

Abstract
It is well known that spatial and temporal regulation of genes is an integral part of governing proper gene expression. Consequently, it is invaluable to understand where and when transcription is taking place within nuclear space and to visualize the relationship between episomes infected within the same cell's nucleus. Here, both immunofluorescence (IFA) and RNA-FISH have been combinedto identify actively transcribing Kaposi's sarcoma-associated herpesvirus (KSHV) episomes. By staining KSHV latency-associated nuclear antigen (LANA), it is possible to locate where viral episomes exist within the nucleus. In addition, by designing RNA-FISH probes to target the intron region of a viral gene, which is expressed only during productive infection, nascent RNA transcripts can be located. Using this combination of molecular probes, it is possible to visualize the assembly of large viral transcription factories and analyze the spatial regulation of viral gene expression during KSHV reactivation. By including anti-RNA polymerase II antibody staining, one can also visualize the association between RNA polymerase II (RNAPII) aggregation and KSHV transcription during reactivation.
Keywords: Microbiology, Issue 131, Viral transcription factory, KSHV, Kaposi's Sarcoma-associated Herpesvirus, transcription, RNA Polymerase II, RNA-FISH, active transcription
Introduction
It has become increasingly clear that the spatiotemporal organization of the nucleus plays an important role in modulating finely-tuned gene expression in eukaryotes. Most tissue specific genes are distributed over many chromosomes and need to be regulated synchronously in order to respond to specific stimuli in a coordinated manner1. Cells construct active chromatin hubs (ACH) as a way of bringing together genes and their cis-regulatory components to a specific nuclear space1.
It has also been demonstrated by various studies that although the nucleus seems very dense and viscous, biologically active molecules can traverse the nucleus rather quickly via diffusion2. As a consequence of this ephemeral property, most DNA binding proteins 'jump' from binding site to binding site, feeling their way around the nuclear space, which allows for a highly adaptive and versatile nucleus2.
Despite this dynamic behavior of biomolecules within the nucleus, nuclear bodies without membranes such as (but not limited to) the nucleolus, Cajal bodies, and promyelocytic leukemia nuclear bodies (PML-NB) still exist. It is through a variety of mechanisms such as tandem DNA repeats (nucleolus), rRNA (nucleolus) and structural proteins such as coilin (Cajal bodies) or PML proteins (PML-NB) that hold these constructs together3,4,5. These structures and other congregations such as transcription factories serve as scaffolds that not only increase the local concentration of required components, but also regulate the composition of proteins and nucleic acids within them to ultimately create a central site for efficient cellular functions6.
Visualizing when and where nuclear structures form provides a wealth of information to researchers studying epigenetics. From a virology perspective, the reactivation of latently infected viruses, such as KSHV, significantly alters the landscape of the nucleus and distribution of nuclear enzymes to shift transcription primarily from cellular genes to viral genes, ultimately to produce fully functional viral progeny7,8. How does KSHV manipulate the cellular gene expression machinery to facilitate viral gene expression? Such information could also shed light on temporal cellular gene regulatory mechanisms.
Like all other herpesviruses, KSHV has two life cycles called lytic replication and latency. KSHV primarily resides in the latency stage, in which most of its viral gene expression is silenced, except latency associated genes9,10. During latency KSHV produces latency associated nuclear antigen (LANA), which constitutively binds the viral genomes and tethers viral chromatin to the human chromosome11. Because of LANA's intimate relationship with the viral genome, it is possible to use IFA and DAPI to stain and locate where the viral episomes were in relation to the host chromatin.
To study KSHV reactivation at the single episome level and the association with other viral episomes in an infected cell, a strategy to locate actively transcribing viral chromatin in situ has been established. Accordingly, LANA and RNAPII IFA with intron RNA-FISH were combined, by generating RNA-FISH probes that bind to the intron region (exact probe sequences can be found in the Izumiya lab's most recent publication8) of KSHV K-Rta-the key viral protein that is essential and sufficient for KSHV reactivation-it was possible to identify where transcription was actually taking place8,11,12,13,14,15. This intron RNA-FISH technique enables researchers to visualize where mRNA is being transcribed immediately before it is spliced and exported to cytoplasm16,17.
KSHV can be reactivated by various chemical stimuli including phorbol esters such as 12-O-Tetradeconoyl-phorbol-13acetate (TPA) and histone deacetylase inhibitors such as sodium butyrate, additionally KSHV can be induced to reactivate by overexpression of the viral transcription factor, K-Rta19. Researchers have successfully increased the efficiency of KSHV reactivation by synchronizing the cell's cycles prior to inducing reactivation18. Thus, for these particular studies, cells were synchronized using a double thymidine block (protocol described below) and incubated with TPA and doxycycline (Dox) for a short time. Doxycyline was used because the cell line utilized in these experiments has a doxycycline-inducible K-Rta cassette, which was cloned from cDNA and does not include the intron region of K-Rta. Although it is possible to reactivate KSHV using only induced K-Rta expression, it has been proven by other researchers that due to a variety of different biochemical factors K-Rta expression alone proves to be a feeble reactivation stimuli20. By combining all of these, and by limiting the drug incubations to a short period of time, a robust but not overly artificial KSHV reactivation was achieved for imaging.
After labeling LANA, RNAPII, K-Rta introns, and DNA as described in this paper, 3D fluorescence imaging was performed using a widefield deconvolution microscope. After processing with imaging software, the spatial distribution of active viral episomes can be properly evaluated. Using this technique, the central questions regarding the fundamental nature of the formation of active chromatin hubs and other nuclear structures can be studied. Having identical viral episomes in a single cell that processes the same regulatory elements may represent a unique research tool to deepen the understanding of spatiotemporal gene regulatory mechanisms.
A limitation of imaging multiple cell specimens fixed at different time points to characterize an inherently dynamic molecular process is that subtle or small-scale changes in fluorescence distribution are undetected or deemed insignificant. This is true unless in the rare instance, every cell observed displays the same subtle change. Thus, the full spatiotemporal relationship of active viral transcription and other nuclear structures cannot be critically evaluated using fixed imaging. To address these technical challenges, the best approach is to image live cells that have marked viral episomes and to follow the location of key cellular enzymes over time.
Protocol
CAUTION: Cell lines used in this procedure contain infectious viruses, be careful and only proceed in level 2 BSL facility or higher.
1. Cell Preparation and Maintenance
Culture TREx-K-RTA BCBL-1 cells (or another KSHV infected PEL cell lines) in RPMI1640 medium supplemented with 15% fetal bovine serum (FBS) and 1% penicillin streptomycin glutamine solution.
Grow cells at 37 °C with 5% carbon dioxide (CO2), making sure to continuously grow and to split cells by a ratio of 1:4 every 2–4 days.
Monitor cell cultures with a light microscope every day to confirm good cell growth and morphology, otherwise it is recommended to restart the cell culture or to adjust the cell culture conditions accordingly.
2. Thymidine Synchronization and Lytic Induction
In a 10 cm Petri dish, plate TREx-K-Rta BCBL-1 cells in freshly prepared media (1 x 106 cells/2 mL of media)
Add 200 mM thymidine (final concentration: 2 mM) to the dish, mix and incubate for 18 h.
Centrifuge the cells for 5 min at 500 x g. Wash the cells with sterile 1x phosphate-buffered saline (PBS), centrifuge again, and resuspend cells in freshly prepared media. Repeat for a total of 3 washes. Resuspend in freshly prepared media.
Allow the cells to exit S-phase for 8 h.
Add thymidine (final concentration: 2 mM), mix and incubate for 16 h.
Centrifuge the cells for 5 min at 500 x g. Wash the cells with sterile 1x phosphate-buffered saline (PBS), centrifuge again, and resuspend cells in freshly prepared media. Repeat for a total of 3 washes. Resuspend in freshly prepared media.
To induce viral reactivation, add 12-O-tetradecanoyl-phrobol-13-acetate (TPA, final concentration 20 ng/mL) and doxycycline (DOX, final concentration 100 ng/mL) to the cell culture, mix and incubate for 4 h. For cell lines without a K-Rta inducible cassette, stimulate reactivation using TPA (final concentration 20 ng/mL and sodium butyrate (1 mM)).
Centrifuge the cells for 5 min at 500 x g. Wash the cells with sterile 1x phosphate-buffered saline (PBS), centrifuge again, and resuspend cells in complete culture media. Repeat for a total of 3 washes. Resuspend in complete culture media.
Allow the cells to grow for 24 h.
3. Fixation and Permeabilization
NOTE: Before proceeding, ensure fixing solution (3.7% formaldehyde in DEPC-treated PBS), diethyl pyrocarbonate-treated phosphate-buffered saline (DEPC-PBS), glycine DEPC PBS (final concentration of glycine: 100 mM) and permeabilizating solution (50% acetone, 50% methanol) are prepared. Typically, each slide will require at least 0.5 million cells, multiply if necessary and include extra cells for back up especially if cells are not healthy.
Collect cells into a 1.5 mL microcentrifuge tube.
Centrifuge the cells for 2 min at 200 x g. Wash the cells 3 times with 1 mL of sterile DEPC PBS. Resuspend in 1.2 mL of DEPC PBS.
Place the appropriate number of coverslips (determined by the experimental setup) into the bottoms of a 6-well plate.
Pipette 200 µL of the cell DEPC PBS mixture onto each coverslip, ensure that at least 0.5 million cells are on each slide. Allow the cells to settle for 2 min, and aspirate excess DEPC PBS leaving only a thin layer of cells. It is normal for cells to be removed in this process. Caution: PFA is toxic, wear appropriate protection.
Carefully add 1 mL of fixing solution (3.7% formaldehyde in DEPC PBS) to each cover slip. Allow the cells to fix for 10 min.
Wash coverslips 3 times with DEPC PBS.
Carefully add 1 mL of glycine DEPC PBS (final concentration of glycine: 100 mM) to each cover slip. Allow the cells to quench for 5 min.
Add 1.5 mL of DEPC PBS and either place on a shaker for 5 min or shake firmly but gently in hand for 1 min. Aspirate the excess DEPC PBS. Be careful not to disrupt the cells. Repeat this step for a total of 3 washes.
Carefully add 1 mL of permeabilizing solution (50% acetone, 50% methanol) to each cover slip. Allow the cells to permeabilize for 15 min. NOTE: At this point, the coverslips can be stored in a -20 °C freezer for no longer than a week. Avoid freezing or minimize the time spent in the freezer as image quality can degrade quickly. Make sure that when in the freezer that the cells remain wet and covered in methanol/acetone, and to ensure that they properly rehydrate with DEPC PBS after removal from the freezer.
Add 1.5 mL of DEPC PBS and either place on a shaker for 5 min or shake firmly but gently in hand for 1 min. Aspirate the excess DEPC PBS, be careful not to disrupt the cells. Repeat this step for a total of 3 washes.
4. IFA and RNA-FISH
NOTE: Regarding RNA FISH Probe labeling and preparation: For the K-Rta intron which is 959 base pairs, 31 different probes were generated that were 20 base pairs each, containing a GC content of about 50%. Working and stock solutions of the probes were aliquoted into 100 µM and 2.5 µM concentrations, respectively.
Label microscope slides with necessary experimental information or some sort of distinguishable label.
Line the bottom of a sealable plastic container with a moist paper towel.
- Prepare the primary antibody solution (1.5 µL of LANA antibody, 0.75 µL of RNA Pol II antibody, 3 µL of baker's yeast tRNA, DEPC PBS up to 30 µL), and multiply recipe for each coverslip present. Place 30 µL of primary antibody solution onto each labeled microscope slide.
- Titrate the primary antibody of interest first and use the optimal amount. Be sure to use purified IgG, as serum (ascites fluid) may contain large amount of RNase.
Place the coverslip (cells facing the solution) onto the labeled microscope slides and move the slides into the container with the moist paper towel. Be careful not to introduce bubbles. Cover the exterior of the container with plastic wrap. Move the container into an incubator set to 37 °C for 1 h.
Prepare a FISH wash buffer (2x saline sodium citrate buffer (SSC) (final), 10% formamide, 90% DEPC dH2O).
Prepare 2x Hybridization Buffer (4x SSC, 20% dextran sulfate).
Prepare the secondary antibody and FISH intron probe buffer (20 µL of 2x hybridization buffer (final 1x Hyb Buffer), 4 µL of baker's yeast tRNA (final 10%), 4 µL of formamide (final 10%), intron K-Rta probe (final 125 nM), 0.8 µg of secondary antibody, DEPC dH2O up to 40 µL). Multiply recipe for every slide needed.
After incubation, wash cells 3 times with DEPC PBS in a 6 well plate for 5 min each.
Wash with FISH wash buffer 3 times for 5 min each. Keep coverslip in FISH-wash buffer until finish preparing mixture of secondly antibody and intron probe (step 4.7).
Clean glass slides used for primary antibody incubation for hybridization.
Place 40 µL of the secondary antibody and FISH intron probe containing solution onto each glass slide.
Place the coverslips with the cells onto the slides with the cell side facing down on top of the buffer, be careful not to introduce bubbles.
Place the slides into a plastic container with a moist paper towel on the bottom. Wrap the plastic container in first plastic wrap and then aluminum foil.
Incubate the container with the slides at 37 °C for 16-24 h.
After incubation, carefully slide the coverslip from the edge of the glass slide, and put back into 6 well plates facing up, and wash cells with FISH-wash buffer 3 times in a 6 well plate for 5 min each.
Wash the cells 2 times with 2x SSC.
Add DAPI (1:1,000) into the 2x SSC and allow to sit for 5 min at room temperature.
Wash the cells 2 times with 2x SSC.
Clean the glass slides used for hybridization, and add 10 µL of mounting solution onto the glass slides.
Place the coverslips face down onto the mounting solution. Be careful not to introduce bubbles.
With a laboratory tissue, gently remove the excess mounting solution, being careful not to squash the cells.
Using nail polish, apply an ample layer around the edge of the coverslips and allow to dry.
Proceed to perform fluorescence microscopy.
5. 3D Fluorescence Microscopy
NOTE: While protocols will vary with the type of fluorescence microscope system used, the following steps will help to ensure the acquisition of optimal image data for quantitative analysis.
Pretreat fixed samples and mount in anti-fade media to delay fluorescence photobleaching during imaging.
Use a high quality objective (such as a 60X 1.42 N.A oil-immersion lens), which can capture a whole cell (or cell nucleus) in the field-of-view.
Set exposure parameters (e.g., excitation power, exposure time) for each fluorescence color channel, using positive- and negative-control samples. Acquire fluorescence images. NOTE: Positive control samples that contain only one fluorescent label should also be used to determine the level of "crosstalk" between color channels. For imaging fixed cell specimens, fluorescence excitation power can be reduced (with slightly longer exposure times) to minimize photobleaching.
Once the imaging protocol has been established, maintain the same exposure parameters consistent for all samples in a study — so that image data can be accurately analyzed and compared.
Perform post-acquisition image processing and 3D reconstruction. Identify the K-Rta transcripts by RNA-FISH signal (red), LANA molecules by immunofluorescence (green), and nuclear chromatin (where applicable) stained with DAPI (blue). NOTE: Regions of the cell nucleus, in which K-Rta and LANA are co-clustered, are thus believed to be associated with viral transcription factories and replication complex.
Representative Results
A slightly abridged protocol was performed where BCBL-1 cells were used and only LANA and K-Rta RNA were stained (Figure 1). This experiment allows us to examine where actively transcribing viral episomes are and the heterogeneity of response to KSHV reactivation stimuli in a population of cells. In the cell indicated by the arrow in Figure 1, the distinct K-Rta fluorescence in regions that closely match the distribution of viral episomes (marked with LANA) is evidence for active transcription taking place close to KSHV genomes. In the same sample, cells such as the one signified by the arrowhead in Figure 1 can be observed, which exhibit much weaker and diffuse K-Rta fluorescence that does not significantly overlap with LANA. This illustrates the general finding that within a population of cells there is considerable variation in the degree of response to reactivation stimuli.
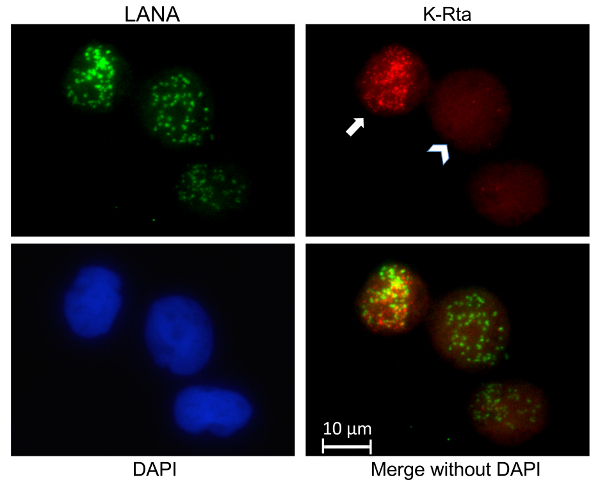
Figure 1: Visualizing active transcription of viral episomes. IFA and RNA-FISH was performed on BCBL-1 cells. The BCBL-1 cells were not synchronized but treated with TPA and sodium butyrate for 4 h. Using LANA immunostaining, viral genomes were visualized in green, while RNA-FISH was performed targeting K-Rta introns to located active transcription in red. Cellular chromatin was stained post fixation and permeabilization with DAPI. K-Rta intron and LANA stains were merged together. The full arrow points to a cell that has fully reactivated and is forming VTFs. While the arrowhead is pointing towards a cell that is only beginning to reactivate, as suggested by the weak K-Rta signal. Please click here to view a larger version of this figure.
Next the effectiveness of thymidine synchronization was examined by visualizing the expression of viral protein K-Rta (Figure 2). After a double thymidine block was performed to synchronize TREX BCBL-1 cells at the G1/S phase transition, the protocol previously described was performed (without LANA and RNAPII staining). By examining the K-Rta staining, it is clearly evident that synchronized cells responded more to the reactivation stimuli than the unsynchronized population. From previous studies conducted in the Izumiya lab (data not shown), it has been validated that RNAPII also colocalizes more frequently with K-Rta after cell cycle synchronization.
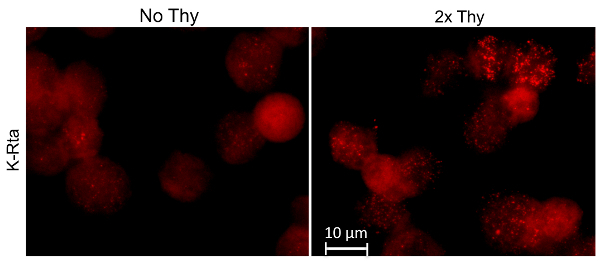
Figure 2: Thymidine effectiveness. RNA-FISH was performed on TREX K-Rta BCBL-1 cells. The cells (indicated by the "2x Thy" title) were synchronized with a double thymidine block. Both unsynchronized and synchronized cell populations were treated with TPA and DOX for 4 h. K-Rta introns were hybridized to RNA FISH probes. Cells that are uniformly bright red are not overexpressing K-Rta, instead they are dead, validated with DAPI staining (not shown). Please click here to view a larger version of this figure.
As a general note, it is very important to stain cells with DAPI when performing this protocol. Apototic cells can easily be recognized by nuclear fragmentation and blebbing. Given the heavy induction stimuli and general cell culture practices, it is common for some dead cells to be seen. It is important to always stain with DAPI as a primary method to discern between live and dead cells. Sometimes antibodies or RNA probes will get caught in the apoptotic cells and produce signals, consequently it is important to exclude those cells from analyses. DAPI staining can serve several additional functions, for example in Figure 3, it is clear that the object indicated by the arrow is actually three separate cells instead of one.
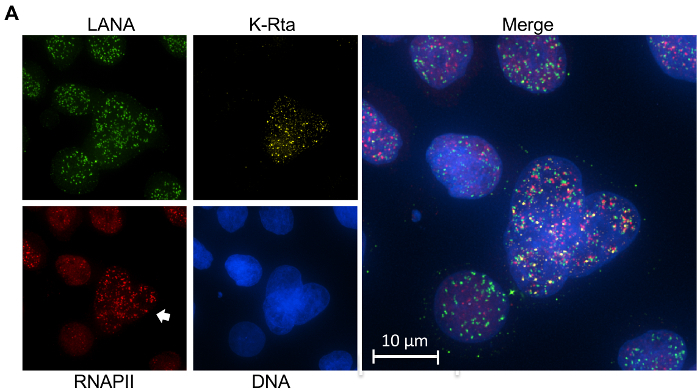
It is important to note that the images shown in Figure 3 and Figure 4 were deconvolved thus creating a more punctate pattern. By examining the RNAPII stain, the difference between the cells indicated by the arrow and the other surrounding cells can be seen. It is noteworthy to mention that the RNAPII signal is much more pronounced than normal due to deconvolution.
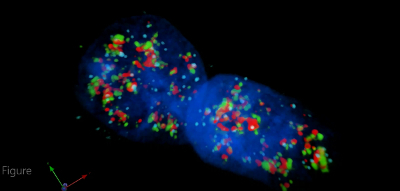
Using 3D microscopy alongside this technique allows us to interrogate the spatial relationship between RNAPII, LANA, and K-Rta. For example, in Figure 4, ring-like RNAPII structures can be seen with KSHV genomes dotted on the periphery. In general, LANA typically colocalizes with RNAPII in cells developing transcription factories. However, not all of the LANA dots colocalize with RNAPII, studies including the 4th dimension (time) would be necessary to clarify this phenotype. It is important to add that the few LANA dots that do not colocalize with K-Rta signals represent the episomal heterogeneity in the response to reactivation stimuli even within an individual cell (Figure 4).
Figure 3: Combined IFA and intron RNA-FISH. IFA and RNA-FISH was performed on TREX K-Rta BCBL-1 cells. Viral genomes were located with immunostaining of LANA (green), active transcription was visualized with RNA-FISH of the intron region of K-Rta mRNA (yellow), RNAPII (red) congregations were labeled with IFA, and the cells were finally stained using DAPI to image DNA (blue). TREx K-RTA BCBL-1 cells were synchronized using a double thymidine block, then cells were reactivated via incubation with TPA and DOX for 4 h, and 24 h later the cells were fixed, permeabilized, and further prepared for imaging. Please click here to view a larger version of this figure.
Figure 4: 3D Visualization of Viral Transcription Factories. Intron RNA-FISH using K-Rta intron probes (light blue), immunostaining of RNAPII (red) and LANA (green), and DAPI DNA staining (dark blue). Deconvolved 3D rendering of a Z stack taken on the microscope, processed and constructed on commercial imaging software (see the table of materials). TREx K-Rta BCBL-1 cells double thymidine synchronized and then treated with TPA and DOX for 4 h. Cells were processed 28 h after reactivation was stimulated. Please click here to view this video. (Right-click to download.)
Discussion
There are some aspects of the protocol that can be altered to accommodate unusual circumstances. The choice of fixation and permeabilization buffers can also be altered. For fixation buffers, paraformaldehyde is also effective and ethanol can be used for permeabilization.
Quenching the formaldehyde with glycine PBS is recommended as it prevents formaldehyde from disabling the antibodies in absence of bovine serum albumin (BSA), but if the coverslips are washed thoroughly enough, the glycine PBS step can be skipped. In the protocol, avoid using bovine serum albumin as a blocking solution. Pilot studies conducted in the Izumiya lab showed weaker RNA-FISH signals, presumably due to RNA degradation. If blocking for the primary antibody is needed, it is recommended to use RNase free BSA. From past experience, it was noticed that if the antibody is specific, inclusion of BSA is not necessary in the reaction. However, it is recommended to always include an excess amount of yeast tRNA in all incubation steps to prevent RNA degradation. It has been noticed that using tRNA as blocking agent increased specific RNA-FISH signals.
Cell cycle synchronization has been implemented to produce a more efficient and synchronous KSHV reactivation. For cell cycle synchronization, hydroxyurea or serum starvation might be alternative approaches. It has been confirmed that hydroxyurea is as effective as using a thymidine block, although incubation with hydroxyurea alone reactivates KSHV weakly.
How can this technique be applied to other studies? In the Izumiya lab's most recent publication, it was shown that colocalization of actively transcribing KSHV episomes and RNA pol II, an essential enzyme for RNA transcription. However, it is known that there are a number of co-activators, co-repressors, and cellular transcriptional factors involved in KSHV gene regulation. Quantitative RT-PCR has historically been used on a population (mixture of both reactivating and latent) of cultured cells, quantifying viral transcripts, and assessing the effects on the viral gene expression. Using the approach described above, analyses can be narrowed down to the single episomal level to examine viral transcription. Thus, the spatiotemporal regulation of other cellular and viral enzymes can be examined to gain insight into their association with KSHV reactivation. It is also noteworthy to mention that because the RNA intron region's transient nature is dependent on their degradation that RNA FISH probes will hybridize and typically localize where active transcription is taking place, however if the introns are not degraded immediately, FISH probes can present signals that are not always located near active transcription.
At the moment, it is difficult to study the relationship between the formation of cellular transcription factories and subsequent gene expression due to their genomic sizes and complexity of configuration of cellular promoters. In this regard, KSHV episomes can be an ideal tool as a result of their relatively small genome size, defined reactivation mechanisms with clear RNA Pol II aggregate formations. By using KSHV's manipulable viral mini-chromosome, herpesvirology can contribute to the cellular epigenetics research field from a unique angle.
Disclosures
The authors of this paper have no financial ties or competing conflicts of interests.
Acknowledgments
This research was supported by National Institutes of Health grant (R01-DE025985) and by an American Cancer Society Research Scholar Award (RSG-13-383-MPC). This work was also supported by grants from the U.S. Department of Agriculture (2015-67015-23268 and 2014-67015-21787) and New Research Initiative Grant from University of California, Davis.
References
- de Laat W, Grosveld F. Grosveld Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11(5):447–459. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science. 2001;291(5505):843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8(12):1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Cioce M, Lamond AI. Cajal bodies: a long history of discovery. Annu Rev Cell Dev Biol. 2005;21:105–131. doi: 10.1146/annurev.cellbio.20.010403.103738. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8(7):574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Sutherland H, Bickmore WA. Transcription factories: gene expression in unions. Nat Rev Genet. 2009;10(7):457–466. doi: 10.1038/nrg2592. [DOI] [PubMed] [Google Scholar]
- Schmid M, Speiseder T, Dobner T, Gonzalez RA. DNA virus replication compartments. J Virol. 2014;88(3):1404–1420. doi: 10.1128/JVI.02046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, et al. Kaposi's Sarcoma-Associated Herpesvirus Hijacks RNA Polymerase II To Create a Viral Transcriptional Factory. J Virol. 2017;91(11) doi: 10.1128/JVI.02491-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest. 2010;120(4):939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10(10):707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M, et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J Biol Chem. 2012;287(8):5806–5818. doi: 10.1074/jbc.M111.289496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guito J, Lukac DM. KSHV Rta Promoter Specification and Viral Reactivation. Front Microbiol. 2012;3:30. doi: 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darst RP, Haecker I, Pardo CE, Renne R, Kladde MP. Epigenetic diversity of Kaposi's sarcoma-associated herpesvirus. Nucleic Acids Res. 2013;41(5):2993–3009. doi: 10.1093/nar/gkt033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CS, et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5(8):192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155(3):606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- Borah S, Darricarrere N, Darnell A, Myoung J, Steitz JA. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog. 2011;7(10):1002300. doi: 10.1371/journal.ppat.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, et al. Histone demethylase JMJD2A regulates Kaposi's sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J Virol. 2011;85(7):3283–3293. doi: 10.1128/JVI.02485-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister SC, Hansen SG, Messaoudi I, Nikolich-Zugich J, Moses AV. Increased efficiency of phorbol ester-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus during S phase. J Virol. 2005;79(4):2626–2630. doi: 10.1128/JVI.79.4.2626-2630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv Cancer Res. 2007;97:81–109. doi: 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- Guito J, Lukac DM. KSHV Rta Promoter Specification and Viral Reactivation. Front Microbiol. 2012;3:30. doi: 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]