

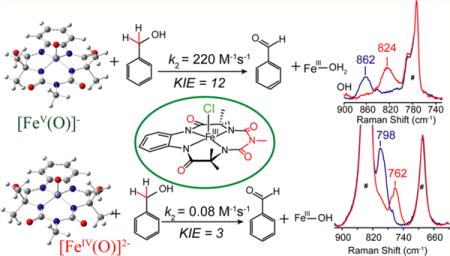
Abstract
In this report we compare the geometric and electronic structures and reactivities of [FeV(O)]− and [FeIV(O)]2− species supported by the same ancillary nonheme biuret tetraamido macrocyclic ligand (bTAML). Resonance Raman studies show that the Fe=O vibration of the [FeIV(O)]2− complex 2 is at 798 cm−1, compared to 862 cm−1 for the corresponding [FeV(O)]− species 3, a 64 cm−1 frequency difference reasonably reproduced by density functional theory calculations. These values are, respectively, the lowest and the highest frequencies observed thus far for nonheme high-valent Fe=O complexes. Extended X-ray absorption fine structure analysis of 3 reveals an Fe=O bond length of 1.59 Å, which is 0.05 Å shorter than that found in complex 2. The redox potentials of 2 and 3 are 0.44 V (measured at pH 12) and 1.19 V (measured at pH 7) versus normal hydrogen electrode, respectively, corresponding to the [FeIV(O)]2−/[FeIII(OH)]2− and [FeV(O)]−/[FeIV(O)]2− couples. Consistent with its higher potential (even after correcting for the pH difference), 3 oxidizes benzyl alcohol at pH 7 with a second-order rate constant that is 2500-fold bigger than that for 2 at pH 12. Furthermore, 2 exhibits a classical kinteic isotope effect (KIE) of 3 in the oxidation of benzyl alcohol to benzaldehyde versus a nonclassical KIE of 12 for 3, emphasizing the reactivity differences between 2 and 3.
Graphical abstract
INTRODUCTION
Monomeric iron-oxo units have been proposed as intermediates involved in either C−H bond cleavage or O atom transfer reactions for several oxidases and oxygenases.1 For nonheme iron-containing enzymes, both iron(IV)- and iron(V)-oxo species have been proposed as active intermediates.2 Understanding the role of the iron oxidation state in the thermodynamics and kinetics of C−H bond cleavage is important not only for understanding the underlying principles that guide these natural enzymes but also for designing synthetic catalysts that mimic the action of these enzymes. It has been shown that both the redox potential of the high-valent species and the basicity of the terminal oxo unit contribute to the reactivity of C−H bond abstraction by metal oxo complexes.3,4 In previous reports,5 reactivity comparisons of model complexes of Compound II (Cpd II; oxoiron(IV)) and Compound I (Cpd I; isoelectronic to oxoiron(V)) have been reported, but no correlation of this intrinsic difference to their spectroscopic properties has been provided. Theoretical studies on the H atom abstraction capabilities of Cpd I and Cpd II mimics have also found sluggish oxidative properties for the latter, with H atom abstraction barriers of 2−5 kcal/mol higher than those computed for the former.6 For nonheme systems, the geometric and electronic structures as well as reactivities of oxoiron(IV) and oxoiron(V) species having the same ancillary nonheme ligand have, to date, not been experimentally compared, primarily due to the paucity of systems with reasonably stable oxoiron(V) species. However, note that computational investigations on hypothetical iron-oxo models have been performed by Neese et al. to correlate the electronic properties and reactivity of these high-valent iron-oxo complexes.6d,e In their study, the superior reactivity of the hypothetical [FeV(O)(NH3)4(OH)axial]2+ over its one-electron reduced species [FeIV(O)(NH3)4(OH)axial]4+ in the oxidation of ethane was established based on the H atom abstraction barrier, the radical character of the iron-oxo bond, and the approach of C− H bond. Experimentally, the reactivities of two pairs of nonheme oxoiron(V) and oxoiron(IV) complexes have been compared in the literature, but no rate measurements have been reported.6f,g However, the fleeting nature of these iron(V) species renders it difficult to obtain structural and mechanistic insights into their inherent differences.
However, oxoiron(V),7a oxoiron(IV),7b,c and (μ-oxo)diiron-(IV)7d complexes of the tetraamido macrocyclic ligand (TAML) have been obtained in greater than 95% yield and characterized. Thus, Fe-TAML represents an exciting system, where the spectroscopic properties and reactivities of the high-valent oxoiron species can be studied and correlated. To date, however, reactivity comparisons between the oxoiron(V) and oxoiron(IV) TAML complexes have not been reported.7d,e A likely reason is the instability of a completely characterized oxoiron(V) complex at temperatures above −40°C, which precluded the use of a common temperature and solvent system to study their reactivity toward substrates.
The synthesis of a room-temperature stable oxoiron(V)-(bTAML) (bTAML = biuret tetraamido macrocycle ligand) complex (bTAML depicted in Figure 1) in greater than 95% yield has been recently reported.8a This oxoiron(V) complex is also stable in solvent containing up to 70% water.8b Additionally, the oxoiron(IV) complex can be generated under the same conditions. Thus, we are able to prepare both the oxoiron(IV) and the oxoiron(V) complexes in very high purity at room temperature using the same solvent system, which facilitates reactivity comparisons using a common substrate. In this report, we compare resonance Raman, X-ray absorption, and electrochemical data for the oxoiron(IV) and oxoiron(V) complexes of the bTAML ligand. Their room-temperature reactivity difference in the oxidation of benzyl alcohol (BnOH) and its correlation with spectroscopic properties supported by density functional theory (DFT) calculations are also discussed in this manuscript.
Figure 1.
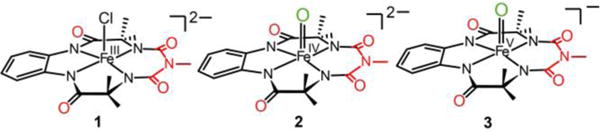
bTAML complexes discussed in this report with tetraethylammonium cations as counterions for all complexes: 1 = chloroiron(III) complex; 2 = oxoiron(IV) complex, and 3 = oxoiron(V) complex.
RESULTS AND DISCUSSION
Synthesis
The complexes [(bTAML)FeIV(O)]2− (2) and [(bTAML)FeV(O)]− (3) were obtained from [(bTAML)-FeIII(Cl)]2− (1). On the one hand, complex 3 was synthesized by addition of 1.2 equiv of sodium hypochlorite (NaOCl) into a solution of 1 in acetonitrile (CH3CN) as reported previously; it exhibits a visible spectrum with bands at 445 and 613 nm (Supporting Information Figure S1 inset).8a,b On the other hand, the corresponding FeIV(O) complex 2 could be obtained by two methods, either by treating the CH3CN solution of 3 with 3 equiv of tetrabutylammonium hydroxide or by the addition of NaOCl to 1 in water (pH 12). Complex 2 exhibits a visible spectrum with a λmax of 429 nm in CH3CN and 460 nm in water pH 12 (Supporting Information Figure S1). Its Mössbauer spectrum shows a doublet with an isomer shift (δ) of −0.21 mm/s and a quadrupole splitting (ΔEQ) of 3.89 mm/s (Supporting Information Figure S2a), similar to those found for the corresponding FeIV(O)2− species supported by the related TAML ligand.7b,c On the basis of their Mössbauer spectra, the purities of both 2 and 38a,b complexes were determined to be greater than 95%.
Resonance Raman Spectroscopy
Resonance Raman data for 2 and 3 were obtained in acetonitrile solvent using 476.5 nm excitation. The Fe=O vibration of 2 is observed at 798 cm−1 (Figure 2a, blue), which shifts to 762 cm−1 (Figure 2a, red) upon 18O substitution (Δ18O/16O = 36 cm−1). On the one hand, the Fe=O vibration of 2 is the lowest value reported for a nonheme FeIV=O species9a,b and reflects the highly basic nature of the bTAML ligand. On the other hand, the corresponding Fe=O vibration for 3 is observed at 862 cm−1 (Figure 2b, blue) and downshifts to 824 cm−1 (Figure 2b, red) upon 18O substitution (see Supporting Information Figure S2b for full spectra), the observed Δ18O/16O of 38 cm−1 being in excellent agreement with that calculated by Hooke’s Law for an Fe=O unit. The ν(Fe=O) of 3 is higher than any other Fe=O vibration measured to date,9a,b presumably because of the FeV oxidation state. The next-highest ν(Fe=O) was observed by IR spectroscopy at 856 cm−1 for the recently reported [FeIV(Osyn)-(TMC)(OTf)]+ complex (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane).9c The 64 cm−1 difference observed between 3 and 2 represents the first time the vibrations of FeV=O and FeIV=O units supported by a common ligand can be compared. Clearly, the oxidation of the Fe(IV) center to Fe(V) results in significant strengthening of the Fe=O bond. For comparison, the ν(Fe=O) values of FeIV=O and FeIII−O units supported by trianionic urea-based tripodal ligand (tris(tert-butylureaylethylene)aminato, [H3buea]3−) reported by Borovik exhibit a frequency difference of 128 cm−1 and an Fe−O bond length difference of 0.15 Å.10
Figure 2.
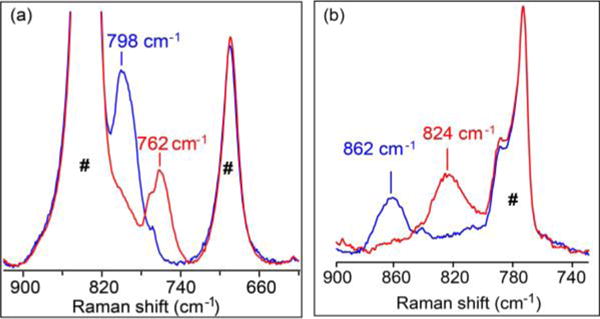
Resonance Raman spectra of 2 in CH3CN at room temperature (left) and 3 in CD3CN at 77 K (right). Blue and red lines represent16O- and 18O-labeled samples, respectively. λex = 476.5 nm; power ≈ 40 mW. (#) Solvent-derived features.
X-ray Absorption Spectroscopy
X-ray absorption spectroscopic (XAS) data were collected at the Fe K-edge for 1, 2, and 3 in CH3CN to gain insight into their iron coordination environments. The X-ray absorption near-edge structure (XANES) region provides information on the oxidation state and symmetry of an iron center. The Fe K-edge of 1 (Supporting Information Figure S2c) was observed at 7122.1 eV, with a pre-edge peak area of 14.1 units (Supporting Information Table S1), consistent with a five-coordinate ferric center (Figure 3, left panel).11 Analysis of the extended X-ray absorption fine structure (EXAFS) region of 1 gives ironscatterer distances that correspond to four N/O atoms at 1.87 Å, one Cl atom at 2.37 Å, six C atoms at 2.83 Å, and multiple scattering pathways involving the carbonyl groups of the ligand at 3.99 Å (Figure 4, top row; Supporting Information Table S2), distances congruent with those from the crystal structure of 1.8c
Figure 3.
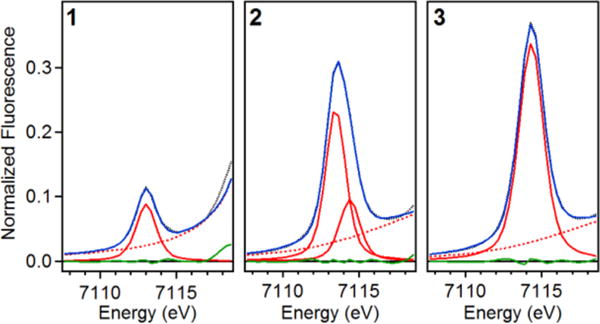
Observed X-ray absorption pre-edge regions of 1 (left), 2 (middle), and 3 (right). Experimental data are represented by black dotted lines, with the best fits as blue solid lines, the modeled baselines as red dashed lines, the fitted component peaks as red solid lines, and the residuals as green solid lines.
Figure 4.
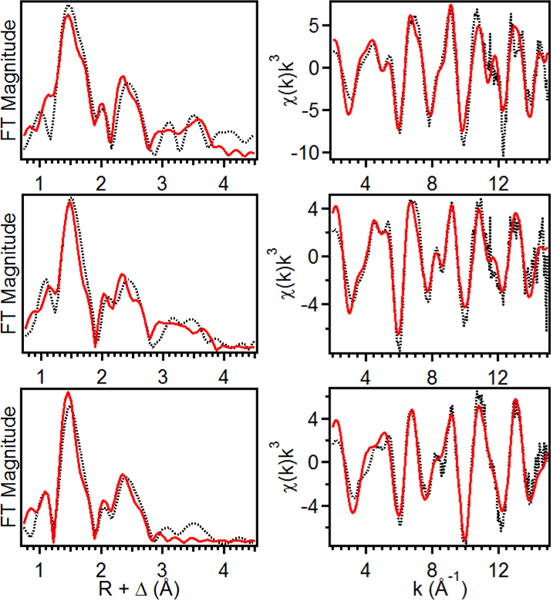
(left) Fourier transforms of the EXAFS data (black dotted) with best fit (solid red) for 1 (top), 2 (middle), 3 (bottom), k range = 2−15 Å−1. (right) Unfiltered EXAFS data (black dotted) with best fit (red solid) for 1 (top), 2 (middle), and 3 (bottom).
Oxidation of 1 to 2 results in an upshift of the K-edge energy to 7124.2 eV (Supporting Information Figure S2c), which is comparable to that reported for [FeIV(O)(TAML)]2− (7124.5 eV)7c and consistent with its assignment as an FeIV center. However, the K-edge energy of 2 is 2.1 eV higher than for that for 1. This larger difference in K-edges is likely due to the bound Cl in 1, as chloride ligands are known to decrease the K-edge energies of the metal centers to which they are bound.7f Complex 2 exhibits a pre-edge area of 52 units (Figure 3, middle panel; Supporting Information Table S1), which is larger than that found for [FeIV(O)(TAML)]2− (41 units).7c EXAFS analysis of 2 shows one O/N atom at 1.64 Å, four N/O at 1.86 Å, six C at 2.82 Å, and scatterers at 4.01 Å involving ligand carbonyl groups (Figure 4, middle row; Supporting Information Table S3). The 0.05 Å shorter Fe=O bond distance found for 2 compared to that of [(TAML)FeIV(O)]2− (1.69 Å)7c may rationalize the larger pre-edge area observed for 2.
Complex 3 exhibits a K-edge at 7125.4 eV (Supporting Information Figure S2c), which is very close to that of the related [(TAML)FeV(O)]− complex reported by Collins (K-edge = 7125.3 eV).7a Complex 3 also gives rise to a pre-edge feature at 7114.3 eV with a very large peak area of 65 units (Figure 3, right panel; Supporting Information Table S1), which is comparable in size to that of [(TAML)FeV(O)]− (∼70 units).7a These quite large values reflect a high degree of distortion from centrosymmetry that is matched only by that estimated for the aqueous oxoiron(IV) complex described by Pestovsky et al.12 Analysis of the EXAFS region of 3 gives ironscatterer distances that correspond to one O/N atom at 1.59 Å, four N/O atoms at 1.86 Å, and six C atoms at 2.82 Å (Figure 4, bottom row; Supporting Information Table S4), which agree with the results for [FeV(O)(TAML)]− (0.7 O/N at 1.58 Å, four N/O at 1.87 Å, and five C at 2.82 Å).7a Within error, the structures of the two FeV(O) complexes are identical. The significant shortening of the axial Fe=O bond in what is postulated to be a square pyramidal complex is very likely the factor that gives rise to the much larger pre-edge area observed for 3 versus that of 2,11 as well as the 64 cm−1 higher Fe=O stretching frequency of 3.
Density Functional Theory
DFT calculations are used to obtain hypothetical three-dimensional models for species 2 and 3. The optimized geometries for 2 and 3 show Fe−O bond lengths of 1.64 and 1.59 Å, respectively, consistent with results from the EXAFS analysis. The predicted 0.05 Å shortening of the Fe−O bond in 3 relative to that of 2 is complemented by an increase in the computed νFe−O from 895 to 964 cm−1, the calculated difference of 69 cm−1 approaching the experimentally observed difference of 64 cm−1. The molecular orbital (MO) diagram (Figure 5) of 2 indicates the presence of a strong σ interaction between the Fe and the O 2pz orbitals (Figure 5, left, contour second from top). For an S = 1 ground state, this orbital is unoccupied. Additional strong π interactions between the singly occupied Fe 3dxz/yz, and O 2px/y orbitals (Figure 5, left, contour at the bottom) result in an Fe−O formal bond order of 2 (1 σ + 2 half π). Note that the strong equatorial ligand field of bTAML raises the energy of the above that of the orbital, which is σ antibonding with respect to the strong axial oxo ligand. This is in contrast to most known nonheme Fe systems, where the strong σ interaction of the oxo ligand places orbitals higher in energy than the .7b,c,g,h,j Rather, the higher orbital energy, as observed here, is a common feature of heme systems, where the porphyrin ligand provides a strong equatorial ligand field.6a,7i The same situation has been encountered recently in an Fe(IV)=O species having a tetracarbene macrocycle in the equatorial plane.7g
Figure 5.
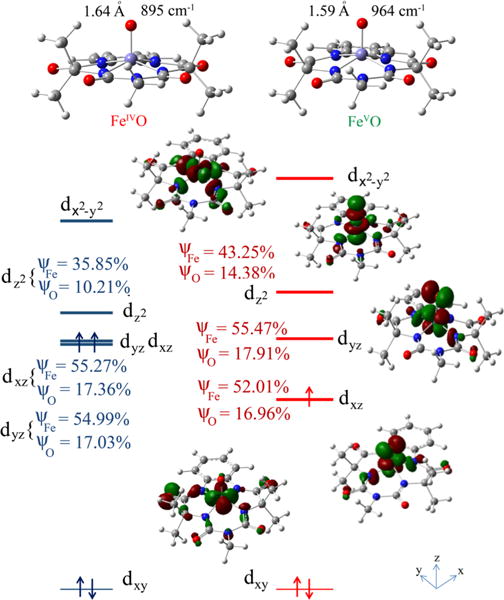
Optimized geometries (top) and MO diagrams (bottom) for [(bTAML)FeIV(O)]2− (2), left and [(bTAML)FeV(O)]− (3), right. ψFe and ψo indicate contributions of Fe and oxo centers in the individual orbitals. Note that only d orbitals of 3 are shown for clarity, and the energy of the nonbonding dxy orbital is set to zero for 2 and 3.
The calculations show that there is strong charge transfer from the tetraanionic equatorial ligand in 2, which appears to result in a decrease in the oxo ligand character of the Fe=O bond. Thus, the coefficients in the Fe−O σ (10% in ) and π (17.36% , 54.99% dxz; Figure 5 left) in 2 are much smaller than those encountered for other nonheme Fe(IV)=O complexes with weaker equatorial ligands, for example, 32% in and 36% 54% dxz(Fe).7h–j These differences may be interpreted as a decrease in the bond covalency of the Fe=O unit in 2 relative to these other complexes, as reflected by the lower Fe=O frequency it exhibits.
The [FeV(O)]− species is one-electron-oxidized, which results in depopulation of one of the two singly occupied dxz/yz orbitals (∼55% metal 3d contribution, Figure 5 right), indicating that the oxidation is metal-based and not ligand-based as is the case for high-valent heme systems (e.g., Compound I6a,7i). Thus, the oxidation leads to depopulation of an Fe−O π* orbital and results in an increase in the Fe−O bond order to 2.5 (1 σ + 1.5 π) in 3. The increase in the Fe−O bond order upon oxidation is reflected in the shorter Fe−O bond found by EXAFS and the higher νFe−O value in 3 relative to 2 observed in the Raman data.
Electrochemistry
The electrochemical properties of the bTAML complexes 1−3 were investigated in water. Cyclic voltammetry (CV) of 1 was performed between pH 11 and 12.5 using a scan window from 0.2 to 0.9 V versus normal hydrogen electrode (NHE; Supporting Information Figure S4). Note that [(bTAML)FeIIICl]2− in water converts to the diaqua complex [(bTAML)FeIII(H2O)2]−.13,14 The first pKa of the coordinated water has been determined to be ∼10.3, leading to the formation of [(bTAML)FeIII(OH)(OH2)]2−.14 A quasireversible one-electron wave at E1/2 = 0.44 V versus NHE (with a peak-to-peak separation of ΔEp = 66 mV and ip,a/ip,c ≈ 0.8) was observed by CV of chemically synthesized 2 at pH 12 (Figure 6A). This feature corresponds to the FeIV(O)/FeIII(OH)(OH2) couple. Furthermore, controlled potential electrolysis of a pH 12 solution of 1 at 0.55 V versus NHE afforded the well-characterized deep red [(bTAML)FeIV(O)]2− complex described before in this work. The FeIV/III electrochemical wave is pH-dependent between 10.5 and 12.2 with the E1/2 versus pH decreasing by 58 mV per pH unit (Supporting Information Figure S5), indicating a proton-coupled electron transfer (PCET) step.15 On the basis of all these observations, we propose that the FeIV/III couple is consistent with the following electrochemical process between pH 10.5 and 12.2:
Figure 6.
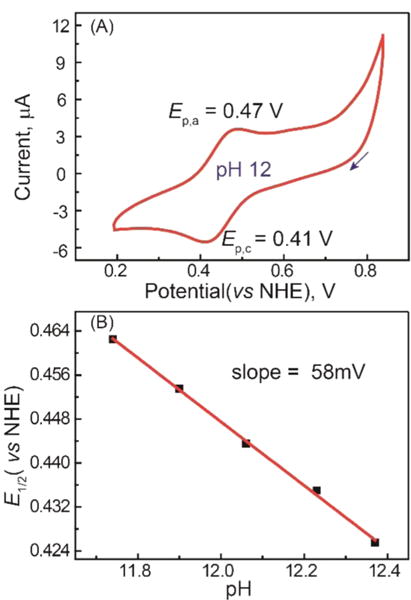
(A) CV of 2 synthesized from 1 and NaOCl in a pH 12 aqueous solution (conditions: GC working electrode, Pt counter electrode, 0.2 M KNO3 as supporting electrolyte, scan rate 50 mV s−1; arrow indicates the direction of potential scanning). (B) Plot of E1/2 vs pH for 1 in water.
Below pH 10, a [FeIV(O)]2−/[FeIII(OH2)2]− couple corresponding to a two-proton/one-electron transfer process was observed, as recently reported.16a
The redox potential for the [FeV(O)]−/[FeIV(O)]2− couple in H2O has been determined by square-wave voltammetry experiments at pH 7 to be E0′= 1.19 V versus NHE.16a There is an apparent difference in potential of 750 mV between the redox couples [FeV(O)−/FeIV(O)2−] at pH 7 and [FeIV(O)2−/FeIII(OH)2−] at pH 12. After correcting for the pH difference based on the observation of a [FeIV(O)]2−/[FeIII(OH2)2]− couple between pH 7 and 10, we estimate a redox potential of 0.9 V versus NHE at pH 7, decreasing the redox potential gap to ∼300 mV. For comparison, the corresponding heme analogues that serve as models of Cpd I and Cpd II exhibit a difference of ∼250 mV.16b
Reactivity
To compare the reactivity of these related iron-oxo species toward C−H bonds, the choice of substrate was important. The calculated bond dissociation energy (BDE) of the FeIII(O−H) bond that corresponds to one-electron reduced [(bTAML)FeIV(O)]2− has a value of only 83 ± 2 kcal/mol,17 obtained using the modified Bordwell equation.18 The expected inability of the oxoiron(IV) complex 2 to oxidize strong C−H bonds is confirmed by its unchanged UV−vis spectrum upon addition of cyclohexane, toluene, and cumene. This moderate BDE of [FeIII(bTAML)O−H]2− (∼83 kcal/mol) is similar to that of nonheme iron complexes such as [FeIII(N4Py)O−H]2+ (∼78 kcal/mol)18 (N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine) but lower than their heme analogues ([FeIII(TMP)O−H] (∼88 kcal/mol)5i (TMP = 5,10,15,20-tetramesitylporphyrin) and [FeIII(TMPS)O−H] (∼90 kcal/mol); TMPS = 5,10,15,20-tetramesitylporphyrinoctasulfonate).16b,19a On the other hand, the oxoiron(V) complex 3 is known to react with strong C−H bonds like cyclohexane.8a The FeIVO−H BDE of [(bTAML)FeIVOH]− is estimated to be about 99 kcal/mol,20 which is roughly the same as that for the Compound I mimic [(4-TMPyP+·)FeIV(O)] (4-TMPyP = 5,10,15,20-tetrakis(N-methyl-4-pyridinium)porphyrin tetracation) with an FeIVO−H BDE of 100 kcal/mol. We therefore chose benzyl alcohol (BnOH) as the substrate as it has a C−H bond with a BDE of <80 kcal/mol.19b
The reaction of 3 with benzyl alcohol in CH3CN was recently shown to be a two-electron process involving abstraction of a C−H bond in the rate-determining step.21a The electron transfer/proton transfer (ET/PT) mechanism after H−atom abstraction from the benzylic position over rebound of hydroxyl radical was proved by using cyclobutanol as a radical clock probe.21a To compare the reactivity of both oxoiron (bTAML) complexes under the same conditions, their reactions with benzyl alcohol were performed under pseudo-first-order conditions in 80% CH3CN/20% H2O as the solvent, which is compatible with both oxoiron(V) and oxoiron(IV) complexes (Scheme 1).
Scheme 1.
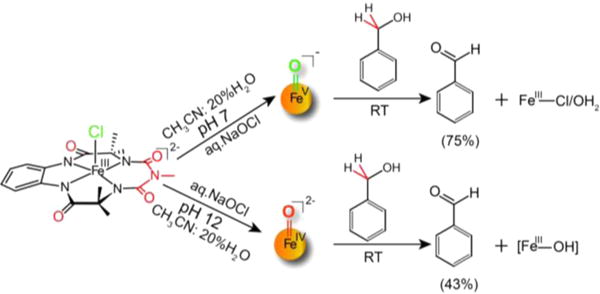
Reactivity of 2 and 3 with BnOH
The kinetics of BnOH oxidation to benzaldehyde by 2 in an 80:20 acetonitrile−water solvent mixture pH 12 at room temperature (RT) was studied under pseudo-first-order conditions. A decrease in the characteristic [FeIV(O)]2− absorption band at 460 nm was observed (Supporting Information Figure S7A) due to oxidation of BnOH to benzaldehyde (yield ≈ 43%; Supporting Information Figures S8 and S9). The less than 50% yield and generation of [FeIII− OH] 2− at the end of the reaction indicate a nonrebound mechanism similar to that reported for other nonheme oxoiron(IV) complexes.21c Analysis of the pseudo-first-order reactions as a function of substrate concentration gave a second-order rate constant k2 of 0.08(0.01) M−1 s−1 (Figure 7A, Supporting Information Figure S7B) and a classical kH/kD kinetic isotope effect (KIE) of ∼3 (Table 1). For comparison, the reaction of 3 with BnOH in an 80:20 acetonitrile−water (pH 7) solvent mixture was monitored by UV−vis absorption spectroscopy as a function of BnOH concentration as recently reported by us (yield of benzaldehyde ≈ 75% yield).21a Resulting kobs (Supporting Information Figure S6) values from pseudo-first-order fits correlated linearly with substrate concentration to give a second-order rate constant k2 (220 ± 20 M−1 s−1; Figure 7B). A nonclassical KIE kH/kD ≈ 12 was determined (Table 1). Thus, 2 has a BnOH oxidation rate that is significantly lower (∼2500-fold) than that of 3. Note that oxidation of benzyl alcohol was performed by 2 and 3 at pH 12 and 7, respectively. The pH of the medium can have a crucial role in hydrogen atom abstraction, as has been observed for [FeIV(O)N4Py]2+.21b
Figure 7.
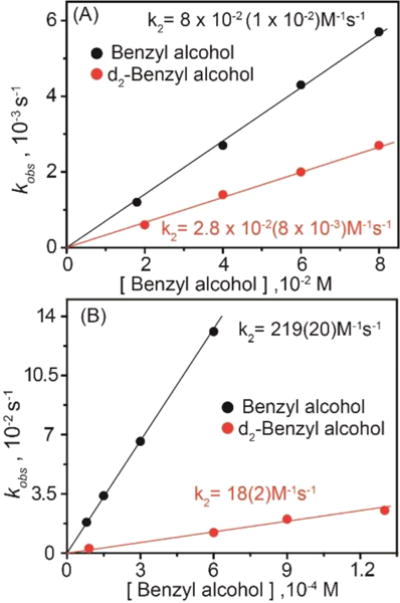
Plots of kobs vs benzyl alcohol concentration for reactions with (A) 2 (1.5 × 10−4 M) and (B) 3 (5 × 10−5 M) performed in a 80:20 acetonitrile−water solvent mixtures at room temperature.
Table 1.
Comparison of Properties of 2 and 3
| properties | 2 | 3 |
|---|---|---|
| ν(Fe=O) (cm−1) | 798 | 862 |
| r(Fe=O) (Å) | 1.64 | 1.59 |
| XAS pre-edge area units | 52 | 65 |
| E0′(V vs NHE) | 0.44 (pH 12) | 1.19 (pH 7) |
| k2 (BnOH) @ RT | 0.08(1) M−1 s−1 | 220(20) M−1 s−1 |
| KIE (BnOH oxidation) | ∼3 | ∼12 |
| ΔH‡ (kcal/mol) | 10.7 ± 0.7 | 7.9 ± 0.5 |
| ΔS‡ (cal/mol/K) | −27 ± 2 | −23 ± 2 |
| TΔS‡ (kcal/mol) | −8.1 ± 0.6 | −6.9 ± 0.6 |
| ΔG‡ (kcal/mol) | 19 ± 1.2 | 15 ± 1 |
To gain better insight into the differential reactivity of 2 and 3 toward BnOH, an Eyring analysis was performed to obtain their activation enthalpies (ΔH‡) and entropies (ΔS‡; Table 1 and Supporting Information Figure S10). For both of the oxidants, the negative entropy of activation observed is indicative of a tightly bound transition state involving both the iron oxo and the substrate. The contribution of ΔH‡ to the free energy of activation is greater than that of the TΔS‡ term for both 2 and 3, indicating that the process is enthalpically driven. However, for 3, the difference between ΔH‡ and TΔS‡ term is much smaller (∼1 kcal/mol). These results are in contrast to alcohol oxidation by Cpd I and II mimics reported by van Eldik, where the process is entropically controlled for the Cpd I mimic but enthalpically controlled for the Cpd II mimic.22 The difference in reactivity of benzyl alcohol oxidation to benzaldehyde between both the Cpd I and II mimics reported by van Eldik is only ∼100-fold.22 Groves et al.5j have also reported a highly reactive Cpd I analogue [(4-TMPyP+•)-FeIV=O]+ in comparison to its one-electron reduced Cpd II analogue [(4-TMPyP)FeIV=O] toward xanthene oxidation albeit with a low KIE (2.1). This large reactivity difference of the highly cationic compound I analogue was rationalized by considering a low-lying a2u porphyrin highest occupied molecular orbital (HOMO) that facilitated spin-state crossing in the course of the reaction. In contrast, studies on the [Mn(H3buea)] complexes reported by Borovik showed a higher reactivity toward dihydroanthracene (DHA) for the monomeric [MnIII(O)]2− complex relative to the [MnIV(O)]− complex, which was attributed to the much enhanced basicity of the [MnIII(O)]2− complex.23a
Before concluding, two important points are worth noting. First, for C−H bond abstraction of benzyl alcohol by 2 and 3, the events accompanying formation of the transition state are not only determined by the electron affinity of the oxidant (which is manifested in the huge difference in their redox potentials reported earlier) but also by the proton affinities of the one-electron reduced species formed after electron transfer. The pKa of the one-electron reduced species generated from 2 (i.e., [FeIII(O)]3−) should be significantly higher than for the one-electron reduced species of 3 (i.e., [FeIV(O)]2−; pKa ≈ 10) but could not be determined experimentally (Supporting Information Figure S3). The pKa of a related [FeIII(O)-(H3buea)]2− species has been determined by Borovik to be greater than 20.10 This high pKa of the putative FeIII(O) would offset the reactivity of 3 in comparison to 2 that would be expected only due to the 300 mV redox potential difference.
The second relates to the respective KIEs of 3 and 12 observed for the reactions of 2 and 3 with BnOH. These values indicate that H atom abstraction is likely an important contributor to the rate determining step for both these complexes.24 Interestingly, the pattern of KIE values observed for 2 and 3 is the reverse of what is observed for nonheme oxoiron(IV) complexes supported by neutral N4 or N5 ligands. For the latter, nonclassical KIE values are observed for S = 1 FeIV(O) complexes,3,24c but classical KIE values are found for catalytic alkane hydroxylation by related nonheme iron complexes for which a powerful FeV(O)(OH) oxidant has been implicated.24f–h The KIE differences could arise from differences in electronic structure between 2 and other S = 1 nonheme FeIV(O) complexes. As shown for 2 in Figure 5, the orbital lies well above the orbital due to the strong equatorial field of the bTAML ligand, resulting in a configuration for the excited quintet (S = 2) state instead of the more typical configuration associated with the other nonheme FeIV=O complexes supported by neutral polydentate ligands. A consequence for 2 is that its S = 2 excited state is calculated to be 20−26 kcal/mol higher in energy than its ground triplet state (S = 1), depending on which functional (UBP86 or UB3LYP) is used (Supporting Information Table S5). A similarly large energy gap is calculated for the FeIV=O(tetracarbene) complex reported by Neese and Meyer,7g but the triplet−quintet energy gap calculated for the prototypical [FeIV(O)(TMC) (NCCH3)]2+ complex is much smaller at 3−5 kcal/mol.24e,i On the one hand, for 2 and the tetracarbene complex, the presence of an unpaired electron in the orbital of the excited S = 2 state significantly weakens the Fe=O bond, thereby destabilizing this spin state. On the other hand, our calculations on 3 show that its doublet (S = 1/2) spin state is more stable than the quartet (S = 3/2) by 10−14 kcal/mol (Supporting Information Table S5a). Future work will focus on gaining further insight into the consequences of these differences in electronic structure on the reactivity of the high-valent oxoiron complexes supported by tetraanionic TAML ligands versus its neutral counterparts.
SUMMARY
In summary, the resonance Raman and XAS properties of [FeIV(O)]2− (2) and [FeV(O)]− (3) species have been characterized, allowing the first detailed comparison of complexes with oxoiron(IV) and oxoiron(V) units supported by the same ancillary bTAML ligand under the same conditions. These results show significant strengthening and shortening of the Fe=O bond upon oxidation from FeIV to FeV due to an increase in π bond order. This structural change translates into a significant difference in redox potential between 2 and 3 and a 2500-fold greater reactivity of 3 over its one-electron reduced analogue 2 in oxidizing benzyl alcohol. DFT calculations provide a computational basis for rationalizing the observed differences in properties.
EXPERIMENTAL SECTION
Materials
(Et4N)2[FeIII(Cl)(bTAML)] 1 was synthesized as described before.8c Aqueous sodium hypochlorite (reagent grade, Aldrich, available chlorine 4.00−4.99%) was used as received and quantified by iodometry. Acetonitrile (LCMS grade, Aldrich) was used by passing through an activated neutral alumina column and then dried as described elsewhere.25 Benzyl alcohol (Aldrich, 99.8%) was passed through activated neutral alumina and distilled prior to use; its purity was checked by gas chromatography-mass spectrometry (GC-MS). All reactions were performed without any special precautions under atmospheric conditions unless otherwise specified. Deionized water was used to make all of the stock solutions for the reaction and kinetic runs. Enriched (18O) water (98%) was procured from the Shanghai Research Institute of Chemical Industry (China).
Generation of [FeIV(O)]2− (2) and [FeV(O)]− (3) Samples
On the one hand, a 0.5 mM solution of [(bTAML)FeIIICl]2− (1) was converted to 3 by using 1.2 equiv of sodium hypochlorite (NaOCl) in acetonitrile.8b On the other hand, the corresponding FeIV(O) complex 2 was obtained by two methods, either by treating the CH3CN solution of 3 with 3 equiv of tetrabutylammonium hydroxide or by addition of NaOCl to 1 in water (pH 12). In both cases, nearly quantitative conversion was confirmed by UV−vis spectroscopy.
Instrumentation
UV−vis spectral studies were performed using an Agilent diode array 8453 spectrophotometer attached with a Peltier temperature controller. CV experiments were performed on a CHI-660 potentiostat. Solutions of 1 were placed in one-compartment three-electrode cells. Glassy carbon (3 mm of diameter) was used as the working electrode, silver/silver chloride (3 M KCl) as reference electrode (unless explicitly mentioned), and Pt wire as counter electrode. Working electrode pretreatment before each measurement consisted of polishing with 0.05 μm alumina paste, rinsing thereafter with water/acetone, and finally blow-drying. All redox potentials in the present work are reported versus NHE by adding 0.21 V to the measured potential. Mössbauer spectra were recorded with two spectrometers using a Janis Research Super Varitemp dewar. The isomer shift was reported relative to Fe metal. The Mössbauer spectrum of 2 was simulated with least-squares fitting using the program SpinCount and the standard spin Hamiltonian.
GC was performed on a PerkinElmer Arnel Clarus 500 instrument equipped with a hydrogen flame ionization detector and HP-5 (30 m × 0.32 mm × 0.25 μm) column. Helium was used as carrier gas at a flow rate of 30 mL min−1. GC-MS analyses were performed on an Agilent 5977A mass-selective detector interfaced with an Agilent 7890B GC in similar conditions using an HP-5-Ms capillary column (30 m × 0.32 mm × 0.25 μm, J & W Scientific).
X-ray Absorption Spectroscopy
The XAS sample of 1 was prepared by dissolving isolated solid in CH3CN to make a 5 mM solution at 20 °C, which was then transferred to an XAS cup and frozen in liquid nitrogen. The XAS sample of 3 was prepared by treating a 5 mM solution of 1 in anhydrous acetonitrile at 20 °C with 1.2 equiv of NaOCl to generate 3 in near-quantitative yield. The green solution was transferred to an XAS cup, and the sample was frozen in liquid nitrogen. The sample of 2 was prepared by generating a 5 mM solution of 3 at 20 °C, then adding 3 equiv of tetrabutylammonium hydroxide to form the FeIV(O) complex.
Iron K-edge X-ray absorption spectra for 1, 2, and 3 were collected on SSRL beamline 9−3 using a 100-element solid-state Ge detector (Canberra) with a SPEAR storage ring current of ∼500 mA at a power of 3.0 GeV. The incoming X-rays were unfocused using a Si(220) double crystal monochromator, which was detuned to 40% of the maximal flux to attenuate harmonic X-rays. Seven, nine, and eight scans of the fluorescence excitation spectra for 1, 3, and 2, respectively, were collected from 6882 to 8000 eV at a temperature (10 K) that was controlled by an Oxford Instruments CF1208 continuous-flow liquid-helium cryostat. An iron foil was placed in the beam pathway prior to the ionization chamber I0 and scanned concomitantly for an energy calibration, with the first inflection point of the edge assigned to 7112.0 eV. A 6 μm Mn filter and a Soller slit were used to increase the signal-to-noise ratio of the spectra. Photoreduction was monitored by scanning the same spot on the sample twice and comparing the first-derivative peaks associated with the edge energy during collection, but none was observed in the present study.
The detector channels from the scans were examined, calibrated, averaged, and processed for EXAFS analysis using EXAFSPAK26a to extract χ(k). Theoretical phase and amplitude parameters for a given absorber−scatterer pair were calculated using FEFF 8.4026b and were utilized by the “opt” program of the EXAFSPAK package during curve fitting. Parameters for 1, 2, and 3 were calculated using a model based on the available crystal structure of the 1 complex.8b In all analyses, the coordination number of a given shell was a fixed parameter and was varied iteratively in integer steps, while the bond lengths (R) and mean-square deviation (σ2) were allowed to freely float. The amplitude reduction factor S0 was fixed at 0.9, while the edge-shift parameter E0 was allowed to float as a single value for all shells. Thus, in any given fit, the number of floating parameters was typically equal to (2 × num shells) + 1. The k range of the data is 2−15 Å−1.
The pre-edge analysis was performed on data normalized in the “process” program of the EXAFSPAK package, and pre-edge features were fit between 7108 and 7117 eV for 1 and between 7108 and 7118 eV for 2 and 3 using the Fityk26c program with pseudo-Voigt functions composed of 50:50 Gaussian/Lorentzian functions.
Resonance Raman Spectroscopy
Resonance Raman spectra were obtained with excitation at 476.5 nm (40 mW at source, Ar+ laser, Spectra-Physics). Data were obtained on samples at room temperature in flat-bottomed NMR tubes using a 90° backscattering arrangement (parallel to the slit direction) and at 77 K on frozen samples in NMR tubes using a 135° backscattering arrangement. The collimated Raman scattering was collected using two Plano convex lenses (f = 12 cm, placed at an appropriate distance) through a holographic super notch filter (Kaiser Optical Systems, INC) into an Acton AM-506M3 monochromator equipped with a Princeton Instruments ACTON PyLON LN/CCD-1340 × 400 detector. The detector was cooled to −120 °C prior to the experiments. Spectral calibration was performed using the Raman spectrum of acetonitrile/toluene 50:50 (v/v).27a Each spectrum was accumulated, typically 60 times with 5 s acquisition time, resulting in a total acquisition time of 5 min per spectrum. The collected data were processed using Spekwin32,27b and a multipoint baseline correction was performed for all spectra.
Raman samples of 2 were prepared by treating a 0.5 mM solution of 3 generated using Na16/18OCl with 6 equiv of tetrabutylammonium hydroxide at 20 °C. Na18OCl was prepared by dilution of 10% NaOClin H218O in 1:9 ratio.27c Raman samples of 3 were generated by addition of 1.2 equiv of Na16/18OCl to a 0.5 mM solution of 1 in CH3CN at 20 °C with stirring.
Kinetic Experiments
The kinetics for the oxidation of benzyl alcohol by 2 were monitored in the kinetic mode of the spectrophotometer using 1 cm quartz cell at 395 nm at the isosbestic points of FeIV species and FeIII at 30.0 ± 0.5 °C as well as other temperatures. All the kinetic experiments were performed in a premixed 80% CH3CN/20% water solvent. For kinetic measurements, 3 was synthesized by using 1.2 equiv of sodium hypochlorite as terminal oxidant. The concentration of 3 (5 × 10−5 M) was kept constant, while substrate concentration was varied. The pseudo-first-order rate constants kobs (calculated by monitoring changes at 395 nm) were obtained from nonlinear curve fitting [(At = Aα − (Aα − Ao)e(−kobst)]. Resulting kobs values correlated linearly with substrate concentration to afford the second-order rate constant k2.28,29 For the kinetic studies of benzyl alcohol oxidation by 2, the complex was generated in 80% CH3CN/20% pH 12 water (adjusted by 5 mM NaOH) by using NaOCl (0.5 equiv) at 30.0 ± 0.5 °C. The reaction kinetics was monitored at various temperatures by the decay in the characteristic absorbance of 2 at 460 nm to extract the pseudo-first-order rate constant (kobs) from nonlinear curve fitting [(At = Aα − (Aα − Ao)e(−kobst)]. Resulting kobs values correlated linearly with substrate concentration to afford the second-order rate constant k2.28,29 The reaction of 3 with benzyl alcohol has been monitored at 395 nm, which is the isosbestic point of FeIII (1) and the FeIV−O−FeIV byproduct formed during the reaction.21a In short, the reaction of benzyl alcohol with 3 forms benzaldehyde and regenerates the starting FeIII (1). Fast comproportionation of 1 and 3 leads to the formation of FeIV−O−FeIV. Hence monitoring the kinetics at 380 nm (monitoring FeIII formation) or at 613 nm (monitoring the decay of the FeV(O) complex) remains complicated. For 2, the reaction kinetics were measured by monitoring the decay of 2 to FeIII−OH at 460 nm.
Product Quantification
Products were first analyzed by GC-MS methods. GC was used for product quantification. To a solution of 3 or 221 (1 × 10−4 M) in CH3CN/water was added benzyl alcohol (1000 equiv, 1 × 10−1 M) at room temperature. After completion of reaction (determined by UV−vis absorption spectroscopy) the products were passed through a short alumina plug and immediately quantified by GC.
Computational Details
DFT-based methods were employed to compare the electronic structures of 2 and 3. All the minima reported in this study were fully optimized at the UBP86/6-311+G* or UB3LYP/6-311+G* level of theory30 using the Gaussian 09 suite of quantum-chemical programs.31 The stationary points on the potential energy surface were characterized by evaluating the vibrational frequencies.
Supplementary Material
Acknowledgments
The work performed at the CSIR-National Chemical Laboratory was supported by SERB, New Delhi (Grant No. SERB/EMR/2014/0016 to SSG). The work performed at the Univ. of Minnesota was supported by the U.S. National Science Foundation (Grant No. CHE-1361773 to L.Q.). XAS data were collected on Beamlines 7-3 and 9-3 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of Beamlines 7-3 and 9-3 is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). S.P. and K.K.S. thank UGC-India for a fellowships. S.P. and S.S.G. thank Dr. A. Das (CSIR-NCL) for use of the electrochemical facility at CSIR-NCL.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.7b00448.
Mossbauer and UV−vis absorption spectra and pKa determination of [(bTAML)FeIV(O)]2−, quantification of benzaldehyde under single turnover conditions, Eyring plots for benzyl alcohol oxidation by 2 and 3, UV−vis absorption spectral scanning and kobs at different substrate concentration of benzyl alcohol of Fe(O) reaction with benzyl alcohol, electrochemistry of [(bTAML)FeIII(OH2)2−] at higher pH, XAS data and EXAFS fitting details, and computational calculation details (PDF)
ORCID
Lawrence Que Jr.: 0000-0002-0989-2813
Abhishek Dey: 0000-0002-9166-3349
Sayam Sen Gupta: 0000-0002-3729-1820
Notes
The authors declare no competing financial interest.
References
- 1.(a) Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]; (b) Ortiz de Montellano PR. Hydrocarbon Hydroxylation by Cyctochrome P450 Enzymes. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Groves JT. Models and Mechanisms of Cytochrome P450 Action. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3rd. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 2.(a) Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;1996:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]; (b) Nam W. High-valent iron(IV)-oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (c) Rittle J, Green MT. Cyctochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; (d) Meunier B, de Visser SIP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; (e) Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen Activation at Mononuclear Non-heme Iron Active Sites: Enzymes, Models, and Intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (f) Abu-Omar MM, Loaiza A, Hontzeas N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- 3.Sastri CV, Lee J, Oh K, Lee YJ, Lee J, Jackson TA, Ray K, Hirao H, Shin W, Halfen JA, Kim J, Que L, Jr, Shaik S, Nam W. Axial ligand tuning of a nonheme iron(IV)-oxo unit for hydrogen atom abstraction. Proc Natl Acad Sci U S A. 2007;104:19181–19186. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borovik AS. Role of metal-oxo complexes in the cleavage of CH bonds. Chem Soc Rev. 2011;40:1870–1874. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Groves JT, Gross Z, Stern MK. Preperation and Reactivity of Oxoiron(IV) Porphyrins. Inorg Chem. 1994;33:5065–5072. [Google Scholar]; (b) Nam W, Lee HJ, Oh SY, Kim C, Jang HG. First success of catalytic epoxidation of olefins by an electron-rich iron(III) porphyrin complex and H2O2: imidazole effect on the activation of H2O2 by iron porphyrin complexes in aprotic solvent. J Inorg Biochem. 2000;80:219–225. doi: 10.1016/s0162-0134(00)00085-4. [DOI] [PubMed] [Google Scholar]; (c) Nam W, Park SE, Lim IK, Lim MH, Hong J, Kim J. First direct evidence for stereospecific olefin epoxidation and alkane hydroxylation by an oxoiron(IV) porphyrin complex. J Am Chem Soc. 2003;125:14674–14675. doi: 10.1021/ja0368204. [DOI] [PubMed] [Google Scholar]; (d) Nehru K, Seo MS, Kim J, Nam W. Oxidative N-Dealkylation Reactions by Oxoiron(IV) Complexes of Nonheme and Heme Ligands. Inorg Chem. 2007;46:293–298. doi: 10.1021/ic0614014. [DOI] [PubMed] [Google Scholar]; (e) Park MJ, Lee J, Suh Y, Kim J, Nam W. Reactivities of Mononuclear Non-Heme Iron Intermediates Including Evidence that Iron(III)-Hydroperoxo Species Is a Sluggish Oxidant. J Am Chem Soc. 2006;128:2630–2634. doi: 10.1021/ja055709q. [DOI] [PubMed] [Google Scholar]; (f) Jeong YJ, Kang Y, Han AR, Lee YM, Kotani H, Fukuzumi S, Nam W. Hydrogen Atom Abstraction and Hydride Transfer reactions by Iron(IV)−Oxo Porphyrins. Angew Chem, Int Ed. 2008;47:7321–7324. doi: 10.1002/anie.200802346. [DOI] [PubMed] [Google Scholar]; (g) Fukuzumi S, Kotani H, Lee YM, Nam W. Sequential Electron-Transfer and Proton-Transfer Pathways in Hydride-Transfer reactions from Dihydronicotinamide Adenine Dinucleotide Analogues to Non-heme Oxoiron(IV) Complexes and p-Chloranil. Detection of Radical Cations of NADH Analogues in Acid-Promoted Hydride-Transfer Reactions. J Am Chem Soc. 2008;130:15134–15142. doi: 10.1021/ja804969k. [DOI] [PubMed] [Google Scholar]; (h) Ji L, Franke A, Brindell M, Oszajca M, Zahl A, van Eldik R. Combined Experimental and Theoretical Study on the Reactivity of Compounds I and II in Horseradish Peroxidase Biomimetics. Chem – Eur J. 2014;20:14437–14450. doi: 10.1002/chem.201402347. [DOI] [PubMed] [Google Scholar]; (i) Fertinger C, Hessenauer-Ilicheva N, Franke A, van Eldik R. Direct Comparison of the reactivity of Model Complexes for Compounds 0, I, and II in Oxygenation, Hydrogen−Abstraction, and Hydride−Transfer Processes. Chem -Eur J. 2009;15:13435–13440. doi: 10.1002/chem.200901804. [DOI] [PubMed] [Google Scholar]; (j) Bell SR, Groves JT. A Highly Reactive P450 Model Compound I. J Am Chem Soc. 2009;131:9640–9641. doi: 10.1021/ja903394s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Altun A, Shaik S, Thiel W. What is the Active Species of Cytochrome P450 during Camphor Hydroxylation? QM/MM Studies of Different Electronic States of Compound I and of Reduced and Oxidized Iron-Oxo Intermediates. J Am Chem Soc. 2007;129:8978–8987. doi: 10.1021/ja066847y. [DOI] [PubMed] [Google Scholar]; (b) Tahsini L, Bagherzadeh M, Nam W, de Visser SP. Fundamental Differences of Substrate Hydroxylation by High-Valent Iron(IV)-Oxo Models of Cytochrome P450. Inorg Chem. 2009;48:6661–6669. doi: 10.1021/ic900593c. [DOI] [PubMed] [Google Scholar]; (c) Ricciardi G, Baerends EJ, Rosa A. Charge Effects on the Reactivity of Oxoiron(IV) Porphyrin Species: A DFT Analysis of Methane Hydroxylation by Polycationic Compound I and Compound II Mimics. ACS Catal. 2016;6:568–579. [Google Scholar]; (d) Berry JF, DeBeer George S, Neese F. Electronic structure and spectroscopy of “superoxidized” iron centers in model systems: theoretical and experimental trends. Phys Chem Chem Phys. 2008;10:4361–4374. doi: 10.1039/b801803k. [DOI] [PubMed] [Google Scholar]; (e) Geng C, Ye S, Neese F. Does a higher metal oxidation state necessarily imply higher reactivity toward H-atom transfer? A computational study of C−H bond oxidation by high−valent metal− oxo and − nitrido complexes. Dalton Trans. 2014;43:6079–6086. doi: 10.1039/c3dt53051e. [DOI] [PubMed] [Google Scholar]; (f) Lyakin OY, Bryliakov KP, Talsi EP. EPR, 1H and 2H NMR, and Reactivity Studies of the Iron-Oxygen Intermediates in Bioinspired Catalyst System. Inorg Chem. 2011;50:5526. doi: 10.1021/ic200088e. [DOI] [PubMed] [Google Scholar]; (g) Oloo WN, Feng Y, Iyer S, Parmelee S, Xue G, Que L., Jr Cyclohexene as a substrate probe for the nature of the high-valent iron-oxo oxidant in Fe(TPA)-catalyzed oxidations. New J Chem. 2013;37:3411–3415. [Google Scholar]
- 7.(a) de Oliveira FT, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Bominaar EL, Münck E, Collins TJ. Chemical and spectroscopic evidence for an FeV-oxo complex. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]; (b) Popescu DL, Vrabel M, Brausam A, Madsen P, Lente G, Fabian I, Ryabov AD, van Eldik R, Collins TJ. Thermodynamic, Electrochemical, High-Pressure kinetics, and Mechanistic studies of the Formation of Oxo FeIV-TAML Species in Water. Inorg Chem. 2010;49:11439–11448. doi: 10.1021/ic1015109. [DOI] [PubMed] [Google Scholar]; (c) Chanda A, Shan X, Chakrabarti M, Ellis WC, Popescu DL, Tiago de Oliveira F, Wang D, Que L, Collins TJ, Münck E, Bominaar EL. (TAML)FeIV=O Complex in Aqueous Solution: Synthesis, Spectroscopic and Computational Characterization. Inorg Chem. 2008;47:3669–3678. doi: 10.1021/ic7022902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ghosh A, Tiago de Oliveira F, Yano T, Nishioka T, Beach ES, Kinoshita I, Münck E, Ryabov AD, Horwitz CP, Collins TJ. Catalytically Active μ-Oxodiiron(IV) Oxidants form Iron(III) and Dioxygen. J Am Chem Soc. 2005;127:2505–2513. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]; (e) Kundu S, Thompson JV, Ryabov AD, Collins TJ. On The Reactivity of Mononuclear Iron(V)oxo Complexes. J Am Chem Soc. 2011;133:18546. doi: 10.1021/ja208007w. [DOI] [PubMed] [Google Scholar]; (f) Sarangi R. X-ray absorption near-edge spectroscopy in bioinorganic chemistry: Application to M-O2 systems. Coord Chem Rev. 2013;257:459–472. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ye S, Kupper C, Meyer S, Andris E, Navratil R, Krahe O, Mondal B, Atanasov M, Bill E, Roithova J, Meyer F, Neese F. Magnetic Circular Dichroism Evidence for an Unusual Electronic Structure of a Tetracarbene-Oxoiron(IV) Complex. J Am Chem Soc. 2016;138:14312–14325. doi: 10.1021/jacs.6b07708. [DOI] [PubMed] [Google Scholar]; (h) Decker A, Rohde JU, Que L, Solomon EI. Spectroscopic and Quantum Chemical Characterization of the Electronic Structure and Bonding in a Non-Heme FeIV=O Complex. J Am Chem Soc. 2004;126:5378–5379. doi: 10.1021/ja0498033. [DOI] [PubMed] [Google Scholar]; (i) Decker A, Solomon EI. Comparison of FeIV=O Heme and Non-heme Species: Electronic Structure, Bonding, and Reactivities. Angew Chem, Int Ed. 2005;44:2252–2255. doi: 10.1002/anie.200462182. [DOI] [PubMed] [Google Scholar]; (j) Solomon EI, Light MK, Liu VL, Srnec M, Wong DS. Geometric and Electronic Structure Contributions to Function in Non-heme Iron Enzymes. Acc Chem Res. 2013;46:2725–2739. doi: 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ghosh M, Singh KK, Panda C, Weitz A, Hendrich MP, Collins TJ, Dhar BB, Sen Gupta S. Formation of a Room Temperature Stable FeV(O) Comples: reactivity Toward Unactivated C-H Bonds. J Am Chem Soc. 2014;136:9524–9527. doi: 10.1021/ja412537m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh KK, Tiwari MK, Ghosh M, Panda C, Weitz A, Hendrich MP, Dhar BB, Vanka K, Sen Gupta S. Tuning the Reactivity of FeV(O) toward C-H bonds at Room temperature: Effect of Water. Inorg Chem. 2015;54:1535–1542. doi: 10.1021/ic502535f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Panda C, Ghosh M, Panda T, Banerjee R, Sen Gupta S. Fe(III) complex of biuret-amide based macrocyclic ligand as peroxidase enzyme mimic. Chem Commun. 2011;47:8016–8018. doi: 10.1039/c1cc12686e. [DOI] [PubMed] [Google Scholar]
- 9.(a) McDonald AR, Que L., Jr High-valent nonheme iron-oxo complexes: synthesis, structure, and spectroscopy. Coord Chem Rev. 2013;257:414–428. [Google Scholar]; (b) Klein JEMN, Que L. Encyclopedia of Inorganic and Bioinorganic Chemistry (EIBC) John Wiley & Sons, Ltd; 2016. Biomimetic High-Valent Mononuclear Nonheme Iron-Oxo Chemistry. [DOI] [Google Scholar]; (c) Prakash J, Rohde GT, Meier KK, Münck E, Que L., Jr Upside Down! Crystallographic and Spectroscopic Characterization of an [FeIV(Osyn) (TMC)]2+ Complex. Inorg Chem. 2015;54:11055–11057. doi: 10.1021/acs.inorgchem.5b02011. [DOI] [PubMed] [Google Scholar]
- 10.(a) Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. Formation, Structure, and EPR Detection of a High Spin FeIV-Oxo Species Derived from Either an FeIII-O or FeIII-OH Complex. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. O2 activation by nonheme iron complexes: A monomeric Fe(III)-oxo complex derived from O2. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 11.(a) Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L., Jr X-ray absorption spectroscopy of iron-tyrosinate proteins. J Am Chem Soc. 1984;106:1676–1681. [Google Scholar]; (b) Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A Multiple Analysis of Fe K-Edge 1s→ 3d Pre-Edge Features of iron complexes. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]; (c) Chandrasekaran P, Stieber SCE, Collins TJ, Que L, Jr, Neese F, DeBeer S. Prediction of high-valent iron K-edge absorption spectra by time-dependent Density Functional Theory. Dalton Trans. 2011;40:11070–11079. doi: 10.1039/c1dt11331c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Jr, Bakac A. Aqueous FeIV=O: Spectroscopic Identification and Oxo-Group Exchange. Angew Chem, Int Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh A, Ryabov AD, Mayer SM, Horner DC, Prasuhn DE, Sen Gupta S, Vuocolo L, Culver C, Hendrich MP, Rickard CEF, Norman RE, Horwitz CP, Collins TJ. Understanding the Mechanism of H+-Induced Demetalation as a Design Strategy for Robust Iron(III) Peroxide-Activating Catalysts. J Am Chem Soc. 2003;125:12378–12379. doi: 10.1021/ja0367344. [DOI] [PubMed] [Google Scholar]
- 14.Warner GR, Mills MR, Enslin C, Pattanayak S, Panda C, Panda TK, Gupta SS, Ryabov AD, Collins TJ. Reactivity and Operational stability of N-Tailed TAMLs Through Kinetic Studies of the Catalyzed Oxidation of Orange II by H2O2: Synthesis and X-ray Structure of an N-Phenyl TAML. Chem - Eur J. 2015;21:6226–6233. doi: 10.1002/chem.201406061. [DOI] [PubMed] [Google Scholar]
- 15.(a) Huynh MHV, Meyer TJ. Proton-Coupled Electron Transfer. Chem Rev. 2007;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Proton-Coupled Electron Transfer. Chem Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 16.(a) Pattanayak S, Chowdhury RD, Garai B, Singh KK, Paul A, Dhar BB, Gupta SS. Electrochemical formation of FeV(O) and mechanism of its reaction with water during O-O bond formation. Chem - Eur J. 2017;23:3414–3424. doi: 10.1002/chem.201605061. [DOI] [PubMed] [Google Scholar]; (b) Liu M-H, Su YO. Selective electrocatalysis of alkene oxidations in aqueous media. Electrochemical and spectral characterization of oxo-ferryl porphyrin, oxo-ferryl porphyrin cation radical and their reaction products with alkenes at room temperature. J Electroanal Chem. 1998;452:113–125. [Google Scholar]
- 17.BDE(FeIII−OH) = 23.06 × E0′(FeIV/III) + 1.37pH + 57 = 23.06 × 0.44 V + 1.37 × 12 + 57 = 83 ± 2 kcal/mol.
- 18.Wang D, Zhang M, Bühlmann P, Que L., Jr Redox Potential and C-H Bond Cleaving Properties of a Nonheme FeIV=O Complex in Aqueous Solution. J Am Chem Soc. 2010;132:7638–7644. doi: 10.1021/ja909923w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Wolak M, van Eldik R. Mechanistic Studies on Peroxide Activation by a Water-Soluble Iron(III)-Porphyrin: Implications for O-O Bond Activation in Aqueous and Nonaqueous Solvents. Chem - Eur J. 2007;13:4873–4883. doi: 10.1002/chem.200601148. [DOI] [PubMed] [Google Scholar]; (b) Brandi P, Galli P, et al. Kinetic Study of the Hydrogen Abstraction Reaction of the Benzotriazole-N-Oxyl Radical (BTNO) with H-Donar Substrates. J Org Chem. 2005;70:9521–9528. doi: 10.1021/jo051615n. [DOI] [PubMed] [Google Scholar]
- 20.BDE(FeIV−OH) = 23.06 × E0′(FeV/IV) + 1.37pKa + 57 = 23.06 × 1.19 V + 1.37 × 10.05 + 57 = 99 ± 2 kcal/mol.
- 21.(a) Ghosh M, Nikhil YLK, Dhar BB, Sen Gupta S. Mechanism of Alcohol Oxidation by FeV(O) at Room Temperature. Inorg Chem. 2015;54:11792–11798. doi: 10.1021/acs.inorgchem.5b01937. [DOI] [PubMed] [Google Scholar]; (b) Park J, Lee YM, Nam W, Fukuzumi S. Brønsted Acid-Promoted C-H Bond Cleavage via Electron Transfer from Toluene Derivatives to a Protonated Nonheme Iron(IV)-Oxo Complex with No Kinetic Isotope Effect. J Am Chem Soc. 2013;135:5052–5061. doi: 10.1021/ja311662w. [DOI] [PubMed] [Google Scholar]; (c) Cho KB, Hirao H, Shaik S, Nam W. To rebound or dissociate? This is the mechanistic question in C-H hydroxylation by heme and nonheme metal-oxo complexes. Chem Soc Rev. 2016;45:1197–1210. doi: 10.1039/c5cs00566c. [DOI] [PubMed] [Google Scholar]
- 22.Oszajca M, Franke A, Drzewiecka-Matuszek A, Brindell MG, Stochel GY, van Eldik R. Temperature and Pressure Effects on C-H Abstraction Reactions Involving Compound I and II Mimics in Aqueous Solution. Inorg Chem. 2014;53:2848–2857. doi: 10.1021/ic402567h. [DOI] [PubMed] [Google Scholar]
- 23.(a) Parsell TH, Yang MY, Borovik AS. C-H Bond Cleavage with Reductants: Re-Investigation the Reactivity of Monomeric MnIII/IV-Oxo Complexes and the Role of Oxo Ligand Basicity. J Am Chem Soc. 2009;131:2762–2763. doi: 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh KK, Tiwari MK, Dhar BB, Vanka K, Sen Gupta S. Mechanism of Oxygen Atom Transfer from FeV(O) to Olefins at Room Temperature. Inorg Chem. 2015;54:6112–6121. doi: 10.1021/ic503053q. [DOI] [PubMed] [Google Scholar]; (c) Schröder D, Shaik S, Schwarz H. Two-State reactivity as a New Concept in Organometallic Chemistry. Acc Chem Res. 2000;33:139–145. doi: 10.1021/ar990028j. [DOI] [PubMed] [Google Scholar]; (d) Shaik S, Danovich D, Fiedler A, Schröder D, Schwarz H. Two-State Reactivity in Organometallic Gas-Phase Ion Chemistry. Helv Chim Acta. 1995;78:1393–1407. [Google Scholar]; (e) Shaik S, Filatov M, Schröder D, Schwarz H. Electronic Structure Makes a Difference: Cytochrome P-450 Mediated Hydroxylations of Hydrocarbons as a TWO-State Reactivity Paradigm. Chem - Eur J. 1998;4:193–199. [Google Scholar]; (f) Shaik S, Hirao H, Kumar D. Reactivity of High-Valent Iron-Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Acc Chem Res. 2007;40:532–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- 24.(a) Mayer JM. Hydrogen Atom Abstraction by Metal-Oxo Complexes: Understanding the Analogy with Organic Radical Reactions. Acc Chem Res. 1998;31:441–450. [Google Scholar]; (b) Shaik S, Kumar D, de Visser SP. A Valance Bond Modeling of Trends in Hydrogen Abstraction Barriers and Transition States of Hydroxylation Reactions Catalyzed by Cytochrome P450 Enzymes. J Am Chem Soc. 2008;130:10128–10140. doi: 10.1021/ja8019615. [DOI] [PubMed] [Google Scholar]; (c) Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr Nonheme FeIVO Complexes That Can Oxidize the C-H Bond of Cyclohexane at Room Temperature. J Am Chem Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]; (d) Hirao H, Kumar D, Que L, Shaik S. Two-State reactivity in Alkane Hydroxylation by Non-Heme Iron-Oxo Complexes. J Am Chem Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]; (e) Mandal D, Ramanan R, Usharani D, Janardanan D, Wang B, Shaik S. How Does Tunneling Contribute to the Counterintuitive H-Abstraction reactivity of Nonheme Fe(IV)O Oxidants with Alkanes? J Am Chem Soc. 2015;137:722–733. doi: 10.1021/ja509465w. [DOI] [PubMed] [Google Scholar]; (f) Chen K, Que L., Jr Stereospecific Alkane hydroxylation by Non-Heme Iron Catalysts: Mechanistic Evidence for an FeV=O Active Species. J Am Chem Soc. 2001;123:6327–6337. doi: 10.1021/ja010310x. [DOI] [PubMed] [Google Scholar]; (g) Chen K, Costas M, Kim J, Tipton AK, Que L., Jr Olefin cis-Dihydroxylation versus Epoxidation by Non-Heme Iron Catalysts: Two Faces of an FeIII-OOH Coin. J Am Chem Soc. 2002;124:3026–3035. doi: 10.1021/ja0120025. [DOI] [PubMed] [Google Scholar]; (h) Oloo WN, Que L., Jr Bioinspired Nonheme Iron Catalysts for C-H and C=C Bond Oxidation: Insight into the Nature of the Metal Based Oxidants. Acc Chem Res. 2015;48:2612–2621. doi: 10.1021/acs.accounts.5b00053. [DOI] [PubMed] [Google Scholar]; (i) Swart M. A change in the oxidation state of iron: scandium is not innocent. Chem Commun. 2013;49:6650–6652. doi: 10.1039/c3cc42200c. [DOI] [PubMed] [Google Scholar]
- 25.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4th. Elsevier Science; United States: 1997. [Google Scholar]
- 26.(a) George GN. EXAFSPAK: A Suite of Computer Programs for Analysis of X-ray Absorption Spectra. 1990 http://www-ssrl.slac.stanford.edu/exafspak.html.; (b) Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys Rev B: Condens Matter Mater Phys. 1998;58:7565–7576. [Google Scholar]; (c) Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Crystallogr. 2010;43:1126–1128. [Google Scholar]
- 27.(a) ASTM E1840−96. Standard Guide for Raman Shift Standards for Spectrometer Calibration. ASTM International; 2007. [DOI] [Google Scholar]; (b) Menges F. Spekwin32-optical spectroscopy software. Version 1.72.0. 2015 http://www.effemm2.de/spekwin/; (c) Draksharapu A, Codolà Z, Gómez L, Lloret-Fillol J, Browne WR, Costas M. Spectroscopic Analyses on Reaction Intermediates Formed during Chlorination of Alkanes with NaOCl Catalyzed by a Nickel Complex. Inorg Chem. 2015;54:10656–10666. doi: 10.1021/acs.inorgchem.5b01463. [DOI] [PubMed] [Google Scholar]
- 28.Cho K, Leeladee P, McGown AJ, DeBeer S, Goldberg DP. A High-Valent Iron-Oxo Corrolazine Activates C-H Bonds via Hydrogen-Atom Transfer. J Am Chem Soc. 2012;134:7392–7399. doi: 10.1021/ja3018658. [DOI] [PubMed] [Google Scholar]
- 29.Espenson JH. Chemical Kinetics and Reaction Mechanisms. 2nd. McGraw-Hill, Inc.; New York: 1995. [Google Scholar]
- 30.(a) Perdew JP, Chevary SH, Vosko KA, Jackson KA, Pederson MR, Singh DJ, Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys Rev B: Condens Matter Mater Phys. 1992;46:6671. doi: 10.1103/physrevb.46.6671. [DOI] [PubMed] [Google Scholar]; (b) Perdew JP, Chevary SH, Vosko KA, Jackson KA, Pederson MR, Singh DJ, Fiolhais C. Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys Rev B: Condens Matter Mater Phys. 1993;48:4978. doi: 10.1103/physrevb.48.4978.2. [DOI] [PubMed] [Google Scholar]; (c) Perdew JP, Burke K, Wang Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys Rev B: Condens Matter Mater Phys. 1996;54:16533. doi: 10.1103/physrevb.54.16533. [DOI] [PubMed] [Google Scholar]; (d) Adamo C, Barone V. Exchange functionals with improved long-range behaviour and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J Chem Phys. 1998;108:664. [Google Scholar]; (e) Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648. [Google Scholar]; (f) Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B: Condens Matter Mater Phys. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (g) Roothaan CC. Self-Consistent Field Theory for Open Shells of Electronic Systems. Rev Mod Phys. 1960;32:179. [Google Scholar]; (h) McWeeny R, Diercksen G. J Chem Phys. 1968;49:4852. [Google Scholar]; (i) Pople JA, Nesbet RK. J Chem Phys. 1954;22:571. [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision B01. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.