

Abstract
Neurotrauma and neurodegenerative disease often result in lasting neurological deficits due to the limited capacity of the central nervous system (CNS) to replace lost neurons and regenerate axonal pathways. However, during nervous system development, neuronal migration and axonal extension often occur along pathways formed by other cells, referred to as "living scaffolds". Seeking to emulate these mechanisms and to design a strategy that circumvents the inhibitory environment of the CNS, this manuscript presents a protocol to fabricate tissue engineered astrocyte-based "living scaffolds". To create these constructs, we employed a novel biomaterial encasement scheme to induce astrocytes to self-assemble into dense three-dimensional bundles of bipolar longitudinally-aligned somata and processes. First, hollow hydrogel micro-columns were assembled, and the inner lumen was coated with collagen extracellular-matrix. Dissociated cerebral cortical astrocytes were then delivered into the lumen of the cylindrical micro-column and, at a critical inner diameter of <350 µm, spontaneously self-aligned and contracted to produce long fiber-like cables consisting of dense bundles of astrocyte processes and collagen fibrils measuring <150 µm in diameter yet extending several cm in length. These engineered living scaffolds exhibited >97% cell viability and were virtually exclusively comprised of astrocytes expressing a combination of the intermediate filament proteins glial-fibrillary acidic protein (GFAP), vimentin, and nestin. These aligned astrocyte networks were found to provide a permissive substrate for neuronal attachment and aligned neurite extension. Moreover, these constructs maintain integrity and alignment when extracted from the hydrogel encasement, making them suitable for CNS implantation. These preformed constructs structurally emulate key cytoarchitectural elements of naturally occurring glial-based "living scaffolds" in vivo. As such, these engineered living scaffolds may serve as test-beds to study neurodevelopmental mechanisms in vitro or facilitate neuroregeneration by directing neuronal migration and/or axonal pathfinding following CNS degeneration in vivo.
Keywords: Bioengineering, Issue 131, Neural tissue engineering, living scaffolds, neurotrauma, neuroregeneration, cell migration, axon pathfinding, astrocyte networks, neural stem cells
Introduction
The central nervous system (CNS) has a limited capacity to counteract the loss and/or dysfunction of neurons and axonal pathways that accompany conditions such as traumatic brain injury (TBI), stroke, spinal cord injury (SCI), and neurodegenerative disease1,2,3,4,5. Neurogenesis in the CNS is restricted to a limited number of areas in the brain, hampering the restoration of lost neurons6,7. Additionally, regeneration of lost axonal pathways in the CNS is insufficient due to the lack of directed guidance, the presence of outgrowth inhibitors, and reactive astrogliosis following damage to neural tissue2,8,9,10. Astrocytes typically have diverse functions in assisting neurons with ion homeostasis, neurotransmitter clearance, synapse formation, and neurovascular coupling11. Nevertheless, following even mild damage to neural tissue, astrocytes may undergo molecular, structural, and functional changes as they transition to a hypertrophic state11. In response to severe neurotrauma, these changes result in the formation of a scar with a penumbra containing migrating reactive astrocytes and a lesion core that includes leukocytes leaked from the ruptured blood-brain barrier (BBB), microglia, oligodendrocytes, and fibroblasts11,12,13. These reactive astrocytes attain a morphology of filamentous, disorganized processes and exhibit increased expression of intermediate filament proteins and chondroitin sulfate proteoglycans (CSPGs), which hinder neural regeneration12. Even though the glial scar initially helps restore BBB integrity and avoid transmission of the inflammatory response to surrounding healthy tissue, it serves as a physical and biochemical barrier against axon regeneration12,14,15,16. For instance, axons that encounter the glial scar display bulbous dystrophic growth cones and stunted growth12. Furthermore, the disorganization of astrocytic processes after injury impedes the extension of regenerating axons17. The outcome of these inhibitory characteristics is manifested in the often-permanent physical and neurological impairments that patients suffer after severe neurotrauma, including TBI and SCI.
Regardless of the extrinsic challenges facing functional regeneration in the CNS, axons have been shown to possess an intrinsic ability to regenerate. For instance, the dynamic nature of the dystrophic growth cones in contact with the glial scar suggests that these endings retain their capacity to extend12. Consequently, it is believed that a main hindrance to axonal re-growth is the inhibitory environment of the post-injury CNS and that providing a more permissive environment via reducing glial scarring and/or providing regenerative bridges across the scar would be advantageous. Indeed, previous studies have demonstrated that CNS neurons were capable of extending axons through a lesion using peripheral nerve grafts as bridges, which present a more favorable environment for axon regeneration12,18,19. Several other strategies have been pursued to exploit this vestigial regenerative capacity. For example, manipulation of cell growth signaling pathways in various injury models has resulted in axonal regeneration and glial scar reduction10,20,21. Additionally, studies have shown that treatment with chondroitinase ABC, which cleaves the majority of the sugar chains in CSPGs, lessens the inhibitory effect of CSPGs secreted by reactive astrocytes22. Despite encouraging results, these approaches do not provide directed guidance of growth cones, which can potentially result in aberrant regeneration12, and also do not account for the loss of neurons. Cell-based approaches have been utilized in attempts to surmount the effects of the glial scar and to replenish lost cells, particularly neurons. Some groups have dedifferentiated reactive astrocytes into neurons, while others have transplanted neural progenitor cells into CNS lesions to repopulate the injury area and promote axon regeneration23,24,25. However, stem cell transplantation alone is limited by low survival rates, poor integration, and modest retention in the damaged tissue5. Furthermore, these cell-based strategies fail to restore long-distance axonal tracts, especially in a controlled manner. Therefore, biomaterials in combination with other approaches are being explored as delivery vehicles for various neural and progenitor cells and growth factors26. Biomaterial-based approaches feature a high degree of design control to produce constructs that mimic the specific physical, haptotaxic, and chemotaxic cues present in the three-dimensional (3D) microenvironment of the target host tissue27,28,29,30,31,32,33,34. Reproduction of these environmental signals is paramount for transplanted cells to present native-like morphology, proliferation, migration, and signaling, among other neurobiological characteristics29. Despite these advantageous properties, advancement beyond traditional cell seeded biomaterial scaffolds is required to simultaneously promote directed long-distance axonal regeneration and replace lost neurons.
A promising alternative approach is based on neural tissue engineered "living scaffolds", which are distinct from other cell-based approaches due to the presence of living neural cells with a preformed cytoarchitecture that emulates native neuroanatomy and/or developmental mechanisms to facilitate targeted replacement, reconstruction, and regeneration of neural circuitry4,35. Considerations for the design of living scaffolds include the phenotypes and sources of neural cells, as well as the mechanical/physical properties and the biochemical signals dictated by the composition of any accompanying biomaterials35. After fabrication in vitro, these living scaffolds can be implanted in vivo to present cell-adhesion molecules and chemotactic and neurotrophic signals to actively regulate neural cell migration and axon outgrowth depending on the state and progression of regenerative processes35. Glial cells can serve as a basis for the engineered cytoarchitecture of living scaffolds since these cells mediate various developmental mechanisms in vivo. During brain development, new neurons rely on basal processes extended by radial glia from the ventricular zone towards the developing cortical plate as living scaffolds for directed migration36,37. Furthermore, extending growth cones are shown to orient themselves by sensing attractive and repellent signals elicited by glial guidepost cells, and so-called "pioneering" axons are suggested to reach the correct targets by extending along pre-patterned glial scaffolds35,38,39. Thus, glial cells are necessary for the guidance of pioneering axons, which later serve as axon-based "living scaffolds" to direct the projection of "follower" axons. Moreover, glia-mediated growth mechanisms have been shown to persist postnatally, as neuroblasts follow the rostral migratory stream (RMS) to navigate from the subventricular zone (SVZ), one of the few remaining areas of neurogenesis in the adult brain, to the olfactory bulb (OB)40. These neuroblasts in the RMS migrate within the glial tube (Figure 1A-1), which is comprised of longitudinally-aligned astrocytic processes, via direct cell-cell adhesions and localized soluble factors37,41. Finally, while CNS damage in mammals causes disrupted astrocytic process arrangement forming a glial scar that physically impedes axonal regeneration17, many non-mammalian systems lack the formation of a detrimental glial scar. Rather, glial cells of non-mammalian species maintain more organized, aligned patterns that are used as guides through the injured region17,42,43. For instance, in non-mammalian SCI models, axons are shown to grow in close association with glial bridges crossing the lesion, suggesting an important role for organized glial scaffolds as substrates facilitating axonal regeneration and functional recovery (Figure 1A-2)42,44,45. Recapitulation of the neuroanatomical features and the developmental/regenerative mechanisms described above may yield a new class of engineered glial-based living scaffolds that can concurrently drive immature neuronal migration and axonal pathfinding through otherwise non-permissive environments, thereby potentially mitigating the effects of neuronal and axon tract degeneration associated with CNS injury and disease.
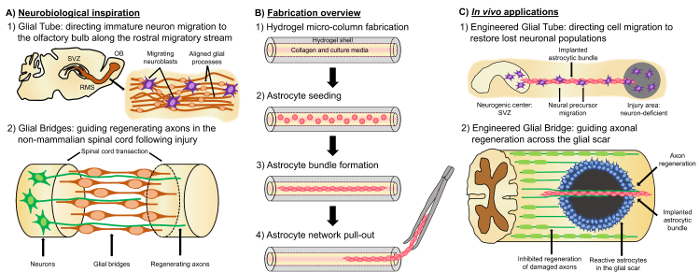
Our research group has previously designed multiple types of living scaffolds for reconstruction and regeneration of axonal tracts in the CNS and the peripheral nervous system (PNS) via micro-tissue engineered neural networks (micro-TENNs) and tissue engineered nerve grafts (TENGs), respectively27,46,47,48. Both strategies are based inherently on biomimicry. Micro-TENNs are anatomically-inspired structures designed to structurally and functionally replace axonal tracts connecting distinct neuronal populations of the brain. TENGs exploit the developmental mechanism of axon-facilitated axonal regeneration, exemplified by "follower" axon growth along "pioneer" axons, to achieve targeted host axonal regeneration35,46,48. We recently capitalized on the versatility of the living scaffold technique using a similar encasement scheme as micro-TENNs and seeking inspiration from the glia-based mechanisms present throughout development. Here, we developed constructs consisting of aligned astrocytic bundles spanning the collagenous lumen of a hydrogel micro-column49. These astrocytic living scaffolds are developed by first filling a capillary tube-acupuncture needle assembly with liquid agarose to create a hollow cylindrical hydrogel with an outer diameter (OD) and inner diameter (ID) corresponding to the diameters of the tube and needle, respectively. Following agarose gelation and extraction of the hydrogel micro-column from the capillary tube, the hollow interior is coated with type I collagen to supply an environment permissive for astrocyte adhesion and aligned bundle formation (Figure 1B-1). Afterwards, the lumen is seeded with cerebral cortical astrocytes isolated from postnatal rat pups (Figure 1B-2). Contrary to two-dimensional (2D) alignment techniques that rely on the application of electric fields, micropatterned grooves, and extracellular matrix (ECM) protein patterning, astrocyte alignment in the living scaffold technique relies on self-assembly according to controllable variables such as substrate curvature (column ID), cell density, and collagen concentration50,51,52. The astrocytes contract and remodel the collagen, and acquire a bipolar, longitudinally-aligned morphology analogous to the natural scaffolds observed in vivo (Figure 1B-3). Indeed, we are actively pursuing the use of these cable-like structures as physical substrates for targeted guidance of migrating immature neurons as well as facilitating axonal regeneration through the unfavorable environment of the damaged CNS, particularly the mammalian glial scar (Figure 1C). This article will present the detailed fabrication method for the astrocytic micro-columns, phase contrast and immunofluorescence images of the expected cytoarchitecture, and a comprehensive discussion on the current limitations and future directions of the technique.
Figure 1: Inspiration, Fabrication Protocol, and Proposed Applications for the Aligned Astrocytic Networks. (A) Neurobiological inspiration: (1) Neuroblasts originating from the neurogenic subventricular zone (SVZ) utilize the longitudinally aligned glial tube in the rostral migratory stream (RMS) for directed migration towards the olfactory bulb (OB); (2) Non-mammals such as amphibians and fish can sustain regeneration after neural tissue damage in part due to the formation of a glial bridge that connects the ends of a lesion (e.g. transected spinal cord) and serves as a scaffold for the guidance of regenerating axons. (B) Fabrication overview: (1) construction of a micron-sized, hollow hydrogel micro-column with the lumen coated with ECM, (2) seeding of primary cortical astrocytes isolated from postnatal rat pups, (3) self-assembly of the longitudinally-oriented bundles in culture, and (4) extraction of the bundle from the biomaterial encasement for future implantation studies. (C) In vivo applications: (1) These living scaffolds may serve as engineered glial tubes for directed neuron migration from neurogenic centers to repopulate neuron-deficient regions; (2) Recapitulation of the developmental mechanism of pioneering axon guidance and the regenerative mechanism of glial bridges in non-mammals may endow these astrocytic scaffolds with the capacity to direct axon regeneration across the non-permissive environment of the mammalian glial scar. Please click here to view a larger version of this figure.
Protocol
All procedures were approved by the Institutional Animal Care and Use Committees at the University of Pennsylvania and the Michael J. Crescenz Veterans Affairs Medical Center and adhered to the guidelines set forth in the NIH Public Health Service Policy on Humane Care and Use of Laboratory Animals (2015).
1. Development of the Agarose Hydrogel Micro-columns
Make an agarose 3% weight/volume (w/v) solution by weighing 3 g of agarose and transferring it to a sterile beaker containing 100 mL of Dulbecco's phosphate buffered saline (DPBS). Add a sterile magnetic bar to the beaker, and place it on the surface of a hot plate/stirrer. Keep the beaker covered to prevent evaporation of its contents in the next step.
Heat the agarose and DPBS at a temperature of 100 °C and stir at 60-120 rpm. Adjust these settings as necessary, and constantly monitor the progression of the dissolution process as the solution changes initially from opaque to a clear appearance that signifies that the agarose has been completely dissolved. Caution: The hot beaker and the solution are hot!
As the agarose solution is heated and stirred, retrieve four empty 10 cm Petri dishes and add 20 mL of DPBS to two of them. Place acupuncture needles (diameter: 300 µm, length: 40 mm), glass microliter capillary tubes (diameter: 701.04 µm, length: 65 mm, capacity: 25.0 µL), and a bulb dispenser inside the biosafety cabinet. When the liquid agarose solution clears out, maintain constant heating at approximately 50 °C and stirring to avoid gelation of the agarose.
Introduce an acupuncture needle into the bottom opening of a bulb dispenser. Insert a capillary tube over the needle exposed to the outside. Secure the capillary tube by entering part of it into the rubber section of the bulb dispenser cylinder.
Transfer 1 mL of liquid agarose with a micropipette to the surface of an empty Petri dish, and place one end of the capillary tube vertically (with the needle inserted) in contact with agarose, while the rubber cap of the bulb dispenser is being pinched inwards. Slowly release the pressure on the bulb dispenser cap to draw agarose into the capillary tube. NOTE: The transfer of liquid agarose into the capillary tubes must be performed rapidly. If the liquid agarose is left to cool on the Petri dish surface for sufficient time (approximately 60 s), it starts to gel, preventing suitable suctioning of the agarose along the capillary tube.
Place each bulb-tube-needle assembly in a free Petri dish, and let the agarose gel inside the capillary tubes solidify for 5 min. Carefully pull the capillary tube with your hands out of the rubber stopper in the bulb dispenser cylinder, leaving the needle and the agarose gel in place inside the tube.
Manually extract the acupuncture needle by slowly pulling it out of the capillary tube; the newly solidified agarose cylinder also slides out of the tube in this procedure, still surrounding the needle. Gently nudge the micro-column along the acupuncture needle with the tip of sterile forceps to move it to the end. Place the needle over an open, DPBS-containing Petri dish and push the micro-column into the DPBS with forceps. NOTE: If the agarose micro-column remains within the glass capillary tube upon removal of the acupuncture needle, slowly push the agarose micro-column out of the capillary tube with a 25-mm gauge needle and into the dish with DPBS.
Sterilize microscalpels or forceps using a hot bead sterilizer. Make 4% w/v agarose by weighing 4 g of agarose and transferring it to 100 mL of DPBS. Heat and stir as explained in step 1.2 to obtain a clear 4% liquid agarose solution. Maintain heating and stirring of the solution throughout the following steps.
Transfer the Petri dishes containing the micro-columns to a dissection hood, and move a micro-column with fine forceps to an empty Petri dish. Utilize the stereoscope for visual guidance and a microscalpel to cut the micro-columns to the desired length. Trim both ends to form beveled ends at 45o angles from horizontal to facilitate handling of the micro-columns during ECM and cell addition.
Repeat the previous step for three more micro-columns in the same Petri dish and line up the four constructs in parallel with a separation of <3 mm between each of the cylinders. Load 50 µL of 4% agarose solution with a micropipette and pour a streak/line of liquid over the micro-column array to connect and bundle the constructs into groups of four (hereafter called "micro-column boats"). Avoid movement of 4% agarose to the ends of the micro-columns, which may clog the interior, by minimizing the distance between the constructs and adding the agarose quickly in a thin line. NOTE: Arranging groups of four micro-columns into "boats" is not required to fabricate the astrocytic scaffolds. Nevertheless, the boats serve to hasten the fabrication process and to offer a safer way to move and handle micro-columns in later steps.
Let the micro-column boat cool for 1-2 min to permit gelation of the added 4% agarose.Pick up the micro-column boat with fine forceps by the connecting 4% agarose, and move the micro-columns to the other DPBS-containing Petri dish prepared in step 1.1.3. Fabricate more boats with the remaining micro-columns as desired.
Move the Petri dishes containing the micro-columns and/or boats to a biosafety cabinet and sterilize with exposure to ultraviolet (UV) light for 30 min. Caution: Wear appropriate protection to prevent exposure to UV.
Store the dishes with the micro-column boats in DPBS at 4 °C, if ECM addition and cell plating will not occur immediately afterwards, up until 1 week. Repeat the micro-column fabrication steps if the constructs are not used during this timeframe.
2. Primary Cell Culture and Isolation
- Cortical astrocyte isolation from rat pups
- In preparation for the following steps, add 20 mL of Hank's balanced salt solution (HBSS) to four 10 cm Petri dishes that will serve as reservoirs for the dissected tissues. Keep all the dishes on ice. Sterilize clean surgical scissors, forceps, micro-spatula, and micro-scalpels in a hot bead sterilizer.
- Prewarm 0.25% trypsin with 1 mM ethylenediaminetetraacetic acid (EDTA) and 0.15 mg/mL deoxyribonuclease (DNase) I in HBSS at 37 °C. In addition, prewarm the astrocyte culture medium at 37 °C, which consists of Dulbecco's Modified Eagle Medium (DMEM) with Ham's F-12 Nutrient Mixture and 10% fetal bovine serum (FBS).
- Anesthetize postnatal day 0 or day 1 Sprague-Dawley rat pups by exposing them to hypothermic conditions, and euthanize by decapitation. Pin the head into a stage using a 19 mm gauge needle placed in the snout anterior to the eyes.
- Make an incision with a scalpel down the middle posterior to anterior, from the base of the neck up to just behind the eyes. Carry out another incision going laterally from directly behind the eyes downwards, forming a T-shape. Use fine forceps to pull the cranial skin/skull off to the side.
- Hold the head by the snout (facing up) by placing one prong of the forceps in the mouth of the animal and the other on the outside surface. Remove the brain with a sterile micro-spatula and place it in one of the Petri dishes filled with chilled HBSS. Place this Petri dish on ice immediately afterwards and at all times except when in use.
- Situate a granite block previously stored at -20 °C below a stereoscope inside a dissection hood. Place the Petri dish with the brain tissue on the surface of the granite block to preserve its low temperature during the dissection procedure. Use the stereoscope as visual guidance throughout the following steps.
- If the olfactory bulbs remain intact, extract them by cutting with forceps or microscalpels. In addition, use a microscalpel to remove the cerebellum and to make a midline incision separating the two cerebral hemispheres. Transfer the hemispheres with forceps to a Petri dish containing fresh, chilled HBSS.
- Dissect the midbrain structures from the inside of the hemispheres with a microscalpel to obtain only the separated cortices. Use fine forceps to peel the meninges, a sheet-like structure, off from the cortical tissue and to transfer the isolated cortices to a new Petri dish with cold HBSS. Use the microscalpel to mince the tissue in order to increase the surface area for trypsin action in the next step.
- Use a Pasteur pipette to transfer the cortices to a 15 mL centrifuge tube containing 4 mL of prewarmed trypsin-EDTA (8 cortices in each tube). Expose the cortical tissue to trypsin-EDTA for 5-7 min at 37 °C.
- With a Pasteur pipette, carefully remove the trypsin-EDTA and add 400 µl of 0.15 mg/mL DNAse I solution to the centrifuge tube. Agitate the tube or vortex until all the tissue is dissociated and there are no remaining tissue pieces in the liquid. If it is not possible to completely dissolve the tissue, remove the remaining fragments with the tip of a Pasteur pipette.
- Centrifuge the tube containing the dissociated cell solution at 200 x g for 3 min. Remove the supernatant with a Pasteur pipette without disturbing the cells. Add 1 mL of astrocyte culture medium (defined in step 2.1.2) to the tube with a micropipette and agitate to resuspend and create a homogeneous solution.
- Transfer 10 mL of astrocyte culture medium with a serological pipette to a T-75 flask. Add 250 µl of the 1 mL cell solution (prepared in step 2.1.11) with a micropipette to the flask to plate one pup brain worth of cells per flask. Distribute the discharged cell solution evenly across the culture medium and gently agitate the flask to further promote an even distribution.
- Culture the plated flasks in a humidified incubator at 37 °C and 5% CO2. After reaching 24 and 72 h in culture, mechanically agitate the flask to detach non-adherent cell types, such as neurons and oligodendrocytes.
- Afterwards, at each of these timepoints, perform a media change. Hold the flask vertically so the culture media lies on the bottom of the flask, not covering the adhered cells. Aspirate the culture medium with a Pasteur pipette, pressing the tip of the pipette against the bottom corner of the flask to avoid extracting the cells. Place the flask in the original horizontal position and gently add 10 mL of astrocyte medium over the cells with a serological pipette.
- Return the flasks to the incubator after each media change. After 90% confluency is achieved, mechanically agitate the flask once more to remove any remaining non-adhering cells.
- Passage the astrocytes by taking out the culture medium with a vacuum and a Pasteur pipette. Add 5 mL of 0.25% trypsin-EDTA with a serological pipette over the adhered cells. Gently agitate the flask to ensure the trypsin covers all the cells, and incubate the flask for 5-7 min at 37 °C and 5% CO2.
- Quench trypsin by adding 1 mL of astrocyte medium to the cells with a serological pipette. Extract the cell solution from the flask with a serological pipette and transfer it to a sterile 15 mL centrifuge tube. Centrifuge the tube at 200 x g for 3 min.
- Remove the supernatant with a serological pipette and resuspend it in 1 mL of astrocyte culture medium. Agitate the tube to ensure the cell solution is homogeneous. Count the number of cells in the solution with a hemocytometer or an automatic cell counter. NOTE: A flask that is 90% confluent typically yields approximately 3 million astrocytes.
- Add 10 mL of astrocyte culture medium to a T-75 flask. Perform a 1:4 dilution by transferring 250 µl of the cell solution (step 2.1.18) with a micropipette to the T-75 flask already containing culture medium. Gently agitate to ensure a homogeneous cell distribution throughout the surface of the flask.
- Repeat steps 2.1.16-2.1.19 each time 90% confluency is achieved to passage the cells.
- Cortical Neuron Isolation from Rat Fetuses
- Follow similar preparations as steps 2.1.1 and 2.1.2, with the exception that the prewarmed media is co-culture media, consisting of Neurobasal medium + 2% B-27 supplement (for neurons) + 1% G-5 serum-free supplement (for astrocytes) + 0.25% L-glutamine.
- Euthanize timed-pregnant embryonic day 18 Sprague-Dawley rats with carbon dioxide asphyxiation and confirm death by decapitation.
- Extract the rat fetuses and dissect the cortices from the rest of the cerebral tissue in Petri dishes containing HBSS on the surface of the chilled granite block, using a stereoscope for visual guidance and sterilized scissors, microscalpel, and forceps53. After the successive dissection of the heads, brains, cerebral hemispheres, and cortices, transfer each tissue to a new HBSS-filled Petri dish.
- Mince the cortical tissue into smaller fragments to increase the surface area for trypsin. Transfer 4-6 cortices with a Pasteur pipette to a tube with 5 mL of prewarmed trypsin-EDTA and maintain at 37 °C to dissociate the tissue. At 5-7 min manually agitate the tube to mix and prevent clumping of the tissue.
- Remove the tube from 37 °C after 10 min. As explained previously in step 2.1, extract the trypsin-EDTA and substitute with 1.8 mL of 0.15 mg/mL DNAse solution. Afterwards, vortex the tissue until the solution appears homogeneous, with no tissue fragments floating in the liquid.
- Centrifuge the dissociated tissue solution at 200 x g for 3 min. After removing the supernatant, resuspend in 2 mL of co-culture medium. Count the number of cells in this solution with a hemocytometer or an automatic cell counter. NOTE: The usual yield is 3.0-5.0 x 106 cells/cortical hemisphere.
- Prepare a new cell solution with a density of 2.0-4.0 x 105 cells/mL in the co-culture media defined above.
3. Development of the Astrocytic Cables Inside the Micro-columns
- ECM core fabrication NOTE: The ECM has to be added to the interior of the hydrogel micro-columns on the same day in which cell seeding will be performed.
- Inside a biosafety cabinet, prepare a 1 mg/mL solution of rat tail type I collagen in co-culture medium in a sterile microcentrifuge tube. Maintain the microcentrifuge tube with the ECM solution on ice at all times except when in use.
- Transfer 1-2 µL of the ECM solution with a micropipette onto a strip of litmus paper to estimate its pH. Add 1 µL of 1 N sodium hydroxide (NaOH) or hydrochloric acid (HCl) with a micropipette to increase or decrease the pH of the ECM solution, respectively, and pipette up and down to homogenize. Verify the new pH with a litmus paper strip and add more acid or base, as needed, until the pH is stable in the 7.2-7.4 range.
- Move the Petri dishes from step 1.2 to a dissection hood, and transfer 4-5 micro-columns or a boat with sterile fine forceps to an empty, sterile 35 or 60 mm Petri dish. Using the stereoscope for guidance, situate the 10 µL tip of a micropipette at one end of each micro-column and suction to empty the lumen of DPBS and air bubbles. Confirm the absence of air bubbles with the stereoscope to ensure that the ECM added in the next step can flow freely across the lumen.
- Charge a P10 micropipette with ECM solution, place the 10 µL tip against one end of the hydrogel micro-columns, and deliver enough solution to fill the lumen (approximately 3-5 µL), observing the entry of ECM with the stereoscope. Pipette a small reservoir (2-4 µL) of ECM solution on either end of the micro-column. NOTE: Always manage 4-5 micro-columns or one boat at a time to prevent prolonged drying of the constructs, as this may result in the crumpling of the micro-column structure. Completely dried micro-columns firmly attach to the surface of the Petri dishes, which prevents their utilization for cell seeding. Excessive amounts of co-culture media (as a hydration measure) cannot be added to the micro-columns because this may cause the ECM to flow out during the incubation period described below.
- Pipette co-culture media in a ring around the Petri dish to provide a humidity sink to prevent the columns from drying out during incubation. Incubate the Petri dish containing the ECM-containing micro-columns at 37 °C and 5% CO2 for 1 h to promote polymerization of collagen before adding the neurons and/or astrocytes. NOTE: The ECM should form a layer coating the inner surface of the hollow lumen, rather than a solid ECM core encompassing the interior, both of which can be observed using the stereoscope. If the ECM forms a core, continue the incubation period until only the layer is left. With this layer formed, the astrocyte cell solution can fill the interior of the micro-column in the plating steps.
- During the incubation period, prepare the astrocyte cell solution (as described below).
- Astrocyte and Neuron Seeding in the Micro-Columns
- Passage the plated astrocytes (between the fourth and twelfth passage) as explained in steps 2.1.16-2.1.19. After counting the number of cells in the flask, prepare cell solution at a density of 9.0-12.0 x 105 cells/mL solution suspended in cell-culture media.
- Using a stereoscope, place the tip of a P10 micropipette at one end of the micro-columns, and transfer sufficient cell solution (~5 µL) into the lumen to completely fill it. As done above with the ECM, add small reservoirs of cell solution to both ends of each micro-column.
- Incubate the plated micro-columns on the Petri dishes at 37 °C and 5% CO2 for 1 h to promote the attachment of astrocytes to the ECM. If neurons will not be added to micro-columns, proceed to step 3.2.5.
- Following the initial incubation period, add 1-2 µL of the cortical neuron solution obtained in step 2.2.7 into both ends of the micro-columns with a micropipette, while observing under the stereoscope. Ensure that sufficient media is present in the dishes to avoid drying, and incubate again for 40 min at 37 °C and 5% CO2 to allow for the adhesion of neurons. NOTE: Cortical neuron solution can also be added 1-2 days after bundle formation, performing step 3.2.4 after carefully removing the culture media from the Petri dish with a micropipette.
- After the incubation period, carefully fill the Petri dishes containing the plated micro-columns with 3 or 6 mL of co-culture media for 35 or 60 mm Petri dishes, respectively. Maintain the plated micro-columns in culture at 37 °C and 5% CO2 to promote the self-assembly of the aligned astrocytic bundles, which should form a bundled, cable-like structure after 6-10 h.
- Stabilization of Astrocyte Cable Architecture NOTE: After formation of bundles, approximately 6-12 h of culturing the constructs, perform the following steps to prevent the collapse of the aligned structure of the astrocytic scaffolds.
- In a sterile microcentrifuge tube, prepare a 3 mg/mL rat tail collagen I solution in co-culture media. Adjust the pH of the solution to 7.2-7.4 following the procedure outlined in step 3.1.2. Maintain the collagen stock and co-culture media on ice when not in use, and place the prepared collagen solution on ice at all times.
- Remove the media from the Petri dishes containing the astrocytic scaffolds with a micropipette, leaving some media on the sides of the dish to create a humidity sink that ensures the hydration of the micro-columns. Place the dish under a stereoscope to aid in visualization.
- With a micropipette, take 2-3 µL of collagen solution and discharge to each end of the micro-columns, using the stereoscope for visual guidance. Make sure the dish has sufficient media around the sides to act as a humidity sink around the edges of the dish. Incubate the Petri dishes with the micro-columns for 30 min at 37 °C and 5% CO2 in a humidified incubator to promote the gelation of the newly added collagen.
- Slowly add 3 or 6 mL of co-culture media (for 35 or 60 mm Petri dishes, respectively) to the Petri dishes with a pipette, and culture the dishes in a humidified incubator at 37 oC and 5% CO2.
4. Extraction of the Astrocytic Bundles from the Hydrogel Interior
Sterilize glass coverslips in an autoclave. Prepare a 20 µg/mL solution of poly-L-lysine (PLL) in sterile cell culture grade water.
Inside a biosafety cabinet, manually transfer the sterilized glass coverslips to a sterile 10 cm Petri dish, and add sufficient PLL solution to cover the coverslips.
Incubate the coverslips, covered with the PLL solution, for 30 min at 37 °C to coat the surface. Remove the PLL solution with a Pasteur pipette after 30 min. Rinse three times by adding cell culture grade water to the coverslip and removing it with a Pasteur pipette.
After the formation of the aligned glial bundle, transfer the micro-column delicately to a sterile Petri dish with sterile fine forceps, and add a small pool of 10 µL of co-culture media with a micropipette to prevent dehydration and ensure bundle health. Extract the astrocytic bundle from the hydrogel micro-column by gently pulling from one end with sterile surgical forceps, using a stereoscope for visual guidance.
Holding the astrocytic bundle with forceps, mount it on a PLL-coated coverslip. Fix and stain with the protocol below.
5. Immunocytochemistry for In Vitro Studies
NOTE: For this study, the primary antibodies were rabbit anti-glial acidic fibrillary protein (GFAP) (1:500), mouse anti-β-tubulin III (1:500), rabbit anti-collagen I (1:500), mouse anti-nestin (1:200), and rabbit anti-vimentin (1:100). The secondary antibodies were donkey anti-mouse 568, donkey anti-rabbit 568, donkey anti-rabbit 488, and donkey anti-mouse 568 (1:500 for all). In all instances, add enough volume of each solution to entirely cover the micro-columns.
Prepare a 4% volume/volume (v/v) formaldehyde solution in 1x DPBS inside a chemical fume hood. CAUTION: formaldehyde is a toxic compound known to be carcinogenic, and must be disposed of in a separate container. Always manipulate this compound inside a chemical fume hood and utilize personal protective equipment (PPE) such as laboratory coat, safety glasses, and gloves.
In the case of the micro-columns, discard the culture media from the Petri dishes containing the constructs with a Pasteur pipette without accidentally suctioning the hydrogel cylinders. Add sufficient formaldehyde solution to cover the micro-columns or the mounted coverslips (both of which remain on the Petri dishes) and incubate for 35 min at 18-24 °C.
Extract the 4.0% formaldehyde solution from the fixed micro-columns or the coverslips with a serological pipette. Rinse the fixed micro-columns three times with PBS by quickly adding and removing PBS twice and then letting them soak for 10 min a third time.
Dissolve normal horse serum (NHS) in PBS for a concentration of 4% v/v. Prepare a solution with non-ionic detergent at a concentration of 0.3% v/v using the NHS solution as the solvent.
Remove the PBS from the Petri dishes containing the micro-columns or the coverslips with a pipette. Add sufficient 0.3% detergent solution to cover the micro-columns/coverslips for 60 min at 18-24 °C to permeabilize the cells.
Take out the detergent solution, and rinse quickly two times with PBS and three times by soaking for 5 min. Calculate the required volume of 4% NHS and each of the primary antibodies to prepare a solution with the desired antibody concentrations.
Remove the PBS from the Petri dishes and add enough primary antibody (diluted in 4% NHS-DPBS) solution to cover the micro-columns or the extracted astrocytic bundles. Incubate overnight (12-16 h) at 4 °C.
Take out the primary antibody solution and quickly rinse two times with PBS and three times by soaking for 5 min each. Prepare the secondary antibody solution, in the dark, with each antibody present at a dilution of 1:500 in 4.0% NHS solution.
Incubate cells with the secondary antibody solution for 2 h at 18-24 °C. Throughout the entire time of incubation, cover the Petri dishes containing the micro-columns with aluminum foil to avoid exposure to light.
Remove the secondary antibody solution and add Hoechst solution (1:10,000) for 10 min at 18-24 °C to stain the nuclei. CAUTION: Hoechst is a known mutagen that should be treated as a carcinogen. Therefore, it must be disposed of in a separate container and PPE should be used at all times when manipulating this agent.
Rinse with PBS five times, each time for 5-10 min. Afterwards, store the stained micro-columns at 4 °C in PBS and cover with aluminum foil. In the case of the mounted astrocytic bundles, add a drop of aqueous mounting medium to the bundle, place another glass coverslip over the fixed bundle, and seal both coverslips with nail polish for long-term storage and subsequent imaging.
6. Viability Assay with Live-dead (Calcein-AM/ethidium Homodimer) Staining
Prepare the reagent solution by adding 5 μl calcein AM (4 mM in anhydrous dimethyl sulfoxide (DMSO)) and 20 μl ethidium homodimer-1 (EthD-1) (2 mM in DMSO/H2O 1:4 v/v) to 10 mL of 1x DPBS. Cover the solution with aluminum foil or store in the dark to protect it from the light.
Aspirate media from the Petri dish containing the plated micro-columns with a Pasteur pipette, and add enough solution (prepared above) to cover the constructs. Incubate for 30 min at 37 °C and 5% CO2, keeping the Petri dish covered to protect the solution from the light.
Remove the solution with a micropipette or Pasteur pipette. Rinse 2-3 times with DPBS as explained in step 5.3. Flood the Petri dishes with DPBS according to the size of the dish. Image cells immediately afterwards using epifluorescence or confocal imaging. NOTE: Conversion of the membrane-permeable calcein AM to calcein by metabolically active cells yields the green fluorescence of calcein. Dead cells are marked as membrane-compromised cells, which permit the entry of EthD-1 into the cell and its binding to nucleic acids, causing red fluorescence.
Representative Results
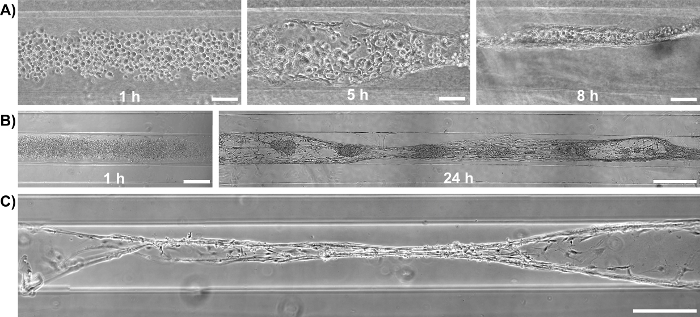
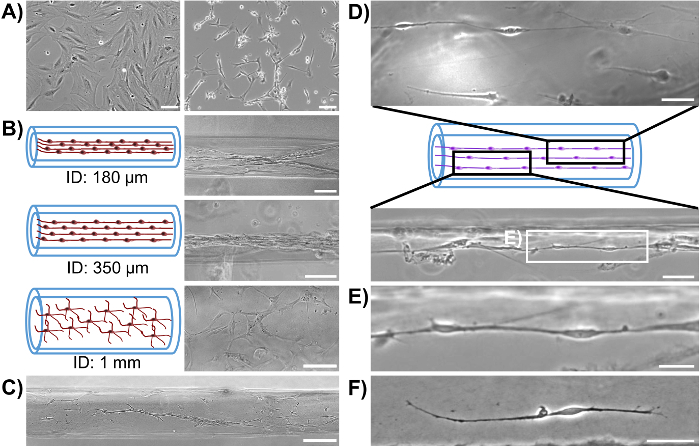
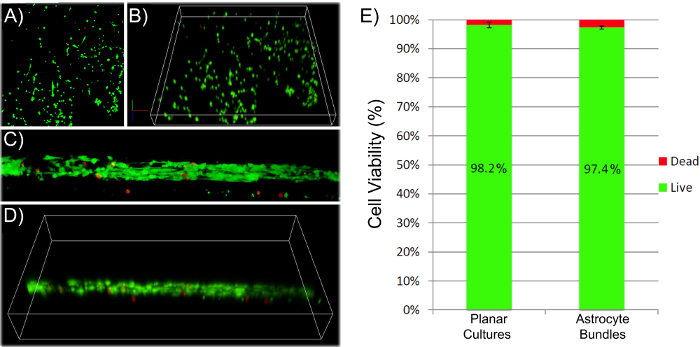
Initially, phase-contrast microscopy was used to monitor the progression of astrocyte adhesion and bundle formation and the overall stability of the cytoarchitecture as a function of time. At 1 h after plating, astrocytes were found throughout the lumen of the micro-columns with a spherical morphology (Figure 2A). At 5 h, astrocytes started extending processes and contracting, while by 8 h cells exhibited a complete process-bearing morphology and formed cable-like structures oriented along the longitudinal axis of the micro-column (Figure 2A). Notably, compared to the initially dispersed presence of cells across the micro-columns, astrocytes appeared to have contracted to form a network of dense bundles along the interior(Figure 2B). After the initial cell alignment, the astrocyte networks often pulled together along the longitudinal axis, resembling a zipper closing, to form a dense bundle in the center of the micro-column (Figure 2C). The formation of this bundled architecture was due to a self-assembly process, in part dictated by the choice of micro-column ID and cell density. Astrocytes aligned and acquired a bipolar morphology when seeded in micro-columns with an ID less than 350 µm (Figure 3B, top and middle, and 3D-3F)49. On the contrary, these cells demonstrated random directionality when an ID of 1 mm was utilized (Figure 3B, bottom), which is similar to the appearance of 2D astrocyte cultures exposed to the serum-free, co-culture medium (Figure 3A, right). Furthermore, even though astrocytes still display alignment at low seeding densities (2.0-3.0 x 105 cells/mL or 2.0-3.0 x 102 cells/mm3) (Figure 3C), dense networks are formed only at the high densities (9.0-12.0 x 105 cells/mL or 9.0-12.0 x 102 cells/mm3) within cylindrical micro-columns of similar dimensions (seen in Figure 3B, top), as suggested in the protocol49. Viability in the astrocytic bundles was quantified, using the live/dead assay, as the ratio of the area of cells fluorescing green (calcein AM) relative to the total area of cells fluorescing green (calcein AM) and red (EthD-1). The viability of the astrocytic bundles was 97.4%, which is similar to the 98.2% viability of 2D astrocyte culture controls (Figure 4A-4B), proving that the environment within the micro-columns is suitable for astrocyte vitality (Figure 4E). The 3D reconstructions of the live and dead cells in the astrocytic scaffolds also demonstrate the overall cell alignment within the constructs (Figure 4C-4D). Additionally, ultra-long astrocytic micro-columns were developed to investigate the stability of the astrocyte bundles with substantial length. Even at the extraordinary distance of 3.5 cm, the cytoarchitecture of aligned, dense, and contracted astrocytic bundles was sustained consistently throughout the length of the micro-column (Figure 5A), as clearly seen in the zoom-in regions of the micro-columns (Figure 5B-5C).

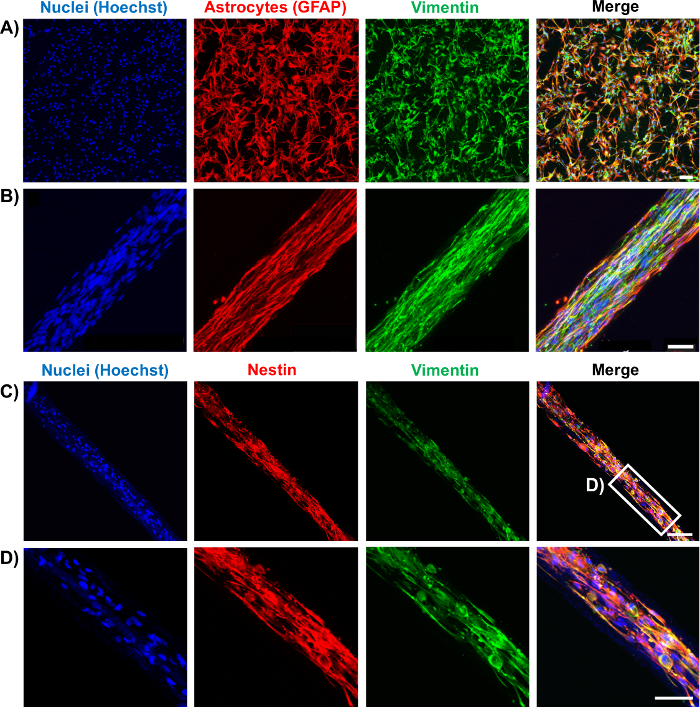
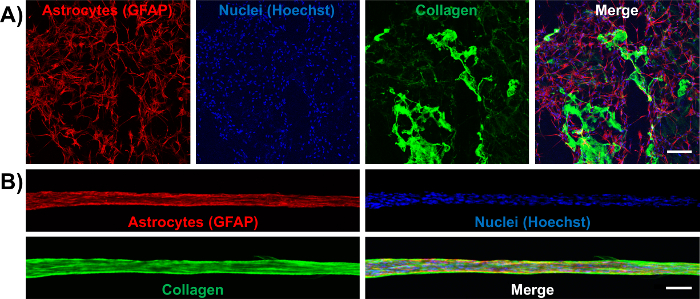
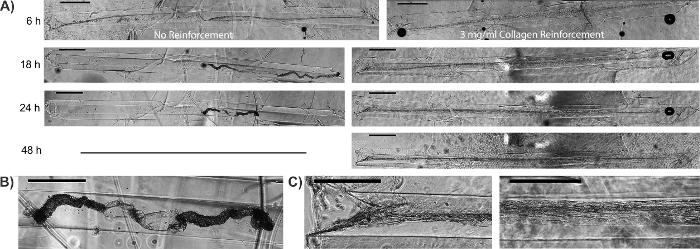
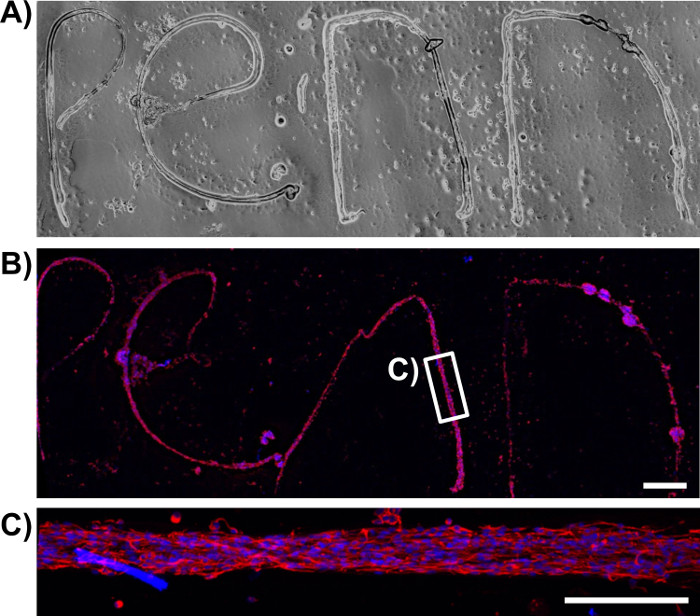
Once formed, the bundles were fixed and stained by immunocytochemistry techniques for intermediate filament proteins characteristic of glial cells, such as GFAP, nestin, and vimentin. The confocal images confirm the process-bearing morphology of astrocytes and longitudinal alignment of processes (Figure 6 and Figure 7). Expression of the intermediate filament proteins can be seen in both the 2D astrocyte cultures (Figure 7A) as well as the astrocyte bundles within the micro-column (Figure 6, Figure 7B-7D). In addition, collagen staining corroborated the presence of this ECM protein in the micro-columns. The astrocytes did not form a continuous structure with the collagen when cultured in a 2D environment (Figure 8A), whereas the astrocytes appeared to adhere to and remodel the collagen present within the lumen when cultured in the micro-columns, allowing for cell contraction and bundle formation (Figure 8B). Overlay of the GFAP and collagen channels demonstrated that the astrocyte cables consisted of tightly associated processes with longitudinal collagen fibers (Figure 8B). Therefore, apart from offering anchorage, collagen fibrils may also have provided additional directional cues for astrocytes to form dense bundles after contraction. In some cases, astrocyte-collagen contraction persisted, resulting in a collapse of the aligned bundle over 12-24 h after formation (Figure 9A). In order to stabilize the cable-like astrocyte architecture within the hydrogel micro-columns, additional collagen matrix was added to the ends of a fully formed bundle to inhibit further contraction and collapse. This reinforcement technique allowed for the survival of the desired cable architecture for at least 4 days, with a longer window of stability expected (Figure 9B). Moreover, representative astrocytic bundles were extracted from the hydrogel micro-column, mounted, and fixed with 4% formaldehyde on glass coverslips to assess their robustness when exposed to tension forces and to the external environment. Even after extraction and physical manipulation into different shapes (Figure 10A, 10B), the astrocytic somata and processes maintained an aligned, bundled micro-scale morphology (Figure 10C), highlighting the resiliency of the self-aligned astrocytic scaffold, once formed, even if the structural support of the hydrogel micro-column is absent.
Primary cortical neurons were also co-cultured with astrocytes inside the micro-columns to explore whether the astrocyte bundles were capable of fomenting neuronal adhesion and neurite outgrowth. GFAP and the microtubule protein β-tubulin III were used as specific markers for astrocytes and neurons, respectively. Figure 11A shows that astrocytes co-seeded with cortical neurons were also capable of exhibiting processes and forming aligned bundles. Neurons appeared to have adhered preferentially to the astrocyte bundles, since all positive β-tubulin III staining co-localized with GFAP (overlays in Figure 11B-11C). To demonstrate the regenerative properties of the astrocytic constructs, cortical neurons (Figure 12B) were plated within the hydrogel micro-column 2 days after astrocytes were plated (Figure 12A). Neurons attached to the astrocyte bundle, and extended neurites directly along the astrocytic tracts. Further inspection showed neurite outgrowth oriented in the direction of the astrocytic cable, whereas in regions where the astrocyte tract was not present, neurites were seen growing in a disorganized manner (Figure 12C). These observations demonstrate that the protocol results in aligned, viable, and robust astrocytic networks that have the potential to act as implantable living pathways promoting neuron adhesion and neurite outgrowth.
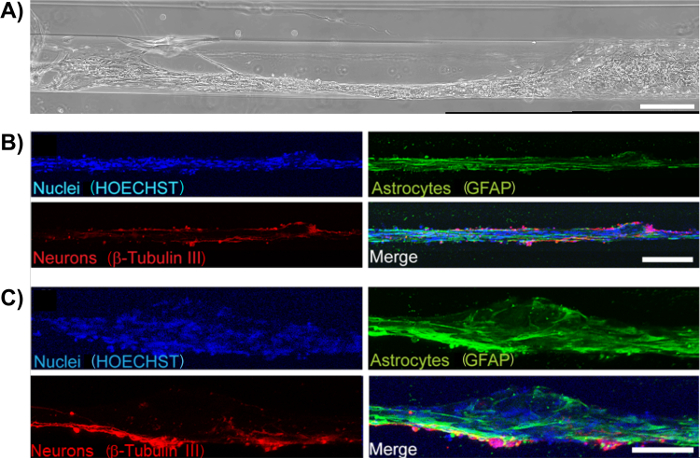
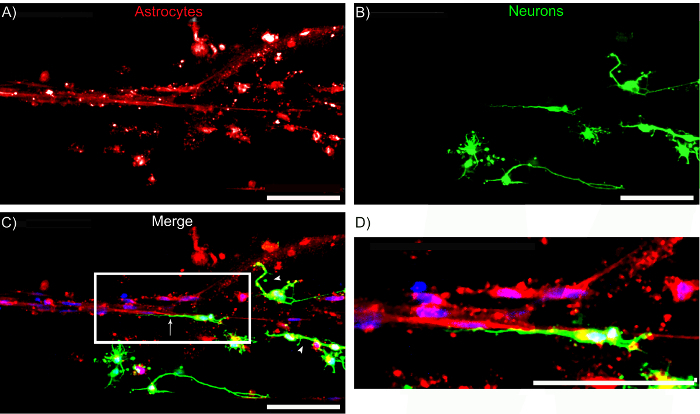
Figure 2: Brightfield Microscopy Shows Formation of Astrocyte Bundles within Hydrogel Micro-Columns over Time. (A) Astrocyte morphology as a function of time after plating: (1 h) astrocytes are spherical in shape and dispersed throughout the construct; (5 h) astrocytes initiate process extension and contraction; (8 h) astrocytes have aligned and contracted. Scale bars = 100 µm. (B) Bundle formation over time in an additional representative scaffold: (1 h) initially the astrocytes are spherical and do not exhibit processes; (24 h) a dense bundle of astrocytic processes is formed. Scale bars = 300 µm (left) and 500 µm (right). (C) Fully formed astrocytic bundle 24 h after plating, showing a "zippering" effect, where the ends of the cable attach to distinct ends of the walls of the lumen. Scale bar = 1000 µm.
Figure 3: Micro-Column Inner Diameter and Cell Density Influence the Self-Assembly of Aligned Bundles of Bipolar Astrocytes. (A) 2D astrocyte cultures with serum-containing media (left) and serum-free co-culture media (right) show a non-process and multi-process bearing morphology, respectively. Scale bars = 100 µm. (B) Astrocytes seeded at high density (9.0-12.0 x 105 cells/mL) in micro-columns with an ID of 180 and 350 µm form aligned bundles relative to the longitudinal axis, while an ID of 1 mm results in astrocytes displaying multiple, unaligned processes throughout the diameter of the lumen. Scale bars = 100 µm (top, middle) and 150 µm (bottom). (C) Astrocytes seeded in a 350 µm ID micro-column at low density (2.0-4.0 x 105 cells/mL) are aligned, but not bundled. Scale bar = 100 µm. (D) When plated in a 350 µm ID micro-column at low or medium cell density, astrocytes acquire a bipolar morphology. Scale bars = 300 µm. (E) Box in D showing a zoom-in of the chains of bipolar astrocytes that form inside the hydrogel micro-columns. Scale bar = 300 µm. (F) Example of an individual bipolar astrocyte. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 4: Astrocytes Remain Viable after Bundle Formation within the Hydrogel Micro-Column. (A-B), (C-D) 3D confocal reconstructions showing the viability of cells throughout planar cultures and aligned astrocytic bundles, respectively, as assayed with calcein-AM/ethidium homodimer staining (green-live cells; red-dead cells). (E) Quantification of live and dead cells resulted in a similar percentage of live/dead astrocytes when cultured on 2D surfaces and in the micro-columns. Error bars represent standard deviation. Please click here to view a larger version of this figure.
Figure 5: Phase-Contrast Image of a Representative Ultra-Long Aligned Astrocytic Bundle within the Hydrogel Micro-Column. (A) Bundle formation throughout the entire length of a 3.5 cm micro-column. Scale bar = 1,000 µm. (B-C) Higher magnification zoom-ins showing astrocyte alignment in various regions along the entirety of the construct, corresponding to the boxes in A. Scale bars = 500 µm. Please click here to view a larger version of this figure.
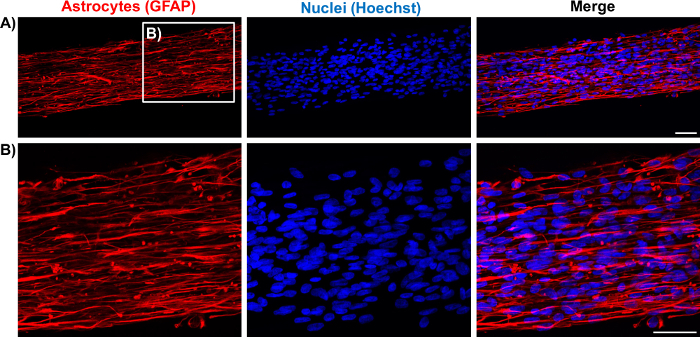
Figure 6: Immunolabeling of the Astrocytic Scaffolds Demonstrates the Longitudinal Alignment of Astrocytic Processes. (A) Confocal reconstruction of an astrocytic bundle showing staining for GFAP (red; left) and nuclei (Hoechst; middle) and an overlay of both channels (right). (B) Higher magnification zoom-in of the astrocytic bundle in the boxed region in A permits visualization of the cytoarchitecture of the astrocytes within micro-columns. Scale bars = 50 µm. Please click here to view a larger version of this figure.
Figure 7: Characterization of Representative Astrocytic Bundles through Immunocytochemistry. (A-B) Astrocytes cultured on 2D surfaces and seeded in hydrogel micro-columns express similar astrocytic markers GFAP (red) and vimentin (green). (A) Confocal reconstruction of a 2D planar astrocyte culture showing the separate and merged channels. Scale bar = 100 µm. (B) Confocal images of an astrocytic bundle, confirming expression of the markers and showcasing astrocyte alignment. Scale bar = 50 µm. (C-D) Astrocytes cultured within micro-columns also express the intermediate filament proteins nestin (red) and vimentin (green). (C) Confocal image of an astrocytic bundle. Scale bar = 100 µm. (D) Higher magnification zoom-in of C. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 8: Astrocytes in the Aligned Bundles Intimately Interact with a Contracted, Longitudinal Collagen Matrix, as Opposed to the Irregular Arrangement of Collagen in 2D Cultures. Cells were immunolabeled to observe astrocytes (GFAP; red), nuclei (Hoechst; blue), and collagen (green). (A) 2D astrocyte cultures with collagen at 1 mg/mL show unaligned astrocytes and randomLy distributed collagen. Scale bar = 250 µm. (B) Collagen is pulled off from the walls of the micro-column lumen and is contracted to form an aligned matrix that is part of the astrocytic bundle. Scale bar = 150 µm. Please click here to view a larger version of this figure.
Figure 9: Additional Collagen can be Used to Stabilize the Structure of Astrocytic Cables. (A) Bundles formed approximately 6 h after plating astrocytes within collagen-coated hydrogel micro-columns. Once bundles were formed, higher concentration (3 mg/mL) collagen was added to the ends of the micro-column to inhibit contraction of the astrocytic cables. In constructs not stabilized with additional collagen, the astrocytic-collagen bundles had begun to contract by 18 h, and the astrocyte cables had completely collapsed by 24 h. This contraction was not observed when the bundles were reinforced with 3 mg/mL collagen. Scale bar = 1,000 μm. (B) Zoom-in of collapsed astrocytic bundle not reinforced with 3 mg/mL collagen. Scale bar = 500 μm. (C) Zoom-in of regions from collagen-reinforced micro-columns. Astrocyte bundle morphology was maintained throughout the entirety of the micro-column. Scale bars = 500 μm. Please click here to view a larger version of this figure.
Figure 10: Extraction of the Astrocytic Bundles from the Micro-Column and Mounting on Poly-L-lysine Coated Glass Coverslips Evidences the Robustness of the Technique. (A) Phase-contrast image of several extracted astrocytic bundles, manipulated into spelling the word "Penn". (B) Confocal reconstruction of the same extracted bundles stained for the astrocytic marker GFAP (red) and cell nuclei (Hoechst; blue). Scale bar = 100 µm. (C) Magnification of the boxed region in B shows that, even when manipulated into irregular shapes, the astrocytic scaffolds retain an aligned, bipolar morphology and a bundled cytoarchitecture. Scale bar = 200 µm. Note that any structural differences between the phase and confocal images is likely an artifact of constructs shifting due to rinsing during the immunocytochemistry procedure. Please click here to view a larger version of this figure.
Figure 11: Neurons Adhere to and Extend Neurites along the Aligned Astrocyte Bundles in Neuron-Astrocyte Co-Cultured Scaffolds. (A) Phase-contrast image of a neuron-seeded astrocytic bundle. Scale bar = 300 µm. (B)-(C) Neurons were observed to adhere to the astrocytic bundles and extend neurites along the direction of the cable (Hoechst-blue; GFAP-green; β-tubulin III-red). Scale bars = 200 and 100 µm, respectively. Please click here to view a larger version of this figure.
Figure 12: Astrocyte Cables serve as a Living Scaffold for Directed Neurite Outgrowth. Confocal reconstructions showing a region of a micro-column consisting of neurons seeded approximately 2 days after bundle formation. (A) GFAP-positive astrocyte bundles, (B) β-tubulin III-positive neurons, and (C) merge of all channels, including HOECHST-stained nuclei (blue). Note that neurons exhibited neurite alignment when adhered to regions with aligned astrocyte processes (arrows), whereas neurons not adhered to aligned astrocytes appeared to exhibit random (i.e. multi-directional) neurite outgrowth (arrowheads). (D) Zoom-in of region shows the directionality of neurite outgrowth. Scale bars = 100 μm. Please click here to view a larger version of this figure.
Discussion
When compared to the more supportive environment of the PNS, the CNS is particularly limited in handling the detrimental consequences of neurotrauma and neurodegeneration. After a serious insult to the mammalian CNS, a glial scar is formed, consisting of a core of fibrotic and inflammatory cells surrounded by a dense meshwork of disorganized reactive astrocytes that secrete axon outgrowth-inhibiting proteoglycans14. This scar acts as a physical and biochemical obstruction against the regeneration of axon endings12. Furthermore, the adult mammalian CNS possesses limited sources of new neurons to restore lost cells following trauma or neurodegeneration, since neurogenic centers are restricted to the SVZ, the hippocampal dentate gyrus, and potentially other usually inactive areas54. An ideal strategy would circumvent both aspects of the limited recuperative capabilities of the CNS, which current approaches fail to address. As such, our research group has developed astrocyte-based scaffolds aimed at presenting living pathways for neural migration towards neuron-deficient areas and guided extension of regenerating axon growth cones. Aligned astrocytic bundles contained within the collagen interior of an agarose micro-column constitute these astrocytic living scaffolds. A considerable distinction from conventional cell- and biomaterial-based strategies used for CNS repair and regeneration is that the cytoarchitecture of these constructs is completely preformed in vitro to simulate several in vivo glia-based guidance and scaffolding architectures. For instance, during embryonic development, radial glia pathways act as living scaffolds for new neurons migrating to the neocortex, and glial cells mediate the targeted extension of pioneering axons posing as patterned substrates and/or guidepost cells35,38,39. Embryonic radial glia in the subventricular zone also produce astrocytes that form longitudinally aligned glial tubes, over which neuroblasts migrate postnatally in a chained-formation from the SVZ to the OB41. Furthermore, it has been demonstrated in some non-mammals that, following CNS damage, a greater degree of regeneration is possible due to the formation of an organized, aligned astrocytic arrangement44. For example, adult zebrafish are capable of regenerating their spinal cords following the migration of immature glial cells to the lesion site, differentiation into a bipolar morphology, and elongation of glial processes to bridge the lesion, forming a path that directs regenerating axons45. Engineered constructs of aligned astrocytic bundles may be capable of recapitulating these mechanisms and presenting the required structural and soluble signals for glia-mediated neuronal migration and axon regeneration35. In addition, due to the inclusion of living astrocytes, these constructs can actively present the required environmental cues based on feedback from the host.
In our technique, astrocyte alignment is a self-assembly process contingent on the physical cues and cell-cell interactions promoted by the micro-column inner diameter, collagen concentration, and seeding density. Another attractive feature of this method is its micron-scale size, which may allow for minimally invasive implantation in damaged areas of the CNS. Moreover, results presented in this manuscript demonstrate that the constructs obtained with the protocol possess high viability and express the intermediate filament proteins GFAP, nestin, and vimentin, regardless of astrocyte age as defined by passage number. While both GFAP and vimentin are expressed in immature and mature astrocytes, nestin is mainly restricted to undifferentiated or immature cells, with maturing glia upregulating the expression of GFAP and decreasing production of nestin45,55. Previous studies have shown that astrocyte behavior changes with age, however consistent expression of intermediate filament proteins, as well bundling was seen across a range of passage numbers (passages 4-12)56. The astrocytic scaffolds also were shown to achieve ultra-long, centimeter-scale lengths in vitro, which suggests that this technique is adaptable to the distinct injury lengths and geometries encountered in vivo. Furthermore, the aligned astrocytic bundles exhibit remarkable structural integrity and stability, as their cable-like cytoarchitecture is maintained even after being subjected to extraction from the micro-columns. Astrocyte-based living scaffolds also support neuron adhesion and neurite outgrowth, reinforcing the potential of this technique to promote directed neuron migration and axon extension for CNS repair.
The aligned astrocytic bundles are contained within the collagenous interior of a protective tubular hydrogel encasement. Fabrication of the astrocytic scaffolds commences with micro-column assembly by inserting an acupuncture needle into a capillary tube and suctioning liquid agarose into it. Agarose was the selected biomaterial because of its inert properties, biocompatibility, and easily controlled mechanical, physical, and biochemical properties49. Agarose concentration in the hydrogel may be an important variable since cells sense mechanical stimuli from their substrate (e.g. stiffness) and respond with intracellular changes57. Design criteria for these micro-columns includes a high enough porosity to allow for adequate mass transport, but a small enough pore size to prohibit lateral process outgrowth; these criteria are satisfied by the nano-scale open-porosity present in agarose at the concentration used in this protocol. The agarose concentration in this protocol was also chosen due to its stability, ease of handling, and similarity in mechanical properties to the brain environment58. A noteworthy consideration is the central placement of the needle within the capillary tube. Oftentimes, the needle may be slightly slanted or displaced during the process, causing the lumen to be located off-center relative to the outer walls following agarose gelation and micro-column removal from the tube. To avoid this, the needle should be straight, and the plunger-tube-needle assembly should be maintained in a vertical position while agarose is being drawn in to retain the needle along the central axis. Precise centralization of the lumen safeguards the aligned astrocytes between the agarose walls; the lumen is more prone to rupturing if located proximal to the outer rim of the cylinder. In addition, sufficient distance between the lumen and the outer wall may offer protective benefits during and after transplantation (provided the astrocytic constructs are not removed from the micro-column prior to implant). This initial stage of astrocytic scaffold fabrication is paramount for the success of the technique, as the selection of needle diameter is crucial for proper astrocyte soma/process alignment and cable self-assembly. We found that astrocyte alignment was inversely dependent on micro-column ID, with an optimal ID range for cell alignment and the formation of robust astrocyte bundles being 180 to 350 µm (Figure 3). Similarly, it was previously demonstrated that neurons direct their outgrowth along the direction of minimum substrate curvature to lessen process bending59,60, and similar mechanisms are likely at work to affect astrocyte alignment in our system49. In addition to curvature affecting astrocyte alignment, we found that a 180 to 350 µm ID range resulted in astrocytes acquiring a bipolar morphology49, contrary to the stellate, multi-polar form observed with an ID of 1 mm or when astrocytes were cultured in 2D (Figure 3). It is likely that these profound morphological changes to a bipolar astrocyte morphology alter their physiological characteristics as well; for instance, these astrocytes may express alternative secretable factors or cell surface markers that may be advantageous for regeneration, but this warrants further characterization. Overall, our findings demonstrate that suitable selection of physical cues, such as surface curvature, are imperative to emulating native cytoarchitecture in astrocytic living scaffolds.
Construction of the astrocytic scaffolds proceeds with the sequential inclusion of the collagen ECM solution and cortical astrocytes into the hollow interior of the micro-columns. The inner lumen of the micro-column is coated with collagen to support astrocyte survival, attachment, and aligned bundle formation, due to the inability of agarose to independently support neurite outgrowth61. A collagen concentration of 1 mg/mL was chosen because previous studies determined that lower and higher concentrations resulted in a lack of adhesion and instability of the ECM, respectively49. Thus, as expected, collagen concentration has dramatic effects on the effectiveness of this technique. Furthermore, the ECM solution consists of only type I collagen. When compared to laminin and Matrigel-coated substrates for 2D astrocytic cultures, astrocytes plated on type I collagen demonstrated optimal adhesion and network formation62. The incubation time used for collagen polymerization prior to cell seeding is also an essential step in the protocol. We previously found that when neurons were delivered concomitantly with unpolymerized ECM to the agarose micro-columns, reduced neurite outgrowth and axonal fasciculation were seen47, and it is possible that these findings would extend to astrocytes as well. Since collagen is required for astrocyte adhesion and bundle formation, the presence of ECM throughout the construct and the absence of air bubbles or empty pockets should be visualized and confirmed with microscopic techniques.
The protocol concludes with astrocyte plating, an incubation period to ensure cell adhesion to collagen, and short-term culture for the formation of the target cytoarchitecture. Astrocytes between passage number 4-12 were used, as they exhibited robust astrocytic bundling and collagen reformation. These astrocytes also presented a mature phenotype consisting of >95% pure cultures63,64 with little to no neuronal contamination. In contrast to ordinary astrocyte cultures, which employ serum-containing medium (Figure 3A, left), our technique utilizes serum-free medium to allow astrocytes to adopt a process-bearing morphology (Figure 3A, right) and form bundles of aligned somata and processes within the micro-columns62,65,66. In terms of cell seeding concentration, higher concentrations in the range of 9.0-12.0 x 105 cells/mL, with an ID of 350 µm, resulted in the formation of tightly packed bundles with accompanying collagen remodeling, while astrocytes were more dispersed, but still aligned, when lower concentrations were utilized (Figure 3C). Thus, the effect of seeding concentration on cell-cell interactions influences the degree of astrocyte alignment and the formation of the observed cable-like structures. The injection of the cell solution into the lumen is generally visualized using a stereoscope to ensure homogenous delivery across the micro-column length, which is crucial for continuous bundle formation. After seeding, the preliminary incubation time prior to flooding with culture media is indispensable to secure the anchorage of astrocytes to the ECM. The duration may be modified if astrocytes escape from the constructs, provided that the constructs are sufficiently humidified to preclude drying. Phase-contrast images of the astrocytic scaffolds as a function of time showed that by 8 h, a network of aligned astrocytic bundles was in the process of being formed. In addition, the detachment of ECM from the lumen wall, its ensuing contraction, and the tight association between cells and the contracted collagen was observed when constructs were labeled for collagen and GFAP as shown in Figure 8. This phenomenon is possibly a consequence of astrocytic contractile forces detaching the collagen from the walls, based on prior reports of the fibroblast-like capacity of glial cells to generate forces that distort flexible substrates and the matrix-remodeling ability of astrocytes in astrocyte-only and neuron-astrocyte co-cultures28,49,67,68. Specifically, one of the consequences of astrocyte motility and the integrin-mediated interaction with collagen fibrils is the generation of forces sufficient to contract the fibrils17,69. Overall, implementation of the described protocol will produce living scaffolds comprised of self-assembled networks of dense, aligned astrocytic cable-like structures that recapitulate physical substrates present at several stages of CNS development.
In addition to individual issues that could arise throughout the protocol, the astrocytic scaffold technique possesses other hindrances that may affect its capacity to repair the CNS. For instance, the ultimate success of this technique is contingent on nervous system plasticity and functional integration of the astrocytic scaffold with the host circuitry. Related to the implantation of these scaffolds is the possibility of an inflammatory response that may counter the pro-regenerative attributes of the astrocytic bundles. All transplant-based techniques have the capacity to rupture the blood-brain barrier and cause infiltration of inflammatory cells, due to the proximity between neurons and capillaries in the tissue47. In this case, the hydrogel micro-column or another encasement scheme for the astrocytic scaffolds may be utilized as a vehicle for the controlled release of anti-inflammatory factors to lessen the inflammatory response. Initially, the aligned astrocytic bundle was not stable inside the micro-column, as the forces that compel cell alignment and matrix remodeling ultimately led to a collapse of the desired cytoarchitecture after 1-2 days in culture. However, additional contraction and bundle collapse was impeded by providing additional reinforcement for the bundles at the ends of the micro-column by adding higher concentration collagen in order to hold the bundle in place. An additional tactic could be to employ a secondary encapsulation, whereby a new hydrogel coating is applied to the aligned astrocytic bundles as they are pulled from the micro-columns. It is likely that the delivery of the aligned astrocyte networks into the brain would also stabilize the constructs due to cell-cell adhesions between host and construct cells along the entire length, although this will need to be tested directly in future studies.
Accordingly, future efforts will be aimed at developing an effective transplantation method that ensures stabilization and protection of the astrocytic bundle during and after in vivo delivery in order to allow for neuronal migration, repair, and regeneration in the injured CNS. We have previously demonstrated successful implantation of neuronal/axonal-based micro-TENNs, which share the encasement scheme of the astrocytic constructs, in corticothalamic pathways, resulting in survival and structural integration with the host tissue at least until 1 month46,47. Thus, the robustness of the cables after extraction from the micro-columns can be exploited. In this case, the agarose hydrogel would serve as the initial culture bed to induce self-assembly and then the astrocytic cable would be removed from the encasement and directly injected into the brain using a thin-walled needle. Astrocyte deformation due to mechanical forces during extraction from the hydrogel micro-column has not been observed, and is not expected. Previous work studying the effects of mechanical deformation on astrocyte reactivity suggests that any resulting deformation would not be applied rapidly enough to induce astrocytosis or process elongation62,70. To minimize the damage to brain tissue and the possible inflammatory response, the bundles within the hydrogel could be implanted using the needle-less technology developed previously for micro-TENNs. In this methodology, the hydrogel is coated with a thin layer of carboxymethylcellulose that allows for needle-less entry into the brain which may diminish the disruptive and inflammatory footprint of implantation47. Afterwards, transplantation studies may be performed to assess the ability of these scaffolds to survive, integrate with the host tissue, and promote neural migration and axon guidance. Particularly, these constructs may be implanted with one end emanating from a neurogenic region such as the SVZ to access its pool of neural stem cells and neuroblasts, in direct mimicry of the RMS, and the other end connected to an injury site to potentially drive neural migration and repopulate the area in a metered fashion.
Subsequent to transplant protocol optimization, the technique can be improved in other areas. The micro-columns may be fabricated specifically with immature astrocytes (e.g. radial glia, RMS-type cells) or mature phenotypes that are later dedifferentiated to augment the regenerative-inducing capacity of these constructs49. In in vitro models of the glial scar, mature astrocytes have not had the capability of forming pathways for axon outgrowth71. In contrast, immature astrocytes have been shown to promote neurite outgrowth in vitro and axonal regeneration in vivo when transplanted concomitantly with chondroitinase ABC to address the high levels of CSPGs in the glial scar71,72. To coincide with the advent of personalized medicine, autologous astrocytic micro-columns may be developed by seeding stem cells from sources such as induced pluripotent stem cells (iPSCs) and human neural stem cells (hNSCs). This modification will permit these scaffolds to function as conduits for individualized and specific pathway repair and regeneration. Furthermore, use of these cell sources may circumvent the possible immunogenic response that astrocyte implantation could cause. Even though immunogenicity differs according to iPSC cell lines, these cells are theoretically capable of averting or diminishing immune responses upon transplantation, as suggested by the absence of deleterious responses after iPSC-derived organ implantation in mice73,74. Furthermore, autologous hNSCs and their astrocytic derivatives repress allogeneic immune responses75. Consequently, fabricating these aligned astrocyte networks with autologous stem cells may be a critical future step in making this technique more readily applicable in the clinical setting.
Apart from the application as conduits for CNS repair and regeneration, astrocytic living scaffolds may be used as in vitro test-beds, with or without the hydrogel encasement based on the goals of a particular testing paradigm. For instance, these astrocytic constructs may be used to investigate neurobiological phenomena such as neuron migration and neurite extension mediated by astrocytes, exploiting the controllable properties and the 3D environment of living scaffolds as models of in vivo conditions. The protocol discussed in this manuscript detailed the fabrication of self-assembled aligned astrocytic bundles in a collagenous matrix within a hydrogel micro-column. By recapitulating features of brain neuroanatomy, developmental/regenerative mechanisms, and neuroblast migration pathways, these implantable astrocytic scaffolds have the potential to offer supportive pathways for directed neural migration and axon guidance in the otherwise inhibitory environment of the damaged CNS.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Financial support was provided by the National Institutes of Health [U01-NS094340 (Cullen) & F31-NS090746 (Katiyar)], Michael J. Fox Foundation [Therapeutic Pipeline Program #9998 (Cullen)], Penn Medicine Neuroscience Center Pilot Award (Cullen), National Science Foundation [Graduate Research Fellowships DGE-1321851 (Struzyna)], Department of Veterans Affairs [RR&D Merit Review #B1097-I (Cullen)], and the U.S. Army Medical Research and Materiel Command [#W81XWH-13-207004 (Cullen) & W81XWH-15-1-0466 (Cullen)].
References
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407(6807):963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006;7(8):617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani L, Petrinovic MM. Targeting Axonal Regeneration: The Growth Cone Takes the Lead. J. Neurosci. 2014;34(13):4443–4444. doi: 10.1523/JNEUROSCI.0320-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struzyna LA, Harris JP, Katiyar KS, Chen HI, Cullen DK. Restoring nervous system structure and function using tissue engineered living scaffolds. Neural Regen. Res. 2015;10(5):679–685. doi: 10.4103/1673-5374.156943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Katsanevakis E, Liu X, Zhang N, Wen X. Engineering neural stem cell fates with hydrogel design for central nervous system regeneration. Prog. Polym. Sci. 2012;37(8):1105–1129. [Google Scholar]
- Lie DC, Song H, Colamarino SA, Ming G, Gage FH. Neurogenesis in the Adult Brain: New Strategies for Central Nervous System Diseases. Annu. Rev. Pharmacol. Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- Gao Y, Yang Z, Li X. Regeneration strategies after the adult mammalian central nervous system injury-biomaterials. Regen. Biomater. 2016;3(2):115–122. doi: 10.1093/rb/rbw004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner EA, Strittmatter SM. Axon Regeneration in the Peripheral and Central Nervous Systems. Results Probl. Cell Differ. 2009;48:339–351. doi: 10.1007/400_2009_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Yin Y. Combinatorial Treatments for Promoting Axon Regeneration in the CNS: Strategies for Overcoming Inhibitory Signals and Activating Neurons’ Intrinsic Growth State. Dev. Neurobiol. 2007;67(9):1148–1165. doi: 10.1002/dneu.20515. [DOI] [PubMed] [Google Scholar]
- Kyungsuk K, Liu K, et al. Promoting Axon Regeneration in the Adult CNS by Modulation of the PTEN / mTOR Pathway. Science. 2008;322(5903):963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015;18(7):942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J. Functional regeneration beyond the glial scar. Exp. Neurol. 2014;253:197–207. doi: 10.1016/j.expneurol.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010;79(2):77–89. doi: 10.1016/j.bcp.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Toy D, Namgung U. Role of Glial Cells in Axonal Regeneration. Exp. Neurobiol. 2013;22(2):68–76. doi: 10.5607/en.2013.22.2.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East E, de Oliveira DB, Golding JP, Phillips JB. Alignment of astrocytes increases neuronal growth in three-dimensional collagen gels and is maintained following plastic compression to form a spinal cord repair conduit. Tissue Eng. Part A. 2010;16(10):3173–3184. doi: 10.1089/ten.tea.2010.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal Elongation into Peripheral Nervous System "Bridges" after Central Nervous System Injury in Adult Rats. Science. 1981;214(4523):931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- Benfey M, Aguayo AJ. Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature. 1982;296(11):150–152. doi: 10.1038/296150a0. [DOI] [PubMed] [Google Scholar]
- Fry EJ, Chagnon MJ, López-Vales R, Tremblay ML, David S. Corticospinal tract regeneration after spinal cord injury in receptor protein tyrosine phosphatase sigma deficient mice. Glia. 2010;58(4):423–433. doi: 10.1002/glia.20934. [DOI] [PubMed] [Google Scholar]
- Lin B, Xu Y, Zhang B, He Y, Yan Y, He M-C. MEK inhibition reduces glial scar formation and promotes the recovery of sensorimotor function in rats following spinal cord injury. Exp. Ther. Med. 2014;7(1):66–72. doi: 10.3892/etm.2013.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Carter LM. Manipulating the glial scar: Chondroitinase ABC as a therapy for spinal cord injury. Brain Res. Bull. 2011;84(4-5):306–316. doi: 10.1016/j.brainresbull.2010.06.015. [DOI] [PubMed] [Google Scholar]
- Vadivelu S, Stewart TJ, et al. NG2+ Progenitors Derived From Embryonic Stem Cells Penetrate Glial Scar and Promote Axonal Outgrowth Into White Matter After Spinal Cord Injury. Stem Cells Transl. Med. 2015;4:401–411. doi: 10.5966/sctm.2014-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Natsume A, et al. Interferon-beta delivery via human neural stem cell abates glial scar formation in spinal cord injury. Cell Transplant. 2013;22(12):2187–2201. doi: 10.3727/096368912X657882. [DOI] [PubMed] [Google Scholar]
- Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell. 2014;14(2):188–202. doi: 10.1016/j.stem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam RY, Fuehrmann T, Mitrousis N, Shoichet MS. Regenerative therapies for central nervous system diseases: a biomaterials approach. Neuropsychopharmacology. 2014;39(1):169–188. doi: 10.1038/npp.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Tang-Schomer MD, et al. Microtissue engineered constructs with living axons for targeted nervous system reconstruction. Tissue Eng. Part A. 2012;18(21-22):2280–2289. doi: 10.1089/ten.tea.2011.0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Wolf JA, Vernekar V, Vukasinovic J, LaPlaca MC. Neural tissue engineering and biohybridized microsystems for neurobiological investigation in vitro (Part 1) Crit. Rev. Biomed. Eng. 2011;39(3):201–240. doi: 10.1615/critrevbiomedeng.v39.i3.30. [DOI] [PubMed] [Google Scholar]
- Irons HR, Cullen DK, Shapiro NP, Lambert NA, Lee RH, Laplaca MC. Three-dimensional neural constructs: a novel platform for neurophysiological investigation. J. Neural Eng. 2008;5(3):333–341. doi: 10.1088/1741-2560/5/3/006. [DOI] [PubMed] [Google Scholar]
- Morrison BI, III, Cullen DK, LaPlaca M. In Vitro Models for Biomechanical Studies of Neural Tissues. Stud. Mechanobiol. Tissue Eng. Biomater. 2011;3:247–285. [Google Scholar]
- Vukasinovic J, Cullen DK, Laplaca MC, Glezer A. A microperfused incubator for tissue mimetic 3D cultures. Biomed. Microdevices. 2009;11(6):1155–1165. doi: 10.1007/s10544-009-9332-6. [DOI] [PubMed] [Google Scholar]
- Cullen DK, Lessing MC, Laplaca MC. Collagen-dependent neurite outgrowth and response to dynamic deformation in three-dimensional neuronal cultures. Ann. Biomed. Eng. 2007;35(5):835–846. doi: 10.1007/s10439-007-9292-z. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Vernekar VN, Shoemaker JT, Cullen DK. Three-dimensional neuronal cultures. Methods Bioeng. 3D Tissue Eng. 2010.
- Chwalek K, Tang-Schomer MD, Omenetto FG, Kaplan DL. In vitro bioengineered model of cortical brain tissue. Nat. Protoc. 2015;10(9):1362–1373. doi: 10.1038/nprot.2015.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struzyna LA, Katiyar K, Cullen DK. Living scaffolds for neuroregeneration. Curr. Opin. Solid State Mater. Sci. 2014;18(6):308–318. doi: 10.1016/j.cossms.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL. The basics of brain development. Neuropsychol. Rev. 2010;20(4):327–348. doi: 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko N, Marín O, et al. New neurons clear the path of astrocytic processes for their rapid migration in the adult brain. Neuron. 2010;67(2):213–223. doi: 10.1016/j.neuron.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo A, Booth GE. Glia dictate pioneer axon trajectories in the Drosophila embryonic CNS. Development. 2000;127(2):393–402. doi: 10.1242/dev.127.2.393. [DOI] [PubMed] [Google Scholar]
- Chotard C, Salecker I. Neurons and glia: Team players in axon guidance. Trends Neurosci. 2004;27(11):655–661. doi: 10.1016/j.tins.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Wang C, Liu F, et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 2011;21(11):1534–1550. doi: 10.1038/cr.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretto P, Giachino C, Aimar P, Fasolo A, Bonfanti L. Chain formation and glial tube assembly in the shift from neonatal to adult subventricular zone of the rodent forebrain. J. Comp. Neurol. 2005;487(4):407–427. doi: 10.1002/cne.20576. [DOI] [PubMed] [Google Scholar]
- Zukor KA, Kent DT, Odelberg SJ. Meningeal cells and glia establish a permissive environment for axon regeneration after spinal cord injury in newts. Neural Dev. 2011;6 doi: 10.1186/1749-8104-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier PJ. Penetration of grafted astrocytic scars by regenerating optic nerve axons in xenopus tadpoles. Brain Res. 1979;164(1-2):61–68. doi: 10.1016/0006-8993(79)90006-4. [DOI] [PubMed] [Google Scholar]
- Lee-Liu D, Edwards-Faret G, Tapia VS, Larraín J. Spinal cord regeneration: Lessons for mammals from non-mammalian vertebrates. Genesis. 2013;51(8):529–544. doi: 10.1002/dvg.22406. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Sztal TE, Jusuf PR, Hall TE, Nguyen-Chi M, Currie PD. Fgf-Dependent Glial Cell Bridges Facilitate Spinal Cord Regeneration in Zebrafish. J. Neurosci. 2012;32(22):7477–7492. doi: 10.1523/JNEUROSCI.0758-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struzyna LA, Wolf JA, et al. Rebuilding Brain Circuitry with Living Micro-Tissue Engineered Neural Networks. Tissue Eng. Part A. 2015;21(21-22):2744–2756. doi: 10.1089/ten.tea.2014.0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JP, Struzyna LA, Murphy PL, Adewole DO, Kuo E, Cullen DK. Advanced biomaterial strategies to transplant preformed micro-tissue engineered neural networks into the brain. J. Neural Eng. 2016;13(1):16019–16037. doi: 10.1088/1741-2560/13/1/016019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JH, Cullen DK, et al. Long-Term Survival and Integration of Transplanted Engineered Nervous Tissue Constructs Promotes Peripheral Nerve Regeneration. Tissue Eng. Part A. 2009;15(7):1677–1685. doi: 10.1089/ten.tea.2008.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CC, Katiyar KS, et al. Transplantable living scaffolds comprised of micro-tissue engineered aligned astrocyte networks to facilitate central nervous system regeneration. Acta Biomater. 2016;38:44–58. doi: 10.1016/j.actbio.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JK, Fuss B, Colello RJ. Electric field-induced astrocyte alignment directs neurite outgrowth. Neuron Glia Biol. 2006;2(2):93–103. doi: 10.1017/S1740925X0600010X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao TW, Tresco PA, Hlady V. Astrocytes alignment and reactivity on collagen hydrogels patterned with ECM proteins. Biomaterials. 2015;39:124–130. doi: 10.1016/j.biomaterials.2014.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseeva T, Katechia K, Robertson M, Riehle MO, Barnett SC. Long-term neurite orientation on astrocyte monolayers aligned by microtopography. Biomaterials. 2007;28(36):5498–5508. doi: 10.1016/j.biomaterials.2007.08.034. [DOI] [PubMed] [Google Scholar]
- Pacifici M, Peruzzi F. Isolation and culture of rat embryonic neural cells: a quick protocol. J. Vis. Exp. 2012. [DOI] [PMC free article] [PubMed]
- Conway A, Schaffer DV. Biomaterial microenvironments to support the generation of new neurons in the adult brain. Stem Cells. 2014;32(510):1220–1229. doi: 10.1002/stem.1650. [DOI] [PubMed] [Google Scholar]
- Barry D, McDermott H. Differentiation of radial glia from radial precursor cells and transformation into astrocytes in the developing rat spinal cord. Glia. 2005;50(3):187–197. doi: 10.1002/glia.20166. [DOI] [PubMed] [Google Scholar]
- Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007;101(3):794–805. doi: 10.1111/j.1471-4159.2006.04369.x. [DOI] [PubMed] [Google Scholar]
- Discher DE, Janmey P, Wang Y-L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science. 2005;310(5751):1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Balgude AP, Yu X, Szymanski A, Bellamkonda RV. Agarose gel stiffness determines rate of DRG neurite extension in 3D cultures. Biomaterials. 2001;22(10):1077–1084. doi: 10.1016/s0142-9612(00)00350-1. [DOI] [PubMed] [Google Scholar]
- Smeal RM, Tresco PA. The influence of substrate curvature on neurite outgrowth is cell type dependent. Exp. Neurol. 2008;213(2):281–292. doi: 10.1016/j.expneurol.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Smeal RM, Rabbitt R, Biran R, Tresco PA. Substrate curvature influences the direction of nerve outgrowth. Ann. Biomed. Eng. 2005;33(3):376–382. doi: 10.1007/s10439-005-1740-z. [DOI] [PubMed] [Google Scholar]
- Jain A, Kim Y-T, McKeon RJ, Bellamkonda RV. In situ gelling hydrogels for conformal repair of spinal cord defects, and local delivery of BDNF after spinal cord injury. Biomaterials. 2006;27(3):497–504. doi: 10.1016/j.biomaterials.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Katiyar KS, Winter CC, Struzyna LA, Harris JP, Cullen DK. Mechanical elongation of astrocyte processes to create living scaffolds for nervous system regeneration. J. Tissue Eng. Regen. Med. 2016. [DOI] [PMC free article] [PubMed]
- McCarthy KD, De Vellis J. Preparation of Separate Astroglial and Oligodendroglial Cell Cultures from Rat Cerebral Tissue. J. Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Stabenfeldt SE, Simon CM, Tate CC, LaPlaca MC. In Vitro Neural Injury Model for Optimization of Tissue-Engineered Constructs. J. Neurosci. Res. 2007;85:3642–3651. doi: 10.1002/jnr.21434. [DOI] [PubMed] [Google Scholar]
- Kim SU, Stern J, Kim MW, Pleasure DE. Culture of purified rat astrocytes in serum-free medium supplemented with mitogen. Brain Res. 1983;274(1):79–86. doi: 10.1016/0006-8993(83)90522-x. [DOI] [PubMed] [Google Scholar]
- Morrison RS, de Vellis J. Growth of purified astrocytes in a chemically defined medium. Proc. Natl. Acad. Sci. U. S. A. 1981;78(11):7205–7209. doi: 10.1073/pnas.78.11.7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak D, Harris AK. Connective tissue morphogenesis by fibroblast traction. I. Tissue culture observations. Dev. Biol. 1982;90(2):383–398. doi: 10.1016/0012-1606(82)90388-8. [DOI] [PubMed] [Google Scholar]
- Hsiao TW, Swarup VM, Kuberan B, Tresco PA, Hlady V. Astrocytes specifically remove surface-adsorbed fibrinogen and locally express chondroitin sulfate proteoglycans. Acta Biomater. 2013;9(7):7200–7208. doi: 10.1016/j.actbio.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JB, Bunting SCJ, Hall SM, Brown RA. Neural tissue engineering: a self-organizing collagen guidance conduit. Tissue Eng. 2005;11(9):1611–1617. doi: 10.1089/ten.2005.11.1611. [DOI] [PubMed] [Google Scholar]
- Cullen DK, Simon CM, LaPlaca MC. Strain rate-dependent induction of reactive astrogliosis and cell death in three-dimensional neuronal-astrocytic co-cultures. Brain Res. 2007;1158:103–115. doi: 10.1016/j.brainres.2007.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Miller JH, Coulson-Thomas YM, Horn KP, Alilain WJ, Silver J. Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev. Neurobiol. 2010;70(12):826–841. doi: 10.1002/dneu.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson S, Strömberg I. Guidance of dopaminergic neuritic growth by immature astrocytes in organotypic cultures of rat fetal ventral mesencephalon. J. Comp. Neurol. 2002;443(3):237–249. doi: 10.1002/cne.10119. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Han Y, Cao X. Induced pluripotent stem cell (iPSCs) and their application in immunotherapy. Cell. Mol. Immunol. 2014;11(1):17–24. doi: 10.1038/cmi.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cao J, et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov. 2015;1 doi: 10.1038/celldisc.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolmer-Solberg N, Cederarv M, Falci S, Odeberg J. Human neural stem cells and astrocytes, but not neurons, suppress an allogeneic lymphocyte response. Stem Cell Res. 2009;2(1):56–67. doi: 10.1016/j.scr.2008.06.002. [DOI] [PubMed] [Google Scholar]