

Abstract
Brain development is a complex process, which is controlled in a temporo-spatial manner by gradients of morphogens and different transcriptional programs. Additionally, epigenetic chromatin modifications, like histone methylation, have an important role for establishing and maintaining specific cell fates within this process. The vast majority of histone methylation occurs on the flexible histone tail, which is accessible to histone modifiers, erasers, and histone reader proteins. In contrast, H3K79 methylation is located in the globular domain of histone 3 and is implicated in different developmental functions. H3K79 methylation is evolutionarily conserved and can be found in a wide range of species from Homo sapiens to Saccharomyces cerevisiae. The modification occurs in different cell populations within organisms, including neural progenitors. The location of H3K79 methylation in the globular domain of histone 3 makes it difficult to assess. Here, we present methods to isolate and culture cortical progenitor cells (CPCs) from embryonic cortical brain tissue (E11.5-E14.5) or cerebellar granular neuron progenitors (CGNPs) from postnatal tissue (P5-P7), and to efficiently immunoprecipitate H3K79me2 for quantitative PCR (qPCR) and genome-wide sequencing.
Keywords: Neuroscience, Issue 131, Cerebellar cell culture, cortical cell culture, cortex development, cerebellar development, H3K79 methylation, DOT1L, ChIP, DOT1L inhibition
Introduction
The sensory, motor, and cognitive functions of the brain are highly complex and susceptible to physical and environmental changes. The brain consists of three general parts the hind-, mid-, and forebrain, which are deeply connected. Within the forebrain, the telencephalon can be divided into a dorsal telencephalon (DT) and a ventral telencephalon (VT). The DT of mice consists of six cortical layers which are formed between E11.5 and E18.5 in an "inside-out" manner1. The VT includes the ganglionic eminences in development, which later form the basal ganglia2,3. Several cell types can be classified in the mammalian central nervous system such as neurons, astrocytes, or oligodendrocytes4, which develop in a temporo-spatial manner5. First, the neural progenitor cells (NPCs) give rise to different kinds of neurons, interneurons in the VT, and projection neurons in the DT, and later on to glial cells (e.g., astrocytes6). During cortical development, the most superficial layer (layer I), which contains Cajal-Retzius cells, is formed first. Then, between E12.5 and E14.5, NPCs generate deeper neuronal layers (VI, V) while between 14.5 and 16.5, progenitors give rise to upper layer (IV-II) neurons7,8. Neuronal identity is specified by different morphogen-induced temporo-spatial transcriptional programs and additionally by epigenetic programs2.
The cerebellum, which is implicated in motor coordination, is located in the hindbrain and develops between E10 and roughly P20 in mice9. It contains the cerebellar cortex and the cerebellar nuclei10. The adult cerebellar cortex consists of three layers, the outermost molecular layer, the Purkinje cell layer, and the innermost granular layer containing granular neurons10. The cerebellar granule cells are the smallest neurons and represent about 80% of all neurons in the vertebrate brain11. They develop from precursors located in the external germinal zone and migrate through the Purkinje cell layer to their destination12. Like in the telencephalon, the development of the cerebellum is regulated by several important morphogens, which have specific time- and space-dependent functions and initiate defined transcriptional programs10.
The development of cortical and cerebellar layers is controlled by transcriptional expression of specific morphogens and, thus, by the chromatin state of the DNA. In a simplified view, chromatin states can be divided into euchromatin as transcriptionally active and heterochromatin as transcriptionally silent regions. The nucleosome as the basic unit of chromatin contains two copies of each core histone H2A, H2B, H3, and H4, surrounded by 147 base pairs of DNA13. Histones are highly post-translationally modified by methylation, acetylation, phosphorylation, ubiquitination, sumoylation, ADP-ribosylation, deamination, and proline isomerization14,15. Histone lysine methylation is considered to be the most stable histone modification that controls transcription, replication, recombination16, DNA-damage response17, and genomic imprinting18. Lysines can be mono-, di-, or tri-methylated19 and appear not only on the accessible histone tails but also within the globular domain of histones20. Specific methylations at H3K4 and H3K36 are mainly associated with euchromatin, specific methylations at H3K9, H3K27, or H4K20 are mainly found in heterochromatic regions, although all residues are located within the histone tail14,19,21. H3K79 methylation is located within the histone globular domain and has been associated with transcriptional activity, but also with transcriptionally inert genomic regions22. The modification is evolutionarily conserved since it has been observed in yeast, calf thymus, chicken, and human23. H3K79 mono, di, and trimethylation (H3K79me1, me2, me3) are catalyzed by the histone methyltransferases DOT1L24,25 and the Nuclear SET Domain-Containing Protein 2 (Nsd2)26. DOT1L is implicated in proliferation, DNA repair, and cellular reprograming27. Loss of Dot1l in mice leads to a prenatal death around the developmental stage E10.528,29. During heart development and in myocardiocyte differentiation, DOT1L is essential for gene expression regulation30. In the central nervous system, DOT1L function might be implicated in neural tube development31, it is involved in suppressing Tbr1-expression during forebrain development32, and may function in the regulation of ER-stress response genes33. The context-dependent activating or repressing action of H3K79me, especially with in vivo situations like the development of the central nervous system, is to date only partially understood32. Since H3K79 methylation is located in the globular domain of histone 3, it is sterically less accessible in comparison to modifications on the flexible histone tails23. To understand the function of H3K79 methylation, reliable and reproducible analysis methods to determine its location and genomic environment are needed. In this methods paper, we present isolation methods of different neural progenitors (CPCs for the cortex and CGNPs for the cerebellum), effective DOT1L inhibitor treatment, and a ChIP method to analyze H3K79 methylation via qPCR or sequencing at different time points during cortical and cerebellar development. For an overview of the protocol and its possibilities, see Figure 1.
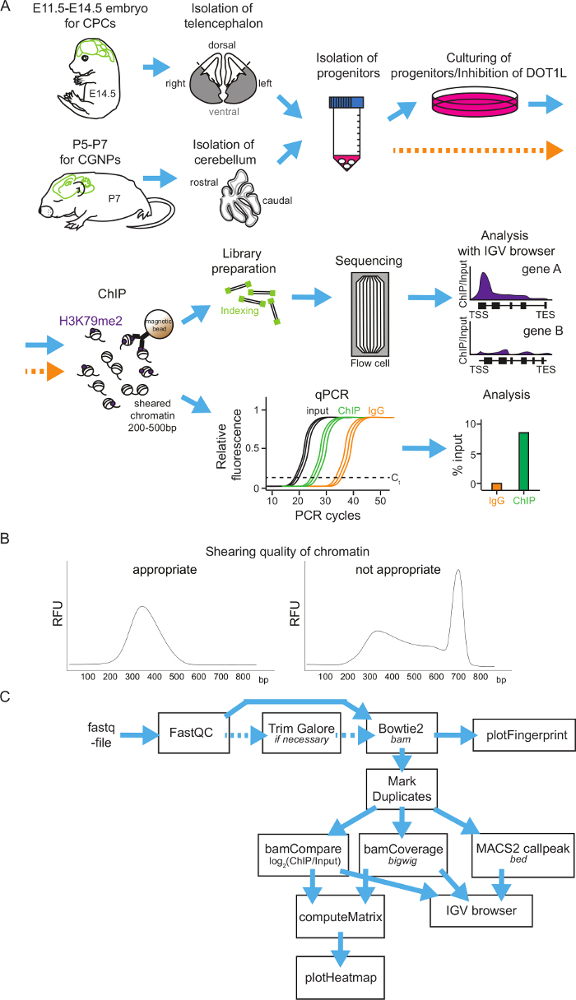
Protocol
Animal welfare committees of the University of Freiburg and local authorities approved all animal experiments (G12/13, G16/11) mentioned in the following protocol.
1. Preparations
- Preparations for CPCs isolation
- Set up timed mating to obtain embryos at different stages of cortex development (between E11.5 and E14.5). Use mice of the strain NMRI (Naval Medical Research Institute) which are at least 8 weeks old. After mating, consider a positive vaginal plug at E0.5.
- To have enough material, use one litter of NMRI mice for one H3K79me2 ChIP from cortices of E12.5 or E14.5. For later embryonic stages, use one litter for more ChIP analyses (average litter size NMRI: 10 embryos).
- Precool Hank´s Balanced Salt Solution (HBSS) and phosphate-buffered saline (PBS) to 4 °C (long-term storage at RT).
- Equilibrate the 4 °C stored Trypsin-EDTA (0.05 % w/v) to 37 °C.
- Prepare cortical-cell medium (CCM): Supplement the medium for neurons (see Table of Materials) with final concentrations of 2% (v/v) B-27 supplement, 5 µg/mL Apo-Transferrin, 1 µg/mL Glutathione, 0.5 mM L-glutamine, 0.8 µg/mL Superoxide dismutase, and 1% (v/v) Penicillin-Streptomycin-Neomycin antibiotic mixture. Store it at 4 °C. For the experiment, equilibrate the medium to 37 °C.
- Thaw fetal calf serum (FCS), aliquot it (50 mL each), and equilibrate one aliquot to 37 °C (long-term storage at -20 °C).
- Prepare stocks of DNAse 1 (10 mg/mL) in H2O for cell culture. Store aliquots at -20 °C.
- If needed, dissolve the specific DOT1L inhibitors EPZ-5676 or SGC0946 in DMSO to a stock concentration of 100 mM. Apply an end concentration of 1-5 µM to the cells at day 0 and reapply the inhibitor every two days.
- For CPC cultivation, coat cell culture plates (6 well or 12 well) with poly-L-ornithine hydrobromide (1 mg/mL in 150 mM boric acid pH 8.4) for at least 1 h at RT and afterwards with laminin (1 mg/mL) in CCM medium at 37 °C overnight.
- Preparations for CGNP isolation
- Organize an appropriate mating of mice to generate P5-P7 animals for isolation of the cerebellum (3-5 animals per ChIP and condition).
- Prepare HBSS/Glucose by adding 6 mg/mL glucose to HBSS buffer.
- Prepare the CGNP cell culture medium (CGM) by adding 1 % (v/v) N2 supplement, 1 % (v/v) Penicillin-Streptomycin-Neomycin, 25 mM KCl and 10 % FCS to DMEM-F12. For CGNP cultivation prepare CGM medium without FCS but including 0.6 µg/mL sonic hedgehog (SHH) (CGM-SHH) and equilibrate it to 37 °C when needed.
- Coat 6-well cell culture plates with 100 µg/mL poly-D-lysine for 1-2 h at RT for glia-cell removal. Afterwards wash twice with sterile ddH2O and let the plates dry. Store the plates for maximum one week at 4 °C.
- For cultivation of CGNPs coat 6-well cell culture plates with poly-L-ornithine (0.1 mg/mL) at 4 °C overnight. Afterwards wash twice with H2O for cell culture and let the plates dry. NOTE: The plates can be stored for maximum one week at 4 °C.
- Prepare 0.025 % trypsin (w/v) in HBSS/Glucose and equilibrate it to 37 °C when needed.
- Preparations for ChIP of H3K79me2
- Prepare PFA (1 % in PBS, pH 8) freshly before crosslinking chromatin. To prepare PFA heat 50 mL PBS in the microwave to maximum 65 °C. During constant stirring add 0.5 mg PFA to the PBS. Then add 50 µL 10 M NaOH and wait until PFA is dissolved. Afterwards, add 42.5 µL HCl (37 % v/v) to get a pH of 8. Verify the pH again and then let the 1 % PFA solution reach RT.
- Prepare stocks of RNase (1 mg/mL) and Proteinase K (20 µg/µL) separately in sterile ddH2O. Store the aliquots at -20 °C.
- Prepare the following buffers and reagents: PBS containing 0.02% Tween, Lysis buffer, Glycine, Dilution buffer, ChIP buffer 1, ChIP buffer 2, ChIP buffer 3, and TE buffer and store them at 4 °C. Prepare the Elution buffer freshly each time.
- Prepare PBS containing 0.02% Tween. Store for at most one-week at 4 °C.
- Prepare lysis buffer using 50 mM Tris at pH 8.0, 10 mM EDTA, 1% (w/v) sodium dodecyl sulfate (SDS), and 1x Protease inhibitor.
- Prepare 2.5 M glycine.
- Prepare dilution buffer using 20 mM Tris at pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% (v/v) Triton X-100, 0.25% (w/v) SDS, and 1x Protease inhibitor.
- Prepare ChIP buffer 1 using 20 mM Tris at pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% (v/v) Triton X-100, and 0.2% (w/v) SDS.
- Prepare ChIP buffer 2 using 20 mM Tris pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% (v/v) Triton X-100, and 0.2% (w/v) SDS.
- Prepare ChIP buffer 3 using 20 mM Tris pH 8.0, 250 mM LiCl, 2 mM EDTA, 1% (v/v) NP-40, and 1% (w/v) SDS.
- Prepare TE buffer using 20 mM Tris pH 8.0 and 2 mM EDTA.
- Prepare fresh elution buffer containing 1% (w/v) SDS, 100 mM NaHCO3.
2. Neural Progenitor Isolation of Brain Tissue
- Isolation and optional cultivation of CPCs
- Euthanize the pregnant animals by cervical dislocation and transfer the embryos to ice-cold HBSS.
- Remove the skull with scissors and isolate the brain, then remove the meninges, if applicable, with small forceps and isolate cortices (whole, DT, or VT) of both hemispheres by removing all other brain parts under the binocular scope using small forceps and, if necessary, small scissors. Store the isolated cortices in 5 mL HBSS buffer at 4 °C in a 15 mL tube.
- Centrifuge the isolated brain material for 5 min at 1,000 x g and 4 °C.
- Wash the cortices with 5 mL ice-cold HBSS and homogenize them with a 1 mL pipette tip by pipetting up and down. If necessary, cut the tip of the pipette tip.
- Centrifuge the homogenized sample for 5 min at 1,000 x g and 4 °C. Remove the HBSS.
- Add 3 mL Trypsin and incubate the sample for 5 min at 37 °C.
- Add 1 mL FCS, 5 mL CCM, and 30 µL of DNase 1 to the sample and homogenize the sample with a 5 mL glass pipette by pipetting carefully up and down.
- Centrifuge the isolated cells for 5 min at 1000 x g and 4 °C. Discard supernatant, add 5 mL CCM, and homogenize the sample again.
- Dilute an aliquot of the sample and count it with a Neubauer counting-chamber. An average amount of 4.5 x 106 cells per embryo is expected. For cultivation of the isolated CPCs proceed with step 2.1.10. For ChIP, immediately proceed with step 3.
- For cultivation, plate the CPCs on poly-L-ornithine (0.1 mg/mL) and laminin (1 mg/mL) coated dishes at a density between 8 x 104 and 2.5 x 105 cells/cm2 in CCM medium and incubate them at 37 °C, 5% CO2, and 100% relative humidity.
- Since the CPCs start to differentiate into neurons in cell culture (after approximately 4 days), consider the dissection date as day in vitro 0 (day 0). For longer terms of cultivation, change half of the medium every fourth day with fresh CCM medium.
- Apply 1-5 µM SGC0946 or EPZ5676 dissolved in DMSO at day 0 (4-5 h after cell isolation) and refresh it every second day. Use DMSO as a control treatment. Test the efficiency of inhibitor treatment via standard immunoblot methods, if required.
- Isolation and optional cultivation of CGNPs
- Euthanize P5-7 animals by decapitation using scissors. Remove the scalp skin, open the skull and remove the brain using small scissors and forceps. Isolate the cerebellum and transfer it to ice-cold HBSS/Glucose. Remove all meninges and blood vessels and transfer the cerebella into 15 mL tubes filled with cold HBSS/Glucose.
- Wash the cerebella three times with 10 mL ice-cold HBSS/Glucose (collect them by centrifugation at 650 x g for 5 min at 4 °C) and homogenize cerebella subsequently by gently pipetting up and down with a 1 mL pipette (maximum 2-3 times) to get 0.5-1 mm3 fragments.
- Add 5 mL of 0.025% trypsin in HBSS/Glucose and incubate the tissue under constant stirring in a 37 °C water bath for 15 min. Stop the digestion by adding 5 mL of CGM and collect the tissue by centrifugation at 650 x g for 5 min at 4 °C.
- Remove the supernatant and triturate the tissue with a 1 mL pipet tip in 1 mL CGM and transfer it to a new 15 mL tube. NOTE: It is important to avoid air bubbles.
- Add 5 mL CGM and incubate the mixture for 2 min on ice to settle tissue remnants. Transfer the supernatant into a new 15 mL tube. Add 2 mL CGM to the residual tissue and repeat the trituration procedure.
- Pool the supernatants (without tissue remnants, about 10 mL in total) and centrifuge at 650 x g for 5 min at 4 °C to collect the cerebellar cells. Resuspend the pellet in 10 mL CGM.
- Since astrocytes adhere faster and stronger to poly-D-lysine than CGNPs, plate the cells on 100 µg/mL poly-D-lysine coated 6-well-plates (up to 4 mL per well) and incubate for 20 min at 37 °C to remove the astrocytes.
- Shake the plate and collect the supernatant. Then centrifuge at 650 x g for 5 min at 4 °C in a 15 mL tube. Resuspend the pellet in 10 mL CGM and count the cells with a Neubauer counting chamber. NOTE: CGNPs are round, small, and show a halo when imaged using phase contrast microscopy.
- If required, seed the cells in 37 °C pre-warmed CGM on poly-L-ornithine-coated plates (3 x 106 cells per 6-well) and incubate them at 37 °C, 5% CO2 and 100% relative humidity. NOTE: If seeding is not performed, continue on to step 3.1.
- After 6-12 h, exchange the medium to CGM-SHH. Treat the isolated CGNPs 6 h (or when changing to CGM-SHH) after isolation with DOT1L-inhibitor and DMSO control and renew it every second day (compare with 2.1.12). Test the efficiency of inhibitor treatment via standard immunoblot methods if required. For ChIP, proceed with step 3.3. NOTE: Leaving CGM on the plates (and with that FBS) will result in differentiation of the CGNPs into cerebellar granular neurons (CGNs).
3. Fixation of Cells and Shearing of Chromatin
NOTE: Perform steps 3.1 and 3.2 if cells are not cultured, if they are proceed to step 3.3.
Collect the cells by centrifugation for 4 min at 1,000 x g and add 1.3 mL PBS. Transfer the sample to a 1.5 mL tube. Centrifuge for 4 min at 1,000 x g at 4°C. Wash the sample twice with 1.3 mL PBS.
- Critical step: Add 350 µL 1% PFA to the sample and incubate it for 5 min at 22 °C. To stop the reaction, add 18.4 µL glycine, and 1 mL PBS. Centrifuge the sample for 5 min at 1,000 x g at 4 °C.
- Wash the sample twice with ice-cold PBS. Collect the fixed cells by centrifugation for 5 min at 1,000 x g and 4 °C. Keep the samples on ice from now on. NOTE: The fixation time must be precisely 5 min to get optimal results. Different cell numbers may require time adaptations.
- Critical step: To cultured CGNPs (or CPCs), add up to 1 mL 1% PFA directly to the cell culture plate wells. After a 5 min incubation at 22 °C, add an appropriate amount of glycine and PBS and harvest the cells with a cell scraper.
- Transfer the cells into 1.5 mL tubes and centrifuge the sample for 5 min at 1,000 x g and 4 °C. Wash the sample twice with ice-cold PBS. Collect the fixed cells by centrifugation for 5 minat 1,000 x g at 4 °C. Keep the samples on ice from now on.
- Critical step: Add 700 µL of lysis buffer (+ protease inhibitor) and incubate the sample for 15 min at 4 °C. Vortex the sample every 5 min. Shear the chromatin of the lysed cells 3 x 10 min in the sonicator (30 s pulse, 30 s pause) at maximum power.
- Ensure that the volume does not exceed 350 µL per tube. Vortex the samples every 10 min. Check the lysate for remaining nuclei with a phase-contrast microscope. NOTE: It is important to get optimal shearing results (compare with Figure 1B) for later analysis. In case of using other methods for shearing the chromatin, optimize the shearing to chromatin fragments sized between 200-500 bp.
Pellet cell remnants for 10 min, 13,000 x g, 4 °C. Use the supernatant for the preclearing step 5.2. Freeze the sample at -20 °C for later use, if necessary.
4. Preparation of the Beads and Preclearing
Wash 45 µL protein A magnetic beads/ChIP and 20 µL magnetic beads/sample with 1 mL ice-cold PBS containing 0.02% Tween three times using a magnetic stand. Then, add 1 mL of ice-cold PBS to the beads.
To 1 mL ice-cold PBS and 45 µL beads/ChIP, add 3 µg of antibody/ChIP (H3K79me2 or rabbit IgG). Incubate the samples for 2 h at 4 °C on a rotator to bind the antibodies to the beads. Depending on the antibody, use ~3 µg antibody/ChIP.
Wash the antibody-bound-beads three times with 1 mL ice-cold PBS containing 0.02% Tween. Add the appropriate bead volume (starting volume of magnetic beads used) of ice-cold PBS to the washed antibody-coupled-beads.
Add 600 µL of dilution buffer and 20 µL of washed magnetic beads to the sample for preclearing (total volume 1,320 µL) and incubate for 2 h at 4 °C on a rotator. Remove the beads using a magnetic stand. Take 5% (33 µL) of the lysates as input samples and freeze them at -20 °C.
5. Chromatin Immunoprecipitation
Divide the precleared extract into two tubes (approximately 643.5 µL), add the same volume of dilution buffer and the antibody-bound-beads (45 µL/sample; H3K79me2 or rabbit IgG). Incubate the samples at 4 °C overnight on a rotator.
Wash the beads with ice-cold ChIP buffer 1, ChIP buffer 2, and ChIP buffer 3 for 10 min each at 4 °C on a rotator. Afterwards, wash three times with TE buffer for 5 min at 4 °C on a rotator.
Elute the beads with elution buffer for 1 h at 1,400 rpm in a shaker at room temperature.
Add 10 µg RNase (1 mg/mL) per ChIP sample and 5 µg to the input samples and incubate the samples for 30 min at 37 °C (1,400 rpm).
Add 100 µg Proteinase K (20 µg/µL) per ChIP sample and 50 µg per input and incubate overnight at 65 °C at 1,400 rpm.
6. Purification of ChIP samples
Purify the ChIP and input samples with a DNA purification kit (see Table of Materials) according to the manual. Use more purification columns when more than 5 µg of DNA is expected. Elute the sample with 2 x 15 µL of the DNA elution buffer provided in the kit.
Perform quantification of the samples (use 1 µL) by using a visualizing fluorophore and a fluorospectrometer (see Table of Materials).
7. Analysis of ChIP Samples via qPCR
- For primer design for qPCR analysis, retrieve genomic sequences of interest from the Integrative Genomics Viewer (IGV) browser34. Design primers around the transcriptional start site (TSS) of a gene, since H3K79me2 is likely to be located there. As a control, consider non-coding genomic regions or regions about 10 Kb upstream of the TSS or 10 Kb downstream of the transcriptional end site (TES).
- Generate the primers with https://www.ncbi.nlm.nih.gov/tools/primer-blast/ using a product size between 70 and 200 bp and an optimal temperature preset of 60 °C. For trial purposes, use primers for the genomic regions Aft4, Ddit3, Scd1, Aft3, and Npm1 listed in Table 1.
- Test newly designed primers with the following qPCR program, master mix, and an annealing temperature gradient between 58 °C and 63 °C, and select the annealing temperature with the highest product quantity and quality.
- Apply DNA gel electrophoresis to control the expected product size. For qPCR analysis, use a Real-Time PCR Detection System (see Table of Materials).
- Prepare the master mix using 10 µL DNA polymerase master mix, 0.5 µL primer forward (10 µM), 0.5 µL primer reverse (10 µM), 4 µL nuclease-free water, and 5 µL DNA (1 ng/µL).
- Use a 2-step qPCR program as follows: 5 min at 95 °C, 50x (30 s at 95 °C, 30 s at 58 °C-63 °C), and denaturation gradient constantly at 4 °C.
- Create a dilution series of genomic, sheared, purified DNA samples (2 ng/µL, 1 ng/µL, 0.5 ng/µL, 0.25 ng/µL, and 0.125 ng/µL) and use 5 µL for an efficiency test using qPCR. Determine the slope of the standard curve and calculate the primer efficiency by the following formula: Efficiency (%) = (10^(-1/slope)-1)*100.
- Use primers with efficiencies between 90% and 105% in an ideal case, or at least with efficiencies between 85% and 115%.
To analyze the ChIP and input samples, apply 0.1-1 ng of ChIP DNA mixed with respective forward and reverse primer, nuclease-free water, and DNA polymerase master mix in a final volume of 20 µL for each qPCR reaction.
Determine the threshold cycle (Ct) values of ChIP and input samples and normalize the sample Ct to Ct values of input to calculate enrichment (% input) with the following formula. Use Ct values obtained from IgG control to determine the background level. % input = 100-2^(norm. input − norm. ChIP) norm. input = Ct (input) − log2(dilution factor) norm. ChIP = Ct (ChIP) − log2(dilution factor) NOTE: norm. = normalized
8. Analysis of ChIP samples via Sequencing
Transfer the ChIP samples (input and immunoprecipitated DNA sample) to a sequencing facility.
To prepare a library beforehand, use an appropriate library preparation kit (see Table of Materials) and convert 2 ng of the input DNA and everything of the immunoprecipitated DNA into indexed libraries for next generation multiplex sequencing on the indicated platform (see Table of Materials).
Critical step: Following the kit instructions, ligate sequencing adaptors (with a working concentration of 15 µM) to end repaired and dA-tailed DNA fragments. For sequencing adaptors and PCR primers use appropriate oligos (see Table of Materials). For this amount of input DNA, use 6-9 PCR cycles to enrich adaptor ligated DNA fragments. NOTE: Normally, it is not necessary to perform a size selection of the adaptor ligated DNA. It is best to use as few PCR cycles as possible, since many PCR amplification cycles result in PCR duplicates and a high GC bias.
For a final library check, take 0.5 µL of the total reaction for analysis to visualize size distribution on an appropriate device (see Table of Materials). For quantification, use a visualizing dye in combination with a fluorospectrometer or other methods (see Table of Materials).
Since H3K79me2 appears to be a histone modification, which is broadly spread over larger genomic regions, sequence the samples paired-end with a read length of 50 bp and with a sequencing depth of 75 Mio reads.
Analyze the ChIP-seq data with the Galaxy/Uni Freiburg Server (galaxy.uni-freiburg.de).
Perform quality control with obtained ChIPseq with FastQC and, if appropriate, map them directly with Bowtie2 version 2.2.035 with or without trimming. As reference genome assembly use mm9 or the newest version mm10; and as reference annotation Ensembl FTP release 79.
Optional: Remove read duplicates using Picard MarkDuplicates (http://broadinstitute.github.io/picard) before peak calling.
Call peaks with MACS2 version 2.1.036 using the 'broad' option for H3K79me2.
To obtain a log2 ratio between ChIP and input sample, the log2 of the number of reads of the reads ratio of both samples use BamCoverage and BamCompare.
For all other in-depth ChIP-seq specific analysis, apply deepTools2 version 2.3.5 or 2.4.137, i.e. to generate coverage track files (bamCoverage for the ChIP only, bamCompare for comparison of ChIP to the input), to estimate the ChIP performance use plotFingerprint and to generate a general overview use computeMatrix, plotHeatmap. Select K-mean clustering during the computeMatrix process to cluster genomic regions according to the H3K79me2 distribution.
Use published ChIP-seq data as H3K79me2 of E14.5 DT deposited at NCBI for trial purposes (Accession Number: SRP057733).
Representative Results
General scheme of neural progenitor isolation, cultivation, H3K79me2 ChIP and ChIP analysis methods:Figure 1 shows a flowchart to perform H3K79me2 ChIP of cortical progenitor cells at different time points during embryonic brain development or of cerebellar granular neuron progenitors in postnatal stages. As a first step, the brain has to be isolated and the telencephalon (between E11.5 and E14.5) or the cerebellum (P5-P7) has to be retrieved. It is possible to analyze the different regions of the telencephalon by dividing it into the DT and VT. Afterwards the tissue will be homogenized and the neural progenitors segregated. Now, it is possible to culture the progenitor cells and treat them with DOT1L inhibitors. Another possibility is to fix the progenitors directly and subject them to the ChIP procedure. For ChIP, the chromatin will be sheared into fragments of 200-500 bp and incubated subsequently with magnetic-beads-coupled antibody against H3K79me2. After washing the beads and retrieving the DNA, the precipitated DNA can be analyzed by sequencing or by qPCR (Figure 1A). It is essential to have a good size distribution of the chromatin fragments after shearing, so it is advisable to check the fragment size with DNA gel electrophoresis or with other methods (examples in Figure 1B). When choosing genome-wide sequencing, ChIP-seq-reads for H3K79me2 occupancy will be provided as a fastq-file for further analysis (Figure 1C). The quality of the sequencing reads needs to be assessed by application of FastQC and, if necessary, the fastq-files can be trimmed using TrimGalore. Mapping to the Mus musculus genome mm9 or mm10 can be performed with Bowtie2 and controlled via PlotFingerprint. It is advisable to remove duplicates with MarkDuplicates and to normalize the reads by BamCoverage and to compare the ChIP to the input samples with BamCompare. The ChIP-seq reads can subsequently be visualized genome wide in heatmaps or gene-wise in Integrative Genomics Viewer (IGV) browser sessions.
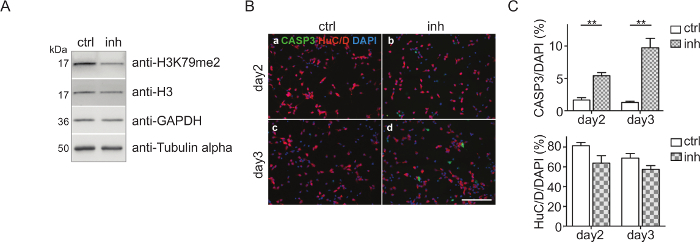
CPC culture and DOT1L inhibition: CPCs were isolated and treated with 5 µM DOT1L inhibitor SGC0946 at day 0 and day 2. The solvent DMSO was used as a control. At day 3, CPCs were harvested, the proteins were isolated, quantified, and analyzed via immunoblot (Figure 2A). Figure 2A shows that H3K79me2 levels are effectively decreased after three days of CPC culturing and DOT1L inhibition ensuring an effective inhibitor treatment scheme. The protein amount of H3, GAPDH, or Tubulin-alpha serving as loading controls was thereby not impaired. Immunostaining of fixed CPCs with antibodies against active caspase 3 (CASP3+) for detecting apoptosis and HuC/D for visualizing neuronal cells indicated that treatment of CPCs with DOT1L inhibitor led to increased cell death after three days in culture, as shown in Figure 2B. Quantification of the cells which are CASP3+/DAPI+ or HuC/D+/DAPI+ within the CPC culture revealed a significant rise of CASP3+ cells revealing programmed cell death upon DOT1L inhibition. The number of HuC/D-positive neurons, however, remained unchanged (Figure 2C).
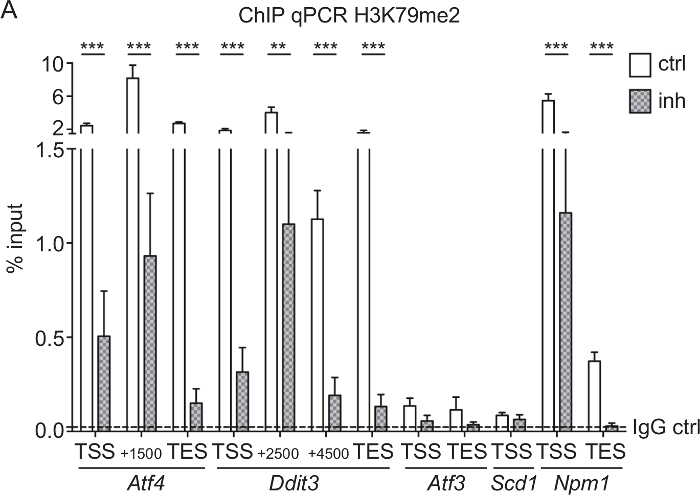
H3K79me2 ChIP-qPCR of Aft4, Ddit3, Scd1, Aft3, and Npm1 from CPCs treated with DOT1L inhibitor: To test if the SGC0946 inhibitor treatment of DOT1L was efficient and led to a reduction of H3K79me2 at specific genes, we performed a ChIP analysis followed by qPCR of CPC treated for 3 days. Rabbit IgG was used as a ChIP control. Since it is known that H3K79me2 is located at the endoplasmatic stress-responsive genes Aft4, Ddit3, Scd1, and Aft3 as well at the nuclear transport gene Npm133,38, we used qPCR primers covering the TSS, the TES, and genomic regions within the gene bodies to determine differences in H3K79me2 distribution after DOT1L inhibition (Figure 3A). Genes with high H3K79me2 levels in the first place such as Atf4, Ddit3, and Npm1 were most responsive to the inhibitor treatment so that three days of inhibition of DOT1L led to a significant decrease of H3K79me2. Genes with a lower H3K79me2 coverage, such as Atf3 and Scd1, showed no significant change in H3K79me2 levels.
Overview of H3K79me2 ChIP-seq analysis: Genome-wide analysis of the H3K79me2 levels in CPCs from E14.5, performed and analyzed according to the presented schemes and protocols (Figure 1), revealed a very high quality ChIP. Whereas the binned counts, sorted according to their frequency of the input reads in the Fingerprint blot (Figure 4A), were evenly distributed, the H3K79me2 counts showed enrichment at specific regions (bins ranked with 1), which displays successful accumulation of chromatin fragments modified at H3K79. A heatmap of H3K79me2 along genes between their TSS and their TES and +/-10Kb upstream or downstream shows (Figure 4B) that H3K79me2 peaks around the TSS and decreased towards the TES in general. There are genes, which are completely covered with H3K79me2, and genes, which have a peak only within the TSS region. Endoplasmatic-stress related genes such as Atf4, Ddit3, and the nucleophosmin Npm1 harbor, for instance, a high H3K79me2 level at the TSS, which is only slightly decreased along the whole gene body (Figure 4C). In contrast, Scd1 has a low H3K79me2 occupancy at the TSS and no H3K79me2 within the gene body. Atf3 as a last example has different sharp and low level H3K79me2 peaks along the gene body.
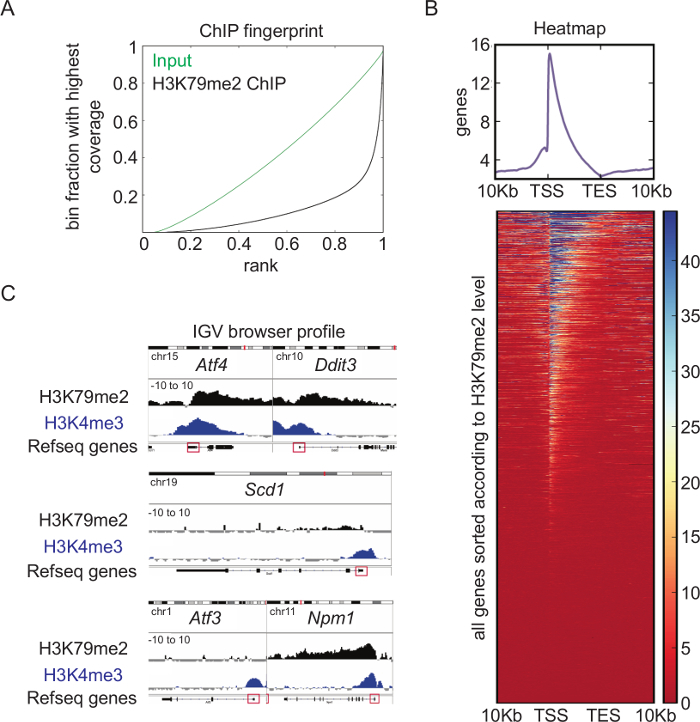
Figure 1: Scheme of H3K79me2 ChIP protocol from isolated CPCs or CGNPs and flowchart of sequencing analysis.(A) Overview of presented protocol. For CPC isolation, first E14.5 brains will be isolated and the cortices can be divided into DT and VT, if needed. For CGNPs, cerebelli have to be retrieved from P5-P7 mice. The tissue will be homogenized, the progenitors segregated, and then the cells can be cultured and treated with an appropriate inhibitor or directly used for ChIP. For ChIP, fixation of the chromatin is followed by shearing and immunoprecipitation with an anti-H3K79me2 antibody and rabbit IgG as control. After the DNA purification, ChIP samples can be analyzed via library preparation and sequencing or can be analyzed via qPCR. (B) Examples of appropriate and inappropriate quality chromatin. The DNA fragment distribution is shown in bp. (C) ChIP-seq results can be subjected to an analysis pipeline on the Freiburg/Galaxy server. After a quality control (FastQC), reads may be trimmed with TrimGalore and then mapped via Bowtie2 to the mouse genome (in the presented cases mm9). PlotFingerprint evaluates the quality of the ChIP. To define peaks for H3K79me2, MACSpeaks can be applied. For normalization BamCoverage and for comparison to the input BamCompare are usable. To visualize the results IGV browser and heatmaps are suitable. Expected file-formats are italicized. Please click here to view a larger version of this figure.
Figure 2: Cortical progenitor culture and DOT1L inhibition.(A) Immunoblots showing H3K79me2 levels in CPC at E14.5 after DOT1L inhibition for 3 days (day 3, n=3-11) in comparison to the DMSO control. H3, GAPDH, and Tubulin alpha were used as loading controls. (B) Immunocytological staining of a CPC culture at days 2 and 3. Activated Caspase 3 (CASP3 green)-positive cells and neurons (HuC/D: red) were stained. DAPI (blue) was used to visualize the cell nuclei. Scale bar: 100 µm. (C) Quantification of the immunostainings presented in (B). The ratio of positive cells for CASP3+/DAPI+ or HuC/D+/DAPI+ in percent is given. For statistical analysis, unpaired Student's t-tests were performed. p<0.01 **. This figure was modified from Roidl et al.33,38 Please click here to view a larger version of this figure.
Figure 3: H3K79me2 ChIP-qPCR of Aft4, Ddit3, Aft3, Scd1, and Npm1 from CPCs treated with DOT1L inhibitor. (A) E14.5-derived CPCs were treated with 5 µM DOT1L inhibitor 4-5 h after isolation and for a second time at day 2 in culture. CPCs were fixed at day 3, which was followed by chromatin extraction, ChIP experiment, and qPCR. Results are represented as the mean (+/-SEM) % input (n=3). IgG was used as a negative control. The mean of the IgG level is depicted as dashed line. For statistical analysis, two-way ANOVA was applied. p<005 *; p<0.01 **, p<0.001 ***; TSS transcriptional start site, TES transcriptional end site. This figure was modified from Roidl et al.33,38 Please click here to view a larger version of this figure.
Figure 4: Overview of H3K79me2 ChIP-seq analysis.(A) Fingerprint of H3K79me2 ChIP-seq from E14.5-derived CPCs compared to input shows an enrichment of DNA fragments for H3K79me2 ensuring a high ChIP-seq quality. (B) H3K79me2 ChIP-seq results plotted in a heatmap. Strongly enriched genomic regions ARE presented in blue. Scaling 0 (dark red) - 45 (dark blue). H3K79me2 peaks near the TSS and declines in the 3´ direction to TES. (C) H3K79me2 ChIP-seq reads were mapped to the mm9 genome and visualized with the integrative genome viewer (IGV). The H3K79me2 occupancy of a gene is displayed as log2 of the number of reads ratio between ChIP and input reads per kilobase per million (Scaling: -10 to 10). Refseq gene structure is represented while a red box indicates the first exon including the TSS. The genes Aft4, Ddit3, Scd1, Aft3, and Npm1 are shown. For comparison, a H3K4me3 signal of the same genomic region is displayed. TSS transcriptional start site, TES transcriptional end site. This figure was modified from Roidl38. Please click here to view a larger version of this figure.
| Gene | Region | Primer forward 5'-3' | Primer reverse 5'-3' |
| Aft4 | TSS | GGACGATCTCTAACGCCACA | GCCCTAAACCCGCCCTTTAT |
| Aft4 | TSS+1500 | CTCCCGAATATGACATGAACCG | TCCATTCGAAACAGAGCATCG |
| Aft4 | TES | CCGTAATAGGGTAGTCAGGTGC | AAAGAATGACACTGAAAACCCACA |
| Ddit3 | TSS | GTACTGGCTCCGTCTAACCC | CAAGAGAGGGCCTGTAAGCA |
| Ddit3 | TSS+2500 | TCAAGCAGCCGGTCTCATAG | CTCAGATCCCCCAATGGCTT |
| Ddit3 | TSS+4500 | GAGCTGGAAGCCTGGTATGA | TCACCTCTTCGTTTCCTGGG |
| Ddit3 | TES | CACCAAGCATGAACAGTGGG | GTACCGTCTATGTGCAAGCC |
| Aft3 | TSS | AACTGAGAGCGCAACTCCTC | GCTGCCGTTCTTAGCTGGTA |
| Aft3 | TES | CTGTTGGCACAAAGTGGCTC | GGGATCTGCCATGGTGGAAA |
| Scd1 | TSS | TAGTGACCACACACAAAAGCTC | CCCAAGTGTAATTTGGATGATTTCC |
| Npm1 | TSS | TCCCCCTCCAGTCAGTTACC | CGTCCTTTCCTTGGCGTGA |
| Npm1 | TES | AGGGACATACTTAAGACAAGCCAG | AGGATTGAGGCAGACTGTCAAT |
| TSS:Transcriptional start site | |||
| TES:Transcriptional end site |
Table 1: List of primers used for ChIP-qPCR.
Discussion
There are two major ways to perform chromatin immunoprecipitation to detect the genomic occupancy of histone modifications, transcription factors, histone code readers, writers, or erasers. One is the native ChIP method using nuclease digested, native chromatin for immunoprecipitation, and the other is the presented method using PFA-fixed, sheared chromatin, in which the nucleosomes and other DNA-attached proteins are covalently bound to the DNA39. Native ChIP with its high antibody detection rate should be applicable for histone methylation since the chromatin and histone methylations are relatively stable throughout the ChIP. But since a large amount of starting material is needed, it is not applicable for studying cortical or cerebellar development, where the number of cells from mouse brains is usually limited. Although antibodies are generally raised against non-fixed material, we could prove that the antibody specifically detects H3K79me2 since specific inhibition of DOT1L leads to a decreased H3K79me2 signal in ChIP-qPCR analysis and in immunoblots (Figure 2A and Figure 3A). ChIP using cross-linked material might be inefficient, but the presented protocol ensures enough specifically enriched material for qPCR and sequencing analysis.
For an effective ChIP, the SDS concentration of the lysis buffer can be crucial, since SDS denatures proteins and makes them accessible for antibody binding. This feature is especially important for H3K79me2, which is a histone modification located within the globular domain of histone 3 and within the nucleosome core of chromatin's smallest unit22. Thus, for H3K79me2, a higher SDS concentration (approximately 0.3% w/v) during the assay is needed to ensure optimal antigene accessibility. With this SDS concentration, we could highly enrich H3K79me2 over IgG controls (Figure 3) and over input (Figure 4). Thus, the quality of the ChIP-material for sequencing was appropriate and the enrichment over input comparable to that of H3K4me3 (Figure 4). We expect that the same SDS concentration would be needed for ChIP of H3K79 mono or trimethylation mark. In general, the more stringent the ChIP buffer is, the less false-positive chromatin fragments are immunoprecipitated, if the quality of the antibody is not affected by the detergent used. For the future, it might be feasible to apply the presented protocol to other histone modifications within the globular domain of histones.
If the antibodies are suitable for ChIP and are stable at higher detergent concentrations, the protocol mainly contains two critical steps. First, the fixation of the cells and the subsequent shearing of the chromatin should be optimized for each cell type and each ultrasonicator. Prolonged cross-linking leads to fixation artifacts and may reduce the antibody detection rate since antigens may be masked. The chromatin shearing needed to lead a size distribution of the DNA between 200 and 500 bp (Figure 1B). Nucleosomes contain 147 bp of DNA. Chromatin fragments that are too lengthy will lead to false positive results in qPCRs. During genome-wide sequencing, an incorrect size distribution will lead to false negative results, since often only small fragments are considered. The second critical step is the library preparation for genome-wide sequencing. A DNA amplification step needs to be performed using 6-9 PCR-cycles. Avoiding numerous PCR cycles is essential since they may lead to a high GC bias and many PCR duplicates. With the presented protocol, enough DNA can be retrieved to keep the number of necessary PCR cycles low.
Brain tissue consists of a very diverse amount of cell types such as neurons, oligodendrocytes, and glia cells4. During development these cells derive from neural stem cells and build specific areas in the brain in a time- and location-dependent manner3. Neurogenesis of the cerebral cortex, for instance, takes place between E11.5 and E18.5 in mice. At earlier stages such as E11.5 and E12.5 almost all cells have progenitor identity1, but later on cellular diversity needs to be taken into account. Therefore, a ChIP of cortical tissue at E14.5 can reveal H3K79me2 or any other histone modification at that specific time point but cannot display cell-type specific chromatin features. This issue can be solved by enrichment of specific cells with Fluorescence-activated cell sorting (FACS) or Magnetic-activated cell sorting (MaCS-sorting)40, which is feasible after brain homogenization. With MaCS-sorting neural progenitor cells carrying specific cell surface markers can be labeled with magnetic beads and then isolated using a magnetic stand40. Afterwards the cells can be cultured or directly used for ChIP. The presented protocol may be used not only for neural progenitor cells but also for other homogeneous cell populations. With addition of FACS or MaCS, the protocol can be applied for all isolatable cells within an organism.
Taken together, the presented protocol makes it possible to investigate the histone modification H3K79me2 at different time points of cortical and cerebellar development in an efficient and reproducible way. It may be applicable to other histone modifications located within the histone core and can be applied to other homogeneous cell types within the organism. Since H3K79me2 is a conserved histone modification23, the protocol may also be suitable to analyze H3K79me2 not only in mice but across different species.
Disclosures
The authors declare that they have no competing financial interests
Acknowledgments
We thank Henriette Bertemes for helping to establish the CGN cultivation protocol within the lab. This method paper was supported by the DFG-funded CRC992 Medical Epigenetics by funding to TV. The authors acknowledge the support of the Freiburg Galaxy Team: Pavankumar Videm, Björn Grüning and Prof. Rolf Backofen, Bioinformatics, University of Freiburg, Germany funded by Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and German Federal Ministry of Education and Research (BMBF grant 031 A538A RBC (de.NBI)).
References
- Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Squire LR. Neuroscience: breaking down scientific barriers to the study of brain and mind. Science. 2000;290:1113–1120. doi: 10.1126/science.290.5494.1113. [DOI] [PubMed] [Google Scholar]
- Götz M, Sommer L. Cortical development: the art of generating cell diversity. Dev. Camb. Engl. 2005;132:3327. doi: 10.1242/dev.01931. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 2010;11:377–388. doi: 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- Davis AA, Temple S. A self-renewing multipotential stem cell in embryonic rat cerebral cortex. Nature. 1994;372:263–266. doi: 10.1038/372263a0. [DOI] [PubMed] [Google Scholar]
- Sauvageot CM, Stiles CD. Molecular mechanisms controlling cortical gliogenesis. Curr. Opin. Neurobiol. 2002;12:244–249. doi: 10.1016/s0959-4388(02)00322-7. [DOI] [PubMed] [Google Scholar]
- McConnell SK, Kaznowski CE. Cell cycle dependence of laminar determination in developing neocortex. Science. 1991;254:282–285. doi: 10.1126/science.254.5029.282. [DOI] [PubMed] [Google Scholar]
- Desai AR, McConnell SK. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Dev. Camb. Engl. 2000;127:2863–2872. doi: 10.1242/dev.127.13.2863. [DOI] [PubMed] [Google Scholar]
- Glickstein M, Strata P, Voogd J. Cerebellum: history. Neuroscience. 2009;162:549–559. doi: 10.1016/j.neuroscience.2009.02.054. [DOI] [PubMed] [Google Scholar]
- Marzban H, et al. Cellular commitment in the developing cerebellum. Front. Cell. Neurosci. 2015;8 doi: 10.3389/fncel.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo FAC, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009;513:532–541. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48:17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Zhang Q, et al. Histone modification mapping in human brain reveals aberrant expression of histone H3 lysine 79 dimethylation in neural tube defects. Neurobiol. Dis. 2013;54:404–413. doi: 10.1016/j.nbd.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- Sanders SL, et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebede AF, Schneider R, Daujat S. Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. FEBS J. 2015;282:1658–1674. doi: 10.1111/febs.13047. [DOI] [PubMed] [Google Scholar]
- Roidl D, Hacker C. Histone methylation during neural development. Cell Tissue Res. 2014;356:539–552. doi: 10.1007/s00441-014-1842-8. [DOI] [PubMed] [Google Scholar]
- Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006;34:2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p Modulates Silencing in Yeast by Methylation of the Nucleosome Core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- Jones B, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo Park J, et al. RE-IIBP Methylates H3K79 and Induces MEIS1-mediated Apoptosis via H2BK120 Ubiquitination by RNF20. Sci. Rep. 2015;5:12485. doi: 10.1038/srep12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaming H, van Leeuwen F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma. 2016. [DOI] [PubMed]
- Jones B, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, et al. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010;116:4483–4491. doi: 10.1182/blood-2010-03-276501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo P, et al. DOT1L-mediated H3K79me2 modification critically regulates gene expression during cardiomyocyte differentiation. Cell Death Differ. 2016;23:555–564. doi: 10.1038/cdd.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, et al. Histone modification mapping in human brain reveals aberrant expression of histone H3 lysine 79 dimethylation in neural tube defects. Neurobiol. Dis. 2013;54:404–413. doi: 10.1016/j.nbd.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Büttner N, Johnsen SA, Kügler S, Vogel T. Af9/Mllt3 interferes with Tbr1 expression through epigenetic modification of histone H3K79 during development of the cerebral cortex. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7042–7047. doi: 10.1073/pnas.0912041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roidl D, et al. DOT1L Activity Promotes Proliferation and Protects Cortical Neural Stem Cells from Activation of ATF4-DDIT3-Mediated ER Stress In Vitro. Stem Cells Dayt. Ohio. 2016;34:233–245. doi: 10.1002/stem.2187. [DOI] [PubMed] [Google Scholar]
- Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roidl D. Histone modifications during cerebral cortex development. 2015.
- Turner B. Mapping Protein/DNA Interactions by Cross-Linking. Institut national de la santé et de la recherche médicale; 2001. ChIP with Native Chromatin: Advantages and Problems Relative to Methods Using Cross-Linked Material. [PubMed] [Google Scholar]
- Dincman TA, Beare JE, Ohri SS, Whittemore SR. Isolation of cortical mouse oligodendrocyte precursor cells. J. Neurosci. Methods. 2012;209:219–226. doi: 10.1016/j.jneumeth.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]