

Abstract
Rationale:
Progressive restriction of the spinal bio-mechanics is not-uncommon deformity encountered in spine clinics. Congenital spinal fusion as seen in Klippel-Feil-anomaly, progressive non-infectious anterior vertebral fusion, and progressive spinal hyperostosis secondary to ossification of the anterior longitudinal spinal ligament are well delineated and recognized.
Patient concerns:
A 24-year-old girl has history of osteoporosis since her early childhood, associated with multiple axial and appendicular fractures and scoliosis. Recently she presented with episodes of severe back pain, spinal rigidity/stiffness with total loss of spine biomechanics.
Diagnoses:
She was provisionally diagnosed as having osteogenesis imperfecta and was investigated for COL1A1/A2 mutations which have been proven to be negative. Autosomal recessive type of osteogenesis imperfecta was proposed as well, no mutations have been encountered. A homozygous for CTSA gene mutation, the gene associated with Galactosialidosis was identified via whole exome sequencing (Next-Generation Sequencing projects) has been identified.
Interventions:
Early in her life she had a history of frequent fractures of the long bones since she was 4 years which was followed by vertebral fractures at the age of 12 years. She manifested lower serum 25OH-D levels and were associated with lower LS-aBMD Z-scores with higher urinary bone turnover indexes (urinary NTX/Cr).
Outcomes:
Lysosomal storage diseases (LSD) have a strong correlation with the development of osteoporosis. LSD causes skeletal abnormalities results from a lack of skeletal remodeling and ossification abnormalities owing to abnormal deposition of GAGs (impaired degradation of glycosaminoglycans ) in bone and cartilage. 3D reconstruction CT scan of the spine showed diffuse hyperostosis of almost the entire spine (begins at the level of T4- extending downwards to involve the whole thoraco-lumbar and upper part of the sacrum) with total diffuse fusion of the pedicles, the transverse and articular processes, the laminae and the spinous processes.
Lessons:
This is the first clinical report of adult patient with a history of osteoporosis and fractures with the late diagnosis of Galactosialidosis. Osteogenesis imperfecta (autosomal dominant and recessive) were the first given diagnoses which proven negative. The pathophysiology of the spine ankylosis in our current patient and its correlation with LSD, antiresorptive medications, vitamin D3 and supplemental calcium is not fully understood. Therefore, further studies are needed to elucidate this sort of correlation.
Keywords: ankylosed, backbone, COL1A1/A2, CTSA, osteoporosis
1. Introduction
Progressive reduction in spine biomechanics can result from a long list of congenital and acquired etiologies. The embryologic development of the vertebral column is a complex and rapid process. The complete anatomic pattern is formed in mesenchyme during the first 6 weeks of intrauterine life. Defects of formation or segmentation of the primitive vertebrae may occur during this period. Once the abnormal mesenchyme mold is established, the cartilaginous and bony stages follow that pattern, and the vertebral anomalies are then fully established at birth.[1]
Other spine mal-development that needs to be considered are progressive noninfectious anterior vertebral fusion (Copenhagen syndrome), which results in block vertebrae associated with progressive loss of the physiological spine mobility.[2]
Forestier disease is another form of abnormality that consists of anterolateral, peri-vertebral, and ligament ossification, and is known as diffuse idiopathic skeletal hyperostosis (DISH) or the vertebral ankyloses hyperostosis syndrome. In Forestier disease, there is progressive spine stiffness and decreased range of movement of the spinal column.[3,4]
Galactosialidosis (combined neuraminidase and beta-galactosidase deficiency), the infantile type, can present with fetal hydrops, hepatosplenomegaly, inguinal hernia, telangiectasia, and sometimes periosteal cloaking.[5,6] The late infantile type has coarse facial features, organomegaly, dysostosis, and often a cherry red macular spot as the main features. Our current patient and her siblings all manifested osteoporosis and fractures as the chief clinical presentation. None showed organo.megaly and or cherry red macular spots. The condition is caused by a deficiency of carboxypeptidase-L protective protein.[7]
2. Case report
2.1. Chief presenting complaint
A 24-year-old girl had a history of osteoporosis, multiple fractures, and scoliosis since her early childhood. Recently, she presented with episodes of severe back pain and spinal rigidity/stiffness with a total loss of spine biomechanics.
2.2. Past medical history
Early in her life, she had a history of frequent fractures of the long bones since she was 4 years, which was followed by vertebral fractures at the age of 12 years. She manifested lower serum 25OH-D levels that were associated with lower LS-aBMD Z-scores with higher urinary bone turnover indexes (urinary NTX/Cr). She was provisionally diagnosed as having osteogenesis imperfecta and was investigated for COL1A1/A2 mutations, which have been proven to be negative. Autosomal recessive type of osteogenesis imperfecta was proposed and no mutations have been encountered. To expedite the relief of her osteoporotic bones and frequent fractures, bisphosphonates (intravenous pamidronate therapy for 2 years in, cycles of 1 mg/kg daily over 3 consecutive days at a mean cycle interval of 3.8 months) have been administered for 2 successive years along with supplemental calcium and vitamin D. The aim was to increase the size of the vertebral bodies and to remodify the pre-existing vertebral compressive fractures, to lessen the rate of fractures of the long bones and to maintain serum 25-hydroxyvitamin-D levels above 50 nmol/L (20 ng/mL). Progressive rigidity of the osteoporotic spine was the outcome. The patient experienced gradual loss of her physiological biomechanical spine mobility. To elicit the reason behind this progressive deformity, 3D reconstruction computed tomography (CT) scan was organized. Surprisingly, it showed diffuse hyperostosis of almost the entire spine, turning the spine into an ankylosed concrete-like structure. We wonder if osteoporotic patients with a history of metabolic errors of metabolism are susceptible to develop spine ankyloses when treated with anti-resorptive medications. It might be possible that the long-term administration of osteoporotic patients with pamidronate, supplemental calcium, and vitamin D might lead to mal-deposition of excess bone along the tilted, fractured spine.
Clinical examination revealed that height was 158 cm with a body mass index of 17.9 kg/m2 (underweight). Abnormal craniofacial contour associated with mild coarse facial features and massive gingival hyperplasia. All her spinal movements were markedly reduced. Movements of both hips were severely restricted and bilateral elbow fixed flexion deformities were present as well. She manifested a high tower head with wide frontal area and discoloration of teeth. She has short trunk but with disproportionately long limbs, mitral regurgitation, and kyphoscoliosis. She had normal intelligence with no neurological findings and no macular cherry-red spots were encountered. No hepatosplenomegaly was elicited as well. She experienced mild respiratory problems, but her pulmonary functions were to certain extent within normal values. She manifested 65° Cobbs angle, which did not respond sufficiently to early life application of Boston brace. Musculoskeletal examination showed greater reduction in thoracic movements and in lumbar movements associated with pain and extreme spine rigidity. In addition, movements of both hips were severely restricted and she showed positive Patrick test (FABER test, which is a passive screening test for musculoskeletal pathologies of the lumbar, hip, S1 joint dysfunction, and to identify iliopsoas spasm). FABER test stands for the assessment of flexion, abduction, and external rotation. The value of these 3 motions is to localize a clinical pain provocation test and to find pathologies at the hip, lumbar, and sacroiliac region. Anterior and posterior impingement tests were negative and ROM (range of motion); right 85°/left 110°, extension right 15°/left 20° were elicited. Abduction, right = 20°/left = 30°, adduction right hip 28°, and left 40°. She was extensively investigated for liver function tests, TSH, PTH, 25-hydroxyvitamin D level, serum protein electrophoresis, 24-hour urine calcium/creatinine, serum homocyteine level, and triglycerides. Her blood tests for hematology, biochemistry, ESR, hormonal, and rheumatoid arthritis were normal. Calcium was 7.2 mEq/L (n.v 8.5–10.3 nEq/L), parathyroid hormone was 35.2 pg/mL (n.v 10.0–65 pg/mL), and 25-hydroxyvitamin-D measurement with a DiaSorin radioimmunoassay was 8.1 ng/mL (n.v 9.0–37.6 ng/mL). Inflammatory markers were normal and HLA-B27 was negative. Two years after discontinuation of intravenous pamidronate, the BMC Z-scores remained above the pretreatment levels but below normal levels. Her cortical width was assessed via transiliac bone biopsy (tetracycline-based histomorphometric study) and it showed that the cortical width of the iliac bone has been doubled during the treatment with pamidronate, supplemental calcium, and vitamin D. Biochemical markers of bone turnover such as bone-specific alkaline phosphatase (BSALP), osteocalcin (as a marker of bone turnover), and vitamin K (plays role in the y-carboxylation of osteocalcin). These biomarkers were used to determine whether the therapy by pamidronate became ineffective. There was almost 30% reduction in these biomarkers of resorption. It is well known that failure to show reduction in resorption markers might indicate poor compliance to pamidronate therapy. These biomarkers have been applied to offer more advantages over using BMD in order to assess the effectiveness of antiresorptive treatment.
2.3. Diagnosis
We performed skeletal survey and tomographic studies to elucidate the reason behind the progressive restriction of the spine biomechanics: Anteroposterior pelvis radiograph showed severe osteopenia, no features of sacro-iliitis, spherical capital femoral epiphyses with markedly short femoral necks, and bilateral narrowing of the joint spaces overwhelmed by severe osteoporosis (Fig. 1). 3D reformatted CT scan of the spine (anterior view) showed severe/diffuse ossification of the anterior longitudinal spinal ligaments on both sides of the spine interspersed with massive hyperostosis covering the whole spine. Areas of hyper and hypo-mineralization all over the axial and the appendicular skeleton are notable (Fig. 2). 3D reconstruction CT scan of the posterior aspect of the spine showed diffuse hyperostosis of almost the entire spine (begins at the level of T4- extending downwards to involve the whole thoracolumbar and upper part of the sacrum) with total diffuse fusion of the pedicles, the transverse and articular processes, the laminae, and the spinous processes (Fig. 3). 3D reconstruction CT scan of the cranium showed no trace for the sagittal suture (sclerosed/synostosed sagittal suture – see arrow) but persistence of the coronals and substantial diffuse gingival hyperplasia of the upper gum (Fig. 4). In this girl, a homozygous mutation for CTSA gene mutation, the gene associated with Galactosialidosis, was identified via whole exome sequencing in this patient and her other 3 siblings (Courtesy of Dr. Lindsay C. Burrage/ Brendan Lee Laboratory, Next-Generation Sequencing projects, Baylor College of Medicine- Molecular and Human Genetics-USA- enzyme activity was not performed because of logistical reasons).
Figure 1.
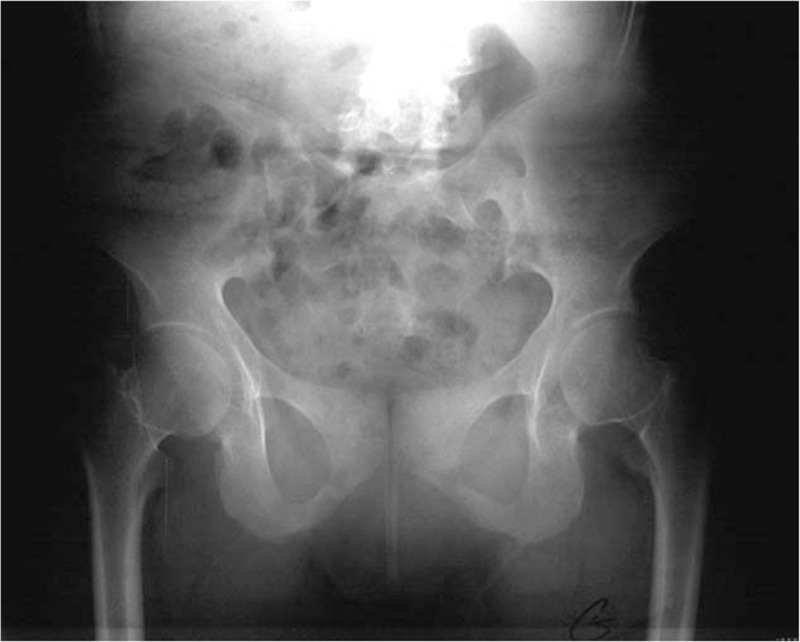
Anteroposterior pelvis radiograph showed severe osteopenia, no features of sacro-iliitis, spherical capital femoral epiphyses with markedly short femoral necks, and bilateral narrowing of the joint spaces overwhelmed by severe osteoporosis.
Figure 2.

3D reformatted CT scan of the spine (anterior view) showed severe/diffuse ossification of the anterior longitudinal spinal ligaments on both sides of the spine interspersed with massive hyperostosis covering the whole spine. Note areas of hyper- and hypo-mineralization all over the axial and the appendicular skeleton.
Figure 3.
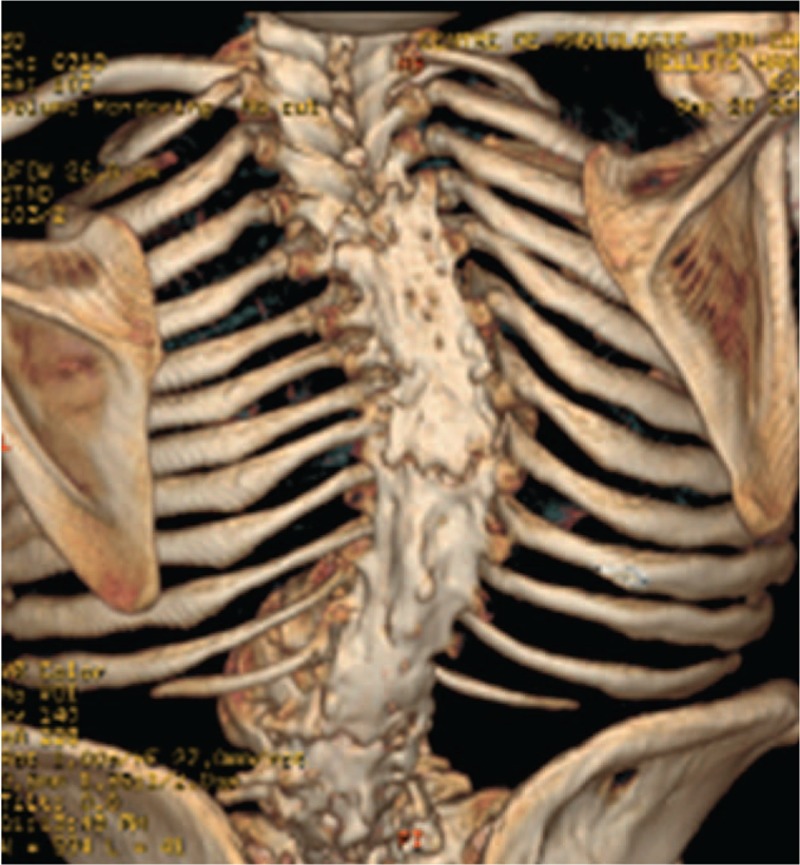
Three-dimensional reconstruction CT scan of the posterior aspect of the spine showed diffuse hyperostosis of almost the entire spine (begins at the level of T4, extending downwards to involve the whole thoracolumbar and upper part of the sacrum) with total diffuse fusion of the pedicles, the transverse and articular processes, the laminae, and the spinous processes.
Figure 4.

3D reconstruction CT scan of the cranium showed no trace for the sagittal suture (sclerosed/synostosed sagittal suture-arrow head) but persistence of the coronals (arrow).
3. Discussion
Osteoporosis is a major public health problem, affects millions of people in Europe and in the United States. Fractures, the most important consequence of osteoporosis, are associated with enormous costs and substantial morbidity and mortality. The prevention and treatment of this disease are therefore of prime importance.[8,9] Osteoporotic patients are characterized by bone resorption that exceeds bone formation. Thereby, the administer of antiresorptive agents to restore skeletal balance by reducing bone turnover process, primarily at the tissue level is the main goal to achieve. The mechanism of antiresorptive agents is to reduce the incidence of fracture by enhancing and stimulating new bone formation in osteoporosis and to rebuild skeletal losses. Bone modeling, in contrast to bone remodeling, is a process that leads to changes in the size and shape of the bones. Osteoblasts and osteoclasts are key cellular components of bone modeling, but they are not coupled to each other as they are in bone remodeling. Although bone modeling in the adult human skeleton is not a primary mechanism of skeletal homeostasis, it does contribute to bone mass and strength. As is the case for bone remodeling, the precise molecular mechanisms that initiate bone modeling are not known, but they may play a role in the actions of anabolic therapies for osteoporosis. Several genetic diseases have been linked to secondary osteoporosis [8–10]
Spinal hyperostosis has been known to be linked to DISH. DISH is a supposedly noninflammatory disease in which the spinal ligaments and entheses slowly become ossified. Three diagnostic criteria for the diagnosis of DISH in humans have been used most frequently: spondylosis, intervertebral osteochondrosis, and ankylosing spondylitis.[3,4] Al Kaissi et al[2,11–13] described several patients with unusual spine hyperosotosis in correlation with variable etiological understandings. The early-onset senile ankylosing vertebral hyperostosis was directly related to congenital spine malformation complex in connection with syndromic associations.
Lysosomal storage diseases (LSDs) are genetic disorders that cause metabolic pathway deficiencies and abnormal accumulation of material within the lysosome, leading to excess storage of substances, such as mucopolysaccharides, glycoproteins, amino acids, and lipids.
Although the LSDs encompass a large group of approximately 50 rare inherited metabolic disorders, certain similarities, and differences may be appreciated between the LSDs based on the type of enzyme deficiency, the type of abnormal substrate that accumulates, and the specific tissue or organs most affected.[14]
Galactosialidosis is an autosomal recessive LSD associated with a combined deficiency of enzymes neuraminidase 1 (NEU1) and β-galactosidase (β-GAL). The early infantile form is the most severe type.[15] Galactosialidosis shares many of the physical findings with other lysosomal storage disorders, such as coarse facies, edema, abnormal bone formation, and cherry red spots on eye examination.[16] Lysosomal storage disorders causes defective bone formation secondary to defects in blastogenesis in the first 6 weeks of fetal life.[17]
Other reports described the strong correlation between lysosomal storage disorders and various skeletal pathologies. The multisystems involvement in connection with Gaucher disease type 1 and 3 (bone complications, including osteopenia/osteoporosis, increased fracture risk, Erlenmeyer flask deformity, bone crises, osteonecrosis, lytic bone lesions, osteosclerosis, cortical thinning, acute osteomyelitis, and growth retardation, affect up to 90% of GD patients, mainly present in type 1 (the chronic non-neurological form) and type 3 (the subacute neurological form). The mucopolysaccharidoses (MPS), the gylcoproteinoses, and pycnodystosis are characterized by a wide spectrum of radiographic pathologies ranging from asymptomatic evidence of bone disorders to overt bone crises as seen in Gaucher, short stature with features of dysostosis multiplex in MPS, or osteopetrosis with pathological fractures (pycnodysostosis). Patel et al[6], Caciotti et al[18], Clarke et al[19] and Rosenbloom et al[20] concluded that the pathophysiology of skeletal disease is only partially understood, which might involves direct substrate storage, inflammation, and other complex alterations of cartilage and bone metabolism.
Panis et al[21] described the correlation of classic galactosemia, which is an autosomal recessive disorder with decreased BMD.
van Erven et al[22] postulated that the pathophysiological mechanism of low bone mass in patients with the classic type of galactosemia is not fully understood. Several factors were proposed such as dietary restriction, primary ovarian insufficiency in women, abnormal glycosylation of collagen or other glycoproteins involved in bone metabolism, and the decreased insulin-like growth factor (IGF-I) and IGF-binding protein-3 concentrations.[22]
Genetic disorders associated with gingival enlargement were classified into 4 main categories according to etiology, clinical presentation, and histopathological findings. The first category is hereditary gingival fibromatosis (HGF). It represents a heterogeneous group of disorders characterized by progressive enlargement of the entire gingiva. HGF may appear as an isolated entity, that is, as autosomal dominant gingival fibromatosis, or as part of a syndrome. The second category is lysosomal storage disorders. It is a group of disorders characterized by deposition of macromolecules anywhere in the body including the gingiva.[23–25]
LSDs causing skeletal abnormalities result from a lack of skeletal remodeling and ossification abnormalities owing to abnormal deposition of GAGs in bone and cartilage. GAGs also accumulate in tendons, ligaments and joint capsules, and patients may develop joint contractures.[26]
Yoshioka et al[27] showed that vitamin D3 induced hypercalcemia or pre-treatment with CaCl2 enhances CCl4-induced hepatotoxicity, presumably via disruption of calcium homeostasis. They demonstrated their evidence that calcium enhances CCl4-induced hepatotoxicity in the early stage in mice. These findings may have relevance to the mechanism of toxicity of other hepatotoxic compounds. The aim of this study was to determine whether calcium potentiates acute carbon tetrachloride (CCl4) induced toxicity. Elevated calcium levels were induced in mice by pre-treatment with cholecalciferol (vitamin D3; V.D3), a compound that has previously been shown to induce hypercalcemia in human and animal models.[27]
4. Conclusion
Osteoporosis is a metabolic bone disease characterized by loss of bone mass and strength, resulting in an increased risk of fractures. The diagnosis and management of osteoporosis has gained tremendous interests among several medical disciplines. Genetic disorders have a strong correlation with the development of osteoporosis. Lysosomal storage disorders are among a group of heritable bone disease in which osteoporosis is one of the main features. But, neveretheless, the study of osteoporosis through the undrestanding of the cellular processes that regulate bone modelling and remodelling via the administration of anti-resorpotive and anabolic medications, have to be questioned and based in accordance with the clinical phenotype and the genotype of every patient. Antiresorptive agents have been used and considered the most prominent therapeutic advances to combat fractures and to correct and rebuild skeletal losses. The latter statement is on the one hand partly true, but on the other hand, we cannot control optimal rebuilding processes of skeletal losses in osteoporotic patients. It is certain that stimulating the process for new bone formation inside the osteoporotic bones can to certain extent be guaranteed, but we cannot halt such processes to involve other organs. Particularly in patients with LSDs, the skeletal abnormalities and ossification abnormalities are immense due to abnormal deposition of GAGs in bone and cartilage.
The spine for instance, in patients with scoliosis, has immense deficiency in maintaining spine mobility and thereby the immobile, inert spine is susceptible to receive and store the maximal amount of new bone. We published several papers regarding the early-onset vertebral ankyloses in children because of inert, immobile spine in connection with different syndromic entities. The systemic administration of antiresorptive, supplemental calcium and vitamin D agents should be used with caution and strict monitoring is mandatory, particularly in patients with lysosomal storage disorders. Otherwise, drugs that are intended to give more strength to bones might have the opposite effect, and the earlier they are administered, the greater possibility that such deleterious unexpected effects will actualize. We wish to stress that osteoporotic spine in patients with metabolic errors of metabolism treated with antiresorptive, vitamin D, and calcium supplementation might be a possible confounder to develop spine ankyloses. We finally stress that pathophysiology of the spine in our current patient and its correlation with antiresorptive medications, vitamin D3, and supplemental calcium is not fully understood. Therefore, further studies are needed to elucidate this sort of correlation.
Acknowledgment
We wish to thank Dr. Lindsay C. Burrage/Brendan Lee Laboratory, Baylor College of Medicine- Molecular and Human Genetics-Houston, TX.
Author contributions
Patient's documentation and writing the MS, analysis of data, and approval of the final version: A. A. Kaissi, F. B. Chehida, R. Ganger, S. G. Kircher.
Conceptualization: Ali Al Kaissi, Farid Ben Chehida, Susanne Gerit Kircher.
Data curation: Ali Al Kaissi, Rudolf Ganger, Susanne Gerit Kircher.
Formal analysis: Susanne Gerit Kircher.
Methodology: Ali Al Kaissi.
Supervision: Franz Grill.
Writing – original draft: Ali Al Kaissi.
Writing – review & editing: Susanne Gerit Kircher.
Footnotes
Abbreviations: 25OH-D = 25-hydroxy-vitamin-D3, BMD = bone mineral density, BMI = body mass index, CCl4 = carbon tetrachloride, COL1A1/A2 = collagen 1(alpha1,2), CTSA = cathepsin A, GAGs = glycosaminoglycans, LS = lumbar segment, LSD = lysosomal storage disorder, NTX/Cr = to assess the accuracy of urinary collagen type 1 cross-linked N-telopeptide (NTX) excretion.
Given that all investigations and interventions in this report were performed as part of the regular health care whose informed consent was obtained on admission, an ethical approval was obtained.
Patient's consent was obtained.
No funds were received for this study.
The authors declare no conflict of interests.
References
- [1].McMaster JM. Congenital Scoliosis. The Pediatr Spine: Principles and Practice (chapter 9). 1994;New York: Raven Press, 228. [Google Scholar]
- [2].Al Kaissi A, Chehida FB, Ghachem MB, et al. Progressive non-infectious anterior vertebral fusion, split cord malformation and situs inversus visceralis. BMC Musculoskelet Disord 2006;5:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Forestier J, Rotes-Querol J. Senile ankylosing hyperostosis of the spine. Ann Rheum Dis 1950;9:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Resnick D, Shaul RS, Robins JM. Diffuse idiopathic skeletal hyperostosis (DISH): Forestier's disease with extraspinal manifestations. Radiology 1975;115:13–24. [DOI] [PubMed] [Google Scholar]
- [5].Zammarchi E, Donati MA, Morrone A, et al. Early-infantile galactosialidosis: clinical, biochemical, and molecular observations in a new patient. Am J Med Genet 1996;64:453–8. [DOI] [PubMed] [Google Scholar]
- [6].Patel MS, Callahan JW, Zhang S, et al. Early-infantile galactosialidosis: prenatal presentation and postnatal follow-up. Am J Med Genet 1999;85:38–47. [PubMed] [Google Scholar]
- [7].Rudenko G, Bonten E, Hol WGJ, et al. The atomic model of the human protective protein/cathepsin A suggest a structural basis for galactosialidosis. Proc Natl Acad Sci U S A 1998;95:621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Looker AC, Orwoll ES, Johnston CC, Jr, et al. Prevalence of low femoral bone density in older U.S. adults from NHANES III. J Bone Miner Res 1997;12:1761–8. [DOI] [PubMed] [Google Scholar]
- [9].Recker R, Lappe J, Davies KM, et al. Bone remodeling increases substantially in the years after menopause and remains increased in older osteoporosis patients. J Bone Miner Res 2004;19:1628–33. [DOI] [PubMed] [Google Scholar]
- [10].Seeman E, Delmas PD. Bone quality—the material and structural basis of bone strength and fragility. N Engl J Med 2006;354:2250–61. [DOI] [PubMed] [Google Scholar]
- [11].Al Kaissi A, Grill F, Alexander K, et al. Progressive noninfectious anterior vertebral fusion in a girl with axial mesodermal dysplasia spectrum. Clin Dysmorphol 2008;17:65–8. [DOI] [PubMed] [Google Scholar]
- [12].Al Kaissi A, Klaushofer K, Krebs A, et al. Unusual facies, thumb hypoplasia, distinctive spinal fusions and extraspinal mobility limitation, in a pair of monozygotic twins. Clin Dysmorphol 2007;16:151–5. [DOI] [PubMed] [Google Scholar]
- [13].Al Kaissi A, Ben Chehida F, Ben Ghachem M, et al. Occipitoatlantoaxial junction malformation and early onset senile ankylosing vertebral hyperostosis in a girl with MURCS association. Am J Med Genet A 2009;149A:470–4. [DOI] [PubMed] [Google Scholar]
- [14].Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr 2004;144:S3–14. [DOI] [PubMed] [Google Scholar]
- [15].d’Azzo A, Andria G, Strisciuglio P. Scriver CR, Beaudet AL, Sly WS, et al. Galactosialidosis. The Metabolic & Molecular Bases of Inherited Disease. Vol. III (8th edn). New York: McGraw-Hill; 2001. 3811. [Google Scholar]
- [16].Carvalho S, Martins M, Fortuna A, et al. Galactosialidosis presenting as nonimmue fetal hydrops: a case report. Prenat Diagn 2009;29:895–6. [DOI] [PubMed] [Google Scholar]
- [17].Sewell AC, Pontz BF, Weitzel D, et al. Clinical heterogeneity in infantile galactosialidosis. Eur J Pediatr 1987;146:528–31. [DOI] [PubMed] [Google Scholar]
- [18].Caciotti A, Catarzi S, Tonin R, et al. Galactosialidosis: review and analysis of CTSA gene mutations. Orphanet J Rare Dis 2013;8:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Clarke LA, Hollak CE. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract Res Clin Endocrinol Metab 2015;29:219–35. Review. [DOI] [PubMed] [Google Scholar]
- [20].Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Crit Rev Oncog 2013;18:163–75. [DOI] [PubMed] [Google Scholar]
- [21].Panis B, Forget PP, van Kroonenburgh MJ, et al. Bone metabolism in galactosemia. Bone 2004;35:982–7. [DOI] [PubMed] [Google Scholar]
- [22].van Erven B, Römers MM, Rubio-Gozalbo ME. Revised proposal for the prevention of low bone mass in patients with classic galactosemia. JIMD Rep 2014;17:41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bittencourt L, Campos V, Moliterno L, et al. Hereditary gingival fibromatosis: review of the literature and a case report. Quintessence Int 2000;31:415–8. [PubMed] [Google Scholar]
- [24].Gardner DG. The oral manifestations of Hurler's syndrome. Oral Surg Oral Med Oral Pathol 1971;32:46–57. [DOI] [PubMed] [Google Scholar]
- [25].Keith O, Scully C, Weidmann GM. Orofacial features of Scheie (Hurler Scheie) syndrome (alpha-L-iduronidase deficiency). Oral Surg Oral Med Oral Pathol 1990;70:70–4. [DOI] [PubMed] [Google Scholar]
- [26].Weisstein JS, Delgado E, Steinbach LS, et al. Musculoskeletal manifestations of Hurler syndrome: long-term follow-up after bone marrow transplantation. J Pediatr Orthop 2004;24:97–101. [DOI] [PubMed] [Google Scholar]
- [27].Yoshioka H, Usuda H, Miura N, et al. Vitamin D3-induced hypercalcemia increases carbon tetrachloride-induced hepatotoxicity through elevated oxidative stress in mice. PLoS One 2017;12:e0176524. [DOI] [PMC free article] [PubMed] [Google Scholar]