcd for C21H15F4N5O4 [M + H+] 478.1133. Found [M + H+] 478.1125.
(1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)met-hanol (12b)
MP: 175–177 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.41 (d, J = 8.6 Hz, 1H, -ArH), 8.30 (s, 1H, -ArH), 8.08 (d, J = 4.7 Hz, 3H, -ArH), 7.60 − 7.45 (m, 3H, -ArH), 5.70 (s, 2H, -ArCH2-), 5.20 (s, 1H, -OH), 4.89 − 4.67 (m, 2H, -CH2F), 4.63 − 4.48 (m, 4H, -CH2O-&-CH2OH). 13C NMR (101 MHz, DMSO-d6) δ 174.63, 168.29, 159.94, 140.24, 134.57, 129.11, 127.97, 127.08 (d, J = 5.1 Hz), 126.13, 123.32 (d, J = 274.7 Hz), 123.55, 120.31, 118.56 (q, J = 31.3 Hz), 116.24, 115.43, 82.11 (d, J = 167.7 Hz), 69.23 (d, J = 19.2 Hz), 55.47, 52.71. HRMS (ESI) calcd for C21H17F4N5O3 [M + H+] 464.1346. Found [M + H+] 464.1353.
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzaldehyde (13) [18]
To an oven-dried 100 mL round-bottom flask equipped with a stir bar was added CH2Cl2 (32 mL) and DMSO (1.7 g, 21.7 mmol). The reaction mixture was cooled to −78 °C and oxalyl chloride (1.87 g, 14.7 mmol) was added slowly under N2 atmosphere. The reaction mixture was stirred for 30 minutes at which time 9e (2.68 g, 7.0 mmol) was added. The reaction mixture was stirred for another 30 minutes at which time triethylamine (5.67 mL, 56.0 mmol) was added. The cooling bath was removed and the reaction was allowed to warm to room temperature over 2 h. The reaction mixture was concentrated in vacuo and partitioned between ethyl acetate and 1 M HCl. The layers were separated and the aqueous layer was re-extracted with ethyl acetate. The combined organic layers were washed with 1 M HCl, sat. NaHCO3, brine, and dried over MgSO4. After filtration and concentration in vacuo, a pale-yellow solid was obtained. (1.51 g, 76%).
tert-Butyl (1-(4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)piperidin-4-yl)carba-mate (14)
To a round-bottom flask equipped with a stir bar was added aldehyde 13 (380 mg, 1.0 mmol), amine (21 mg, 1.05 mmol), 1,2-dichloroethane (10 mL), sodium triacetoxyborohydride (318 mg, 1.5 mmol), and acetic acid (120 mg, 2.0 mmol). The reaction mixture was stirred overnight at room temperature. Then, the reaction was quenched with saturated NaHCO3 aqueous and extracted with ethyl acetate (20 mL × 3). The combined ethyl acetate layer was washed with brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column to give white solid product (250 mg, 44%). MP: 147−149 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.39 − 8.32 (m, 2H, -ArH), 8.02 (d, J = 7.9 Hz, 2H, -ArH), 7.52 − 7.45 (m, 3H, -ArH), 5.83 (d, J = 6.7 Hz, 1H, -ArH), 4.90 − 4.83 (m, 1H, -CH2F), 4.79 − 4.72 (m, 1H, -CH2F), 4.61 − 4.56 (m, 1H, -CH2O-), 4.55 − 4.49 (m, 1H, -CH2O-), 3.51 (s, 2H, -ArCH2-), 3.41 − 3.27 (m, 1H, -CHNH-), 2.83 − 2.75 (m, 2H, -NCH2-), 2.12 − 1.97 (m, 2H, -NCH2-), 1.83 − 1.75 (m, 2H, -CH2-), 1.55 − 1.43 (m, 2H, -CH2-), 1.34 (s, 9H, -CH3). 13C NMR (101 MHz, Acetone-d6) δ 174.29, 168.62, 159.90, 159.89, 155.00, 143.07, 133.74, 129.26, 127.11, 126.86 (q, J = 5.1 Hz), 125.33, 123.15 (d, J = 273.7 Hz), 119.17 (q, J = 31.3 Hz), 116.65, 114.43, 81.54 (d, J = 169.7 Hz), 77.51, 68.91 (d, J = 20.2 Hz), 62.18, 52.44, 47.89, 32.27, 27.75.
1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)piperidin-4-amine dihydro-chloride (15)
To a round-bottom flask equipped with a stir bar was added the Boc-protected amine 14 (90 mg, 0.16 mmol) and 2 mL of 4 M HCl in 1,4-dioxane. The reaction was stirred 4 h at room temperature, and the precipitate was filtered off, washed with ethyl ether to give a white solid. (51 mg, 59%). MP: 268−269 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 2H, -NH2), 8.42 (d, J = 9.0 Hz, 1H, -ArH), 8.31 (s, 1H, -ArH), 8.14 (d, J = 7.9 Hz, 2H, -ArH), 7.85 (d, J = 7.4 Hz, 2H, -ArH), 7.58 (d, J = 8.8 Hz, 1H, -ArH), 4.90 − 4.81 (m, 1H, -CH2F), 4.78 − 4.70 (m, 1H, -CH2F), 4.63 − 4.56 (m, 1H, -CH2O-), 4.55 − 4.49 (m, 1H, -CH2O-), 4.36 (s, 2H, -ArCH2-), 3.36 − 3.17 (m, 3H, -N-CH2-&-CHNH2), 3.16 − 3.00 (m, 2H, -N-CH2-), 2.22 − 1.95 (m, 4H, -CH2CH-). 13C NMR (101 MHz, DMSO-d6) δ 174.75, 168.26, 159.99, 134.60, 133.60, 132.92, 127.81, 127.36, 127.10 (d, J = 5.1 Hz), 123.32 (d, J = 273.7 Hz), 118.58 (q, J = 31.3 Hz), 116.20, 115.52, 82.13 (d, J = 168.7 Hz), 69.27 (d, J = 18.2 Hz), 58.66, 49.97, 45.68, 27.21.HRMS (ESI) calcd for C23H24F4N4O2 [M + H+] 465.1908. Found [M + H+] 465.1907.
General procedure for the synthesis of 16a–b
To a round-bottom flask equipped with a stir bar was added aldehyde 13 (1.0 eq), amine (1.5 eq), methanol (14 mL/mmol), and acetic acid (0.5 mL/mmol). The reaction mixture was stirred 1 h at which time sodium cyanoborohydride (1.0 eq) was added. The reaction mixture was stirred overnight and diluted with water. The precipitate was filtered off and washed with water to give an off-white solid product.
(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)glycine (16a)
Yield: 24%. MP: 175-176 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.49 − 8.35 (m, 2H, -ArH), 8.12 (d, J = 7.8 Hz, 2H, -ArH), 7.67 (d, J = 7.9 Hz, 2H, -ArH), 7.56 (d, J = 8.7 Hz, 1H, -ArH), 4.98 − 4.75 (m, 2H, -CH2F), 4.71 − 4.54 (m, 2H, -CH2O-), 3.98 (s, 2H, -ArCH2-), 3.42 (s, 2H, -CH2COOH). 13C NMR (101 MHz, Acetone-d6) δ 174.36, 171.35, 168.62, 159.93, 143.12, 133.77, 129.47, 127.27, 126.88 (q, J = 5.1 Hz), 125.65, 123.15 (d, J = 273.7 Hz), 119.19 (q, J = 31.3 Hz), 116.66, 114.47, 81.56 (d, J = 168.7 Hz), 68.92 (d, J = 20.2 Hz), 57.26, 53.12. HRMS (ESI) calcd for C20H17F4N3O4 [M + H+] 440.1228. Found [M + H+] 440.1210.
3-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)amino)propanoic acid (16b)
Yield: 17%. MP: 198–200 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.49 − 8.37 (m, 1H, -ArH), 8.30 (s, 1H, -ArH), 8.17 − 7.98 (m, 2H, -ArH), 7.67 − 7.44 (m, 3H, -ArH), 4.92 − 4.70 (m, 2H, -CH2F), 4.63 − 4.45 (m, 2H, -CH2O-), 3.87 (s, 2H, -ArCH2-), 2.87 − 2.71 (m, 2H, -NHCH2-), 2.45 − 2.31 (m, 2H, -CH2COOH). 13C NMR (101 MHz, DMSO-d6) δ 174.48, 174.15, 168.53, 159.91, 143.50, 134.53, 129.37, 127.52, 127.05 (d, J = 5.1 Hz), 125.10, 123.33 (d, J = 5.1 Hz), 118.56 (d, J = 273.7 Hz), 116.31, 115.43, 82.12 (d, J = 168.7 Hz), 69.24 (d, J = 19.2 Hz), 51.89, 44.51, 34.00. HRMS (ESI) calcd for C21H19F4N3O4 [M + H+] 454.1384. Found [M + H+] 454.1370.
4.2.4. Synthesis of 21a–b
General procedure for the synthesis of 18a–b
To a round-bottom flask equipped with a stir bar was added 17a–b (1.0 eq), Boc2O (1.1 eq), CH2Cl2 (10 mL/mmol), and triethylamine (1.5 eq). The reaction mixture was stirred at room temperature overnight. The reaction was concentrated in vacuo and partitioned between ethyl ether and 1 M HCl. The organic layer were separated, washed with saturated NaHCO3, brine, dried over MgSO4, and concentrated in vacuo to give a white crystalline solid.
tert-Butyl (4-cyanobenzyl)carbamate (18a)
Yield: 93%. MP: 114-116 °C. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 2H, -ArH), 7.39 (d, J = 8.5 Hz, 2H, -ArH), 4.96 (s, 1H, -NH-), 4.37 (d, J = 5.9 Hz, 2H, -CH2-), 1.46 (s, 9H, -CH3).
tert-Butyl (2-((4-cyanobenzyl)oxy)ethyl)carbamate (18b)
Yield: 83%. MP: 100–102 °C. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 2H, -ArH), 7.33 (d, J = 7.9 Hz, 2H, -ArH), 4.53 (s, 2H, -ArCH2-), 3.80 − 3.65 (m, 2H, -OCH2-), 3.49 − 3.30 (m, 2H, -CH2NH-), 1.40 (s, 9H, -CH3).
General procedure for the synthesis of 19a–b
To a round-bottom flask equipped with a stir bar was added 18a–b (1.0 eq), hydroxylamine hydrochloride (2.0 eq), NaHCO3 (4.0 eq), and methanol (10 mL/mmol). The reaction was refluxed and stirred in a pre-heated 75 °C oil-bath for 6 h. After cooling to room temperature, and the precipitate was filtered off and washed with methanol. The filtrate was concentrated in vacuo and further purified on a silica gel column.
tert-Butyl (E)-(4-(N′-hydroxycarbamimidoyl)benzyl)carbamate (19a)
Yield: 65%. MP: 142-145 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H, -OH), 7.61 (d, J = 8.1 Hz, 2H, -ArH), 7.40 (t, J = 6.1 Hz, 1H, -NHCO-), 7.22 (d, J = 8.1 Hz, 2H, -ArH), 5.77 (s, 2H, -NH2), 4.13 (d, J = 6.1 Hz, 2H, -ArCH2-), 1.40 (s, 9H, -CH3).
tert-Butyl (E)-(2-((4-(N′-hydroxycarbamimidoyl)benzyl)oxy)ethyl)carbamate (19b)
Yield: 40%. MP: 138–140 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.87 (s, 1H, -OH), 7.69 (d, J = 7.7 Hz, 2H, -ArH), 7.28 (d, J = 8.1 Hz, 2H, -ArH), 5.45 (s, 2H, -NH2), 4.54 (s, 2H, -ArCH2-), 3.75 (s, 1H, -NHCO-), 3.68 − 3.59 (m, 2H, -OCH2-), 3.40 − 3.24 (m, 2H, -CH2NH-), 1.47 (s, 9H, -CH3).
General procedure for the synthesis of 20a–b
To a round-bottom flask equipped with a stir bar was added acid 6 (1.0 eq), HOBt (0.2 eq), TBTU (1.0 eq), DMF (5 mL/mmol), and DIPEA (3.0 eq). The reaction mixture was stirred for 0.5 h before adding amidoxime 19a–b (1.0 eq). The reaction mixture was stirred for 1 h at room temperature, then heated in a pre-heated 120 °C oil-bath. The reaction mixture was stirred for 4 h and monitored by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column.
tert-Butyl (4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)carbamate (20a)
Yield: 56%. MP: 158–160 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J = 9.0 Hz, 1H, -ArH), 8.30 (s, 1H, -ArH), 8.03 (d, J = 8.0 Hz, 2H, -ArH), 7.58 − 7.47 (m, 2H, -ArH&-NHCO), 7.44 (d, J = 8.0 Hz, 2H, -ArH), 4.88 − 4.82 (m, 1H, -CH2F), 4.76 − 4.70 (m, 1H, -CH2F), 4.61 − 4.55 (m, 1H, -CH2O-), 4.54 − 4.48 (m, 1H, -CH2O-), 4.22 (d, J = 5.9 Hz, 2H, -ArCH2-), 1.41 (s, 9H, -CH3). 13C NMR (101 MHz, DMSO-d6) δ 174.45, 168.52, 159.88, 156.27, 144.53, 134.49, 128.04, 127.54, 127.04 (q, J = 5.1 Hz), 123.33 (d, J = 273.7 Hz), 121.97, 118.56 (d, J = 30.3 Hz), 116.32, 115.40, 82.11 (d, J = 168.7 Hz), 78.39, 69.22 (d, J = 19.2 Hz), 43.66, 28.65.
tert-Butyl (2-((4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethyl)-carbamate (20b)
Yield: 42%. MP: 144–147 °C. 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H, -ArH), 8.35 (d, J = 8.7 Hz, 1H, -ArH), 8.13 (d, J = 8.1 Hz, 2H, -ArH), 7.38 (d, J = 7.8 Hz, 2H, -ArH), 7.18 (d, J = 8.8 Hz, 1H, - ArH), 4.92 − 4.86 (m, 1H, -CH2F), 4.80 − 4.75 (m, 1H, -CH2F), 4.56 (s, 2H, -ArCH2-), 4.49 − 4.44 (m, 1H, -CH2O-), 4.42 − 4.37 (m, 1H, -CH2OAr-), 3.80 − 3.69 (m, 2H, -OCH2-), 3.52 − 3.35 (m, 2H, -CH2NH-), 1.46 (s, 9H, -CH3).
General procedure for the synthesis of 21a–b
To a round-bottom flask equipped with a stir bar was added Boc-protected amine 20a–b (1.0 eq), followed by 4 M HCl in 1,4-dioxane (20 eq). The reaction mixture was stirred for 2 h and concentrated in vacuo. The resulting residue was suspended in a mixture of ethanol and tert-butyl methyl ether (V/V, 3/1), and the resulting solid was filtered to give the white solid product.
(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanamine hydrochloride (21a)
Yield: 77%. MP: 276–277 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 2H, -NH2), 8.39 (d, J = 8.4 Hz, 1H, -ArH), 8.29 (s, 1H, -ArH), 8.10 (d, J = 7.6 Hz, 2H, -ArH), 7.73 (d, J = 7.6 Hz, 2H, -ArH), 7.56 (d, J = 8.7 Hz, 1H, -ArH), 4.91 − 4.67 (m, 2H, -CH2F), 4.62 − 4.46 (m, 2H, -CH2O-), 4.12 (s, 2H, -CH2-). 13C NMR (101 MHz, DMSO-d6) δ 174.69, 168.31, 159.97, 138.18, 134.58, 130.23, 127.69, 127.10 (d, J = 5.1 Hz), 126.37, 123.34 (d, J = 273.7 Hz), 118.56 (d, J = 31.3 Hz), 116.24, 115.50, 82.13 (d, J = 168.7 Hz), 69.27 (d, J = 19.2 Hz), 42.24. HRMS (ESI) calcd for C18H15F4N3O2 [M + H+] 382.1173. Found [M + H+] 382.1169.
2-((4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)oxy)ethan-1-amine (21b)
Yield: 57%. MP: 266–267 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.47 − 8.35 (m, 2H, -ArH), 8.13 − 8.02 (m, 2H, -ArH), 7.65 − 7.49 (m, 3H, -ArH), 4.87 (d, J = 47.8 Hz, 2H, -CH2F), 4.61 (d, J = 28.7 Hz, 2H, -CH2O-), 3.89 (s, 2H, -ArCH2O-), 3.71 − 3.57 (m, 2H, -CH2O-), 2.78 − 2.66 (m, 2H, -CH2NH2). 13C NMR (101 MHz, Acetone-d6) δ 174.27, 168.67, 159.90, 145.17, 133.73, 128.82, 128.49, 127.16, 127.08, 126.86 (q, J = 5.1 Hz), 123.15 (d, J = 273.7 Hz), 119.18 (q, J = 31.3 Hz), 114.43, 81.54 (d, J = 169.7 Hz), 68.91 (d, J = 31.3 Hz), 60.86, 52.94, 51.25. HRMS (ESI) calcd for C20H19F4N3O3 [M + H+] 426.1435. Found [M + H+] 426.1432.
4.3. In vitro S1PRs binding assay
[32P]S1P was firstly prepared by incubating sphingosine and [γ-32P]ATP with sphingosine kinase 1 as we previously reported [20]. [32P]S1P was dissolved in DMSO, and then diluted in the assay buffer (50 mM HEPES-Na with 5 mM MgCl2, 1 mM CaCl2, and 0.5% fatty acid-free bovine serum albumin, pH = 7.5). Compounds were dissolved in DMSO and diluted to different concentrations with assay buffer, followed by adding commercial cell membranes expressing recombinant human S1PRs (1, 2, 3, 4, and 5) in the assay buffer at room temperature in 96-well plate. [32P]S1P solution was then added to give a final volume of 150 μL containing 0.1 nM of [32P]S1P and 1 μg of membrane protein per well. Competitive binding was performed for 60 min at room temperature and terminated by collecting the membranes onto 96-well glass fiber (GF/B) filtration plates (Millipore, Billerica, MA). Each filter was washed with 200 μL of assay buffer for five times. The filter bound radionuclide was measured by a Beckman LS3801 scintillation counter using Cherenkov counting. The reported IC50 values were calculated using the 4 parameter equation, least-square non-linear regression curve-fit, with GraphPad Prism software (GraphPad Software, Inc). Each assay was repeated at least three times with duplicate wells for each compound; the reported values (mean ± SD, nM) are calculated from the average of all assays. Assays for compounds which showed no activity (IC50 > 1000 nM) were only repeated twice.
4.4. Radiochemistry
4.4.1. Synthesis of precursor 25
4-(3-(4-(Hydroxymethyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (22) [18]
To a round-bottom flask equipped with a stir bar was added 4-hydroxy-3-(trifluoromethyl)benzoic acid (1.96 g, 9.51 mmol), HOBt (0.26g, 1.90 mmol), TBTU (3.05 g, 9.51 mmol), DMF (20 mL), and DIPEA (3.69 g, 28.5 mmol). The reaction mixture was stirred for 0.5 h before adding 8e (1.58 g, 9.51 mmol). The reaction mixture was stirred for 1 h at room temperature and refluxed in a pre-heated 120 °C oil-bath. The reaction mixture was stirred for 4 h and monitored by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column. (1.43 g, 45%)
4-(3-(4-(Azidomethyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (23)
To a round-bottom flask equipped with a stir bar was added 22 (1.39 g, 4.13 mmol), diphenyl phosphoryl azide (1.37 g, 4.96 mmol), and toluene (10.0 mL). The mixture was cooled to 0 °C before adding 1,8-Diazabicyclo[5.4.0]undec-7-ene (0.76, 4.96 mmol) dropwise. The reaction was warmed to room temperature slowly and stirred overnight. Then, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 M HCl, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column to give yellow solid (815 mg, 55%). MP: 158-161°C. 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H, -ArH), 8.22 (d, J = 8.5 Hz, 1H, -ArH), 8.17 (d, J = 7.8 Hz, 2H, -ArH), 7.47 (d, J = 7.9 Hz, 2H, -ArH), 7.09 (d, J = 8.6 Hz, 1H, -ArH), 4.44 (s, 2H, -CH2-).
2-(4-(3-(4-(Azidomethyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methylbenzene-sulfonate (24)
To a round-bottom flask equipped with a stir bar was added 23 (500 mg, 1.38 mmol), ethylene ditosylate (767 mg, 2.07 mmol), K2CO3 (953 mg, 6.90 mmol), and CH3CN (10.0 mL). The reaction mixture was refluxed in a pre-heated 90 °C oil-bath for 12 h. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the residue was purified on a silica gel column to give white solid (300 mg, 39%). MP: 169–170 °C. 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H, -ArH), 8.33 (d, J = 8.7 Hz, 1H, -ArH), 8.19 (d, J = 8.2 Hz, 2H, -ArH), 7.81 (d, J = 8.3 Hz, 2H, -ArH), 7.48 (d, J = 8.1 Hz, 2H, -ArH), 7.38 − 7.32 (m, 2H, -ArH), 7.10 (d, J = 8.7 Hz, 1H, -ArH), 4.45 (s, 2H, -ArCH2-), 4.43 − 4.38 (m, 4H, -OCH2CH2O-), 2.45 (s, 3H, -CH3).
2-(4-(3-(4-((4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)methyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methylbenzenesulfonate (25)
To a round-bottom flask equipped with a stir bar was added azide 24 (243 mg, 0.43 mmol) and THF (2.0 mL). Once the azide was dissolved, water (1.1 mL) was added followed by 3-(methoxymethoxy)prop-1-yne (46 mg, 0.46 mmol), sodium ascorbate (8.5 mg, 0.043 mmol), and copper sulfate pentahydrate (6.4 mg, 0.026 mmol). The reaction mixture was stirred for 4 h and monitored by TLC. Then, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, a white solid was obtained (100 mg, 35%). MP: 168–171°C. 1H NMR (400 MHz, CD3OD) δ 8.35 − 8.29 (m, 2H, -ArH), 8.11 (d, J = 7.2 Hz, 2H, -ArH), 8.02 (s, 1H, -ArH), 7.74 (d, J = 7.3 Hz, 2H, -ArH), 7.45 (d, J = 7.5 Hz, 2H, -ArH), 7.36 (d, J = 8.0 Hz, 2H, -ArH), 7.31 (d, J = 8.4 Hz, 1H, -ArH), 5.67 (s, 2H, -ArCH2-), 4.65 − 4.61 (m, 4H, -CH2OCH2O-), 4.43 - 4.34 (m, 4H, -OCH2CH2O-), 3.27 (s, 3H, -OCH3), 2.37 (s, 3H, -ArCH3).
4.4.2. Radiosynthesis of [18F] 12b
[18F]KF (~7.4 GBq) aqueous was added to a vial containing Kryptofix 222 (~6 mg), and dried by azeotropic evaporation with CH3CN (3 × 1 mL) under N2 flow at 110 °C. To the reaction vial was added precursor 25 (2 mg) as a solution in CH3CN (300 μL). The reaction was placed in a 110 °C oil-bath for 15 min. The reaction was removed from the oil-bath, at which time 6 M HCl (150 μL) was added. The reaction mixture was heated in a 110 °C oil-bath for another 5 min. The reaction was removed from the oil-bath and quenched with 6 M NaOH (150 μL) and diluted with 2.4 mL of the HPLC mobile phase (45% CH3CN in 0.1 M ammonium formate buffer, pH = 4.5). The mixture was passed through a Sep-Pak® Alumina N Cartridge (Part No. WAT020510) and injected onto the semi-preparation HPLC column (Agilent SB-C18 250 × 9.6 mm, 5 μm, UV = 254 nm, 4.0 mL/min). The retention time of [18F]12b is around 19–22 min. The HPLC fraction was collected into a water bottle with 60 mL of water and then trapped on a Sep-Pak® C-18 Cartridge (Part No. WAT020515). The activity (~740 MBq) was washed out with 0.6 mL of ethanol and 5.4 mL of 0.9% saline. After sterile filtration into a glass vial, the [18F]12b was ready for quality control (QC) analysis and animal studies. An aliquot of sample was injected onto an analytical HPLC to determine the concentration of tracer. Meanwhile, the tracer was authenticated by co-injected with non-radiolabeled standard 12b sample solution. The HPLC condition are as follows: Agilent Zorbax SB-C18 column (250 × 4.6 mm); UV absorbance at 254 nM; the mobile phase is consisted of 45% CH3CN in 0.1 M ammonium formate buffer, pH = 4.5; the flow rate is 1.0 mL/min; the retention time of [18F] 12b is 5.7 min.
4.5. Ex vivo autoradiography study
An adult male SD rat (~450 g, Charles River) was injected with [18F]12b (~55 MBq) via the tail vein under 2–3% isoflurane/oxygen anesthesia and euthanized at 60 min post-injection. The brain was immediately removed and snap-frozen over dry ice followed by sectioning at 100 μm and mounting on glass slides. Frozen slides were directly exposed to a BAS storage phosphor screen film (BAS-IP-MS-2025) in an imaging cassette (Fuji Photo Film Co., Tokyo, Japan) for 12 h at −80 °C under the dark. The distribution of radioactivity was visualized by a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film Co., Tokyo, Japan).
4.6. In vitro autoradiography study in LPS-treated mice
Adult male C57BL/6 mice (20–25 g, Charles River Inc., Frederick, MD) were used to generate LPS-induced neuroinflammation murine model. The mice (n = 2) received an intraperitoneal injection of LPS from E. coli 055:B5 (Sigma-Aldrich, St. Louis, MO, USA) in saline (3 mg/mL) at dose of 15 mg/kg (5 mL/kg) and then euthanized at 24 h post-injection. Mouse brains were collected and cut into 20 μm sequential sections using a Microm cryotome and mounted on glass slides. For the control study, the slides were incubated with ~6 nM of [18F]12b at room temperature for 60 min. For the blocking studies, slides were incubated with [18F]12b in the presence of 10 μM of SEW2871 at room temperature for 60 min. Following the incubation, the brain sections were washed and exposed to the Storage Phosphor Screen in an imaging cassette overnight in −20° C at dark for 12 h. The distribution of radioactivity were visualized by a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film, Tokyo, Japan). Photo-stimulated luminescence (PSL) from the brain slices was quantified using Multi Gauge v3.0 software (Fuji Photo Film Co., Tokyo, Japan). Data were background-corrected, and expressed as photo-stimulated luminescence signals per square millimeter (PSL/mm2).
4.7. Biodistribution study
A solution of [18F]12b (~2.2 MBq/100 μL) in 10% ethanol in 0.9% saline was injected via the tail vein into adult female Lewis rats (n = 16, 120–140 g, Charles River) under 2–3% isoflurane/oxygen anesthesia. The rats were euthanized under anesthesia at 5, 30, 60, and 120 min post-injection (n = 4 for each group). The whole brains were quickly harvested and dissected into regions of cerebellum, brain stem, cortex, striatum, thalamus, and hippocampus, the remainder of the brain was also collected to determine the total brain uptake. Organs of blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, thymus, and bone were also dissected and collected for counting. All the samples were counted with a dilution of the injectate on an automated well counter (Beckman Gamma 8000 well counter). Tissues were weighed and the uptake was reported as background and decay-corrected percent injected dose per gram (% ID/g).
Supplementary Material
Compound 12b showed high potency (9.7 nM) and selectivity toward S1PR1 over S1PR2-5 (> 100-fold).
[18F]12b was radiosynthesized with good radiochemical yield, high purity, and specific activity.
[18F]12b was able to cross the blood-brain barrier with high brain uptake.
In vitro autoradiography study in LPS-induced neuroinflammation murine model indicated that [18F]12b has specific binding toward S1PR1.
Acknowledgments
This work was supported by the USA National Institutes of Health through the National Institute of Neurological Disorders and Stroke, and the National Institute of Aging [NS075527 and NS103988] and the National Institute of Mental Health [MH092797]. This work also partially supported by USA Department of Energy Training Grant titled “Training in Techniques and Translation: Novel Nuclear Medicine Imaging Agents for Oncology and Neurology” [DE-SC0008432]. Mass spectrometry was generated from the Washington University Mass Spectrometry facility that is supported by National Institutes of Health [8P41GM103422].
Abbreviations
- S1PR1
Sphingosine-1-phosphate receptor 1
- MS
Multiple sclerosis
- FDA
Food and Drug Administration
- PET
Positron emission tomography
- LPS
Lipopolysaccharides
- CNS
Central nervous system
- SAR
Structure activity relationship
- BBB
Blood-brain barrier
- RT
Room temperature
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- HOBt
Hydroxybenzotriazole hydrate
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DIPEA
N,N-Diisopropylethylamine
- DPPA
Diphenyl phosphoryl azide
- Boc
tert-Butyloxycarbonyl
- MW
Molecular weight
- ClogP
Calculated partition coefficient
- TPSA
Topological polar surface area
- HBD
Hydrogen bond donor
- HPLC
High-performance liquid chromatography
- SD
Sprague-Dawley
- TLC
Thin-layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kunkel GT, MacEyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garris CS, Blaho VA, Hla T, Han MH. Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology. 2014;142:347–353. doi: 10.1111/imm.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montrose DC, Scherl EJ, Bosworth BP, Zhou XK, Jung B, Dannenberg AJ, Hla T. S1P1 localizes to the colonic vasculature in ulcerative colitis and maintains blood vessel integrity. J Lipid Res. 2013;54:843–851. doi: 10.1194/jlr.M034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeshita H, Kitano M, Iwasaki T, Kitano S, Tsunemi S, Sato C, Sekiguchi M, Azuma N, Miyazawa K, Hla T, Sano H. Sphingosine 1-phosphate (S1P)/S1P receptor 1 signaling regulates receptor activator of NF-κB ligand (RANKL) expression in rheumatoid arthritis. Biochem Biophys Res Commun. 2012;419:154–159. doi: 10.1016/j.bbrc.2012.01.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karuppuchamy T, Behrens EH, Gonzalez-Cabrera P, Sarkisyan G, Gima L, Boyer JD, Bamias G, Jedlicka P, Veny M, Clark D, Peach R, Scott F, Rosen H, Rivera-Nieves J. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017;10:162–171. doi: 10.1038/mi.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nofer JR, Bot M, Brodde M, Taylor PJ, Salm P, Brinkmann V, van Berkel T, Assmann G, Biessen EA. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2007;115:501–508. doi: 10.1161/CIRCULATIONAHA.106.641407. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Jin H, Han J, Yue X, Yang H, Zayed MA, Gropler RJ, Tu Z. Upregulated sphingosine 1-phosphate receptor 1 expression in human and murine atherosclerotic plaques. Mol Imaging Biol. 2017 doi: 10.1007/s11307-017-1141-3. http://doi.org/10.1007/s11307-11017-11141-11303. [DOI] [PMC free article] [PubMed]
- 10.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 11.Chun J, Hartung HP. Mechanism of action of oral Fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das BK. Development of Positron Emission Tomography (PET): A Historical Perspective. In: Das BK, editor. Positron Emission Tomography: A Guide for Clinicians. Springer India; New Delhi: 2015. pp. 7–11. [Google Scholar]
- 13.Liu H, Jin H, Yue X, Luo Z, Liu C, Rosenberg AJ, Tu Z. PET imaging study of S1PR1 expression in a rat model of multiple sclerosis. Mol Imaging Biol. 2016;18:724–732. doi: 10.1007/s11307-016-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin H, Yang H, Liu H, Zhang Y, Zhang X, Rosenberg AJ, Liu Y, Lapi SE, Tu Z. A promising carbon-11-labeled sphingosine-1-phosphate receptor 1-specific PET tracer for imaging vascular injury. J Nucl Cardiol. 2017;24:558–570. doi: 10.1007/s12350-015-0391-1. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Jin H, Yue X, Han J, Baum P, Abendschein DR, Tu Z. PET study of sphingosine-1-phosphate receptor 1 expression in response to vascular inflammation in a rat model of carotid injury. Mol Imaging. 2017;16:1–7. doi: 10.1177/1536012116689770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasad VP, Wagner S, Keul P, Hermann S, Levkau B, Schäfers M, Haufe G. Synthesis of fluorinated analogues of sphingosine-1-phosphate antagonists as potential radiotracers for molecular imaging using positron emission tomography. Bioorg Med Chem. 2014;22:5168–5181. doi: 10.1016/j.bmc.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Shaikh RS, Schilson SS, Wagner S, Hermann S, Keul P, Levkau B, Schäfers M, Haufe G. Synthesis and evaluation of fluorinated fingolimod (FTY720) analogues for sphingosine-1-phosphate receptor molecular imaging by positron emission tomography. J Med Chem. 2015;58:3471–3484. doi: 10.1021/jm502021d. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg AJ, Liu H, Jin H, Yue X, Riley S, Brown SJ, Tu Z. Design, synthesis, and in vitro and in vivo evaluation of an 18F-labeled sphingosine 1-phosphate receptor 1 (S1P1) PET tracer. J Med Chem. 2016;59:6201–6220. doi: 10.1021/acs.jmedchem.6b00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilmore JL, Sheppeck JE, Watterson SH, Haque L, Mukhopadhyay P, Tebben AJ, Galella MA, Shen DR, Yarde M, Cvijic ME, Borowski V, Gillooly K, Taylor T, McIntyre KW, Warrack B, Levesque PC, Li JP, Cornelius G, D’Arienzo C, Marino A, Balimane P, Salter-Cid L, Barrish JC, Pitts WJ, Carter PH, Xie J, Dyckman AJ. Discovery and structure–activity relationship (SAR) of a series of ethanolamine-based direct-acting agonists of sphingosine-1-phosphate (S1P1) J Med Chem. 2016;59:6248–6264. doi: 10.1021/acs.jmedchem.6b00373. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg AJ, Liu H, Tu Z. A practical process for the preparation of [32P]S1P and binding assay for S1P receptor ligands, Appl. Radiat Isot. 2015;102:5–9. doi: 10.1016/j.apradiso.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery, ACS Chem. Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wager TT, Hou X, Verhoest PR, Villalobos A. Central nervous system multiparameter optimization desirability: application in drug discovery, ACS Chem. Neurosci. 2016;7:767–775. doi: 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- 23.Wager TT. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem. 2015;58:2584–2608. doi: 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- 25.Rankovic Z. CNS physicochemical property space shaped by a diverse set of molecules with experimentally determined exposure in the mouse brain. J Med Chem. 2017;60:5943–5954. doi: 10.1021/acs.jmedchem.6b01469. [DOI] [PubMed] [Google Scholar]
- 26.Toman RE, Spiegel S. Lysophospholipid receptors in the nervous system. Neurochem Res. 2002;27:619–627. doi: 10.1023/a:1020219915922. [DOI] [PubMed] [Google Scholar]
- 27.Ohuchi H, Hamada A, Matsuda H, Takagi A, Tanaka M, Aoki J, Arai H, Noji S. Expression patterns of the lysophospholipid receptor genes during mouse early development. Dev Dyn. 2008;237:3280–3294. doi: 10.1002/dvdy.21736. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura H, Akiyama T, Irei I, Hamazaki S, Sadahira Y. Cellular localization of sphingosine-1-phosphate receptor 1 expression in the human central nervous system. J Histochem Cytochem. 2010;58:847–856. doi: 10.1369/jhc.2010.956409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kono M, Conlon EG, Lux SY, Yanagida K, Hla T, Proia RL. Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nat Commun. 2017;8:1163. doi: 10.1038/s41467-017-01340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong J, Wang H, Wu G, Zhao J, Zhang L, Zuo L, Zhu W, Gong J, Li Y, Gu L, Li J. Oral treatment with SEW2871, a sphingosine-1-phosphate type 1 receptor agonist, ameliorates experimental colitis in interleukin-10 gene deficient mice. Clin Exp Immunol. 2014;177:94–101. doi: 10.1111/cei.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.