

Abstract
Identifying gene-environment interactions is a central challenge in the quest to understand susceptibility to complex, multi-factorial diseases. Developing an understanding of how inter-individual variability in inherited genetic variation alters the effects of environmental exposures will enhance our knowledge of disease mechanisms and improve our ability to predict disease and target interventions to high-risk sub-populations. Limited progress has been made identifying gene-environment interactions in the epidemiological setting using existing statistical approaches for genome-wide searches for interaction. In this paper, we describe a novel two-step approach using omics data to conduct genome-wide searches for gene-environment interactions. Using existing genome-wide SNP data from a large Bangladeshi cohort study specifically designed to assess the effect of arsenic exposure on health, we evaluated gene-arsenic interactions by first conducting genome-wide searches for SNPs that modify the effect of arsenic on molecular phenotypes (gene expression and DNA methylation features). Using this set of SNPs showing evidence of interaction with arsenic in relation to molecular phenotypes, we then tested SNP-arsenic interactions in relation to skin lesions, a hallmark characteristic of arsenic toxicity. With the emergence of additional omics data in the epidemiologic setting, our approach may have the potential to boost power for genome-wide interaction research, enabling the identification of interactions that will enhance our understanding of disease etiology and our ability to develop interventions targeted at susceptible sub-populations.
Keywords: gene-environment interaction, arsenic, gene expression, DNA methylation, genome-wide
INTRODUCTION
Exposure to arsenic is a serious global public health issue. More than 200 million people worldwide, including approximately 77 million in Bangladesh and 17 million in the U.S., consume drinking water contaminated with arsenic at levels associated with adverse health effects and shortened lifespan (1, 2). Additionally, food-derived inorganic arsenic exposure is also an emerging health concern (3–7). Epidemiologic research has established arsenic exposure as a risk factor for cancers of the skin, lung, bladder, kidney, liver, and possibly prostate (8). Arsenic has also been associated with increased risk of cardiovascular diseases (9–11), non-malignant respiratory diseases (12), diabetes mellitus (13–15), and impaired cognitive function (16).
Skin is a major target organ of arsenic, with skin lesions a hallmark characteristic of chronic exposure and an early manifestation of arsenic toxicity (17). A dose-response relationship between arsenic exposure and skin lesions is well-established (18). However, arsenic exposure itself fails to fully explain the presence of arsenical skin lesions in exposed populations, and inter-individual variability in susceptibility due to inherited genetic variation may play an important role in determining risk (19–21). Variation in the 10q24.32 region (containing arsenic methyltransferase; AS3MT) is associated with arsenic metabolism efficiency (22), and these variants show clear additive interaction with arsenic exposure in relation to skin lesion risk (23). However, other than 10q24.32, there are no other established arsenic susceptibility regions (21).
Identifying gene-environment (GxE) interactions is a central challenge to understand susceptibility to complex, multi-factorial diseases (24). Inter-individual variability in the effects of environmental exposures may be influenced by inherited genetic variation (25). Unfortunately, limited progress has been made identifying GxE interactions in the epidemiological setting using genome-wide searches for interaction (26). In the current study, we describe novel functional approaches using existing genome-wide genotype, gene expression, and DNA methylation data from a large Bangladeshi cohort study specifically designed to assess the effect of arsenic exposure on health. We evaluate gene-arsenic interactions by conducting genome-wide searches for genetic variants that modify the effect of arsenic on molecular phenotypes and then test SNP-arsenic interactions in relation to skin lesion status.
METHODS
Participants
The study sample consists of 5,354 Bangladeshi adults with existing data on arsenic exposure (measured in urine), genome-wide SNP genotypes, and clinical phenotype data. Among these participants, 3,364 are from the Health Effects of Arsenic Longitudinal Study (HEALS) and 1,990 are from the Bangladesh Vitamin E and Selenium Trial (BEST). Selected characteristics of the study participants are shown in Table 1. Additional molecular data is available for a subset of BEST participants, with array-based genome-wide gene expression data on 1,800 participants and array-based epigenome-wide DNA methylation data on 400 participants.
Table 1.
Characteristics of genotyped study participants
| HEALS | BEST | BEST subset with gene expression data | BEST subset with DNA methylation data | |
|---|---|---|---|---|
| N | 3364 | 1990 | 1799 | 400 |
| Male (%) | 45.1 | 54.0 | 55.1 | 52.8 |
| Age in years, Mean (SD) | 37.8 (10.7) | 43.4 (10.6) | 43.5 (10.6) | 43.5 (10.2) |
| Urinary total arsenic adjusted for creatinine in μg/g, Mean (SD) | 258.3 (290.8) | 351.0 (482.8) | 336.7 (437.7) | 303.3 (364.9) |
| Skin lesion case, N (%) | 15.0 | 100.0* | 100.0* | 100.0* |
SD, standard deviation
BEST participants all had skin lesions at baseline. These participants were not included in the Step 2 GxE analyses, due to of lack of variation in the skin lesion phenotype in that cohort.
HEALS is a prospective cohort study originally consisting of 11,746 men and women from Araihazar, Bangladesh, a rural area in which the primary source of drinking water is groundwater provided by hand-pumped tube wells. A large proportion of these wells access groundwater that is naturally contaminated with elevated levels of inorganic arsenic. Participants were recruited between October 2000 and May 2002. HEALS was designed to evaluate the long- and short-term effects of arsenic consumed in drinking water and has been described extensively elsewhere (27). Demographic data, lifestyle data, and urine and blood samples were collected at baseline interviews. The size of the HEALS cohort was increased in 2006-2008, with an additional 8,287 participants added.
BEST is a randomized chemoprevention trial of 7,000 participants from Araihazar, Matlab, and surrounding areas. All participants have skin lesions associated with arsenic exposure. The study was created to evaluate vitamin E and selenium supplementation on non-melanoma skin cancer risk (28). Participant randomization was initiated in 2006. Demographic and lifestyle data and blood samples were collected at baseline.
Informed consent was obtained from all participants. All study procedures were approved by the University of Chicago and Columbia University Institutional Review Boards and the Ethical Committees of the Bangladesh Medical Research Council and the International Center for Diarrhoeal Disease Research, Bangladesh (ICDDR,B).
Genotype data
DNA extraction for genotyping was carried out from the whole blood using the QIAamp 96 DNA Blood Kit (cat # 51161) from Qiagen, Valencia, USA. Concentration and quality of all extracted DNA were assessed using Nanodrop 1000. Samples were processed on Illumina HumanCytoSNP-12 v2.1 chips with 299,140 markers and read on the BeadArray Reader. Image data was processed in BeadStudio software to generate genotype calls.
Quality control was conducted as described previously for 5,499 individuals typed for 299,140 SNPs (23, 29). We removed DNA samples with call rates <97% (n = 13), gender mismatches (n = 79), as well as technical duplicates (n = 53). We removed SNPs that were poorly called (<90%) or monomorphic (n = 38,753), and then removed SNPs with call rates <95% (n = 1,045) or HWE p-values<10−10 (n = 634, which produces no HWE p-values <10−7 in a subset of 1,842 unrelated participants). This QC resulted in 5,354 individuals with high-quality genotype data for 257,747 SNPs. The MaCH software (30) was used to conduct genotype imputation using 1,000 genomes reference haplotypes (1KG phase3 v5, which includes South Asian populations). Only high-quality autosomal imputed SNPs (imputation r2>0.5) with MAF>0.01 were included in this analysis, yielding 8,512,165 imputed SNPs.
Gene Expression Data
Genome-wide mRNA expression data has been generated for a subset of 1,799 BEST participants using Illumina’s HumanHT-12-v4 chip (47,231 transcripts covering 31,335 genes). RNA was extracted from mononuclear cells preserved in RLT buffer, stored at −86°C, using Qiagen RNeasy Micro Kit (cat# 74004). Concentration and quality of extracted RNA were assessed on Nanodrop 1000. cRNA synthesis was done from 250 ng of RNA using Illumina TotalPrep 96 RNA Amplification kit.
DNA Methylation Data
Genome-wide DNA methylation data was generated for a subset of 400 BEST participants using Illumina’s HumanMethylation450 BeadChip (485,577 CpG-sites, including consensus coding sequences, miRNA promoter regions, and disease-related and imprinted genes) (31). Bisulphite conversion of genomic DNA was performed using the Zymo’s EZ DNA Methylation Kit. The assay was conducted using 500 ng of bisulfite-converted DNA per sample. This data has recently been used to identify CpG sites at which DNA methylation is associated with arsenic exposure (32).
Arsenic Exposure Data
Urinary total arsenic concentration is a good biomarker of aggregate ingested arsenic exposure, and captures exposure from all sources including water, food, soil, and dust (33). All study participants have existing urinary total arsenic data available, measured from a spot urine sample by graphite furnace atomic absorption spectrometry, with a detection limit of 2 μg/L, in a single laboratory (Trace Metals Core Facilities Laboratory at Columbia University) (34). Urinary creatinine concentration has also been measured in the same laboratory by a colorimetric method based on the Jaffe reaction (35). Urinary total arsenic was divided by creatinine to obtain a creatinine-adjusted urinary total arsenic concentration, expressed as μg/g creatinine.
Analytical Strategy
The general analysis approach is described in Figure 1. As described in detail below, we used a two-step approach in an attempt to 1) identify SNPs that modify the effect of arsenic on molecular (“omic”) phenotypes (i.e., gene expression and DNA methylation phenotypes, measured genome-wide) and 2) use this set of SNPs to test SNP-arsenic interactions in relation to skin lesion status, the classical sign of arsenic toxicity.
Figure 1. Analysis approach.
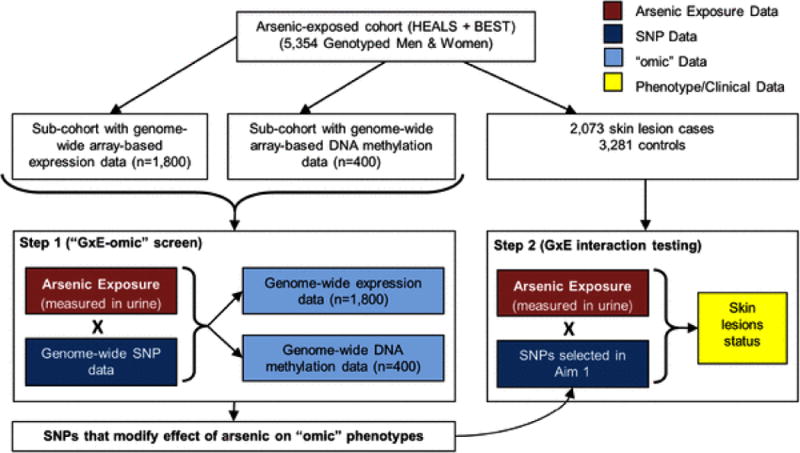
A two-stage “GxE-omics” approach for detecting GxE
Step 1: Identifying SNPs that interact with arsenic to influence molecular “-omic” phenotypes
To identify SNPs that interact with arsenic to influence specific genome features (i.e., individual transcripts or CpGs), we first identified a set of transcripts and a set of CpG methylation sites that show strong evidence of association with arsenic exposure, based on Bonferroni (and/or FDR correction). This was done based largely on our previously reported results on this topic (32). For each arsenic-associated feature (i.e., CpG or transcript), we then conducted a genome-wide search for SNPs that modify the association between arsenic and the selected feature. In order to identify SNPs that modify the effect of arsenic on multiple features, we conducted principal component analysis (PCA) of the arsenic-associated features, and derived arsenic-associated PCs representing the impact of arsenic on the “biological system” represented by the features. These PCs were then used as outcome variables to conduct genome-wide screens for SNPs that modify the effect of arsenic on the biological system, as represented by PCs.
We also conducted genome-wide cis-eQTL (expression quantitative trait loci) and cis-meQTL (methylation QTL) analyses and examined statistical interaction between arsenic exposure and the lead cis-SNP in relation to the transcript or CpG affected by the SNP. To identify eQTLs and meQTLs, we leveraged results from our recent studies of eQTLs and meQTLs in the BEST study (36, 37). At a false-discovery rate (FDR) of 0.01, we observed 7,643 cis-eQTLs and 84,853 cis-meQTLs. Using the lead SNP for each observed eQTL and meQTL, we testing SNP-arsenic interaction in relation to the associated transcript or CpG.
Step 2: Identifying SNPs that interact with arsenic to influence arsenic-related disease
Each SNP identified in step 1 was tested for interaction with arsenic in relation to arsenic-induced skin lesions, the most common sign of arsenic toxicity. This SNP-arsenic interaction analysis was restricted to 503 incident skin lesion cases and 2,493 lesion-free controls from HEALS. For each SNP identified in step 1, we also tested the marginal association between the SNP and skin lesion status, using data on the combined HEALS and BEST studies (2,493 skin lesion cases and 2,861 controls).
Regression Modelling Approach (for both steps)
Our study participants reside in a relatively small geographic area, and some participants are related to other participants. Rather than exclude relatives, we use mixed effects models to account for relatedness. Such models have been developed for the GWA setting, including the software GEMMA. All regression analyses were conducted using the GEMMA software package (38). Regression models for detection of GxE include marginal effects for the genetic variant (coded as 0, 1, or 2 minor alleles) and for arsenic (log-transformed continuous), as well as an interaction term that is the product of these two variables. Arsenic exposure was also modeled as an ordinal and a dichotomous variable in supplementary analyses, to insure our interactions findings are not affected by mis-modelling the effect of the exposure on the outcome. All regression models are adjusted for sex and age (continuous). For binary outcomes (step 2), a linear mixed model treating binary outcomes as continuous variables was used. To approximate the corresponding odds ratio (OR), the beta coefficient was first divided by [x(1 − x)], where x is the proportion of cases in the analysis sample, in order to estimate the beta from a logistic model. This quantity was exponentiated to obtain an OR.
GxE analysis of SNPs that are eQTLs in skin
In addition to the two-step analyses described above, we also studied the effects of SNPs known to be eQTLs in skin on skin lesion risk. Based on eQTL results from the GTEx project (Genotype-Tissue Expression Project) we selected the lead SNP for each eQTL observed in sun-unexposed skin (suprapubic) and sun-exposed skin (lower leg). These tissue types had 5,491and 8,567 eQTLs reported by GTEx respectively, based on FDR<0.05. After combining these two lists, excluding redundant eSNPs, and restricting to SNP with a MAF>5% in our population, we were left with 9,952 SNPs that were eSNPs in skin tissue. Restricting to these SNPs, we conducted SNP-arsenic interaction analyses in relation to incident skin lesions. We modeled urinary arsenic as a continuous exposure (log-transformed), and ordinal variable based on quartiles, as well as a binary variable defined by the median value.
RESULTS
SNP × Arsenic Interaction for Arsenic-associated CpG sites (Step 1)
Based on our prior epigenome-wide association study (EWAS) of arsenic exposure and DNA methylation (32), we selected four CpG sites associated with urinary arsenic with Bonferroni significance (P<1 × 10−7) (Table 2). In genome-wide searches for SNPs showing evidence of interaction with arsenic in relation to these four phenotypes, we identified one region (on chromosome 11) showing modest evidence of SNP-arsenic interaction. SNPs in this region (lead SNP 11:43330815) showed evidence of interaction with arsenic in relation to cg01225779, a CpG on chromosome 5 (Figure 2). For these analyses, arsenic was treated as dummy variable with a cut point at the median, due to inflation in the GxE test statistics observed when arsenic was treated as a continuous variable.
Table 2.
CpG sites identified in epigenome-wide association study of creatinine-adjusted urinary total arsenic concentration
| CpG | Gene* | Beta | P |
|---|---|---|---|
| cg04605617 | PLA2G2C | 0.054 | 3.40 × 10−11 |
| cg01225779 | SQSTM1 | −0.048 | 2.37 × 10−9 |
| cg06121226 | SLC4A4 | −0.059 | 1.16 × 10−8 |
| cg13651690 | IGH | 0.039 | 9.16 × 10−8 |
gene assigned based on Illumina’s annotation file
Figure 2.
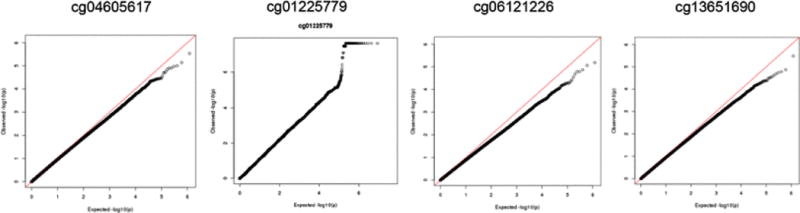
Genome-wide SNPxArsenic analyses for four arsenic-responsive CpG probes
SNP × Arsenic Interaction for Arsenic-associated DNA Methylation Patterns (Step 1)
Following PCA of these four arsenic-associated CpG sites, we observed that the first PC (PC 1) was strongly associated with arsenic exposure (r=0.45; P<0.0001) (Figure 3). Using this PC as a proxy for “epigenetic response to arsenic”, our genome-wide search for SNPs that modify the association of arsenic with this “epigenetic response” variable did not identify any SNPs with clear main effects or GxE effects on the constructed PC (Figure 4).
Figure 3.
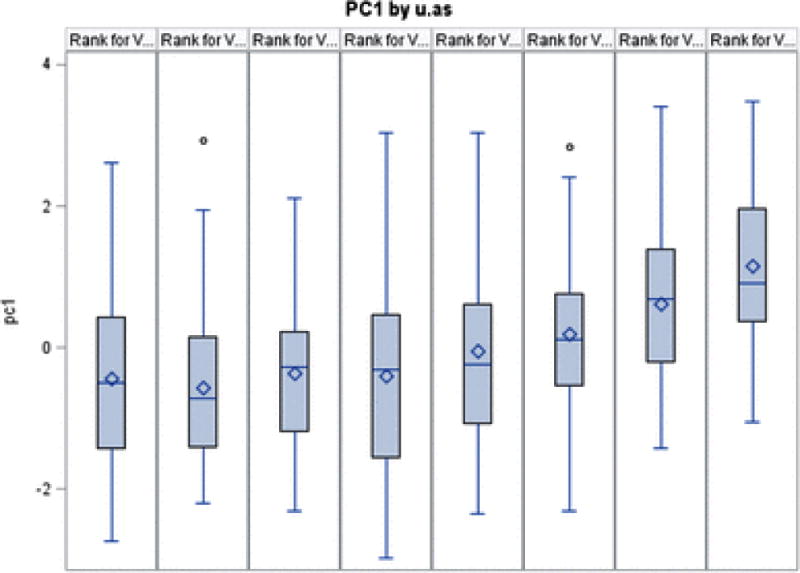
The distribution of the PC representing epigenomic response to arsenic, stratified by arsenic octiles.
Figure 4.
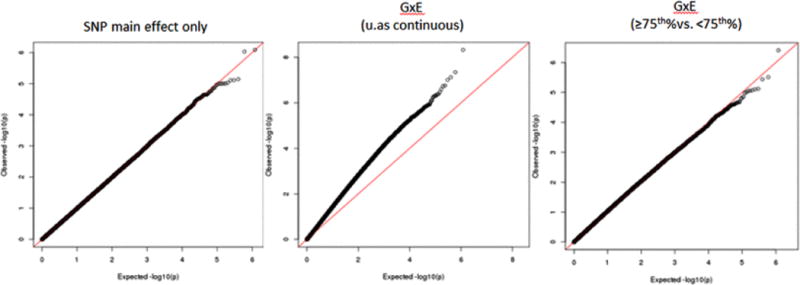
Quantile-quantile plots of p-values for the interaction between genome-wide SNPs and a PC representing the epigenome response to arsenic.
SNP × Arsenic Interaction Analysis for Arsenic-Associated Transcripts (Step 1)
Based on a genome-wide search for association between urinary arsenic and gene expression levels, we selected 4,056 arsenic-associated genes (FDR = 0.05). Following PCA of these arsenic-associated genes, we identified 45 arsenic-associated PCs that we then used as proxies for “transcriptome response to arsenic”. We searched the genome for SNPs that modified the association between urinary arsenic and these PCs, and only three of the PCs showed a P-value<5×10−8 (Figure 5). The lead SNPs best representing these signals were rs17060130 on chromosome 6 and rs12105595 on chromosome 2.
Figure 5.
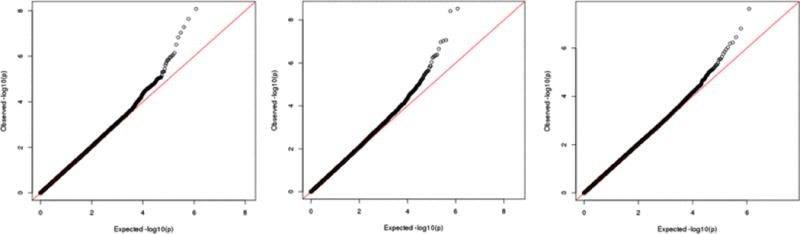
Genome-wide SNP-arsenic interaction analyses for three selected principle components (PCs 51, 64, and 73) representing gene expression patterns.
SNP × Arsenic Interaction for cis-meQTLs and cis-eQTLs (Step 1)
Among the 7,643 cis-eQTLs identified in this dataset, we tested GxE to determine if arsenic modified the association between the lead eSNP and its associated transcript. Among these eQTLs, three eSNPs showed P-values surpassing Bonferroni correction (Figure 6). Among the 84,853 cis-meQTLs identified in this dataset, none showed strong evidence of interaction with urinary arsenic (Figure 6). This null result included the arsenic-associated CpGs, of which three of the four had a meQTL (cg06121226 and two others).
Figure 6.
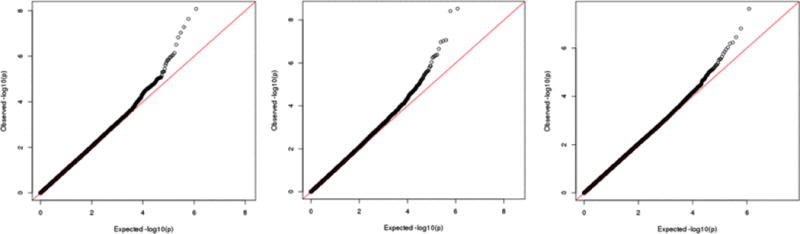
Quantile-quantile plots of p-value for SNP-arsenic interaction for all observed eQTLs (left) and all meQTLs (right). Arsenic was coded as a dummy variable based on the median exposure.
Step 2: Testing identified SNPs for interaction with arsenic in relation to skin lesion status
Despite the fact that none of our analyses in step 1 provided strong evidence for any specific SNP, we selected the six SNPs showing modest evidence of association based on step 1 analyses. We then tested each SNP for SNP-arsenic interaction in relation to skin lesion case/controls status and for marginal association with skin lesions status. In the analyses of interaction (Table 3), none of the SNPs showed a nominally significant P-value of interaction (P<0.05). This was true in both models with exposure modelled as a continuous variable (log-transformed urinary arsenic) and an ordinal variable (urinary arsenic quartiles). Similarly, in analyses of SNPs’ marginal association with skin lesion status, no SNP showed evidence of association.
Table 3.
Results for association and SNP-arsenic interaction in relation to skin lesions status for SNPs selected in Step 1
| SNP Groups | SNP | Chr | minor allele | MAF | OR | 95% CI | Association P (marginal)1 |
Interaction P (log arsenic)2 |
Interaction P (ordinal arsenic)2 |
|---|---|---|---|---|---|---|---|---|---|
| As-associated CpG analysis | rs6672956 | 1 | C | 0.161 | 1.03 | (0.93,1.14) | 0.56 | 0.70 | 0.37 |
|
| |||||||||
| PCA of As-associated gene expression | rs12105595 | 2 | A | 0.015 | 1.04 | (0.77,1.41) | 0.80 | 0.14 | 0.17 |
| rs10508649 | 10 | C | 0.013 | 1.15 | (0.85, 1.55) | 0.37 | 0.26 | 0.31 | |
| rs697216 | 12 | C | 0.499 | 0.96 | (0.89, 1.02) | 0.21 | 0.24 | 0.64 | |
|
| |||||||||
| cis-eQTL analysis | rs2409780 | 8 | C | 0.425 | 1.02 | (0.94, 1.1) | 0.70 | 0.50 | 0.42 |
| rs76363669 | 11 | C | 0.013 | 0.81 | (0.59, 1.11) | 0.20 | 0.28 | 0.29 | |
| rs6858440 | 4 | G | 0.364 | 1.04 | (0.96, 1.12) | 0.34 | 0.25 | 0.46 | |
Marginal associations estimated using 2,493 skin lesion cases and 2,861 controls from HEALS and BEST.
Interactions estimated 503 incident skin lesion cases and 2,493 lesion-free controls from HEALS. Urinary arsenic adjusted for creatinine was treated as natural logarithm transformed continuous variable and quartile ordinal variable. All estimates are from linear mixed model adjusting for age, sex and genotyping batches, accounting for relatedness using GEMMA package.
GxE analysis of SNPs that are eQTLs in skin
We tested SNP-arsenic interaction in relation to incident skin lesions for 9,952 SNPs that are eQTLs in skin tissue based on results from GTEx. However, none of these analyses identified SNPs showing a striking interaction with exposure (Figure 7). When we analyzed all SNPs in the genome for evidence of SNP-arsenic interaction in relation to skin lesion status, we observe a suggestive signal on chromosome 1, with the lead SNP being an intergenic SNP residing between the SNX7 gene and the LPPR5 gene (Figure 8). SNX7 is involved in intracellular trafficking, but its exact function is unknown. LPPR5 is involved in converting phosphatidic acid to diacylglycerol, glycerolipid synthesis, and receptor-activated signal transduction mediated by phospholipase D. Lead SNP rs6659080 is reported as an eQTL in only two tissues (according to GTEx); it is associated with LPPR5 expression in testicular tissue and SNX7 expression in aorta. However, it should be noted that rs6659080 is in strong LD with dozens of nearby SNPs (Figure 8).
Figure 7.
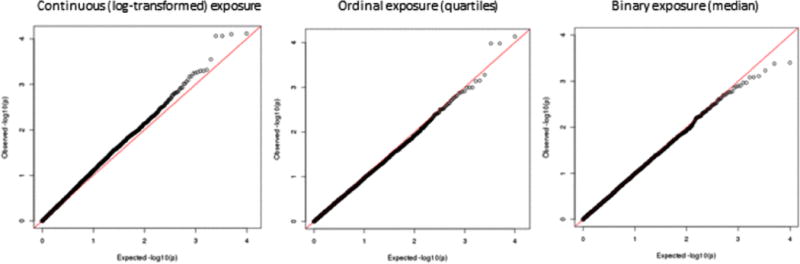
Quantile-quantile plots of the p-values for SNP-arsenic interaction in relation to skin lesion risk for 9,952 SNPs that are eSNPs in skin tissue.
Figure 8.
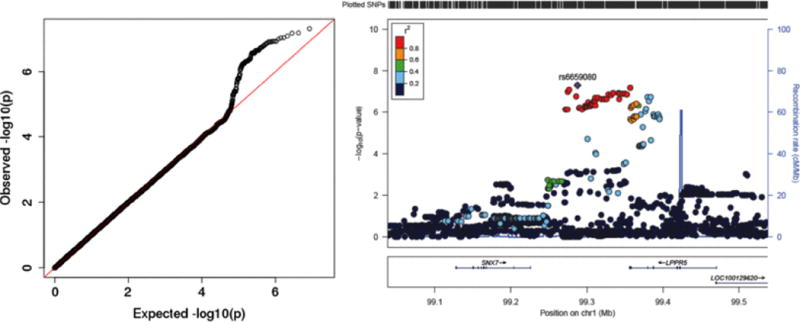
P-values from a genome-wide study of SNP-arsenic interaction in relation to arsenic-induced skin lesions. The left panel is a quantile-quantile plot of the –log10(P-values). The right panel is a regional association plot centered on top SNP rs6659080 which resides on chromosome 1.
DISCUSSION
In this paper, we described a two-step omic approach that addresses the limitations of standard genome-wide interaction approaches. Using existing genome-wide SNP data from a large Bangladeshi cohort study specifically designed to assess the effect of arsenic exposure on health, we searched for gene-arsenic interactions by first conducting a genome-wide search for SNPs that modify the effect of arsenic on molecular phenotypes (gene expression and DNA methylation features). Then, using this set of SNPs that interact with arsenic in relation to molecular phenotypes, we tested SNP-arsenic interactions in relation to skin lesion status, a hallmark characteristic of chronic arsenic toxicity. This approach leverages high-quality measures of arsenic exposure and restricts analyses to SNPs with enhanced probability of interaction with arsenic (by leveraging existing gene expression and DNA methylation data) to overcome the limitations of standard statistical genome-wide interaction approaches.
Evidence has suggested that arsenic exposure itself fails to fully explain the presence of arsenical skin lesions in an exposed population and that inter-individual variation may play an important role in determining sub-populations at higher risk of developing the disease at similarly exposed levels (19). Epidemiologic studies have shown interaction with sex, age, body mass index (39), smoking (40), socioeconomic status (41), nutritional status (42), and genetic variants (as reviewed in (20, 21)). There is known inter-individual variability in the methylation capacity of arsenic (as reviewed in (43)), which has been hypothesized to partly explain the variability in susceptibility to arsenic toxicity and may be attributed in part to genetic variation in genes known to metabolize arsenic. Thus, individuals who do not fully methylate arsenic efficiently could potentially be at increased risk of arsenic-related health effects due to this geneenvironment interaction.
GxE interactions are believed to influence complex diseases, but detecting GxE has proven difficult. The genome-wide association approach has been successful for identifying new susceptibility variants for a wide array of diseases (44). However, variants identified in genome-wide association studies typically do not show strong evidence of interaction with environmental risk factors (45–50). Explicit searches for GxE (as opposed to detection based on marginal effects) may be required to detect interacting variants (51). Limited progress has been made identifying GxE interactions in the epidemiological setting using existing statistical methods for genome-wide searches for interaction. The genome-wide interaction approach suffers from several key limitations, including (1) very large sample size requirements due to the conduct of many statistical tests of interaction and (2) a lack of high-quality exposure data in studies with large-scale genomic data. Considering these limitations, most genome-wide interaction studies that rely on statistical evidence of interaction alone are likely to be underpowered to detect GxE interactions.
New approaches for GxE detection are needed that leverage high-quality exposure measures (52). Genome-wide interaction studies are typically conducted in the context of large genome-wide association consortia studies (53) not designed to assess the impact of specific environmental factors. Thus, exposure measures are often questionnaire-based and/or retrospective, resulting in measurement error that reduces power for genome-wide interaction analyses. Using high-quality, accurate measures of environmental factors with established impact on disease in well-characterized populations will enhance power for GxE detection (51).
Focusing GxE studies on variants that influence molecular, omics phenotypes through GxE can potentially enhance power for GxE detection. Power for genome-wide interaction studies is limited because large numbers of tests are conducted. Focusing analyses on a smaller set of variants with an increased likelihood of GxE can increase power. In cases were transcriptomic or other omics data are available for individuals with exposure data, one could conduct a screen for variants that modify the effect of exposure on these molecular phenotypes. In other words, variants that interact with exposure to influence disease should also influence the cellular/molecular response to exposure.
We believe there is great promise in shifting the focus of GxE interaction research from agnostic genome-wide interaction testing to understanding how genetic variants influence humans’ response to an exposure at the molecular level. Our approach leverages omics data (gene expression and DNA methylation) to first test for gene-arsenic interactions in relation to intermediate molecular phenotypes, with evidence suggesting they represent the underlying mechanistic pathways between chronic arsenic exposure and skin lesion outcome. The major strength to first evaluating interactions with intermediate molecular phenotypes is that power may be enhanced to observe stronger interaction effects for the intermediate phenotype since it is more proximal to exposure than the disease outcome. Additionally, the environmental measure may better reflect the relevant exposure window for the intermediate phenotype as compared to disease outcomes with a longer latency period, also increasing ability to observe GxE interactions. However, the objective of this paper was not to conduct a direct evaluation of our approach in comparison with other gene-environment approaches; therefore, additional methodologic evaluations are needed to formally compare various statistical approaches.
We acknowledge several limitations of our study. While altered gene expression and DNA methylation are associated with our exposure and outcome of interest, there are other pathways that underlie the association between chronic arsenic exposure and skin lesion status. Therefore, the molecular phenotypes evaluated do not represent the only arsenic toxicity pathways. The inclusion of additional intermediate molecular phenotypes (i.e., proteome, microbiome, epigenome) may more comprehensively characterize mechanistic pathways and improve the first-step selection of promising interaction SNPs based on our approach. While previous research has demonstrated significant roles of SNPs on gene expression (eQTL) and DNA methylation (meQTL) as well as arsenic exposure on each of these molecular phenotypes, it is possible that SNPs and arsenic do not interact with respect to these molecular phenotypes. Gene-arsenic interactions may be more apparent for other molecular phenotypes. Also, our study was limited to omics phenotypes measured in blood, while some disease-relevant GxE may be detectable in multiple tissues, the ideal tissue types for detection of GxE will likely be the disease relevant tissues (e.g., skin). We attempted to address this limitation by searching for GxE among SNPs known to be eQTLs in skin tissue, although no clear interactions were identified.
We evaluated genome-wide SNP-arsenic interactions in step 1 only for those molecular traits (differentially methylated CpGs or transcripts) with evidence of marginal/independent association with arsenic exposure. We also evaluate SNP-arsenic interactions in step 1 for SNPs identified from eQTL and meQTL analyses with evidence of marginal/independent association with SNPs. This strategy was employed to reduce the number of multiple comparisons, but is based on the assumption that a marginal effect with either the exposure or gene must be observed for there to be an interaction effect. This assumption may not hold for some GxE interactions.
With the emergence of additional omics data in the epidemiologic setting, our approach may have the potential to boost power for genome-wide interaction research, enabling the identification of interactions that will enhance our understanding of disease etiology and our ability to develop interventions targeted at susceptible sub-populations.
Acknowledgments
This research was funded by the National Institutes of Health grant R21 to B.L.P and M.A.; P42 ES 10349 to J.H.G., and R01 CA 107431 to H.A.
Footnotes
The authors have no conflicts of interest, financial or otherwise.
References
- 1.IARC. http://www.inchem.org/documents/iarc/vol84/84-01-arsenic.html. 2004;84(39).
- 2.Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet. 2010;376(9737):252–8. doi: 10.1016/S0140-6736(10)60481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert-Diamond D, Cottingham KL, Gruber JF, Punshon T, Sayarath V, Gandolfi AJ, et al. Rice consumption contributes to arsenic exposure in US women. Proc Natl Acad Sci U S A. 2011;108(51):20656–60. doi: 10.1073/pnas.1109127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue J, Zartarian V, Wang SW, Liu SV, Georgopoulos P. Probabilistic Modeling of Dietary Arsenic Exposure and Dose and Evaluation with 2003-2004 NHANES Data. Environ Health Perspect. 2010;118(3):345–50. doi: 10.1289/ehp.0901205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.European Food Safety Authority. EFSA Panel on Contaminants in the Food Chain (CONTAM): Scientific opinion on arsenic in food. EFSA Journal. 2009;7:1351. doi: 10.2903/j.efsa.2008.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potera C. U.S. rice serves up arsenic. Environ Health Perspect. 2007;115(6):A296. doi: 10.1289/ehp.115-a296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson BP, Taylor VF, Karagas MR, Punshon T, Cottingham KL. Arsenic, organic foods, and brown rice syrup. Environ Health Perspect. 2012;120(5):623–6. doi: 10.1289/ehp.1104619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, metals, fibres, and dusts. IARC Monogr Eval Carcinog Risks Hum. 2012;100(Pt C):11–465. [PMC free article] [PubMed] [Google Scholar]
- 9.Abhyankar LN, Jones MR, Guallar E, Navas-Acien A. Arsenic Exposure and Hypertension: A Systematic Review. Environ Health Perspect. 2012;120(4):494–500. doi: 10.1289/ehp.1103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, et al. Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol. 2005;162(11):1037–49. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- 11.Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542–55. doi: 10.1007/s11883-012-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guha Mazumder DN. Arsenic and non-malignant lung disease. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2007;42(12):1859–67. doi: 10.1080/10934520701566926. [DOI] [PubMed] [Google Scholar]
- 13.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, et al. Evaluation of the Association between Arsenic and Diabetes: A National Toxicology Program Workshop Review. Environ Health Perspect. 2012 doi: 10.1289/ehp.1104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120(6):779–89. doi: 10.1289/ehp.1104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navas-Acien A, Silbergeld EK, Streeter RA, Clark JM, Burke TA, Guallar E. Arsenic exposure and type 2 diabetes: a systematic review of the experimental and epidemiological evidence. Environ Health Perspect. 2006;114(5):641–8. doi: 10.1289/ehp.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Barranco M, Lacasana M, Aguilar-Garduno C, Alguacil J, Gil F, Gonzalez-Alzaga B, et al. Association of arsenic, cadmium and manganese exposure with neurodevelopment and behavioural disorders in children: a systematic review and meta-analysis. Sci Total Environ. 2013;454–455:562–77. doi: 10.1016/j.scitotenv.2013.03.047. [DOI] [PubMed] [Google Scholar]
- 17.Byrd DM, Roegner ML, Griffiths JC, Lamm SH, Grumski KS, Wilson R, et al. Carcinogenic risks of inorganic arsenic in perspective. Int Arch Occup Environ Health. 1996;68(6):484–94. doi: 10.1007/BF00377874. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T, Yamauchi H, Fan Sun G. Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol. 2004;198(3):243–52. doi: 10.1016/j.taap.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 19.Concha G, Vogler G, Nermell B, Vahter M. Intra-individual variation in the metabolism of inorganic arsenic. Int Arch Occup Environ Health. 2002;75(8):576–80. doi: 10.1007/s00420-002-0361-1. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh P, Banerjee M, Giri AK, Ray K. Toxicogenomics of arsenic: classical ideas and recent advances. Mutat Res. 2008;659(3):293–301. doi: 10.1016/j.mrrev.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez A, Marcos R. Genetic variations associated with interindividual sensitivity in the response to arsenic exposure. Pharmacogenomics. 2008;9(8):1113–32. doi: 10.2217/14622416.9.8.1113. [DOI] [PubMed] [Google Scholar]
- 22.Antonelli R, Shao K, Thomas DJ, Sams R, 2nd, Cowden J. AS3MT, GSTO, and PNP polymorphisms: impact on arsenic methylation and implications for disease susceptibility. Environ Res. 2014;132:156–67. doi: 10.1016/j.envres.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Pierce BL, Tong L, Argos M, Gao J, Farzana J, Roy S, et al. Arsenic metabolism efficiency has a causal role in arsenic toxicity: Mendelian randomization and gene-environment interaction. Int J Epidemiol. 2013;42(6):1862–71. doi: 10.1093/ije/dyt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritz BR, Chatterjee N, Garcia-Closas M, Gauderman WJ, Pierce BL, Kraft P, et al. Lessons Learned From Past Gene-Environment Interaction Successes. Am J Epidemiol. 2017;186(7):778–786. doi: 10.1093/aje/kwx230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabrez S, Priyadarshini M, Priyamvada S, Khan MS, Na A, Zaidi SK. Gene-environment interactions in heavy metal and pesticide carcinogenesis. Mutat Res Genet Toxicol Environ Mutagen. 2014;760:1–9. doi: 10.1016/j.mrgentox.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 26.McAllister K, Mechanic LE, Amos C, Aschard H, Blair IA, Chatterjee N, et al. Current Challenges and New Opportunities for Gene-Environment Interaction Studies of Complex Diseases. Am J Epidemiol. 2017;186(7):753–761. doi: 10.1093/aje/kwx227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahsan H, Chen Y, Parvez F, Argos M, Hussain AI, Momotaj H, et al. Health Effects of Arsenic Longitudinal Study (HEALS): description of a multidisciplinary epidemiologic investigation. J Expo Sci Environ Epidemiol. 2006;16(2):191–205. doi: 10.1038/sj.jea.7500449. [DOI] [PubMed] [Google Scholar]
- 28.Argos M, Rahman M, Parvez F, Dignam J, Islam T, Quasem I, et al. Baseline comorbidities in a skin cancer prevention trial in Bangladesh. Eur J Clin Invest. 2013;43(6):579–88. doi: 10.1111/eci.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierce BL, Kibriya MG, Tong L, Jasmine F, Argos M, Roy S, et al. Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet. 2012;8(2):e1002522. doi: 10.1371/journal.pgen.1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics. 2011;3(6):771–84. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 32.Argos M, Chen L, Jasmine F, Tong L, Pierce BL, Roy S, et al. Gene-specific differential DNA methylation and chronic arsenic exposure in an epigenome-wide association study of adults in Bangladesh. Environ Health Perspect. 2015;123(1):64–71. doi: 10.1289/ehp.1307884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes MF. Biomarkers of exposure: a case study with inorganic arsenic. Environ Health Perspect. 2006;114(11):1790–6. doi: 10.1289/ehp.9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nixon DE, Mussmann GV, Eckdahl SJ, Moyer TP. Total arsenic in urine: palladium-persulfate vs nickel as a matrix modifier for graphite furnace atomic absorption spectrophotometry. Clin Chem. 1991;37(9):1575–9. [PubMed] [Google Scholar]
- 35.Heinegard D, Tiderstrom G. Determination of serum creatinine by a direct colorimetric method. Clin Chim Acta. 1973;43(3):305–10. doi: 10.1016/0009-8981(73)90466-x. [DOI] [PubMed] [Google Scholar]
- 36.Pierce BL, Tong L, Chen LS, Rahaman R, Argos M, Jasmine F, et al. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet. 2014;10(12):e1004818. doi: 10.1371/journal.pgen.1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierce B, Tong L, Argos M, Jasmine F, Rakibuz-Zaman M, Sarwar G, et al. Co-occurring eQTLs and mQTLs: detecting shared causal variants and shared biological mechanisms. bioRxiv. 2016 doi: 10.1038/s41467-018-03209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahsan H, Chen Y, Parvez F, Zablotska L, Argos M, Hussain I, et al. Arsenic exposure from drinking water and risk of premalignant skin lesions in Bangladesh: baseline results from the Health Effects of Arsenic Longitudinal Study. Am J Epidemiol. 2006;163(12):1138–48. doi: 10.1093/aje/kwj154. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y, Graziano JH, Parvez F, Hussain I, Momotaj H, van Geen A, et al. Modification of risk of arsenic-induced skin lesions by sunlight exposure, smoking, and occupational exposures in Bangladesh. Epidemiology. 2006;17(4):459–67. doi: 10.1097/01.ede.0000220554.50837.7f. [DOI] [PubMed] [Google Scholar]
- 41.Argos M, Parvez F, Chen Y, Hussain AZ, Momotaj H, Howe GR, et al. Socioeconomic status and risk for arsenic-related skin lesions in Bangladesh. Am J Public Health. 2007;97(5):825–31. doi: 10.2105/AJPH.2005.078816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zablotska LB, Chen Y, Graziano JH, Parvez F, van Geen A, Howe GR, et al. Protective effects of B vitamins and antioxidants on the risk of arsenic-related skin lesions in Bangladesh. Environ Health Perspect. 2008;116(8):1056–62. doi: 10.1289/ehp.10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tseng CH. A review on environmental factors regulating arsenic methylation in humans. Toxicol Appl Pharmacol. 2009;235(3):338–50. doi: 10.1016/j.taap.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 44.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campa D, Kaaks R, Le Marchand L, Haiman CA, Travis RC, Berg CD, et al. Interactions between genetic variants and breast cancer risk factors in the breast and prostate cancer cohort consortium. J Natl Cancer Inst. 2011;103(16):1252–63. doi: 10.1093/jnci/djr265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012;72(8):2036–44. doi: 10.1158/0008-5472.CAN-11-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hutter CM, Slattery ML, Duggan DJ, Muehling J, Curtin K, Hsu L, et al. Characterization of the association between 8q24 and colon cancer: gene-environment exploration and meta-analysis. BMC Cancer. 2010;10:670. doi: 10.1186/1471-2407-10-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milne RL, Gaudet MM, Spurdle AB, Fasching PA, Couch FJ, Benitez J, et al. Assessing interactions between the associations of common genetic susceptibility variants, reproductive history and body mass index with breast cancer risk in the breast cancer association consortium: a combined case-control study. Breast Cancer Res. 2010;12(6):R110. doi: 10.1186/bcr2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nickels S, Truong T, Hein R, Stevens K, Buck K, Behrens S, et al. Evidence of gene-environment interactions between common breast cancer susceptibility loci and established environmental risk factors. PLoS Genet. 2013;9(3):e1003284. doi: 10.1371/journal.pgen.1003284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Travis RC, Reeves GK, Green J, Bull D, Tipper SJ, Baker K, et al. Gene-environment interactions in 7610 women with breast cancer: prospective evidence from the Million Women Study. Lancet. 2010;375(9732):2143–51. doi: 10.1016/S0140-6736(10)60636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hutter CM, Mechanic LE, Chatterjee N, Kraft P, Gillanders EM, Tank NCIG-ET Gene-environment interactions in cancer epidemiology: a National Cancer Institute Think Tank report. Genet Epidemiol. 2013;37(7):643–57. doi: 10.1002/gepi.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gauderman WJ, Mukherjee B, Aschard H, Hsu L, Lewinger JP, Patel CJ, et al. Update on the State of the Science for Analytical Methods for Gene-Environment Interactions. Am J Epidemiol. 2017;186(7):762–770. doi: 10.1093/aje/kwx228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Austin MA, Hair MS, Fullerton SM. Research guidelines in the era of large-scale collaborations: an analysis of Genome-wide Association Study Consortia. Am J Epidemiol. 2012;175(9):962–9. doi: 10.1093/aje/kwr441. [DOI] [PMC free article] [PubMed] [Google Scholar]