

Abstract
The Sleeping Beauty (SB) transposon is a non-viral integrating system with proven efficacy for gene transfer and functional genomics. To optimize the SB transposon machinery, a transcriptionally regulated hyperactive transposase (SB100X) and T2-based transposon are employed. Typically, the transposase and transposon are provided transiently by plasmid transfection and SB100X expression is driven by a constitutive promoter. Here, we describe an efficient method to deliver the SB components to human cells that are resistant to several physical and chemical transfection methods, to control SB100X expression and stably integrate a gene of interest (GOI) through a "cut and paste" SB mechanism. The expression of hyperactive transposase is tightly controlled by the Tet-ON system, widely used to control gene expression since 1992. The gene of interest is flanked by inverted repeats (IR) of the T2 transposon. Both SB components are packaged in integration defective lentiviral vectors transiently produced in HEK293T cells. Human cells, either cell lines or primary cells from human tissue, are in vitro transiently transduced with viral vectors. Upon addition of doxycycline (dox, tetracycline analog) into the culture medium, a fine-tuning of transposase expression is measured and results in a long-lasting integration of the gene of interest in the genome of the treated cells. This method is efficient and applicable to the cell line (e.g., HeLa cells) and primary cells (e.g., human primary keratinocytes), and thus represents a valuable tool for genetic engineering and therapeutic gene transfer.
Keywords: Molecular Biology, Issue 131, Sleeping Beauty transposon, hyperactive transposase, transcriptional regulation, Tet-ON system, integration defective lentiviral vector, in vitro transposition
Introduction
The hyperactive SB100X transposase coupled to the T2-based transposon has already been used in preclinical gene therapy applications1,2,3, genome modifications4,5, and induced pluripotent stem cell (iPSC) reprogramming6,7. Typically, the SB components are transiently provided by plasmid transfection and a strong constitutive promoter drives the expression of the transposase. However, despite the improved new methods of transfection, delivery of the two-component system remains a challenge for many applications. Thus, the development of a new delivery protocol has increasing interest. Besides transfection methods for plasmid8 and mRNA9, viral delivery based on adenoviral10,11, adeno-associated viral (AAV)12, Baculovirus13, gammaretroviral14 and lentiviral vectors15 has been proposed in the past. Notably, the system of choice must guarantee a transient delivery of the SB components without integration of the viral vector. Nevertheless, the constitutive expression of transposase raises safety concerns due to the risk of uncontrollable rehopping of the transposon.
Therefore, transcriptional regulation of SB100X by a refined Tet-ON system is the first challenge. A modified Tet-responsive promoter, obtained by substituting the original developed minimal cytomegalovirus (CMV) promoter with the minimal promoter (-81) of the Timidine Kinase (TK) gene of Herpes Simplex Virus-1 (HSV-1)16, is cloned upstream of the SB100X cDNA (pTetOTKSB100X). The mutated reverse-tetracycline-transactivator rtTA2s-M217 is expressed under the control of the strong constitutive promoter (phosphoglycerate promoter, PGK) cloned in a different plasmid (pCCL-PGKrtTA2S-M2). When added to the culture medium, dox binds the rtTA2s-M2 modulator and tethers the Tet promoter complex or, resulting in tightly regulated induction of SB100X expression18.
The second major challenge arises from the inefficient transfection of human primary cells by several physical and chemical methods. To efficiently deliver transposase in human cells resistant to transfection, the SB100X and the rtTA2s-M2 expression cassettes are packaged into two integration defective lentiviral vectors (IDLVs)19: IDLVTKSB and IDLVrtTA2s-M2. Viral vectors are transiently produced as third generation vectors in HEK293T cells20 and pseudotyped with the glycoprotein G of the Vescicular Stomatitis Virus (VSV-G), which confers a broad spectrum of infection. Vector particles are concentrated by ultracentrifugation and titrated by HIV-1 Gag p24 immunocapture. To transduce cell lines and primary cells, vector particles are complexed with polybrene and are incubated with target cells in the presence or absence of dox. Drug-dependent activation of SB100X expression in transduced cells is measured by quantitative RT-PCR (qRT-PCR)18.
Once Tet-regulated SB100X expression is demonstrated, transposition of the GOI (e.g., green fluorescent protein, GFP) into the target cell genome follows the molecular events clearly schematized by Bak and Mikkelsen21. A third IDLV vector carrying an expression cassette for the GOI cloned between the T2-transposon IRs (IDLVT2) has to be constructed and packaged as mentioned above. Lastly, the three IDLV vectors can be used to efficiently transduce human cells (e.g., primary keratinocytes) in vitro and integrate the GOI in the presence of doxycycline18.
Protocol
1. Plasmids employed
NOTE: Plasmid pCCL-PGKrtTA2S-M2 was kindly provided by Prof. Zappavigna (University of Modena and Reggio Emilia, Modena, Italy). The pCMVSB100X plasmid carrying the coding sequence of hyperactive transposase and pT2/BH transposon plasmid were kindly provided by Prof Z. Ivics (Paul Ehrlich Institute, Langen, Germany) and Prof. Z. Izvak (Max Delbruck Center for Molecular Medicine, Berlin, Germany).
For all cloning in Escherichia Coli (E. coli), follow the cloning strategy of choice and restriction enzyme procedure or PCR amplification of the fragment to be cloned. NOTE: Table 1 summarizes all of the plasmids employed in this protocol. A description and references for each plasmid are provided.
2. Viral vector production
NOTE: All the viral vectors are produced in a biohazard hood at a BSL2 biosafety containment level, according to institutional rules and regulations.
Culture adherent HEK293T cells in Dulbecco's Modified Eagle Medium supplemented with 10% HyClone serum, 100 U/mL penicillin-streptomycin, and 2 mM glutamine.
Day 0: Prepare five 15 cm plates/viral preparation and seed 5.5x106 cells in 30 mL of the medium.
- Day 1: Transfection
- Before starting transfection, prepare 100 mL of 2x HEPES buffer saline (HBS): NaCl (5 M) 5.6 mL HEPES (1 M) 10.0 mL Na2HPO4 (0.5 M) 0.3 mL Sterile water 84.1 mL Adjust to pH 7.1 with 37% HCl. NOTE: 2x HBS can be stored at 4 °C. An exact pH is extremely important for efficient transfection. The optimal pH range is 7.10 to 7.12.
- Remove medium and add 22.5 mL/plate of fresh medium 4 h before transfection.
- Transfect HEK293T cells with the transfer plasmid, the pMD.Lg/pRRE.D64VInt packaging plasmid, the pMD2.G envelope-encoding plasmid, and the pRSV-Rev (Table 1) according to the following amount/plate: transfer plasmid 25.00 µg pMD.Lg/pRRE.D64VInt 16.25 µg pMD2.G 8.75 µg pRSV-Rev 6.25 µg NOTE: Plasmid DNAs are prepared according to the CsCl protocol described by Maniatis22 or by using an endotoxin-free plasmid purification kit.
- Resuspend 56.25 µg of DNA (mix of 4 plasmids) in 1.125 mL of sterile water and add 125 µL of 2.5 M CaCl2 (solution A).
- Add 1.250 mL of 2x HBS to a sterile 15 mL conical centrifuge tube (solution B).
- Add solution A (1.250 mL) to B (1.250 mL) dropwise while bubbling with a 1 or 2 mL pipette (transfection solution, total volume 2.500 mL).
- Incubate for 20 min at room temperature (RT).
- Using a P1000 micropipette, distribute all of the transfection solution (2.500 mL) to the cell monolayer dropwise.
- Incubate overnight in a cell culture incubator at 37 °C, 5% CO2.
Day 2: Remove the medium and add 15 mL of fresh medium (see step 2.1).
- Day 3: Collect and concentrate the lentiviral particle-containing supernatant.
- Collect supernatant (~70 mL) from all (n=5) transfected cell plates, filter through a 0.45 µm PESS filter and fill 2 polyallomer tubes (1-inch x 3.5 inches)/viral preparation.
- Concentrate the vector particles by ultracentrifugation at 106,000 x g for 2.5 h, at 15 °C with brake.
- Immediately after stopping the ultracentrifugation (to avoid resuspension of the pellet in the tube), gently pour the supernatant into a container containing 10% bleach and suspend the 2 barely visible pellets in 100 µL of 1x PBS (+1% BSA). This is normal as the pellet is clear and very small. Use the concentrated vector freshly prepared, or aliquot and store viral preparations at -80 °C. Caution: The supernatant contains lentiviral particles that MUST be bleached thoroughly before discarding in the biohazard waste.
To titrate the viral vector preparation, use a HIV-1 Gag p24 immunocapture kit according to the manufacturer's protocol. NOTE: To standardize the viral vector production, liposomes-based reagents could be employed. Nevertheless, the viral vector titer is highly dependent on transfection efficiency and purity of the plasmid DNA.
3. Transduction of human cell lines (e.g., HeLa cells)
NOTE: HeLa cells are cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 U/mL penicillin-streptomycin, and 2 mM glutamine. Table 1 summarizes all of the vectors employed in this protocol. A description for each vector is provided. Transduction of HeLa cells allows verification of the transcriptional regulation of SB transposase in the best dose combination of the two IDLV vectors. Variation of IDLV doses may be related to vector particle titration and target cell type.
Day 0: Seed 2x105 cells in 3 mL of medium for each well of a 6-well plate. Prepare 4 wells.
- Day 1: Transduction of HeLa cells.
- For each condition, dilute lentiviral vectors to a final volume of 1 mL medium (see Note above) in the presence of 10 μL of polybrene (final concentration 8 μg/mL): Dilution for well #1: mock transduced cells (negative control). Dilution for well #2: IDLVTKSB vector (2,600 ng of p24). Dilution for wells #3, #4: IDLVTKSB vector (2,600 ng of p24) + IDLVrtTA2s-M2 vector (9,600 ng of p24).
- Remove the medium from each well where HeLa cells were plated at day 0 and add lentiviral vector dilutions to the corresponding well.
- Spinoculate the 6-well plate at 754 x g for 45 min at 20-25 °C, and then transfer the 6-well plate to a cell culture incubator at 37 °C and 5% CO2.
- After 6 h, replace the medium containing vectors with fresh medium in all wells and add doxycycline to 1 μM in well #4. Put the plate into a cell culture incubator at 37 °C for 48 h.
- Day 3: Preparation of cell pellet for RNA extraction.
- Before detaching cells, prepare a trypsin working solution (1x): 2.5% Trypsin (10x) 5.0 mL 500 mM EDTA (final concentration = 5 mM) 0.5 mL 1x PBS 44.5 mL
- Warm trypsin working solution at 37 °C before use. Store trypsin working solution at 4 °C.
- Remove medium and wash cells with 1x PBS. Add 0.5 mL of pre-warmed 37 °C trypsin working solution to each well and incubate at 37 °C for 7 min (in a cell culture incubator).
- After the incubation, add 0.5 mL of serum-containing medium to each well to inactivate trypsin and collect cells in a centrifuge tube. Rinse each well once with 3 mL of 1x PBS, and centrifuge the cell suspension for 5 min at 240 x g.
- Wash cells with 5 mL of 1x PBS, centrifuge for 5 min at 240 x g and discard supernatant. Use freshly collected cell pellets or store at -80 °C. Extract total RNA from pellets to perform semi-quantitative RT-PCR (see step 6).
4. Transduction of human primary cells (e.g., keratinocytes)
NOTE: Human primary keratinocytes are seeded onto lethally irradiated 3T3-J2 cells (feeder layer)23, a kind gift from Yan Barrandon (EPFL, Lausanne, Switzerland).
Grow Swiss mouse 3T3-J2 cells in Dulbecco's Modified Eagle Medium supplemented with 10% donor bovine serum, 50 U/mL penicillin-streptomycin, and 4 mM glutamine (3T3 medium).
Grow keratinocytes plated onto lethally irradiated 3T3-J2 cells in cFAD medium, a Dulbecco's Modified Eagle Medium and Ham's F12 media mixture (3:1) containing fetal bovine serum (10%), penicillin-streptomycin (1%), glutamine (2%), insulin (5 µg/mL), adenine (0.18 mM), hydrocortisone (0.4 µg/mL), cholera toxin (0.1 nM), and triiodothyronine (2 nM).
Day 0: Seed 3x105 lethally irradiated 3T3-J2 cells, previously diluted into 3T3 medium to a final concentration of 1X105 cells/mL, in each well of 6-well plate. Prepare 9 wells.
- Day 1: Transduction of primary keratinocytes NOTE: Transduction of keratinocytes allows quantification of the transcriptional regulation of SB transposase in the best dose combination of the two IDLV vectors (by qRT-PCR) and to verify the integration of the GOI (GFP) in the target cells (by cytofluorimetric analysis).
- Before detaching cells, prepare the trypsin working solution: 0.5% Trypsin-EDTA (10x) 5.0 mL 500mM EDTA (final concentration = 5 mM) 0.5 mL 1x PBS 44.5 mL
- Warm trypsin working solution at 37 °C before use. Store trypsin working solution at 4 °C.
- To detach subconfluent keratinocytes in culture, remove medium, wash cells with 1x PBS and add 1 mL of pre-warmed trypsin working solution to each well. Incubate at 37 °C for 15 min in the cell culture incubator.
- After the incubation, resuspend the cells of a well thoroughly and transfer the cell suspension to a centrifuge tube containing 2 mL of serum-containing medium (if many cells are still attached, incubate for an additional min at 37 °C). Rinse each well once with 3 mL of 1x PBS or fresh medium and add the rinse solution to the centrifuge tube with the cell suspension.
- Centrifuge for 5 min at 580 x g and discard the supernatant.
- Wash cells with 5 mL of 1x PBS, centrifuge for 5 min at 580 x g and discard the supernatant.
- In a 15-mL polypropylene tube, dilute 1.6x105 keratinocytes in 1 mL of cFAD medium, one dilution for each condition.
- Dilute lentiviral vectors in 1 mL of cFAD medium in the presence of 20 μL of polybrene (final concentration of 8 µg/mL at a final volume of 2 mL): Dilutions for wells #1, #2, #3: mock transduced cells (negative control without vectors). Dilutions for wells #4, #5: IDLVTKSB vector (13,000 ng of p24) + IDLVrtTA2s-M2 vector (48,000 ng of p24). Dilutions for wells #6, #7, #8, #9: IDLVTKSB vector (13,000 ng of p24) + IDLVrtTA2s-M2 vector (48,000 ng of p24) + IDLVT2 vector (9,160 ng of p24).
- Transduce cells in suspension by adding each lentiviral vector dilution (1 mL) to the corresponding keratinocyte suspension (1.6x105 keratinocytes in 1 mL).
- Remove the medium from lethally irradiated 3T3-J2 cells seeded at day 0 and plate transduced keratinocytes (1.6x105 keratinocytes in 2 mL) onto them. After transducing a well proceed with the next one.
- Incubate at 25 °C for 30 min, and then transfer the cells to 37 °C in the cell culture incubator for 6 h. NOTE: Do not spinoculate primary keratinocytes, as viability and proliferation will be strongly affected.
- After 6 h, add 1 mL of cFAD medium to each well. Add 1 μM doxycycline (final concentration) in wells #5, #8, and #9. Return cells to 37 °C.
Day 2: Replace cFAD medium with 3 mL of KC medium with 10 ng/mL EGF.
Day 3: Trypsinize and wash cells from wells #1, #4, and #5 as described before (see 4.4.1 to 4.4.6). Freeze cell pellets at -80 °C to extract total RNA for qRT-PCR analysis (see step 7).
Trypsinize cells from well #2, #6, and #8 as described in steps 4.4.1 to 4.4.5 and perform cytofluorimetric analysis for GFP expression (see step 5).
Keep the culture from wells #3, #7, and #9 for at least 3-4 doublings, sufficient to dilute un-integrated IDLVT2 vector (5 days for human primary keratinocytes plated on feeder layer).
Day 6: Approximately 5 days post transduction, trypsinize cells (see steps 4.4.1 to 4.4.5) from wells #3, #7, and #9 to perform cytofluorimetric analysis for GFP expression (see step 5). NOTE: Experiment timing and transduction efficiency are highly influenced by primary cell type and by variability of collected samples.
5. Cytofluorimetric analysis on transduced primary keratinocytes
NOTE: A flow cytometer configured with a blue laser (488 nm), a red laser (633 nm) and filters for detection of GFP and APC fluorescence is employed (see Materials Table).
- To discriminate keratinocytes from 3T3-J2 feeder layer, label transduced keratinocytes detached at day 3 and day 6 (see steps 4.7 and 4.9) with mouse monoclonal APC -conjugated Anti-Feeder antibody.
- Prepare 4 mL of staining buffer for each sample: 5% FBS 200 μL 500 mM EDTA (final concentration = 2.5 mM) 20 μL PBS 1x 3780 μL
- Wash detached cells with 1 mL of staining buffer. Centrifuge for 5 min at 580 x g and discard the supernatant.
- In a tube for flow cytometry acquisition, resuspend 5x104 cells in 100 μL of staining buffer and add 2 μL of Anti-Feeder antibody (1:50). Mix well and incubate for 30 min on ice in the dark.
- After 30 min, wash with 2 mL of staining buffer. Centrifuge for 5 min at 580 x g and discard the supernatant. Resuspend in 200 μL of staining buffer.
- Acquire APC and GFP signals by cytofluorimetric analysis18. The GFP+ cell fraction of the APC- cell population indicates transduced keratinocytes. NOTE: If desired, fix the stained sample before flow cytometry with 2% paraformaldehyde in PBS 1x and store at 4 °C in the dark until the run.
Analyze flow cytometry data by plotting the ratio between the percentage of GFP+cells at the endpoint (5 days) and the percentage of GFP+cells 2 days post transduction, normalized to residual level of the IDLVT2-transduced cells.
6. Semi-quantitative RT-PCR
NOTE: Use a PCR thermal cycler.
Extract total RNA from transduced or control cells using an RNA purification kit, according to the manufacturer's protocol. NOTE: Total RNA can be stored at -80 °C.
Synthesize cDNA in a 20 µL reaction using 100 ng of total RNA and a reverse transcriptase kit, according to the manufacturer's protocol. NOTE: cDNA can be stored at -20 °C.
Design primers specific for SB100X, rtTA2s-M2 and GAPDH (a housekeeping gene). Suggested designs: SB forward primer: 5'-GCCACTCAGCAAGGAAGAAG-3' SB reverse primer: 5'-GTGTTGGAAGACCCATTTGC-3' rtTAM2 forward primer: 5'-GACGACAAGGAAACTCGCTC-3' rtTAM2 reverse primer: 5'-TTACCCGGGAGCATGTCAA-3' GAPDH forward primer: 5'-GACCACAGTCCATGCCATCAC-3' GAPDH reverse primer: 5'-CCACCACCCTGTTGCTGTAG-3'
Perform PCR in a final reaction volume of 50 µL. In particular, assemble in each PCR tube the following reaction: cDNA (1:5 dilution) (from step 6.2) 1.00 µL Forward primer (10 µM stock) 1.00 µL Reverse primer (10 µM stock) 1.00 µL dNTPs (10 µM stock) 1.00 µL Buffer (+ MgCl2) (10x stock) 5.00 µL Taq polymerase (5 U/µL stock) 0.25 µL Sterile water 40.75 µL
Use the following program of the thermal cycler: 1 cycle at 95 °C for 5 min then proceed with 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s for 34 cycles for SB100X, 32 cycles for rtTA2s-M2, and 25 cycles for GAPDH.
Prepare 1% agarose gel. Dissolve 1 g of agarose in 100 mL of TBE 1x (10x stock diluted in sterile water), in a beaker or flask. Melt in a microwave, swirling every minute until the agarose is completely dissolved. Cool the melted agarose until it reaches approximately 50 °C, and then add 5 µL of ethidium bromide (10 mg/mL stock). Pour melted agarose in an assembled gel tray with a comb, for gel casting.
Load samples (10 µL each) on the agarose gel and perform electrophoresis.
If desired, acquire gel picture and perform densitometric analysis on the PCR bands.
7. Quantitative RT-PCR (qRT-PCR)
NOTE: A commercial Sequence Detection System is employed. PCR primers and 6-carboxyfluorescein (FAM) probe for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are purchased commercially.
For retro-transcription, see step 6.2.
Use software to design primers and probe specific for SB100X. Suggested design: SB.2 forward primer: 5'-GAAGAAGCCACTGCTCCAAAA-3', SB.2 reverse primer: 5'-CCCCATGTGCAGTTGCAA-3' probe SB FAM: 5'-CATAAGAAAGCCAGACTACGG-3'
Perform Real-Time PCR in 96-well plates with a PCR Master Mix and primers + probe mix, in a final reaction volume of 25 µL. In particular, consider the following reaction for each well: cDNA (1:10 dilution in sterile water) 1.00 µL 2x Master Mix 12.50 µL Primers + 20x probe mix 1.25 µL Sterile water 10.25 µL NOTE: Perform all reactions in triplicate. To prevent pipetting errors, prepare a mix for at least n+3 reactions (without cDNA). Aliquot using a multichannel pipette.
Run real-time PCR using the following program: 1 cycle at 95 °C for 10 min, then 40 cycles of 95 °C for 15 s and 60 °C for 1 min and hold at 4 °C.
Analyze qRT-PCR data by normalizing the relative expression (RQ value) of the SB100X to the level of GAPDH of the same cDNA sample by the 2-ΔΔCT quantification, using typical data analysis software.
Representative Results
Using the procedure presented here, three IDLV vectors carrying the regulated SB components (SB100X, rtTA2S-M2 and T2-GFP transposon) were packaged and used to efficiently deliver and tightly regulate the SB system in human cells. Figure 1A shows a scheme for in vitro transduction of HeLa cells, which was performed to evaluate the transcriptional regulation of SB transposase with the best dose combination of the two IDLV vectors (reported in Table 2). The transcriptional regulation of SB100X was measured by qRT-PCR (Figure 1B) and plotted as a relative quantity (RQ) value with respect to the off-state (IDLVTKSB alone RQ=1). In transduced HeLa cells, a 8-fold activation (+dox) of SB100X expression over the background (IDLVTKSB) was obtained. Notably, cotransduced HeLa cells in the absence of dox showed transposase expression comparable to the off states, indicating a tight control of TetTK promoter by rtTA2s-M2 modulator.
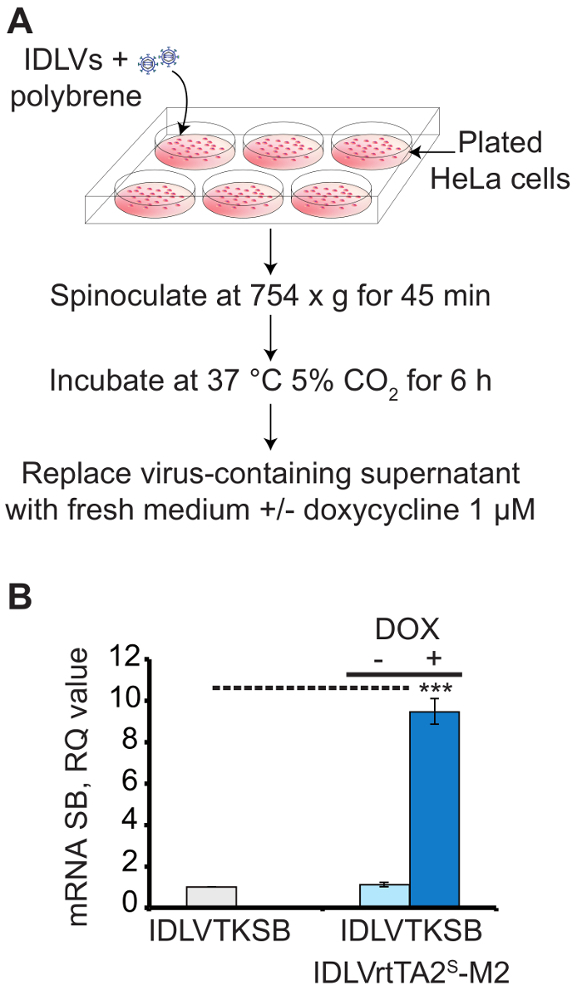
Figure 1: Transduction of HeLa cells for the expression of transcriptionally-regulated SB transposase carried by IDLV vectors.(A) A scheme for in vitro transduction of HeLa cells with IDLVs for the expression of transposase. (B) qRT-PCR analysis of HeLa cells transduced with IDLVTKSB (2,600 ng of p24) in the presence (+) or absence (-) of IDLVrtTA2S-M2 (9,600 ng of p24) and 1 µM doxycycline (dox). The dashed line connects samples showing significantly different values (***P < 0.005). The experiment was performed in triplicate. Mean S.E.M is shown as error bars. (Figure modified from Cocchiarella, F. et al., 201618). Please click here to view a larger version of this figure.
As an example,Figure 2 shows the dose ranging experiment set up to reach the optimal transcriptional regulation of transposase. HeLa cells were transduced with increasing doses (130, 260 , and 2,600 ng of p24) of IDLVTKSB in combination with IDLVrtTA2s-M2 (960 and 9,600 ng of p24), in the presence or absence of dox. Semi-quantitative RT-PCR analysis clearly shows that high doses of both IDLVs were required to strongly induce SB100X expression. Different target cell type and variations in viral vector titration may result in sup-optimal SB expression, and we therefore recommend performing dosage setting experiments.
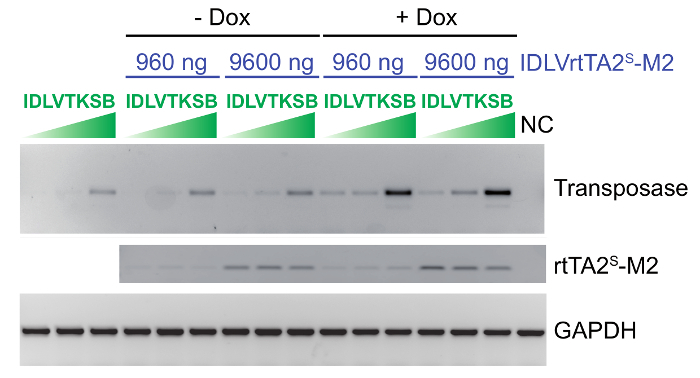
Figure 2: IDLV dose setting experiment in HeLa cells to define the optimal dose combination of the two IDLVs. Semi-quantitative RT-PCR analysis on HeLa cells co-transduced with different doses of IDLVTKSB vector (130, 260, and 2,600 ng of p24) in the absence or presence of IDLVrtTA2s-M2 vector (960 and 9,600 ng of p24) and dox. The GAPDH was amplified as a standard control. SB100X transposase and rtTA2s-M2 expression are reported in all tested conditions; NC = negative control. (Figure modified from Cocchiarella, F. et al., 201618). Please click here to view a larger version of this figure.
The transcriptional regulated SB system could also be used in primary cells, particularly in those cells resistant to several transfection methods, such as human primary keratinocytes. Figure 3 shows a scheme for transduction of primary keratinocytes cultivated on lethally irradiated 3T3-J2 feeder layer, with IDLV vectors carrying the regulated SB components, in the presence or absence of dox to induce SB100X expression.
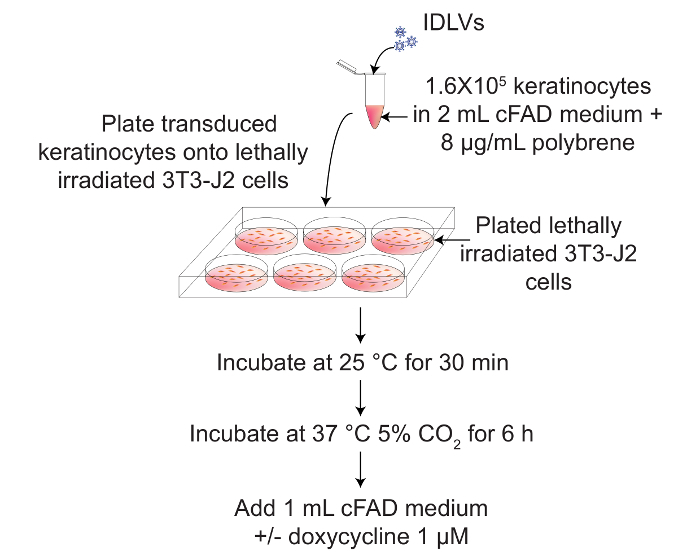
Figure 3: Transduction in suspension of human primary keratinocytes with IDLV vectors for the expression of transcriptionally-regulated SB transposase and transposon. A scheme for in vitro transduction of human primary keratinocytes with IDLVs to express transcriptionally-regulated SB transposase and transposon, in the presence or absence of 1 µM dox. Please click here to view a larger version of this figure.
The qRT-PCR analysis in Figure 4A shows that the best dose combination of transposase and modulator vectors, reported in Table 2, resulted in 15-fold activation of SB expression over the off-state (-dox). The transposition experiment was performed in keratinocytes transduced with the three IDLV vectors (IDLVTKSB, IDLVrtTA2s-M2 and IDLVT2) in the presence or absence of dox. Expression of the GOI (GFP) beared by T2-transposon, in the APC- compartment, was measured by flow cytometer analysis 2 days post-transduction (dpt) and at the endopoint of the culture (5 days for human primary keratinocytes). The endpoint was reached when unintegrated T2-GFP transposon dropped to a barely detectable level (an average of 0.6%) due to cell division-dependent dilution. Figure 4B shows the ratio between the percentage of GFP+ cells in the APC- compartment at the endpoint and 2 days post transduction, normalized to the residual level of the transposon alone. This indicates the percentage of cells hosting at least 1 copy of the expressed GOI stably integrated in their genome, assessed at 12% for human primary keratinocytes.
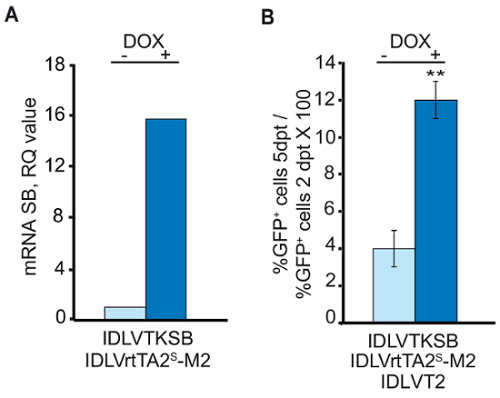
Figure 4: Analysis of the expression of transposase and GFP in transduced human primary keratinocytes. (A) qRT-PCR assay on primary keratinocytes transduced with IDLVTKSB and IDLVrtTA2S-M2 in the presence or absence of 1 µM dox. (B) Co-transduction of primary keratinocytes with IDLVT2 transposon and IDLVs for transposase expression in presence or absence of dox. On Y-axis, %GFP+ cells 5dpt / %GFP+ cells 2 dpt * 100 (mean ± SEM of two experiments). ** show significantly different values (P < 0.05). (Figure modified from Cocchiarella, F. et al., 201618). Please click here to view a larger version of this figure.
| Plasmid | Description | References |
| pCCL-TetOTKSB | Transfer plasmid used to generate IDLVTKSB vector. This plasmid derived from pTetOTKSB, after cloning TetOTKSB in the pCCL lentiviral backbone. | 18, 20 |
| pCCL-PGKrtTA2S-M2 | Transfer plasmid used to generate IDLVrtTA2s-M2 vector. | provided by Prof. V. Zappavigna |
| pCCL-T2GFP | Transfer plasmid used to generate IDLVT2 vector. This plasmid derived from pT2GFP, after cloning the GFP between the T2-transposon IRs in the pCCL lentiviral backbone. | 18, 20 |
| pMD.Lg/pRRE.D64VInt | Packaging plasmid to generate IDLV vector. | 19 |
| pMD2.G | Plasmid coding for VSV-G envelope to generate lentiviral vector. | 20 |
| pRSV-Rev | Rev plasmid to generate lentiviral vector. | 20 |
| Vector | Description | |
| IDLVTKSB | Integration defective lentiviral vector for the expression of the SB100X under the control of Tet-responsive TK promoter. | |
| IDLVrtTA2s-M2 | Integration defective lentiviral vector for the expression of the rtTA2s-M2 modulator under the control of PGK promoter. | |
| IDLVT2 | Integration defective lentiviral vector for the expression of the GOI cloned between the T2-transposon IRs. |
Table 1: Plasmids and vectors employed in this protocol. Descriptions and references are provided.
| Vectors | HeLa (2x105 cells) | Keratinocytes (1.6x105 cells) |
| IDLVTKSB | 2,600 ng p24 | 13,000 ng p24 |
| IDLVrtTA2s-M2 | 9,600 ng p24 | 48,000 ng p24 |
| IDLVT2 | 9,160 ng p24 |
Table 2: Optimized vector doses for transduction of HeLa cells and human primary keratinocytes.
Discussion
Here, we describe a widely accessible methodology to stably integrate a GOI into the genome of target cells by Sleeping Beauty-mediated transposition. Although the SB system was developed to provide a nonviral method for genome editing, efficient delivery of the integration machinery (transposase and T2 transposon) is mandatory. Therefore, to use the SB system on hardly transfectable primary cells, a viral delivery of SB components was pursued in recent years10,12,15. However, none of the viral vectors employed up to now addressed the issue of constitutive expression of transposase that could result in the risk of uncontrollable rehopping of the transposon.
To transcriptionally regulate transposase expression, we took advantage of a modified Tet-ON system16. The minimal TK promoter fused to the TetO element, upon binding to the rtTA2s-M2 modulator in the presence of dox, conferred a fine-tuning of transposase activation over the background (off-state, in the absence of dox). The regulatory performance depends on the appropriate IDLVs (IDLVTKSB and IDLVrtTA2s-M2) vector dose combination and on cell types (e.g., HeLa cells and human primary keratinocytes). Independently from target cells, the vector dose must ensure the highest level of transduction of the target cells without affecting cell viability, thus an escalating dose-combination experiment in HeLa cells (or other cell lines) to define the optimal doses, by semi-quantitative RT-PCR, is strongly suggested.
Transposition of the GOI was assessed in primary cells. Human primary keratinocytes seeded on lethally irradiated 3T3-J2 feeder layer were transduced with 3 IDLVs (IDLVTKSB, IDLVrtTA2s-M2 and IDLVT2) at the experimentally defined doses. A clear dox-dependent activation of the transposase was demonstrated by qRT-PCR. Moreover, stable integration of the GOI was demonstrated by cytofluorimetric analysis of GFP expression in cells transduced in the presence or absence of dox, indicating the possibility to employ the IDLV platform to vehicle the transcriptionally regulated SB components to the primary cells, and the successful integration of a GOI into the genome of target cells.
The critical steps in the protocol are IDLV vector production and transduction of target cells at the optimal efficiency and vector dose combination. Low transfection efficiency of HEK293T cells may result in low vector yield. The definition of doses of IDLVTKSB and IDLVrtTA2s-M2 is necessary to avoid an unbalance of Tet-ON components that may result in high leakiness in the absence of the modulator or in low fold induction of the TetOTK promoter upon dox treatment. These critical steps could be addressed by minor modifications. For instance, the transfection protocol could be standardized using commercial transfection reagents. The transduction protocol should be adapted according to cell type of interest, particularly considering whether spinoculation increases transduction efficiency without affecting cell viability. Vectors and polybrene could be added to the culture medium of the target cells and replaced with fresh medium after 6 h or overnight incubation. It is also worth noting that the percentage of GFP+ cells 5 days after transduction could be affected by the background integration of IDLVs.
A minor limitation of this method is that the IDLV vectors are produced transiently by transfection of HEK293T cells. Depending on the vector doses to be used, repeated vector productions are required. A second limitation is related to the length of the GOI, as the cargo capacity of the IDLV vector is approximately 8 kb.
In conclusion, this method here allows the integration of a GOI through transcriptionally regulated SB components packaged into IDLV vectors, which, among the existing methods to deliver SB system, show a broad range of tropism and high transduction efficiency. Moreover, the modified Tet-ON system permits a transcriptional regulation of the SB100X expression, a relevant issue in the risk of transposon re-hopping or in multiple serial transposition events if they are required. Possible applications of this method include genome engineering of cell lines (e.g., HeLa cells, HEK293 cells) to establish stable packaging cells for viral vectors, and integration of therapeutic genes into clinical relevant primary cells.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Prof. Zappavigna, University of Modena and Reggio Emilia, Modena, Italy for providing us with the pCCL-PGKrtTA2S-M2 plasmid. We also acknowledge Prof Z. Ivics (Paul Ehlrich Institute, Langen, Germany) and Prof. Z. Izvak (Max Delbruck Center for Molecular Medicine, Berlin, Germany) for pCMVSB100X and pT2/BH plasmids. This work was supported by DEBRA international and Italian Ministry of University and Research.
References
- Mates L, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;41(6):753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- Maiti SN, et al. Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013;36(2):112–123. doi: 10.1097/CJI.0b013e3182811ce9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnen S, et al. Sleeping Beauty transposon-mediated transfection of retinal and iris pigment epithelial cells. Invest Ophthalmol Vis Sci. 2012;53(8):4787–4796. doi: 10.1167/iovs.12-9951. [DOI] [PubMed] [Google Scholar]
- Koso H, et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc Natl Acad Sci U S A. 2012;109(44):E2998–E3007. doi: 10.1073/pnas.1215899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belay E, et al. Novel hyperactive transposons for genetic modification of induced pluripotent and adult stem cells: a nonviral paradigm for coaxed differentiation. Stem Cells. 2010;28(10):1760–1771. doi: 10.1002/stem.501. [DOI] [PubMed] [Google Scholar]
- Grabundzija I, et al. Sleeping Beauty transposon-based system for cellular reprogramming and targeted gene insertion in induced pluripotent stem cells. Nucleic Acids Res. 2013;41(3):1829–1847. doi: 10.1093/nar/gks1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kues WA, et al. Derivation and characterization of sleeping beauty transposon-mediated porcine induced pluripotent stem cells. Stem Cells Dev. 2013;22(1):124–135. doi: 10.1089/scd.2012.0382. [DOI] [PubMed] [Google Scholar]
- Turchiano G, et al. Genomic analysis of Sleeping Beauty transposon integration in human somatic cells. PLoS One. 2014;9(11):e112712. doi: 10.1371/journal.pone.0112712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011;18(9):849–856. doi: 10.1038/gt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latella MC, et al. Correction of Recessive Dystrophic Epidermolysis Bullosa by Transposon-Mediated Integration of COL7A1 in Transplantable Patient-Derived Primary Keratinocytes. J Invest Dermatol. 2017;137(4):836–844. doi: 10.1016/j.jid.2016.11.038. [DOI] [PubMed] [Google Scholar]
- Zhang W, et al. Integration profile and safety of an adenovirus hybrid-vector utilizing hyperactive sleeping beauty transposase for somatic integration. PLoS One. 2013;8(10):e75344. doi: 10.1371/journal.pone.0075344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. Hybrid adeno-associated viral vectors utilizing transposase-mediated somatic integration for stable transgene expression in human cells. PLoS One. 2013;8(10):e76771. doi: 10.1371/journal.pone.0076771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen TA, Laakkonen JP, Alasaarela L, Airenne KJ, Yla-Herttuala S. Sleeping Beauty-baculovirus hybrid vectors for long-term gene expression in the eye. J Gene Med. 2014;16(1-2):40–53. doi: 10.1002/jgm.2756. [DOI] [PubMed] [Google Scholar]
- Galla M, et al. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011;39(16):7147–7160. doi: 10.1093/nar/gkr384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldt B, et al. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol Ther. 2011;19(8):1499–1510. doi: 10.1038/mt.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia A, Perani L, Sartori D, Olgiati C, Mavilio F. Site-specific integration of functional transgenes into the human genome by adeno/AAV hybrid vectors. Mol. Ther. 2004;10:660–670. doi: 10.1016/j.ymthe.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Urlinger S, et al. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci U S A. 2000;97(14):7963–7968. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiarella F, et al. Transcriptionally regulated and nontoxic delivery of the hyperactive Sleeping Beauty Transposase. Mol Ther Methods Clin Dev. 2016;3:16038. doi: 10.1038/mtm.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Munoz RJ, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12(3):348–353. doi: 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak RO, Mikkelsen JG. Mobilization of DNA transposable elements from lentiviral vectors. Mob Genet Elements. 2011;1(2):139–144. doi: 10.4161/mge.1.2.17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Molecular Cloning: A laboratory Manual. 4th edition 2012.
- Dellambra E, et al. Corrective transduction of human epidermal stem cells in laminin-5-dependent junctional epidermolysis bullosa. Hum Gene Ther. 1998;9:1359–1370. doi: 10.1089/hum.1998.9.9-1359. [DOI] [PubMed] [Google Scholar]