

Abstract
The circadian pacemaker circuit orchestrates rhythmic behavioral and physiological outputs coordinated with environmental cues, such as day/night cycles. The molecular clock within each pacemaker neuron generates circadian rhythms in gene expression, which underlie the rhythmic neuronal functions essential to the operation of the circuit. Investigation of the properties of the individual molecular oscillators in different subclasses of pacemaker neurons and their interaction with neuronal signaling yields a better understanding of the circadian pacemaker circuit. Here, we present a time-lapse fluorescent microscopy approach developed to monitor the molecular clockwork in clock neurons of cultured Drosophila larval brain. This method allows the multi-day recording of the rhythms of genetically encoded fluorescent circadian reporters at single-cell resolution. This setup can be combined with pharmacological manipulations to closely analyze real-time response of the molecular clock to various compounds. Beyond circadian rhythms, this multipurpose method in combination with powerful Drosophila genetic techniques offers the possibility to study diverse neuronal or molecular processes in live brain tissue.
Keywords: Neuroscience, Issue 131, Neuroscience, circadian rhythms, neuronal communication, time-lapse imaging, ex vivo brain culture, fluorescent reporter, Drosophila
Introduction
Circadian clocks help organisms to adapt to the periodic environmental changes generated by the 24 h rotation of the Earth. The interlocked transcriptional-translational feedback loops commonly underlie the molecular machinery of circadian clocks across species1. The circadian pacemaker circuit composed of clock-containing neurons integrates time-of-day information conveyed by the environmental cues, such as light/dark (LD) and temperature cycles, to orchestrate the rhythms of a plethora of daily physiological and behavioral processes2,3. The coordination of molecular rhythms with neuronal inputs and outputs is critically important for the operation of the circadian circuit but remains only partially understood.
In Drosophila, at the core of the molecular clock, the CLOCK/CYCLE (CLK/CYC) heterodimer activates the transcription of period (per) and timeless (tim). PER and TIM form a complex and enter the nucleus, where they inhibit transcriptional activity of CLK/CYC and consequently their own transcription. Post-transcriptional and post-translational regulations cause delays between CLK/CYC-mediated transcription and repression by PER/TIM, ensuring the generation of circa 24-h molecular oscillations1,3,4. About 150 neurons containing these molecular clocks form a circuit to control circadian behavior of adult flies5. A much simpler yet fully functional circadian circuit consisting of 3 groups of clock neurons - 5 ventral Lateral Neurons (LNvs; 4 PDF-positive LNvs and one PDF-negative LNv, see below), 2 Dorsal Neuron 1s (DN1s) and 2 Dorsal Neuron 2s (DN2s) - is present in the larval brain6,7.
The simple larval circadian circuit offers an excellent model to study the interactions between inter-neuronal communication and the molecular clockwork. Using our newly developed fluorescent reporter PER-TDT, which mimics the levels of PER protein and its subcellular location, we sought to characterize the dynamics of the molecular clockwork in different clock neuron subgroups in the larval circadian circuit8. Furthermore, knowing the key role of the neuropeptide pigment-dispersing factor (PDF) produced by 4 LNvs in regulating circadian rhythms at the neuronal level9,10,11, we wanted to examine the direct effect of PDF on the molecular clocks. To this end, we developed a method to monitor circadian gene expression rhythms in larval brain explant over multiple days by time-lapse confocal microscopy. The protocol was also adapted for pharmacological assays to test the effect of PDF or other compounds on the level of PER-TDT. Thus, the adaptation consists of making the brain explant culture accessible to drug application, increasing temporal resolution and imaging for a shorter duration.
Ex vivo culture of Drosophila brains of different developmental stages have been previously established12,13,14,15,16,17,18. Whereas these protocols have been used for imaging various biological phenomena, some of them are not compatible with imaging at single-cell resolution or do not support the culture for more than several hours. Alternative methods to perform long-term live imaging of circadian neurons in Drosophila include bioluminescence imaging of molecular rhythms19,20,21 and fluorescence imaging of calcium indicator with light sheet microscopy22,23. Although bioluminescence imaging can achieve higher temporal resolution and light sheet microscopy can be adaptable for in vivo imaging, they are limited on the spatial resolution and require specialized microscope systems.
The method described here is tailored to visualize fluorescent signals in the whole brain culture at single-cell resolution over multiple days. This facile and versatile method could be adapted to image cultured adult fly brains and for pharmacological experiments to study many different problems in Drosophila neurobiology.
Protocol
1. Preparation of Stock Solutions Under a Culture Hood
Prepare 400 mL of 1x Schneider Active Medium (SAM) optimized for ex vivo culture of larval brains (modified from reference24,25) (Table 1). Aliquot to 5 mL and flash freeze in liquid nitrogen (LN2), store at - 80 °C.
Prepare 1x Dissecting Saline Solution (DSS) by diluting 10x stock solution (modified from reference26) (Table 2).
Aliquot fibrinogen to 10 mg and store at 4 °C for a single use. Caution: Weigh fibrinogen under laminar flow, wearing gloves, as it is harmful.
Prepare thrombin stock solution in SAM (1500 NIH unit/mg) and aliquot to 10 µL, flash freeze in LN2 and keep at -80 °C. Caution: Wear gloves when handling thrombin as it is harmful.
Aliquot stock solution of 100x Penicillin-Streptomycin. Keep at -20 °C.
Cut circles of polytetrafluoroethylene (PTFE) membrane (see Materials Table) to the size that fits the inside of a glass bottom dish. Rinse them in 70% EtOH and then in autoclaved dH2O. Sterilize and dry under laminar flow with UV. Keep in a sterile Petri dish. Single use. NOTE: PTFE is a porous flexible membrane that allows gas exchange and prevents medium from evaporating during long-term recording.
2. Time-lapse Microscopy Setup
NOTE: The following setup is for a tandem scanner inverted confocal microscope (see Materials Table) equipped with a resonant scanner (8,000 Hz). The system includes a highly sensitive photo-multiplier detector, which together with the resonant scanner prevents phototoxicity and photobleaching. Temperature and humidity are controlled within a microscope environmental chamber (see Materials Table) that covers most of the microscope.
Place a stage-top humidity chamber.
Turn on the temperature and humidity controllers to equilibrate the microscope chamber to 25 °C and 80% humidity.
To perform experiments in constant darkness (DD), cover the microscope environmental chamber with an opaque cloth (unless the microscope chamber is made of an opaque material).
If using a water-immersion objective, position a Water Immersion Micro Dispenser on top of the objective and program a protocol with the Autoimmersion Objective Controller software to dispense water at a constant rate during time-lapse imaging. NOTE: Water-immersion objectives together with a water dispenser or dry objective lens are recommended for long-term live imaging, as other type of immersion medium would dry out.
Place a Petri dish holder on the stage, inside of the humidity chamber.
Launch the microscope software and select the resonant scanner mode from the first dialogue box. Select OK to initialize the stage when prompted. Go to Configuration tab and to Hardware configuration to switch on the relevant laser lines.
Go to Acquire tab and to XY panel to select bidirectional mode and set image acquisition parameters. NOTE: The described imaging setup presented here (Figure 2, Figure 3 and Movie 1) is optimized to observe circadian expression of specific fluorescent reporters. The following parameters were chosen to best fit our specifications: 40X water immersion objective, 8X line average with 512 × 512 image resolution. For multicolor imaging, sequential image acquisition is required when the emission wavelength of one fluorophore and the excitation wavelength of another overlap (here, wavelengths of Yellow Fluorescent Protein (YFP) emission and Tomato excitation overlap). Choose the sequential mode "in between stack" as it is the fastest and the movement of clock neurons is negligible during the acquisition of a Z-stack. Microscopy settings should be adapted to suit the aim of each experiment.
3. Setup of Larval Brain Explant Culture
NOTE: The whole procedure from dissection to embedding of brain explants takes ~30 min.
- Preparation of reagents under a culture hood.
- Thaw an aliquot of thrombin and keep at room temperature until the embedding step (step 3.3). Single use.
- Thaw quickly an aliquot of SAM at 37 °C and keep on ice for a single use.
- Add SAM to a 10 mg aliquot of fibrinogen to 10 mg/mL, flick gently and incubate at 37 °C for at least 30 min and during the embedding step (step 3.3). Single use. NOTE: Do not incubate the fibrinogen solution more than 2 h as it starts to polymerize.
- Thaw an aliquot of 100x stock Penicillin-Streptomycin. Prepare 2.5 mL of SAM containing 1x Penicillin-Streptomycin (SAM/antibiotics). Keep at room temperature. NOTE: Store the remaining 100x stock Penicillin-Streptomycin at 4 °C up to 1 month.
- Prepare an aliquot of 500 µL from the remaining SAM. Keep on ice.
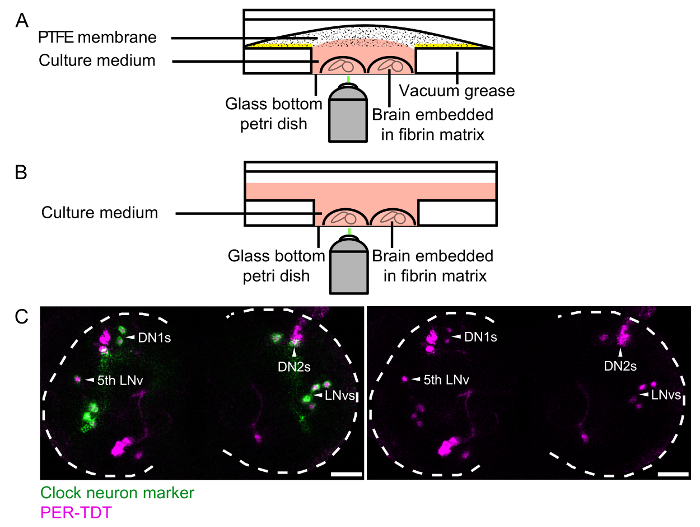
- For long-term imaging, apply autoclaved vacuum grease to the plastic part of the bottom of the glass bottom dish (Figure 1A) and sterilize under laminar flow with UV.
- Dissection of the central nervous system and ventral nerve cord.
- Clean the forceps and 2 x three-well glass dissection dishes with 70% EtOH. Pour 400 µL of DSS into 4 wells and 400 µL of SAM in one well. Keep on ice.
- Scoop out fly medium containing larvae and pick non-wandering 3rd instar larvae (L3) with forceps. Rinse them successively in 2 DSS-filled wells. Keep them in the 3rd DSS-filled well on ice that anesthetizes the larvae. NOTE: L3 lasts approximately 2 days at 25 °C. To observe the brain in physiologically relevant timeframe we chose to use non-wandering L3 larvae. Other subgroups of clock neurons, such as l-LNvs and DN3s, start to be formed at wandering L3 stage but they are not necessarily cycling yet (data not shown). Therefore, although it is possible to image cycling clock neurons at this stage (data not shown), it is important to carefully distinguish the already functional larval clock neurons from developing ones for data analysis. Non-wandering L3 appear approximately 4 to 4.5 days after egg laying at 25 °C. To use this protocol for studying circadian rhythms, synchronize (entrain) the developing larvae in 12 h/12 h LD cycles at 25 °C for 3 days before collecting L3 larvae.
- Dissect larval brains with fine forceps in the 4th DSS-filled well, which has not been used for rinsing larvae, using the inside-out method27.
- Briefly, cut the larva in two, gently pinch the mouth hooks with a pair of forceps and roll up inside-out the body wall using the other pair of forceps, thus exposing the brain. Free the brain by cutting out all the trachea and nerves that attach it to the rest of the body.
- Aspirate the brain in about 3 µL of DSS with a 20 µL pipette (pre-rinsed with DSS) and dispense in the SAM-filled well. Repeat the procedure for up to 7 brains. NOTE: Dissection should be performed on a cold surface to keep the larvae anesthetized and the brain chilled. For example, place the dissection dish on a cooling block connected to a pump to circulate ice-chilled water. The dissection procedure takes ~30 s per brain. It is important to keep the eye/antennal imaginal discs attached to the brains and the ventral nerve cord intact. Tearing off these parts will damage the brain and reduce the quality of the culture. Also, avoid exposing the brains to the air. Before aspirating the first brain, pipette up and down the DSS containing parts of the dissected larvae, as it prevents the brains from sticking to the wall of the tip.
- Embedding of brain explants in a 3D matrix and mounting (~20 min).
- Submerge a dissected brain in fibrinogen working solution (fibrinogen final concentration ~3.3 mg/mL, 10.5 µL per brain) as follows:
- Aspirate 3.5 µL of SAM/antibiotics (with the same tip used in section 3.2.3). Aspirate a dissected brain from the dissection well into the same pipette tip without dispensing the SAM/antibiotics medium in the tip.
- Dispense the brain and the medium on the lid of a sterile Petri dish. Add another 3.5 µL of SAM/antibiotics and 3.5 µL of warm fibrinogen/SAM 10 mg/mL solution on the drop containing the brain. Mix the solution around the brain gently by pipetting with a 20 µL pipette tip. NOTE: Using a pipette rather than forceps in this step avoids stressing the brain.
- Take 2 µL of the fibrinogen working solution from the drop and apply it on the coverslip of the glass bottom dish as a small circle. Add 0.8 µL of thrombin and mix very quickly with a pipette tip. The fibrinogen is converted to fibrin and starts to polymerize.
- Take the brain that sits in the fibrinogen working solution (step 3.3.1) between a pair of forceps and position it on the matrix once a white matrix of fibrin is visible. Orient the brain anterior side down (for imaging clock neurons). Push it down as close as possible to the cover glass. Fibrin clot is composed of layers of fibrin. Pick one layer and fold it on top of the brain with small and gentle movements without detaching the matrix.
- Repeat 2 to 3 times from different parts of the clot to stabilize the brain. NOTE: When handling the brain with forceps, be careful not to dry it or impose it a physical stress.
- Add 20 µL of SAM/antibiotics on top of the embedded brain to stop the action of thrombin.
- Repeat the procedure to mount the remaining brains. It is possible to embed up to 5 to 6 brains on a glass bottom dish.
- For long-term live imaging, add 600 µL of SAM/antibiotics in the inner well (the glass part). Position a pre-cut PTFE membrane on top of the medium and stick it to the vacuum grease applied on the plastic part of the bottom (3.1.5) (Figure 1A).
- For pharmacological assays, fill the dish with 2 mL of SAM/antibiotics (Figure 1B). NOTE: It is very important to avoid evaporation of the medium, as the osmolality of the medium is critical to the viability of neurons. Therefore, for long-term imaging experiments, it is necessary to seal the culture dish with PTFE membrane. Whereas for short-term experiments, sealing membrane is not required as long as the dish is filled with 2 mL of medium and monitored in a humidified chamber.
4. Time-lapse Image Acquisition
Place the glass bottom dish on the Petri dish holder inside the stage-top humidity chamber.
Go to z-stack panel in the Acquire tab and set the z-section steps from the microscope software (2 µm × ~40 in the experiments shown in Figure 2 and Figure 3, and Movie 1). Set the beginning of a z-stack at the plane farthest from the coverslip and the end at the surface of the brain explant, close to the coverslip. Though brain movements are negligible, add extra z-sections at the beginning and end of the z-stack.
Go to Mark & Find panel and set-up multi-position image acquisition. With a 40X objective, 1 brain hemisphere can be captured within a field of view
Go to Acquisition mode panel and select xyzt (z-stack time-lapse). Go to Time Acquisition panel and set the preferred time interval and frequency parameters. NOTE: Acquire time-lapse images with desired frequency. Note that long-term live imaging (24-72 h) with time lapse interval shorter than 2 h tends to results in fewer cycling neurons, probably because it reduces the viability of neurons. The data volume expands as the rate of image acquisition increases, which makes data analysis and storage more challenging.
5. Pharmacological Treatment of brain Explants with Short-term Live Imaging
NOTE: The following procedure is essentially the same as that for long-term imaging but does not require a PTFE membrane. To avoid exposing the brains to blue light when the chemical is added to the culture, the microscope should be placed in the dark room with red light.
Go to Time Acquisition panel and set the desired time interval and number of scan to obtain the baseline data before drug application. Start Scan.
After the baseline scan is completed, lift the scan head of the microscope system. Remove the lid of the stage-top humidity controller and very carefully remove the lid of the glass bottom dish without moving it.
Remove the appropriate amount of culture medium if necessary. Add the experimental solution in the medium and, carefully and very gently, mix with a sterile Pasteur pipette without touching the brain explants. NOTE: For bath application of PDF, use 2 µM PDF stock solution (in 10% DMSO).
Replace the lids of the glass bottom dish and the humid chamber, and the scan head. If using, reposition the black opaque cloth around the microscope chamber.
Check if the brains are at their original positions in live scan mode. Adjust if necessary.
Go to Time Acquisition panel and set the desired time interval and number of scan. Start Scan. NOTE: Here we tested time-lapse images acquired every hour for up to 10 h.
Representative Results
Here, we show the representative results of the long-term recording of a circadian fluorescent reporter in ex vivo larval brain culture, and the live imaging results of PDF bath application on reporter expression.
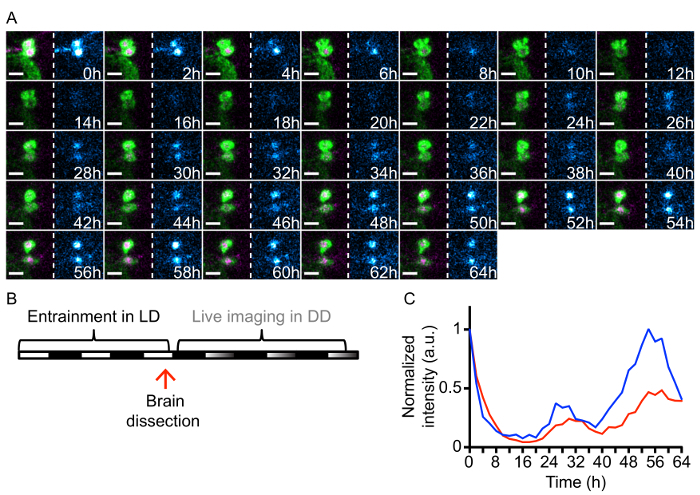
Non-wandering L3 larvae expressing the molecular clock reporter PER-TDT and UAS-mCD8::YFP driven by a clock neuron driver Clk(856)-gal4 (Figure 1C) were entrained in LD. Brains were dissected and set up for long-term culture during the last light phase of the LD cycle just prior the dark phase (Figure 1A and 2B). Time-lapse imaging was performed in DD and brain explants were imaged every 2 h for 64 h. A SUM-stack containing the neuron of interest was created and the mean fluorescence intensity of the area corresponding to the neuron was measured after background subtraction8. To determine rhythmicity of the reporter expression, both Maximum Entropy Spectral Analysis (MESA) and manual inspection were used8. Two DN2s that display circadian rhythms with a period of ~ 26 h in PER-TDT levels are presented here (Figure 2A, 2C, and Movie 1).
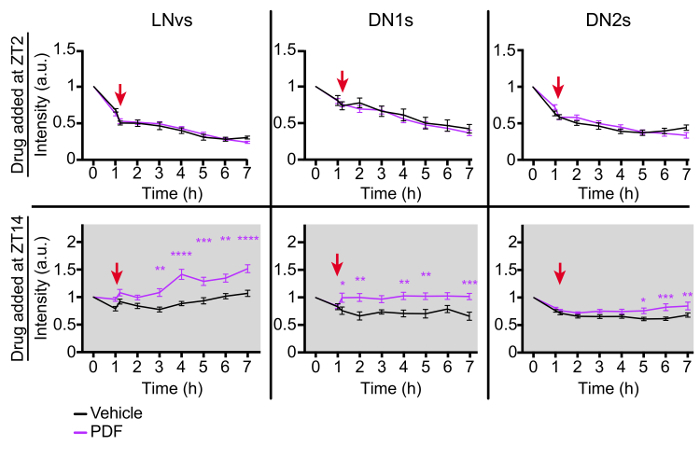
Using essentially the same culture setup (Figure 1B), the effect of the neuropeptide PDF on the expression of PER-TDT was studied. The brains of non-wandering larvae of the same genotype as described above were dissected either at ZT1 (Zeitgeiber Time 1, 1 h after lights on in LD) or ZT13 (1 h after lights off). The vials containing larvae were immediately chilled on ice after taken out of the incubator. At ZT13, the vials were also wrapped in aluminum foil to avoid blue-light exposure. The brain dissection was performed on ice and dissected brains were kept on ice. The brains taken at ZT13 were covered with aluminum foil whenever possible. Thus, although this procedure was performed in the lab lit with natural light, the effect of light exposure on macromolecular synthesis and metabolism was minimum. The brains were scanned twice with 1 h interval to get the baseline data. PDF or vehicle (DMSO) was added to the culture medium and time-lapse images were acquired every 1 h for subsequent 6 h. PER-TDT expression profile showed a time-of-day-dependent increase in fluorescence intensity upon PDF addition at ZT13 in the LNvs and DN1s, and to a lesser extent on the DN2s (Figure 3)8.
These results indicate that the imaging technique and the culture method described here enabled the recording of PER-TDT rhythms at single-cell resolution without noticeable phototoxicity, photobleaching or focus drift.
Note: Images were analyzed and processed using Fiji28. 10X iterative deconvolution was performed with AutoQuant and Imaris. Minor drifts in the time-lapse images were corrected with Correct 3D drift plugin of ImageJ.
Figure 1: Anatomy of clock neurons in the L3 larval brain and brain explant culture setup.(A and B) Schematics of the brain culture setup for long-term imaging (A) and for pharmacological assays with short-term imaging (B). In (A), vacuum grease (yellow) was applied on the plastic part of the glass bottom dish to attach the PTFE membrane. (C) A representative image of a LD-entrained L3 larval brain at ZT2. The expression of the molecular clock reporter PER-TDT (magenta) is visible in clock neurons, which are labeled by UAS-mCD8::YFP(green) driven by Clk(856)-gal4. There are 3 groups of differentiated clock neurons in L3: 5 LNvs (including the 5th LNv, which does not express PDF), 2 DN1s and 2 DN2s, indicated by white arrowheads. Note the expression of PER-TDT in the nuclei of clock neurons. YFP-negative PER-TDT-positive cells are glia and other subgroup of clock neurons that are still developing. Scale bar = 25 µm. Please click here to view a larger version of this figure.
Figure 2: Representative results of long-term live imaging of larval clock neurons.(A) Magnified view of DN2s from the long-term time-lapse imaging of a cultured brain. Brain explants were prepared from the LD-entrained L3 larvae dissected at ZT11. Images were taken every 2 h from CT17 (Circadian Time 17, 5 h after subjective lights off in DD) for 64 h in DD. Merged images of PER-TDT (magenta) and UAS-mCD8::YFP driven by Clk(856)-gal4 (green) are on the left of each panel and PER-TDT levels represented as heatmap with blue gradient are on the right. Scale bar = 10 µm. For representation only, images were processed with 3D correction drift and a 10X iterative deconvolution. (B) Timing of the experiment. (C) Graph representing the relative fluorescence intensity normalized at t = 0 of two DN2s (blue and red). Please click here to view a larger version of this figure.
Figure 3: Representative results of live-imaging for pharmacological experiments. LD-entrained larval brains were dissected and mounted at ZT1 or ZT13, and PDF (2 µM) or DMSO was applied at ZT2 or ZT14. PER-TDT expression in brain explants was monitored every hour by time-lapse microscopy. Mean fluorescence intensity ± SEM normalized to the value at the start of imaging (corresponding to ZT1 or ZT13) is shown. Red arrows indicate the time of PDF or vehicle application (ZT2 or ZT14). *p <0.05, **p <0.01, ***p <0.001 and ****p <0.0001 by two-way ANOVA with Sidak's correction for multiple comparisons. A minimum of 32 LNvs, 18 DN1s and 16 DN2s were analyzed for each data point. This figure has been modified from reference8. Please click here to view a larger version of this figure.
 Movie 1: Long-term live imaging of larval DN2 clock neurons. Time-lapse movie of cultured larval brain dissected at ZT11 and imaged in DD. Magnified views of the DN2s in a single hemisphere expressing PER-TDT and UAS-mCD8::YFP driven by Clk(856)-gal4 are shown. Images were acquired every 2 h for 64 h. PER-TDT (magenta) and YFP (green) are on the left and PER-TDT levels represented as a heatmap (blue gradient) are on the right. Scale bar = 10 µm. Please click here to view this video. (Right-click to download.)
Movie 1: Long-term live imaging of larval DN2 clock neurons. Time-lapse movie of cultured larval brain dissected at ZT11 and imaged in DD. Magnified views of the DN2s in a single hemisphere expressing PER-TDT and UAS-mCD8::YFP driven by Clk(856)-gal4 are shown. Images were acquired every 2 h for 64 h. PER-TDT (magenta) and YFP (green) are on the left and PER-TDT levels represented as a heatmap (blue gradient) are on the right. Scale bar = 10 µm. Please click here to view this video. (Right-click to download.)
| Steps | Reagents | Concentration |
| 1. Mix reagents. | KH2PO4 | 4.18 mM |
| CaCl2 | 1.05 mM | |
| MgSO4.7H2O | 0.7 mM | |
| NaCl | 116 mM | |
| NaHCO3 | 0.7 mg/ml | |
| Glucose | 2 mg/ml | |
| Trehalose | 2 mg/ml | |
| α-Ketoglutaric acid | 0.35 mg/ml | |
| Fumaric acid | 60 μg/ml | |
| Malic acid | 0.6 mg/ml | |
| Succinic acid | 60 μg/ml | |
| Yeast extract | 2 mg/ml | |
| NON heat-inactivated FCS | 20% | |
| dH2O, autoclaved | ||
| 2. Sterilize with 0.22 μm filter. | ||
| 3. Incubate in an incubator for cell culture at 25 °C in the dark for 72 h. Incubator has to be devoid of bacterial or fungal contamination. | ||
| 4. Add reagents. | Insulin | 2 μg/ml |
| Bis-Tris pH 6.8, sterilize with 0.22 μm filter | 5 mM | |
| 5. Adjust pH to 6.8-6.9. | NaOH (10 N) | ~8 drops |
| 6. Sterilize with 0.22 μm filter. | ||
| 7. Aliquot to 5 ml Flash freeze in LN2 Store at -80 °C. Use whithin 60 days. | ||
| Note: For large volumes, sterilize with filter top bottle by vacuum, otherwise use a syringe filter. Open aliquots in a culture hood. Single use. |
Table 1: Preparation of SAM medium (1x) for Drosophila brain culture. Medium to culture brain explant. All steps should be performed in a culture hood except for the incubation of the medium.
| Steps | Reagents | Concentration |
| 1. Mix reagents. | HEPES-KOH, pH 7.4 | 99 mM |
| NaCl | 1.37 M | |
| KCl | 54 mM | |
| NaH2PO4 | 1.7 mM | |
| KH2PO4 | 2.2 mM | |
| Glucose | 33 mM | |
| Sucrose | 438 mM | |
| autoclaved dH2O | ||
| 2. Sterilize with 0.45 μm filter. | ||
| 3. Store at 4 °C up to 6 months. | ||
| Note: For experimental use: dilute to 1X with autoclaved dH2O and sterilize with 0.45 μm filter top bottle by vacuum or with filter syringe. Keep at 4°C. Open in a culture hood, multiple use. |
Table 2: Preparation of DSS (10x). Saline solution to dissect Drosophila brains. Prepare in a culture hood.
Discussion
Here we described the method for long-term fluorescence time-lapse microscopy of cultured larval brains. The success of this type of experiments depends on multiple factors, such as the health of the culture, method for immobilization of the brain explant, fluorescence intensity and signal-to-noise ratio of the reporter, temporal and spatial resolution, and accessibility to the explant. These factors can be mutually exclusive. For example, increasing time-lapse frequency affects the health of the culture, and the immobilization of the brain explant might impede gas exchange. Thus, finding a happy medium (no pun intended) for the phenomena under study is key to a successful protocol. The method described here is optimized to monitor the expression of fluorescent circadian reporters in the scale of hours to days. It is also applicable to study other subjects in Drosophila neurobiology as long as critical processes are unaltered and executed with care.
In this protocol, brain explants are immobilized in a fibrin clot, which creates a 3D matrix. Although other methods for immobilization exist, such as poly-lysine or laminin coating of the cover glass, these slow down the development of larval brains29 and may thus compromise their physiological robustness. By its nature, fibrin provides an excellent matrix that is loose enough to allow nutrients to reach the brain explant and sticky enough for the brain to be close to the cover glass without squashing it16. Owing to these advantages, fibrin clot method has been employed in various protocols for cell culture and brain explant culture14,17,29,30,31,32,33,34. We recommend using a fibrin 3D matrix for larval brain culture even for relatively short-term experiments that last more than a few hours. Larval brain culture without 3D matrix gives erratic results (data not shown), probably due to the lack of physical support required for developing brains.
It is important to carefully observe the morphology of the explant and the cells in the tissue before and during time-lapse imaging to detect any sign of distress. In the experiments presented here, expression of the membrane-bound mCD8::YFP showed intact morphology of the neurons and PER-TDT was detected in the cytoplasm and the nucleus as expected, indicating that the brain explant remained viable and healthy throughout the duration of imaging. Circadian rhythmicity was studied for up to 72 h but we observed that brain explants can be cultured for up to 5 days without deteriorating the integrity of the neurons and the brain (data not shown). If the neurites become dotty or the cell body of the neurons burst, these are generally signs of phototoxicity and easily prevented by adapting microscope settings accordingly. For example, try reducing laser power while increasing the pinhole size, smart gain and the window of detection. However, generally, fluorescence intensity of the reporter is the limiting factor for obtaining images of good signal-to-noise ratio without high-intensity laser illumination. Therefore, the design of the reporter, choice of the fluorophore, and the copy number of the reporter are the first things to consider when troubleshooting viability issues. (In the experiment presented here, 4 copies of per-tdt reporter were used.)
Other signs of a compromised brain culture include the burst of a hemisphere during time-lapse imaging and movement of fluid out of the brain explant. This is likely due to micro-punctures caused during dissection of the brain. Removing the imaginal discs attached to the larval brain is the main source of punctures, thus we advise against it.
The advantage of fluorescence circadian live imaging over bioluminescence imaging is its high spatial resolution. In addition, the ease of using cellular markers together with the rhythm reporter facilitates the analysis of cell-type specificity. In contrast, bioluminescence has superior temporal resolution and excellent signal-to-noise ratio19,20,21. Nevertheless, overall number of cycling neurons per brain imaged by bioluminescence is not superior compared to the number obtained with our method8,19,21. In most cases, the rhythms of the fluorescent reporters detected with our method are synchronized within the same cluster of clock neurons (see Figure 2C), as shown with bioluminescence19. Phase differences between clock neuron clusters were observed in both fluorescence and bioluminescence circadian imaging8,19,21. Therefore, both methodologies are equally valid in observing circadian rhythms in cultured brains, and the choice between florescence and bioluminescence should be made according to the aim of the study.
The main technical difficulty of this method is in the embedding step. Because the protocol calls for fast-scanning confocal imaging with an inverted microscope, and light scattering in the brain tissue reduces the fluorescence detection especially when imaging deep in the tissue, brain explants should be mounted as close as possible to the cover glass. Moreover, the reproducibility of the imaging results depends on the consistency of the z-positions of neurons of interests. Therefore, it is important to orient and mount brains always in the same way, which requires practice. To image neuronal clusters located in anatomically distinct positions, it may be required to run separate experiments with different brain explant orientation. There are alternative methods for deep-tissue live imaging that do not require culturing the tissue, such as by two-photon microscopy and light sheet microscopy. The latter has been successfully used to image circadian calcium dynamics in live Drosophila brains22,23. However, light sheet microscopy has lower spatial resolution than confocal microscopy and thus its application to the imaging at single-cell resolution remains a challenge.
In summary, this protocol combines a simple culture system of larval brain explants under conditions that support time-lapse image acquisition of fluorescent reporters over days and quantitative analysis of biological rhythms. Although there are certain limitations as discussed above, the methodology can be adapted to image different reporters in larval and adult Drosophila brains and further modified to increase sensitivity and spatial resolution. For example, axonal projections located deep in the brain or vesicular transport may be detectable by combining our brain explant culture technique with two-photon microscopy35. That being said, it is still possible to perform live-imaging of organelles such as mitochondria in adult brain explants using this protocol (data not shown). We also presented the variation of this protocol for bath application of experimental solution in pharmacological assays. One could extend this protocol to perform "pulse-chase" experiments by manually replacing the culture medium to wash out the drug or using a perfusion chamber18,36.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Michael Rosbash for his mentorship and support during the initial phase of the development of this method. This work was funded by the JST PRESTO program, the Swiss National Science Foundation (31003A_149893 and 31003A_169548), the European Research Council (ERC-StG-311194), Novartis Foundation for Medical-Biomedical Research (13A39) and the University of Geneva.
References
- Sheeba V, Kaneko M, Sharma VK, Holmes TC. The Drosophila circadian pacemaker circuit: Pas De Deux or Tarantella? Crit Rev Biochem Mol Biol. 2008;43(1):37–61. doi: 10.1080/10409230701829128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados-Fuentes D, Herzog ED. The clock shop: coupled circadian oscillators. Exp Neurol. 2013;243:21–27. doi: 10.1016/j.expneurol.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Emery P. Insect molecular biology and biochemistry. San Diego: Elsevier; 2012. Chapter 15 - Molecular and neural control of insects circadian rhythms; pp. 513–551. [Google Scholar]
- Hardin PE. Molecular genetic analysis of circadian timekeeping in Drosophila. Adv Genet. 2011;74:141–173. doi: 10.1016/B978-0-12-387690-4.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii T, Rieger D, Helfrich-Forster C. Two clocks in the brain: an update of the morning and evening oscillator model in Drosophila. Prog Brain Res. 2012;199:59–82. doi: 10.1016/B978-0-444-59427-3.00027-7. [DOI] [PubMed] [Google Scholar]
- Malpel S, Klarsfeld A, Rouyer F. Circadian synchronization and rhythmicity in larval photoperception-defective mutants of Drosophila. J Biol Rhythms. 2004;19(1):10–21. doi: 10.1177/0748730403260621. [DOI] [PubMed] [Google Scholar]
- Mazzoni EO, Desplan C, Blau J. Circadian pacemaker neurons transmit and modulate visual information to control a rapid behavioral response. Neuron. 2005;45(2):293–300. doi: 10.1016/j.neuron.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Sabado V, Vienne L, Nunes JM, Rosbash M, Nagoshi E. Fluorescence circadian imaging reveals a PDF-dependent transcriptional regulation of the Drosophila molecular clock. Sci Rep. 2017;7:41560. doi: 10.1038/srep41560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun S, et al. Drosophila GPCR Han is a receptor for the circadian clock neuropeptide PDF. Neuron. 2005;48(2):267–278. doi: 10.1016/j.neuron.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Lear BC, et al. A G protein-coupled receptor, groom-of-PDF, is required for PDF neuron action in circadian behavior. Neuron. 2005;48(2):221–227. doi: 10.1016/j.neuron.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Mertens I, et al. PDF receptor signaling in Drosophila contributes to both circadian and geotactic behaviors. Neuron. 2005;48(2):213–219. doi: 10.1016/j.neuron.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Ayaz D, et al. Axonal injury and regeneration in the adult brain of Drosophila. J Neurosci. 2008;28(23):6010–6021. doi: 10.1523/JNEUROSCI.0101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prithviraj R, Trunova S, Giniger E. Ex vivo culturing of whole, developing Drosophila brains. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Siller KH, Serr M, Steward R, Hays TS, Doe CQ. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Mol Biol Cell. 2005;16(11):5127–5140. doi: 10.1091/mbc.E05-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschatzsch M, et al. Regulation of branching dynamics by axon-intrinsic asymmetries in Tyrosine Kinase Receptor signaling. Elife. 2014;3:e01699. doi: 10.7554/eLife.01699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabernard C, Doe CQ. Live imaging of neuroblast lineages within intact larval brains in Drosophila. Cold Spring Harb Protoc. 2013;2013(10):970–977. doi: 10.1101/pdb.prot078162. [DOI] [PubMed] [Google Scholar]
- Januschke J, Gonzalez C. The interphase microtubule aster is a determinant of asymmetric division orientation in Drosophila neuroblasts. The Journal of Cell Biology. 2010;188(5):693–706. doi: 10.1083/jcb.200905024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson WR, Hiesinger PR. Preparation of developing and adult Drosophila brains and retinae for live imaging. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Roberts L, et al. Light evokes rapid circadian network oscillator desynchrony followed by gradual phase retuning of synchrony. Curr Biol. 2015;25(7):858–867. doi: 10.1016/j.cub.2015.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehadova H, et al. Temperature entrainment of Drosophila's circadian clock involves the gene nocte and signaling from peripheral sensory tissues to the brain. Neuron. 2009;64(2):251–266. doi: 10.1016/j.neuron.2009.08.026. [DOI] [PubMed] [Google Scholar]
- Sellix MT, Currie J, Menaker M, Wijnen H. Fluorescence/luminescence circadian imaging of complex tissues at single-cell resolution. J Biol Rhythms. 2010;25(3):228–232. doi: 10.1177/0748730410368016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Holy TE, Taghert PH. Synchronous Drosophila circadian pacemakers display nonsynchronous Ca(2)(+) rhythms in vivo. Science. 2016;351(6276):976–981. doi: 10.1126/science.aad3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Holy TE, Taghert PH. A Series of Suppressive Signals within the Drosophila Circadian Neural Circuit Generates Sequential Daily Outputs. Neuron. 2017. [DOI] [PMC free article] [PubMed]
- Kuppers-Munther B, et al. A new culturing strategy optimises Drosophila primary cell cultures for structural and functional analyses. Dev Biol. 2004;269(2):459–478. doi: 10.1016/j.ydbio.2004.01.038. [DOI] [PubMed] [Google Scholar]
- Schneider I. Differentiation of Larval Drosophila Eye-Antennal Discs in Vitro. J Exp Zool. 1964;156:91–103. doi: 10.1002/jez.1401560107. [DOI] [PubMed] [Google Scholar]
- Jiang SA, Campusano JM, Su H, O'Dowd DK. Drosophila mushroom body Kenyon cells generate spontaneous calcium transients mediated by PLTX-sensitive calcium channels. J Neurophysiol. 2005;94(1):491–500. doi: 10.1152/jn.00096.2005. [DOI] [PubMed] [Google Scholar]
- Hafer N, Schedl P. Dissection of larval CNS in Drosophila melanogaster. J Vis Exp. 2006. p. e85. [DOI] [PMC free article] [PubMed]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich D, Mayseless O, Schuldiner O. Long term ex vivo culturing of Drosophila brain as a method to live image pupal brains: insights into the cellular mechanisms of neuronal remodeling. Front Cell Neurosci. 2015;9:327. doi: 10.3389/fncel.2015.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forer A, Pickett-Heaps J. Fibrin clots keep non-adhering living cells in place on glass for perfusion or fixation. Cell Biol Int. 2005;29(9):721–730. doi: 10.1016/j.cellbi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Forer A, Pickett-Heaps JD. Cytochalasin D and latrunculin affect chromosome behaviour during meiosis in crane-fly spermatocytes. Chromosome Res. 1998;6(7):533–549. doi: 10.1023/a:1009224322399. [DOI] [PubMed] [Google Scholar]
- Lukinavičius G, et al. SiR-Hoechst is a far-red DNA stain for live-cell nanoscopy. Nature Communications. 2015;6:8497. doi: 10.1038/ncomms9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medioni C, Ephrussi A, Besse F. Live imaging of axonal transport in Drosophila pupal brain explants. Nat Protoc. 2015;10(4):574–584. doi: 10.1038/nprot.2015.034. [DOI] [PubMed] [Google Scholar]
- Enoki R, Ono D, Hasan MT, Honma S, Honma K. Single-cell resolution fluorescence imaging of circadian rhythms detected with a Nipkow spinning disk confocal system. J Neurosci Methods. 2012;207(1):72–79. doi: 10.1016/j.jneumeth.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Ozel MN, Langen M, Hassan BA, Hiesinger PR. Filopodial dynamics and growth cone stabilization in Drosophila visual circuit development. Elife. 2015;4 doi: 10.7554/eLife.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Macara AM, Lelito KR, Minosyan TY, Shafer OT. Analysis of functional neuronal connectivity in the Drosophila brain. J Neurophysiol. 2012;108(2):684–696. doi: 10.1152/jn.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]