

Abstract
We aimed to identify crucial genes relevant to the development of consecutive trauma-induced sepsis.
A microarray dataset was used to identify genes differentially expressed between peripheral blood samples from consecutive traumatized patients complicated with sepsis and not complicated with sepsis. The dataset GSE12624 was obtained from Gene Expression Omnibus, containing 34 peripheral blood samples from consecutive traumatized patients complicated by sepsis and 36 consecutive traumatized controls. The differentially expressed genes (DEGs) were identified using Linear Models for Microarray Data package. Then, gene ontology (GO) enrichment analysis for DEGs was performed by Onto-Express. Subsequently, the protein–protein interaction (PPI) network was constructed and pathway enrichment analysis was performed by Search Tool for the Retrieval of Interacting Genes (STRING). Furthermore, protein complexes in the PPI network were predicted by ClusterONE and validated through GO and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses, and protein domain analysis.
Totally, 446 upregulated and 447 downregulated DEGs were identified. Some DEGs were related to acyl-CoA binding (eg, ACBD6), chromosome, and centromeric region (eg, CENPN). In the PPI network, some DEGs were enriched in renin-angiotensin system (RAS, eg, AGTR1 and AGTR2). Three predicted protein complexes were validated in the PPI network. Some genes composing protein complex A were associated with cell proliferation (eg, CDC20, CCNB1, MCM4, RPA2, and PRIM2), and several genes composing protein complex F were implicated in regulation of actin cytoskeleton (eg, PFN2, ARPC2, and WASL).
The results suggest that those DEGs may be crucial in the etiology of consecutive trauma-induced sepsis, and they are expected to be therapeutic targets.
Keywords: consecutive trauma, differentially expressed gene, protein–protein interaction, sepsis
1. Introduction
After consecutive trauma, sepsis is a frequent and severe complication leading to increased mortality.[1] In-hospital mortality for all trauma patients with sepsis ranged from 16.2% to 22.0% between 1993 and 2008.[1] During past years, the molecular changes of sepsis have always been concerned by researchers. The most significant variation of sepsis is the activation of the complement system and hyperactivation of cellular innate immune responses associated with an excessive inflammatory response.[2] Toll-like receptor 4-mediated recognition of lipopolysaccharide is thought to be an important trigger of the inflammatory response in sepsis.[3] In the innate immune response, large amounts of the anaphylatoxin C5a are generated,[4] and it acts as a central mediator in sepsis by modulating other systems, including the coagulation cascade, toll-like receptor 4-mediated responses, and the release of cytokines.[5,6] Besides, myeloid-derived suppressor cells,[7] phosphatidylinositol-3-kinase signaling,[8] PPARγ coactivator-1α,[9] and arachidonic acid metabolism[10] were also discovered to markedly vary in sepsis. Furthermore, variations of protein complexes in sepsis have also been investigated in the past years. For instance, a previous proteomic profiling showed that pentraxin 3 formed a complex with some components of neutrophil extracellular traps in sepsis.[11] Besides, inhibition of mammalian target of rapamycin (mTOR) due to variation of specific protein–protein interactions (PPIs) within the mTOR complex 1 (mTORC1) is responsible for the reduced protein synthesis, which can in part result in muscle atrophy in sepsis.[12]
Recently, Shen et al[13] screened some differentially expressed genes (DEGs) from peripheral blood samples of consecutive traumatized patients complicated with sepsis, using the microarray data of GSE12624 deposited by Thierer. They also found that PLAU (urokinase-type plasminogen activator) and MMP8 (matrix metalloproteinase-8) were the most differentially expressed. However, knowledge about sepsis is not enough for the effective clinical control. In this study, the microarray data of GSE12624 deposited by Thierer was used to identify DEGs in sepsis samples. After gene ontology (GO) enrichment analysis for DEGs and pathway enrichment analysis for DEGs in the PPI network, potential protein complexes in the PPI network were predicted and validated. These findings may contribute to a better understanding of the pathogenesis of consecutive trauma-induced sepsis, and the screened crucial genes were expected to be therapeutic targets of consecutive trauma-induced sepsis.
2. Methods
2.1. Microarray data
The gene expression profile data of GSE12624 were downloaded from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), which was based on the platform of GPL4204 GE Healthcare/Amersham Biosciences CodeLinkUniSet Human I Bioarray (GE Healthcare). This dataset contains 34 peripheral blood samples from 13 consecutive traumatized patients complicated by sepsis (group S) and 36 peripheral blood samples from 13 consecutive traumatized patients not complicated by sepsis (group N). In the present study, the patients were categorized as having or not having sepsis by the intensive care physician on duty according to the criteria provided by Bone et al.[2] Here, the peripheral blood samples were collected from sepsis or nonsepsis patients using the PAXgene Blood RNA System (PreAnalytiX, Heidelberg, Germany). All patients have severe injuries in at least 2 body regions or 3 major fractures with an Injury Severity Score of ≥15 points, between 18 and 65 years of age. The duration between the occurrence of the accident and the time of admission to the intensive care unit (ICU) was less than 12 hours, and the survival time of patients was at least greater than 3 days. None of the patients underwent neuro or cardiac surgery. The patients with severe intracranial head injuries, coagulation abnormalities known at the day of admission to the ICU, acute liver failure, renal failure, hemofiltration, or malignant disease in the patient's history were excluded. In particular, blood samples were obtained within 1 hour after patients arriving in the emergency unit or operating room (baseline values) according to the ICU standards. Here, the study was approved by the Ethics Study Board of the University Hospital of Giessen (file number 79/01) and the ethical board of the Radboud University Nijmegen Medical Center (file number AMO 04/064). Informed consent was obtained from patients or, if patients were incapable of giving consent, from their legal custodian.
CEL files and the probe annotation files were downloaded, and the original gene expression dataset were preprocessed via background correction, quantile normalization, and probe summarization using Affy software package of Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/).[14]
2.2. DEGs screening
The genes that were differentially expressed in group S were identified by the Linear Models for Microarray Data package[15] of Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/). The raw P-value was adjusted using this package. Only the genes meeting the cut-off criterion of P-value <.05 were chosen as DEGs.
2.3. GO functional classification of DEGs
To explore what molecular functions changed in group S, GO functional enrichment analysis of DEGs was performed by Onto-Express in Onto-Tools package (http://vortex.cs.wayne.edu/Projects.html).[16] The P-value <.05 was used as the cut-off criterion.
2.4. Construction and pathway enrichment analysis of PPI network
To reveal the interactions of the screened DEGs, the Search Tool for the Retrieval of Interacting Genes (STRING) database (http://www.string-db.org/)[17] was used to analyze the PPIs for DEGs, and the combined score >0.4 was used as the cut-off criterion. Subsequently, the PPI network of DEGs was visualized by Cytoscape (http://cytoscape.org/).[18]
Furthermore, to investigate what pathways altered in group S, pathway enrichment analysis of DEGs was conducted via STRING, and the P-value <.05 was used as the cut-off criterion.
2.5. Prediction of protein complexes
The plugin Clustering with Overlapping Neighborhood Expansion (ClusterONE) (http://www.paccanarolab.org/clusterone),[19] which is an effective method for detecting potentially overlapping protein complexes from PPI data, was used to predict protein complexes in the PPI network. The graph clustering algorithm was chosen to process weighted graphs and construct overlapping clusters.
2.6. Validation of the predicted protein complexes
To validate the accuracy of the above protein complexes prediction, 3 methods were chosen to validate it, including Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis, protein domain analysis, and GO functional enrichment analysis in cellular component. The verification was performed by detecting the enrichment status of genes belonging to the same protein complex via the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/).[20]
3. Results
3.1. Identification of DEGs
After the data preprocessing, 961 probes were obtained. Based on the cut-off criteria, a total of 893 DEGs were screened out from group S, including 446 upregulated ones and 447 downregulated ones.
3.2. GO functional enrichment analysis of DEGs
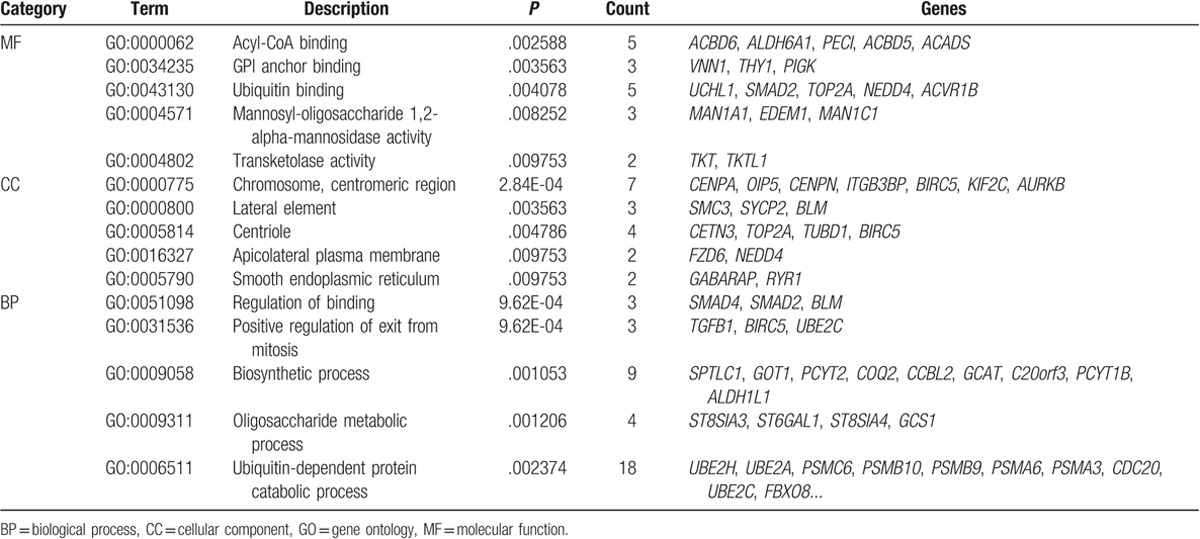
According to GO functional annotation, a set of DEGs in group S were significantly enriched in some molecular functions, such as acyl-CoA binding (eg, ACBD6 and ALDH6A1) and ubiquitin binding (eg, UCHL1 and SMAD2); a series of DEGs were related to some cellular components, such as chromosome, centromeric region (eg, CENPA, OIP5, and CENPN), and centriole (eg, CETN3 and TOP2A); some other DEGs were significantly associated with some biological processes, such as regulation of binding (eg, SMAD4, SMAD2, and BLM) and positive regulation of exit from mitosis (eg, TGFB1, BIRC5, and UBE2C) (Table 1).
Table 1.
The top 5 enriched GO term clusters for the up- and downregulated differentially expressed genes respectively in MF, CC, and BP.
3.3. Analysis of PPI network
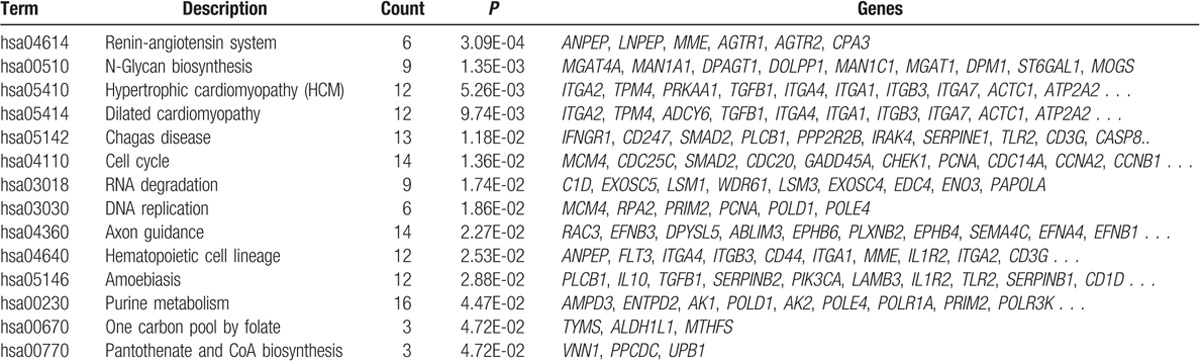
The PPI network contained 2040 PPI pairs, involving 637 DEGs (Fig. 1). According to KEGG pathway enrichment analysis, a total of 14 pathways were enriched for the DEGs in the PPI network, such as renin-angiotensin system (RAS, eg, AGTR1, AGTR2, and LNPEP), N-glycan biosynthesis (eg, MAN1A1, DPAGT1, and MOGS), cell cycle (eg, MCM4, CDC25C, and SMAD2), and hematopoietic cell lineage (eg, ANPEP, FLT3, and ITGA4) (Table 2).
Figure 1.

The PPI network composed of identified DEGs. Red nodes are the genes enriched in the pathway of renin-angiotensin system; purple nodes are the genes enriched in the pathway of N-glycan biosynthesis; green nodes are the genes enriched in the pathway of HCM and dilated cardiomyopathy; the blue node is the gene enriched in the pathway of HCM; and yellow nodes are the genes enriched in the pathway of chagas disease. DEG = differentially expressed gene, HCM = hypertrophic cardiomyopathy, PPI = protein–protein interaction.
Table 2.
The enriched pathways for differentially expressed genes in the protein–protein interaction network.
3.4. Prediction and validation of protein complexes
A total of 9 protein complexes were screened out from the PPI network, namely, protein complex A-I. Pathways enriched by DAVID for DEGs composing these protein complexes were mostly consistent with those predicted by STRING. For example, some DEGs composing protein complex A were enriched in some pathways predicted by STRING, such as cell cycle (hsa04110, eg, CDC25C, CDC20, and CCNB1) and DNA replication (hsa03030, eg, MCM4, RPA2, and PRIM2), and these 2 pathways were also predicted by DAVID. Meanwhile, protein complex A was located at chromosome, nonmembran-bounded organelle, and nuclear lumen, which are associated with DNA replication. Meanwhile, some genes in protein complex F were enriched in axon guidance (hsa04360, eg, EFNB3 and EPHB6) and regulation of actin cytoskeleton (hsa04810, eg, PFN2, ARPC2, and WASL); some genes were related to actin cytoskeleton and Arp2/3 protein complex (Table 3; Fig. 2). Furthermore, pathway and GO enrichment analyses and protein domain analysis of protein complex C were also mostly accordant (Table 3). However, analyses for the remaining 6 complexes were not ideal.
Table 3.
The 3 validated protein complexes in the protein–protein interaction network.
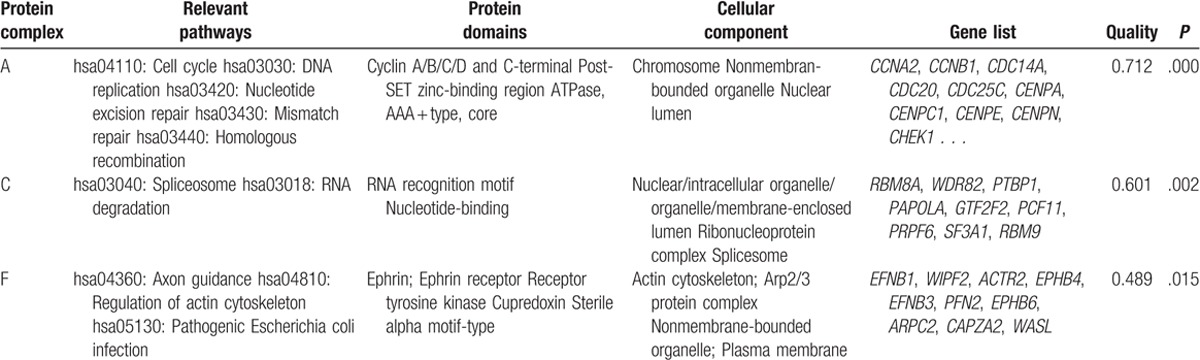
Figure 2.
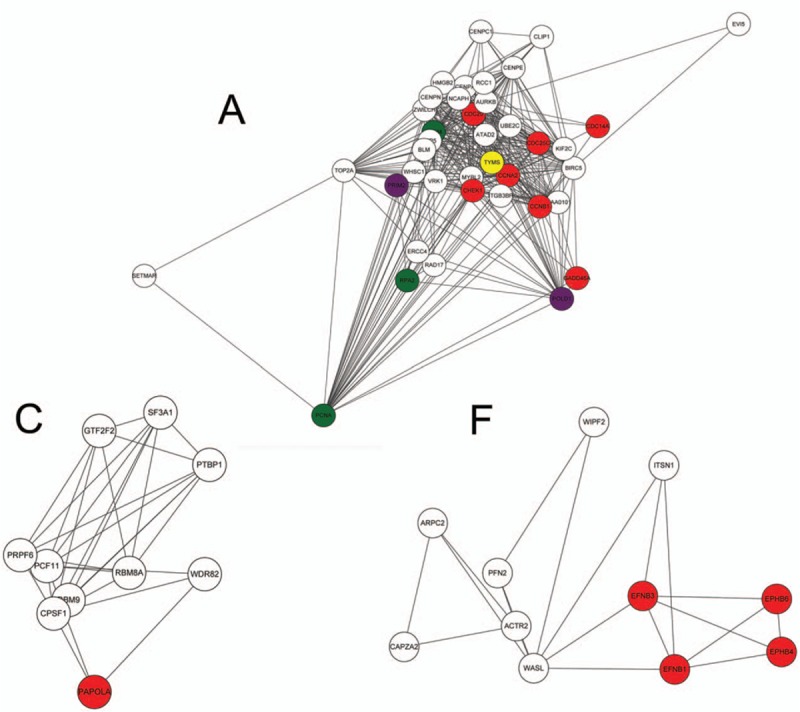
Three validated protein complexes in the protein–protein interaction (PPI) network, protein complex A, C, and F. In protein complex A, red nodes are the genes enriched in the pathway of cell cycle; green nodes are the genes enriched in the pathway of DNA replication; purple nodes are the genes enriched in the pathway of purine metabolism; and the yellow node is the gene enriched in the pathway of one carbon pool by folate. In protein complex C, the red node is the gene enriched in the pathway of RNA degradation. In protein complex F, red nodes are the genes enriched in the pathway of axon guidance.
4. Discussion
In the present study, a total of 893 DEGs were screened out from consecutive trauma patients with sepsis, including 446 upregulated ones and 447 downregulated ones. According to pathway enrichment analysis for DEGs in the PPI network, some DEGs were enriched in RAS (eg, AGTR1 and AGTR2).
There is a difference between results of earlier study conducted by Shen et al[13] and the present study. In the study conducted by Shen et al, the cut-off criterion was set as false discovery rate <0.05 and |log fold change (FC) | >1. Finally, a total of 21 upregulated DEGs and 37 downregulated DEGs were identified. However, in the present study, the cut-off criterion was set as P-value <.05, and the value of log FC was not limited. Therefore, we obtained more DEGs than the earlier study. Notably, the difference of DEGs between the current study and earlier study were also result in the different functions for DEGs. For example, they found that the significantly enriched functions for DEGs were response to wounding (eg, urokinase-type plasminogen activator [PLAU]), endopeptidase inhibitor activity, and complement and coagulation cascade (eg, PLAU). However, our study identified the dramatically enriched functions for DEGs were RAS, N-glycan biosynthesis, cell cycle, and so on.
The RAS is activated in sepsis, and it is associated with microvascular dysfunction in sepsis.[21] A previous study has been reported that angiotensin II (Ang II) and plasma renin activity are elevated in patients with severe sepsis and the degree of elevation is negatively implicated with the rate of microvascular reoxygenation during reactive hyperemia.[21] Besides, the increased plasma level of Ang II has been detected in trauma patients.[22] Recently, Zhang et al[23] have demonstrated that low expression of the RAS is correlated with poor prognosis of patients with severe sepsis. AGTR1 and AGTR2 encode 2 types of Ang II receptor, which have been reported to be downregulated during sepsis.[24,25] There is no evidence that AGTR1 and AGTR2 are involved in consecutive trauma-induced sepsis so far. Therefore, the aforementioned results suggest that AGTR1 and AGTR2 may be crucial in consecutive trauma-induced sepsis through RAS.
Additionally, the validation of predicted protein complexes showed that protein complex A, C, and F might be composed by some DEGs in the PPI network. For instance, based on GO enrichment analysis in cellular component for genes composing protein complex A, these genes were related to chromosome, nonmembran-bounded organelle, and nuclear lumen, which conformed the results of enriched pathways and protein domains. Some genes were correlated with cell cycle (eg, CDC20 and CCNB1) and DNA replication (eg, MCM4, RPA2, and PRIM2) (Table 2). CDC20 (cell division cycle 20), CCNB1 (cyclin B1), MCM4 (minichromosome maintenance complex component 4), RPA2 (replication protein A2), and PRIM2 (primase, DNA, polypeptide 2) all participate in cell proliferation. Sepsis is characterized by an overwhelming production of inflammatory cytokines, which can produce inflammatory responses.[26] T lymphocytes play a role in the control of immune responses,[27] and the expansion of T cells involves cell cycle and DNA replication. CCNB1[28] and MCM4[29] have been demonstrated to be related to immune responses. There are no studies to confirm the role of these cell cycle-related genes in consecutive trauma-induced sepsis so far. Hence, we speculate that some genes composing protein complex A (eg, CDC20, CCNB1, MCM4, RPA2, and PRIM2) may be associated with consecutive trauma-induced sepsis through controlling cell proliferation.
Besides, in protein complex F, genes PFN2, ARPC2, and WASL were enriched in regulation of actin cytoskeleton. Human sepsis is characterized by diffuse microvascular leak and tissue edema.[30] The breakdown in endothelial barrier function plays a pivotal role in the development of sepsis. Jacobson and Garcia[31] have found several agonists to induce dynamic rearrangement of the endothelial cell actin cytoskeleton that corresponds to barrier protection, and thus decrease microvascular permeability in sepsis. Moreover, Goldenberg et al[30] have suggested that reinforcement of the endothelial cytoskeleton is a new therapeutic strategy of sepsis. PFN2 (profilin 2), ARPC2 (actin related protein 2/3 complex, subunit 2), and WASL (Wiskott-Aldrich syndrome-like) are all correlated with actin.[32–34] There is no evidence that these actin-related genes are associated with consecutive trauma-induced sepsis so far. Thereby, these DEGs in protein complex F may be pivotal in the occurrence of consecutive trauma-induced sepsis through regulation of actin cytoskeleton.
However, this study has some limitations. The main limitation in our work was that we did not conduct experiments to validate our predictions. Besides, potential microRNAs and transcription factors targeting DEGs should have been predicted. In further study, we will carry out experimental studies to validate whether these DEGs are relevant to consecutive trauma-induced sepsis or not.
In conclusion, some genes related to RAS (eg, AGTR1 and AGTR2), cell proliferation in protein complex A (eg, CDC20, CCNB1, MCM4, RPA2, and PRIM2), and regulation of actin cytoskeleton in protein complex F (eg, PFN2, ARPC2, and WASL) might play momentous roles in the initiation and development of consecutive trauma-induced sepsis. These findings may be conductive to the better understanding of the etiology of consecutive trauma-induced sepsis and provide theoretical basis for further experimental studies. The screened crucial genes are expected to be therapeutic targets of consecutive trauma-induced sepsis.
Acknowledgments
The authors thank Shanghai Municipal Health and Family Planning Commission project (Grant number: 20124311), project of Science and Technology Commission of Pudong new district, Shanghai (Grant number: PKJ2012-Y25), the academic leader's training plan of health system in Pudong new district, Shanghai (Grant number: PWRd2012-12), Shanghai medical key subject construction project (ZK2012A28), and National Clinical key specialty construction project for the support.
Author contributions
Writing – original draft: L. Dong, H. Li.
Data curation: S. Zhang.
Formal analysis: S. Zhang.
Writing – review & editing: L. Su.
Footnotes
Abbreviations: Ang II = angiotensin II, DAVID = Database for Annotation, Visualization and Integrated Discovery, DEG = differentially expressed gene, GO = gene ontology, ICU = intensive care unit, PPI = protein–protein interaction, RAS = renin-angiotensin system, STRING = Search Tool for the Retrieval of Interacting Genes.
LD and HL contributed equally to this work.
Funding/support: This study is supported by Shanghai Municipal Health and Family Planning Commission project (Grant number: 20124311), project of Science and Technology Commission of Pudong new district, Shanghai (Grant number: PKJ2012-Y25), the academic leader's training plan of health system in Pudong new district, Shanghai (Grant number: PWRd2012-12), Shanghai medical key subject construction project (ZK2012A28), and National Clinical key specialty construction project.
The authors have no conflicts of interest to disclose.
References
- [1].Wafaisade A, Lefering R, Bouillon B, et al. Epidemiology and risk factors of sepsis after multiple trauma: an analysis of 29,829 patients from the Trauma Registry of the German Society for Trauma Surgery. Crit Care Med 2011;39:621–8. [DOI] [PubMed] [Google Scholar]
- [2].Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest J 1992;101:1644–55. [DOI] [PubMed] [Google Scholar]
- [3].Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085–8. [DOI] [PubMed] [Google Scholar]
- [4].Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol 2004;4:133–42. [DOI] [PubMed] [Google Scholar]
- [5].Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol 2006;177:4794–802. [DOI] [PubMed] [Google Scholar]
- [6].Rittirsch D, Flierl MA, Nadeau BA, et al. Functional roles for C5a receptors in sepsis. Nat Med 2008;14:551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cuenca AG, Delano MJ, Kelly-Scumpia KM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med 2011;17:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Recknagel P, Gonnert FA, Westermann M, et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med 2012;9:e1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tran M, Tam D, Bardia A, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 2011;121:4003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bruegel M, Ludwig U, Kleinhempel A, et al. Sepsis-associated changes of the arachidonic acid metabolism and their diagnostic potential in septic patients. Crit Care Med 2012;40:1478–86. [DOI] [PubMed] [Google Scholar]
- [11].Daigo K, Yamaguchi N, Kawamura T, et al. The proteomic profile of circulating pentraxin 3 (PTX3) complex in sepsis demonstrates the interaction with azurocidin 1 and other components of neutrophil extracellular traps. Mol Cell Proteomics 2012;11: M111. 015073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kazi AA, Pruznak AM, Frost RA, et al. Sepsis-induced alterations in protein-protein interactions within mTOR complex 1 and the modulating effect of leucine on muscle protein synthesis. Shock 2011;35:117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shen ZG, Guo JL, Li DS. Screening of differentially expressed genes related to severe sepsis induced by multiple trauma with DNA microarray. Eur Rev Med Pharmacol Sci 2014;18:734–9. [PubMed] [Google Scholar]
- [14].Seo J, Hoffman EP. Probe set algorithms: is there a rational best bet? BMC Bioinformatics 2006;7:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3: [DOI] [PubMed] [Google Scholar]
- [16].Khatri P, Draghici S, Ostermeier GC, et al. Profiling gene expression using onto-express. Genomics 2002;79:266–70. [DOI] [PubMed] [Google Scholar]
- [17].Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011;39(suppl 1):D561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Data Mining in Proteomics: Springer; 2011, 291–303. [DOI] [PubMed] [Google Scholar]
- [19].Nepusz T, Yu H, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods 2012;9:471–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alvord G, Roayaei J, Stephens R, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Doerschug KC, Delsing AS, Schmidt GA, et al. Renin-angiotensin system activation correlates with microvascular dysfunction in a prospective cohort study of clinical sepsis. Crit Care 2010;14:R24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ganter MT, Cohen MJ, Brohi K, et al. Angiopoietin-2, marker and mediator of endothelial activation with prognostic significance early after trauma? Ann Surg 2008;247:320–6. [DOI] [PubMed] [Google Scholar]
- [23].Zhang W, Chen X, Huang L, et al. Severe sepsis: low expression of the renin-angiotensin system is associated with poor prognosis. Exp Ther Med 2014;7:1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bucher M, Ittner K-P, Hobbhahn J, et al. Downregulation of angiotensin II type 1 receptors during sepsis. Hypertension 2001;38:177–82. [DOI] [PubMed] [Google Scholar]
- [25].Bucher M, Hobbhahn J, Kurtz A. Nitric oxide-dependent down-regulation of angiotensin II type 2 receptors during experimental sepsis. Crit Care Med 2001;29:1750–5. [DOI] [PubMed] [Google Scholar]
- [26].Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003;348:138–50. [DOI] [PubMed] [Google Scholar]
- [27].Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005;201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dudhgaonkar S, Thyagarajan A, Sliva D. Suppression of the inflammatory response by triterpenes isolated from the mushroom Ganoderma lucidum. Int Immunopharmacol 2009;9:1272–80. [DOI] [PubMed] [Google Scholar]
- [29].Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Mol Cell Proteomics 2010;9:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goldenberg NM, Steinberg BE, Slutsky AS, et al. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med 2011;3:88ps25. [DOI] [PubMed] [Google Scholar]
- [31].Jacobson JR, Garcia JG. Novel therapies for microvascular permeability in sepsis. Curr Drug Targets 2007;8:509–14. [DOI] [PubMed] [Google Scholar]
- [32].Show MD, Anway MD, Zirkin BR. An ex vivo analysis of Sertoli cell actin dynamics following gonadotropic hormone withdrawal. J Androl 2004;25:1013–21. [DOI] [PubMed] [Google Scholar]
- [33].Snapper SB, Rosen FS. The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu Rev Immunol 1999;17:905–29. [DOI] [PubMed] [Google Scholar]
- [34].Welch MD, DePace AH, Verma S, et al. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol 1997;138:375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]