

Abstract
The human cortex is highly expanded and exhibits a complex structure with specific functional areas, providing higher brain function, such as cognition. Efforts to study human cerebral cortex development have been limited by the availability of model systems. Translating results from rodent studies to the human system is restricted by species differences and studies on human primary tissues are hampered by a lack of tissue availability as well as ethical concerns. Recent development in human pluripotent stem cell (PSC) technology include the generation of three-dimensional (3D) self-organizing organotypic culture systems, which mimic to a certain extent human-specific brain development in vitro. Currently, various protocols are available for the generation of either whole brain or brain-region specific organoids. The method for the generation of homogeneous and reproducible forebrain-type organoids from induced PSC (iPSC), which we previously established and describe here, combines the intrinsic ability of PSC to self-organize with guided differentiation towards the anterior neuroectodermal lineage and matrix embedding to support the formation of a continuous neuroepithelium. More specifically, this protocol involves: (1) the generation of iPSC aggregates, including the conversion of iPSC colonies to a confluent monolayer culture; (2) the induction of anterior neuroectoderm; (3) the embedding of neuroectodermal aggregates in a matrix scaffold; (4) the generation of forebrain-type organoids from neuroectodermal aggregates; and (5) the fixation and validation of forebrain-type organoids. As such, this protocol provides an easily applicable system for the generation of standardized and reproducible iPSC-derived cortical tissue structures in vitro.
Keywords: Developmental Biology, Issue 131, Induced pluripotent stem cells, neurobiology, cerebral organoids, human cortical development, cortical malformations, disease modeling
Introduction
The human brain is clearly one of the most complex organs and is responsible for all human intellectual abilities. Thus, a deeper understanding of human-specific brain development is a critical prerequisite for the understanding of human cognitive abilities. Traditionally, transgenic animals served as model organisms to study brain development. These models provided fundamental insight into the principles of brain development. We now know that a common feature of brain development in all mammals is a precise choreography of progenitor proliferation, neurogenesis, and neuronal migration. There are, however, significant structural differences between the brains of model organisms, such as rodents and humans, especially in the neocortex. The primary mechanisms that have been proposed to contribute to primate cortical evolution are an increased proliferation of stem and progenitor cells as well as the generation of outer radial glia cells (oRGCs), which are only very rarely found in rodents1,2,3.
Methods to model human cerebral cortex development include the generation of PSC-derived telencephalic progenitor cells and cerebral cortex projection neurons as monolayer cultures. These standardized differentiation protocols replay certain aspects of human cortical development such as the stereotypical temporal order of cortical neurogenesis4. They, however, fall short when it comes to the recapitulation of developmental processes of organogenesis such as spatial patterning and morphogenesis. More recent developments in stem cell biology led to the establishment of 3D organoid cultures from PSCs, which are revolutionizing the research of in vitro human organogenesis. Utilizing the capacity of PSCs to self-organize into organotypic structures, various organoids, which reflect key structural and functional properties of organs including those of kidney, gut, the eye, and the brain have been established5. Such organoids contain multiple organ-specific cellular subtypes, which group together and spatially organize very similar to the developing organs in vivo5,6. In addition, cell composition, lineage relationship, and gene network studies using single-cell RNA sequencing revealed that human cerebral organoids faithfully recapitulate major aspects of human fetal neocortex development such as gene expression programs7,8. One major drawback, which prevented their broad application so far, was, however, the large batch-to-batch variations and organoid-to-organoid heterogeneity9.
Here, we provide a detailed protocol for a simple and standardized forebrain-type organoid culture system. The key feature of this system is that it efficiently and reproducibly generates PSC-derived organoids of almost exclusive dorsal telencephalic identity. The protocol is based on the methods used in our recent Cell Reports paper10. It combines the self-organization capacity of iPSCs with selective induction of cortical neuroepithelium and can robustly generate homogeneous cultures of early dorsal telencephalic tissue within 3 weeks. The protocol builds on the previously reported SMAD signaling and Wnt inhibition strategy that guides the differentiation of PSC towards the anterior neuroectodermal lineage11,12 in combination with matrix embedding, which promotes the formation of large and continuous neuroepithelial structures13. We have successfully used the described method on various iPSC lines, with several clones per individual. We showed that this system is suitable for downstream applications in which reproducibility and homogeneity are of major importance such as disease modeling. When applying the protocol to iPSCs derived from patients suffering from a severe cortical malformation, we were able to recapitulate pathological hallmarks of the disease in vitro and to identify new molecular mechanisms leading to the phenotypic changes10. We suggest that the described organoid protocol can be utilized to close the gap between reductionist PSC-derived cortical monolayer cultures and in vivo studies, and that it represents a reliable and stable cell-based model system to simulate early human cortical development in health and disease outside the human body.
Protocol
1. Generation of iPSC Aggregates
- Generation of single-cell monolayer cultures from iPSC colonies
- Prepare a basement membrane extract (BME) coated 6-well plate. Thaw BME on ice at 4 °C for 2-3 h, dilute it with cold Dulbecco's Modified Eagle Medium F12 (DMEM-F12; 1:50 dilution), cover the plate with 1 mL/well of the diluted BME solution, and store the plate overnight at 4 °C.
- Aspirate the medium and wash intact iPSC colonies of at least 2 wells of a 6-well plate with 0.5 mM EDTA in phosphate buffered saline (PBS) twice before incubating colonies with 0.5 mM ethylenediaminetetraacetic acid (EDTA) in PBS for 4 min at room temperature (RT). Aspirate EDTA solution and gently detach the colonies by washing them off the bottom of the dish with 5 mL of DMEM-F12 medium. Collect them in a 15-mL tube and pellet them by centrifugation (4 min at 1,200 x g at RT). Note: iPSCs reprogrammed from commercially available fibroblasts has been successfully used10.
- Aspirate the supernatant and incubate the iPSC colonies with 500 µL of cell-dissociation reagent for 6 min at 37 °C.
- Pipet the cell suspension several times gently up and down with a 1 mL pipette to break remaining cell clusters into single cells.
- Add 4 mL of DMEM-F12 to the cell suspension to dilute the cell-dissociation reagent.
- Spin the cells down at 1,200 x g for 4 min at RT.
- Resuspend the cells in 2 mL of iPSC medium supplemented with 5 µM Y-27632 and seed cells into one well of a BME coated 6-well plate. Note: Use the iPSC medium specified in the Table of Materials for a single-cell monolayer iPSC cultures.
- On the next day, replace the medium with fresh iPSC medium lacking Y-27632. From this point, continue to culture the cells, changing the medium every day until iPSCs are 100% confluent. Depending on the cell line and the confluency of the starting wells, this will take between 2-4 days.
- Once confluent, passage the iPSCs. Aspirate medium and apply 500 µL of cell-dissociation reagent on the cells.
- Incubate the cells for 5-10 min at 37 °C. Gently rock the plate to detach the cells.
- Wash the cells from the well using 2 mL of DMEM-F12 and collect them in a 15-mL tube. Add DMEM-F12 for a total volume of 5 mL.
- Spin down the cells at 1,200 x g for 4 min at RT and aspirate the supernatant.
- Seed the cells in iPSC medium supplemented with 5 µM Y-27632 in a 1:2 to 1:4 ratio on a BME coated 6-well plate (2 mL/well of medium).
- Continue to culture the cells for 2-5 days and change iPSC medium lacking Y-27632 every day. Once confluent, passage iPSCs (steps 1.1.4-1.1.8). Note: Culture the iPSCs for at least 2 passages as a monolayer in the iPSC medium specified in the Table of Materials before using them for the generation of iPSC aggregates to allow the cells to adapt to the culture conditions.
- Use monolayer iPSC cultures when they are 70-90% confluent for the generation of iPSC aggregates. Note: iPSC adapted to the culture conditions as single cells are less prone to stress that leads to cell death during the dissociation and aggregation procedure. Monolayer iPSC cultures need to display typical pluripotent morphology with no evidence of differentiation. Test the cultures on a regular basis for mycoplasma contamination. Work only with mycoplasma-free iPSC cultures.
- Aspirate the culture medium from one well of a 6-well plate and apply 500 µL of cell-dissociation reagent on the cells.
- Incubate the cells for 5-10 min at 37 °C. Gently rock the plate to detach the cells.
- Wash cells from the well using 2 mL of DMEM-F12 and collect them in a 15-mL tube. Add DMEM-F12 for a total volume of 10 mL.
- For cell counting, take 25 µL from the cell suspension and mix it with 25 µL of trypan blue to mark dead cells. Count the living cells using a counting chamber.
- Collect enough cells (4,500 cells per iPSC aggregate) from the cell suspension in a 15 mL tube.
- Spin down the cells at 1,200 x g for 4 min at RT and aspirate the supernatant.
- Resuspend the cells in an appropriate volume of iPSC medium supplemented with 50 µM Y-27632 to obtain 4,500 live cells per 150 µL. Note: Using a high concentration of Y-27632 (50 µM) is critical for the survival of the iPSCs.
- Plate 150 µL in each well of a low-attachment 96-well U-bottom plate and place it in the incubator at 37 °C and 5% CO2. Note: When generating iPSC aggregates, consider that at least 6 organoids are needed for quality control at day 20 (see Section 5).
2. Induction of Anterior Neuroectoderm
Closely monitor morphology changes of the iPSC aggregates every day under the tissue culture microscope using a 4X or 10X power lens. Observe at day 1, cell aggregates with clear borders. Continue to culture iPSC aggregates in the incubator at 37 °C and 5% CO2. Note: A certain number of dead cells/well is normal and will not affect organoid generation.
Feed the iPSC aggregates starting from day 2 and continuing with every other day by gently aspirating approximately 2/3 of the medium without disturbing the cell aggregates at the bottom. Add an additional 100 µL of iPSC medium lacking Y-27632. Note: Within 4-6 days the cell aggregates will be 350-450 µm in diameter and exhibit smooth edges.
At this stage, pool the cell aggregates with a cut 100 µL pipette tip into a low-attachment 6 cm dish (maximum of 20 aggregates/dish) in cortical induction medium containing DMEM-F12 with N2 supplement (1:200), B27 supplement (1:100), glucose (0.2 mg/mL), cyclic adenosine monophosphate (cAMP; 0.15 µg/mL), 0.5% non-essential amino acids (NEAA), 1% L-alanyl-L-glutamine, heparin (10 µg/mL), and the compounds LDN-193189 (180 nM), A83-01 (500 nM), and inhibitor of Wnt response-1 (IWR-1) (10 µg/mL). Feed the cell aggregates by changing the cortical induction medium 3 days after transferring them to the 6-cm dish.
Closely monitor morphological changes during cortical induction under the tissue culture microscope using a 4X power lens. Note: After 4-5 days in cortical induction medium, edges of the cell aggregates should begin to brighten at the surface, indicating neuroectodermal differentiation. At this stage, radial organization of a pseudostratified epithelium emerges. Proceed to Section 3 to embed the cell aggregates in a BME matrix.
3. Embedding of Neuroectodermal Aggregates in a Matrix Scaffold
Thaw BME on ice at 4 °C for 2-3 h. Aliquot undiluted BME in sufficient amounts.
Prepare a plastic paraffin film sheet for the embedding procedure. Cut the plastic paraffin film using sterile scissors in 4 cm x 4 cm large pieces, put a piece of the plastic paraffin film over an empty 100 µL tip tray for 100 µL tips, and press with a gloved finger so that small dimples in the plastic paraffin film sheet are created (1 dimple/cell aggregate needed). Clean the plastic paraffin film with 70% ethanol and irradiate it with UV light (power: 15 watts, wavelength: 435 nm) under the closed sterile bench for 30 min.
Transfer each cell aggregate to one dimple of the plastic paraffin film sheet using a 100 µL cut tip with 1.5-2 mm opening in diameter. In the case that two cell aggregates are fused, do not separate them but transfer them together to one dimple.
Gently aspirate the medium surrounding the cell aggregates using an uncut 100 µL pipette tip. Note: Be careful not to suck the cell aggregates into the tip, as this will damage the aggregates.
Add 40 µL of undiluted BME to each cell aggregate.
Position each cell aggregate in the middle of the BME drop using an uncut 100 µL pipette tip. Note: Be very careful not to harm the developing neuroepithelium with the pipette tip.
Carefully transfer the plastic paraffin film sheet using sterile forceps into a 10 cm Petri dish (or another sufficient cell culture dish) and place the dish in the incubator for 15-20 min to allow the BME to solidify.
Meanwhile, prepare a low-attachment 6 cm dish containing 5 mL of cortical induction medium.
After the polymerization of BME, remove the droplets containing the cell aggregates from the plastic paraffin film sheet. Turn the plastic paraffin film over using sterile forceps and gently squeeze the cell aggregates into the slightly tilted (about 30 °) low-attachment 6 cm dish until the droplets fall off the plastic paraffin film sheet. Transfer a maximum of 16 cell aggregates into one 6 cm dish.
Continue to incubate the cell aggregates at 37 °C.
4. Generation of Forebrain-type Organoids from Neuroectodermal Aggregates
One day after embedding in a BME matrix, place organoid culture dishes on a rocking cell culture shaker with a tilting angle of 5 ° and 14 rpm, installed in a cell culture incubator.
Monitor organoids every day. Homogeneous neuroepithelial loop-like structures gradually develop after embedding in BME matrix.
When neuroepithelial loop structures are visible, change the medium to organoid differentiation medium containing DMEM-F12 with N2 supplement (1:200), B27 supplement (1:100), glucose (0.2 mg/mL), cAMP (0.15 µg/mL), 0.5% NEAA, 1% L-alanyl-L-glutamine, insulin (2.5 µg/mL)
Replace organoid differentiation medium every 3-4 days until the desired differentiation time point is reached. Then fix organoids (see Section 5). Note: Occasionally, the tissue may exhibit buds of optically translucent tissue without loop structures. Although this is not ideal, this is not affecting the development of cortical structures. The organoids can be cultured in organoid differentiation medium for up to 40 days. For extended culture periods (40-100 days) the organoid differentiation medium can be supplemented with 1:50 BME to increase tissue complexity and with BDNF and GDNF to allow neuronal survival and maturation14,15.
5. Fixation and Validation of Forebrain-type Organoids
For the validation, collect 6 organoids at day 20. Use 3 organoids for mRNA isolation and PCR analyses. Transfer the additional 3 organoids to a 24-well plate containing PBS using a cut 1 mL pipette tip with an opening of 3-3.5 mm for fixation and sequential immunocytochemical analyses.
Wash the organoids twice by carefully aspirating the PBS and replacing it with fresh PBS using a 5-mL pipette. Fix the organoids for 15 min in cold 4% paraformaldehyde (PFA) (pH 7.4). Caution: Beware that PFA is a known human carcinogen. All work must be done in a chemical fume hood wearing nitrile gloves. It is recommended to wear safety glasses. Formaldehyde can cause irreversible damage to the cornea.
Aspirate the PFA carefully and wash three times using room temperature PBS for 10 min.
Replace the PBS with a 30% sucrose solution (wt/vol, PBS-based) and store the samples at 4 °C to allow organoids to dehydrate. Organoids can be stored for up to 7 days until further processing.
One day after fixation, pre-stain the dehydrated organoids by adding trypan blue at approximately 1:50 for 10 min to allow the visualization of the organoids during the cryosectioning procedure.
Prepare the embedding medium containing 10% sucrose, 7.5% gelatin wt/vol in PBS, and warm it to 75 °C until it is liquid.
Replace 30% sucrose solution with embedding medium and transfer the 24-well plate onto a heating plate (60 °C) for 15 min to equilibrate the organoids.
Cover the bottom of the embedding molds with a layer of embedding media and place them on ice to polymerize.
Transfer the organoids from the 24-well plate to the embedding molds containing the polymerized embedding medium, add additional embedding medium on top so that the organoids are covered, and place the mold quickly in a 100% ethanol/dry ice freezing bath (temperature should be between -30 to -50 °C) for at least 1 min to shock-freeze.
Place the mold with forceps on dry ice temporarily and either store them at -80 °C or directly proceed with cryosectioning.
Cryosection organoids at 20 µm thickness and collect the sections on microscope slides, keeping track of the order of sections on the slides (sequential uptake). Allow sections to dry on slides for several hours, before storing them at -80 °C or directly performing immunocytochemical staining.
Representative Results
The standardized forebrain-type organoid protocol described here typically generates highly homogenous organoid cultures of almost exclusively dorsal cortical identity from human iPSCs within 20 days of cultivation (protocol outlined in Figure 1A). It is recommended to perform several quality control steps during the time course of the protocol, defined here as: 'go' (continue the differentiation process) and 'no-go' (suboptimal cultures, it is recommended to terminate the batch) (Figure 1). It is also advisable to document each quality control step well by taking images and notes.
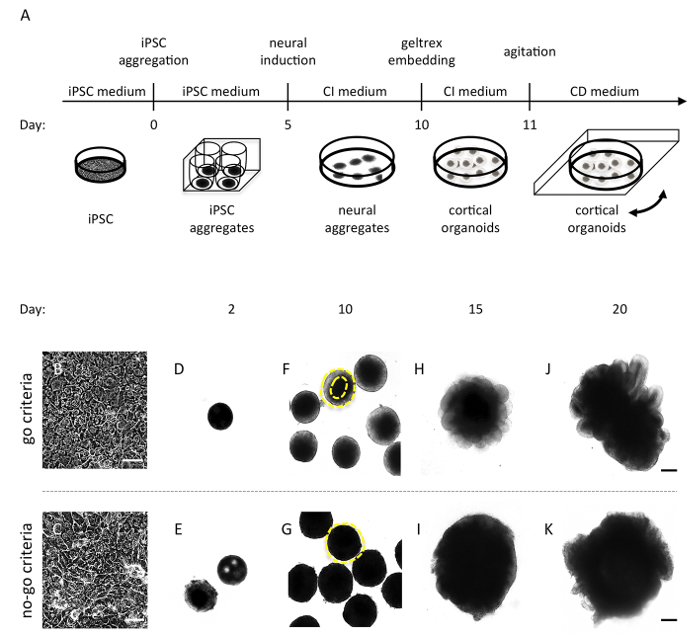
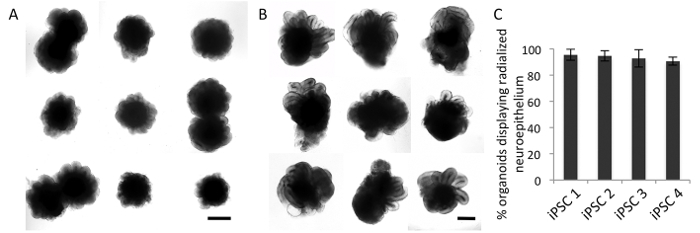
The first critical step in generating forebrain-type organoids is to start with high-quality iPSC cultures. It is important that iPSCs do not contain larger fractions of differentiated cells. Only use iPSC cultures that present as a homogeneous monolayer of undifferentiated cells at the starting population (Figures 1B, C). In addition, it is crucial to start with the given cell number for iPSC aggregation. The first detailed inspection of the iPSC aggregates should be performed on day 2. At this stage, the aggregates should have formed compact cell buds with smooth edges ('go') whereas irregular appearing aggregates or aggregates with cavities should be discarded ('no-go') (Figures 1D, E). The next quality control step should be performed at day 10 of the protocol. At this time point, the cell aggregates should show smooth and optically translucent tissue on the outer surface representing induction of neuroectoderm ('go') whereas the absence of such tissue indicates suboptimal neural induction ('no-go') (Figures 1F, G). Only those aggregates that exhibit a translucent surface (Figure 1F) should be embedded into BME matrix. Once embedded, the cortical organoids will develop continuous neuroepithelial loop-like structures, which will expand quickly. Analyze the efficiency of the cortical induction at day 15 and day 20 by investigating whether the organoids have developed polarized neural ectoderm as demonstrated in Figure 1H, J ('go'). In the case that organoids have not developed such neuroepithelial buds ('no-go' as illustrated in Figure 1I, K), critically revise the performed quality control steps for troubleshooting. When tightly following the protocol, highly standardized organoid batches will be generated (Figures 2A, B), which will show ≥ 90% homogeneity in polarized neural ectoderm formation within and across batches (Figure 2C). A common mistake leading to variable efficiency of polarized neural ectoderm formation is to increase the starting cell number for iPSC aggregation, or to start with low-quality iPSC cultures such as mycoplasma contaminated cultures or cultures that contain differentiated cells.
A detailed validation of the dorsal telencephalic identity of the generated organoids should be performed at day 20. To that end, 3 organoids should be fixed and used for immunofluorescence analyses. Stratified neuroepithelial loops (Figure 3A) express the neural stem cell marker Sox2 (Figure 3B, D), the forebrain markers Pax6 and Otx2 (Figures 3C, E), and the dorsal cortical marker Emx1 (Figure 3F). These cortical loops are further characterized by an apical localization of N-cadherin and ZO-1 (Figures 3G, H), ventricular zone radial glia cell (vRGC)-derived microtubule, which spans from the apical to the basal side of the structures (Figure 3I), and apical located dividing cells that stain positive for phosphorylated vimentin (p-Vimentin, Figure 3J). Cell death might be present inside the organoid structures. Central apoptosis is normal and does not affect the development of cortical tissue. Additionally, 3 organoids should be used to assess the homogeneity of the protocol by gene expression analyses. Forebrain-type organoids show expression of the dorsal forebrain markers (FoxG1, Otx2, Emx1), while expression of the midbrain (FoxA2, Pax5) and the hindbrain (HoxB2, HoxA4, HoxB4, HoxB6) markers is not detectable (Figure 3K).
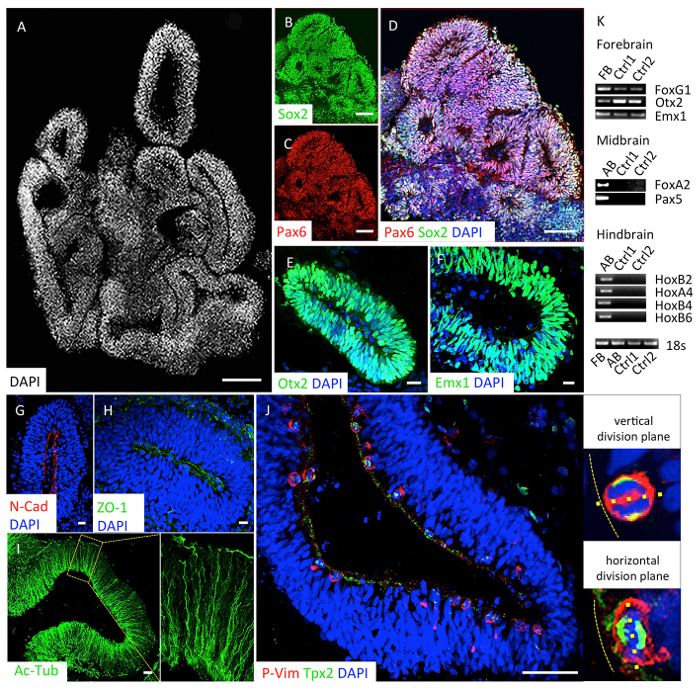
Batches of quality-controlled organoids can be used for various applications like the analyses of the division plane of apical radial glial cells. To that end, we suggest performing double staining using antibodies against p-Vimentin and Tpx2 (Figure 3J). P-Vimentin is phosphorylated by CDK1 during mitosis and is located in the nucleus, thus marking all nuclei in the mitotic phase16. Tpx2 is a microtubule associated protein that can visualize the mitotic spindle and the apical processes during interkinetic nuclear migration17,18. Using these markers, three aspects of vRGC division can in principle be analyzed: (I) whether cell division takes place at the apical side, (II) whether the division plane is aligned vertical (indicating symmetric cell division), horizontal, or oblique (indicating asymmetric cell division) to the apical surface, and (III) whether microtubule organizing centers are formed normally.
Organoids can be also further differentiated into more complex organized and stratified cortical tissue structures. Cortical structures within day 35 ± 2 organoids are composed of a ventricular zone (VZ)-, an inner and outer subventricular zone (SVZ), as well as a cortical plate (CP)-like area. Within the VZ and the inner and outer SVZ, vRGCs, intermediate progenitors (IPs), and cells reminiscent of oRGCs can be identified. In addition, the initial formation of a layered cortex can be observed in the CP-like area with deep cortical neurons expressing Tbr1 and Ctip2 inside and upper cortical neurons expressing Satb2, as well as Reelin-expressing cells in the outer regions10.
Figure 1: Schematic overview of the organoid protocol and illustration of 'go' and 'no-go' criteria. (A) Schematic overview of the protocol. CI medium: cortical induction medium; CD: cortical differentiation medium. (B-C) Image of an optimal 90% confluent iPSC monolayer culture (B) and a non-suitable iPSC culture exhibiting differentiation (C). (D-E) An iPSC aggregate optimal in size, cell density, and surface appearance (D) and two 'no-go' cell aggregates exhibiting either cell spares cavities (E, upper aggregate) or irregular edges (E, lower aggregate) two days following cell aggregation. (F-G) Cell aggregates exhibiting translucent and smooth edges (F) and cell aggregates lacking optical clearing (G). The yellow line is visualizing the area of interest. (H-K) An optimal organoid with continuous neuroepithelial loops (H, J) and an organoid that failed to develop radially organized neuroectoderm (I, K) imaged at day 15 and day 20, respectively. Scale bars, B-C 500 µm; D-K 200 µm. Please click here to view a larger version of this figure.
Figure 2: Homogeneity and reproducibility of the forebrain-type organoid protocol. (A-B) Representative bright-field images of organoids from one batch at day 15 (A) and day 26 (B). (C) Quantitative analyses of organoids at day 20. Organoids which display at the outer surface a neuroepithelium, recognizable in bright-field as optically clear superficial tissue with a clear border and evidence of radial cellular architecture were quantified (n = 3 per iPSC line with at least 16 organoids per experiment). Scale bars, A-B: 500 µm. Error bars ± SD. Please click here to view a larger version of this figure.
Figure 3: Validation of forebrain-type organoids at day 20. (A-J) Immunocytochemical characterization of organoids. Organoids organize in multiple neuroepithelial loops (A, counterstained with DAPI). Stratified organized cells within the neuroepithelial loops express the neural stem cell marker Sox2 (B, D), the forebrain markers Pax6 (C, D) and Otx2 (E), as well as the dorsal forebrain marker Emx1 (F). Cortical loop structures exhibited a fine adherent junction belt at the most apical side with the accumulation of N-cadherin (G) and zona occludens protein 1 (ZO-1; H). Ventricular RGCs' microtubule networks (stained by acetylated α-tubulin, Ac-Tub) extend from the apical to the basal side of the loop structures (I). Proliferating cells expressing p-vimentin (p-Vim) are located at the apical surface. Mitotic spindles are stained by Tpx2. Representative higher magnification image of a vertical and a horizontal division plane are shown on the right (J). (K) RT-PCR analysis for the region-specific transcription factors at day 20 of two independent sets of organoids derived from 2 different iPSC lines. FB: fetal brain control; AB: adult brain control. Scale bars, A-D 200 µm; E-I 10 µm. Please click here to view a larger version of this figure.
| Epitope | Dilution |
| Sox2 | 1 - 300 |
| Pax6 | 1 - 500 |
| Otx2 | 1 - 500 |
| Emx1 | 1 - 50 |
| N-cadherin | 1 - 500 |
| ZO-1 | 1 - 100 |
| P-Vimentin | 1 - 1000 |
| Tpx2 | 1 - 500 |
| Acetylated α-tubulin | 1 - 500 |
| Alexa488 anti ms | 1 - 1000 |
| Alexa488 anti rb | 1 - 1000 |
| Alexa555 anti ms | 1 - 1000 |
| Alexa555 anti rb | 1 - 1000 |
Table 1: Antibodies for quality control of organoids at day 20.
| Primer | Sequence |
| Otx2 forward | tgcaggggttcttctgtgat |
| Otx2 reverse | agggtcagagcaattgacca |
| FoxG1 forward | ccctcccatttctgtacgttt |
| FoxG1 reverse | ctggcggctcttagagat |
| Emx1 forward | agacgcaggtgaaggtgtgg |
| Emx1 reverse | caggcaggcaggctctcc |
| FoxA2 forward | ccaccaccaaccccacaaaatg |
| FoxA2 reverse | tgcaacaccgtctccccaaagt |
| Pax5 forward | aggatgccgctgatggagtac |
| Pax5 reverse | tggaggagtgaatcagcttgg |
| HoxB2 forward | tttagccgttcgcttagagg |
| HoxB2 reverse | cggatagctggagacaggag |
| HoxA4 forward | ttcagcaaaatgccctctct |
| HoxA4 reverse | taggccagctccacagttct |
| HoxB4 forward | acacccgctaacaaatgagg |
| HoxB4 reverse | gcacgaaagatgagggagag |
| HoxB6 forward | gaactgaggagcggactcac |
| HoxB6 reverse | ctgggatcagggagtcttca |
| 18s forward | ttccttggaccggcgcaag |
| 18s reverse | gccgcatcgccggtcgg |
Table 2: Primer and primer sequences for gene expression profile.
Discussion
Brain organoids represent a powerful tool for studying human brain development in vitro as they provide the relevant species background and the complex 3D arrangement of cells in a tissue context. With that, they bridge the gap between non-human animal models and reductionist human two-dimensional monolayer cell culture techniques. Their applications are, however, hampered by a lack of reproducibility9. We have developed a forebrain-type organoid protocol, which overcomes the large sample-to-sample variability by combining the self-organizing capacity of iPSC with their amenability to patterning factors. Specifically, iPSCs were aggregated to promote self-organization and subsequently inhibit TGF-ß/SMAD signaling to promote dorsal cortex differentiation by exposing the cultures to a BMP (LDN-193189) and a TGF-β type I receptor inhibitor (A83-01). Additionally, a compound inhibiting the Wnt pathway (IWR) to prevent posteriorization was applied. In contrast to 'intrinsic' cerebral organoid protocols19, which are based on self-assembly without external control giving rise to rather heterogeneous brain organoids and exhibiting large batch variations (measured by the efficiencies of polarized neural ectoderm formation15), the protocol described here reproducibly generates homogeneous forebrain-specific organoids from human iPSCs.
These forebrain-type organoids can be used for a variety of applications such as neurodevelopmental studies, evolutionary studies including gene function studies, disease modeling and, potentially, drug testing and therapeutic purposes. The protocol is, however, most suitable to examine early aspects of human cortical development. We have for example used the forebrain-type organoids to examine human-specific aspects of vRGC behavior. More specifically, pathophysiological changes associated with a severe form of lissencephaly, a human cortical malformation characterized by a near absence of cortical folding, was addressed. Only certain aspects of this disease can be modeled in mice as the mouse brain is naturally lissencephalic. When applying the organoid system to lissencephaly patient-derived iPSCs, we could reliably recapitulate human-specific aspects of the disease and identify underlying mechanisms. More specifically, we could demonstrate that patient-derived organoids show a significant reduction in size caused by a switch from symmetric to asymmetric cell division of vRGCs. This switch was associated with alterations in the organization of vRGCs' microtubule network, a disruption of the architecture of the VZ niche and altered expression of cell adhesion molecules, leading to an impaired activation of the N-cadherin/β-catenin signaling axis10. Of note: β-catenin-dependent regulation of vRGC division modes was suggested to be human-specific as overexpression of β-catenin in mice leads to tangential cortex expansion and subsequently cortical folding20. Thus, our data highlight that the forebrain-type organoid system represents a promising tool to study in a quantifiable manner the human-specific aspects of early cortical development in vitro.
A major challenge for the future is to maintain the homogeneity of the organoids across extended time periods in order to achieve more mature neuronal phenotypes. This might be realized by one or more of the following: culturing the organoids in a bioreactor system14, applying floating scaffolds15, supplementing the differentiation medium with neural growth, or neuronal survival factors. Finally, a controlled increase in brain complexity might be achieved by fusing the forebrain-type organoids with cerebral organoids of different regional identity21,22.
Taken together, the forebrain-type organoid protocol presented here offers an easily applicable and reliable tool for the generation of early cortical structures in vitro. The protocol gives rise to highly homogeneous early cortical tissue across multiple iPSC lines and can be utilized to reliably generate individual-specific cortical tissue. Thus, the system is particularly suitable for applications that require a high degree of homogeneity and reproducibility such as disease modeling.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The work was supported by the Ministry of Innovation Science and Research of North Rhine-Westphalia (Junior Research Group) and by the ERA-NET NEURON, JTC 2015 Neurodevelopmental Disorders, STEM-MCD.
References
- Otani T, Marchetto MC, Gage FH, Simons BD, Livesey FJ. 2D and 3D Stem Cell Models of Primate Cortical Development Identify Species-Specific Differences in Progenitor Behavior Contributing to Brain Size. Cell Stem Cell. 2016;18(4):467–480. doi: 10.1016/j.stem.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz SA, et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13(6):690–699. doi: 10.1038/nn.2553. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464(7288):554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15(3):477–486. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- Eiraku M, Sasai Y. Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr Opin Neurobiol. 2012;22(5):768–777. doi: 10.1016/j.conb.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Camp JG, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A. 2015;112(51):15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545(7652):48–53. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Lancaster MA. Stem Cell Models of Human Brain Development. Cell Stem Cell. 2016;18(6):736–748. doi: 10.1016/j.stem.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Iefremova V, et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017;19(1):50–59. doi: 10.1016/j.celrep.2017.03.047. [DOI] [PubMed] [Google Scholar]
- Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A. 2013;110(50):20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165(5):1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, et al. Guided self-organization and cortical plate formation in human brain organoids. Nat Biotechnol. 2017. [DOI] [PMC free article] [PubMed]
- Yamaguchi T, et al. Phosphorylation by Cdk1 induces Plk1-mediated vimentin phosphorylation during mitosis. J Cell Biol. 2005;171(3):431–436. doi: 10.1083/jcb.200504091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidebrecht HJ, et al. p100: a novel proliferation-associated nuclear protein specifically restricted to cell cycle phases S, G2, and M. Blood. 1997;90(1):226–233. [PubMed] [Google Scholar]
- Kosodo Y, et al. Regulation of interkinetic nuclear migration by cell cycle-coupled active and passive mechanisms in the developing brain. EMBO J. 2011;30(9):1690–1704. doi: 10.1038/emboj.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297(5580):365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017. [DOI] [PMC free article] [PubMed]
- Birey F, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]